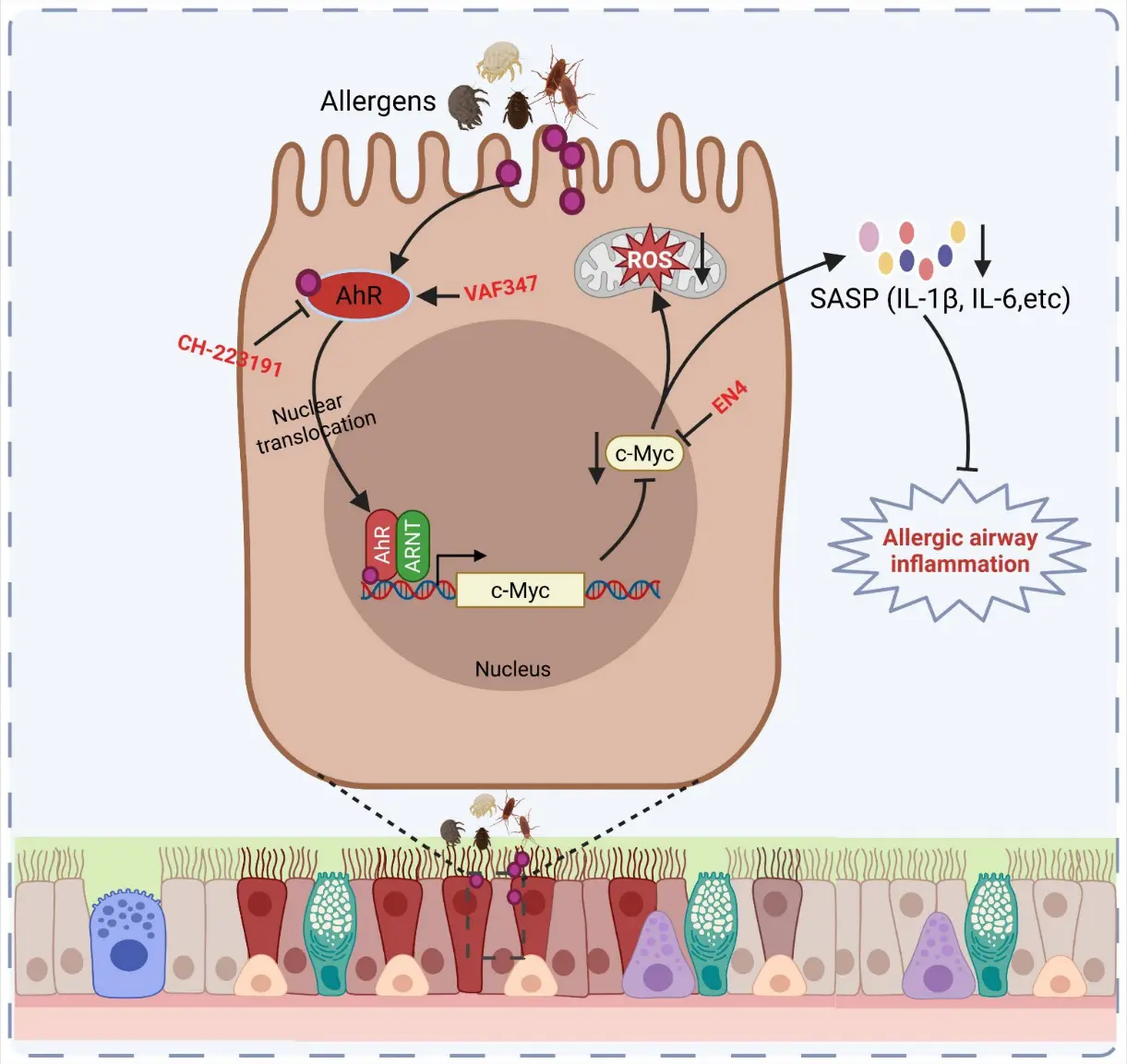
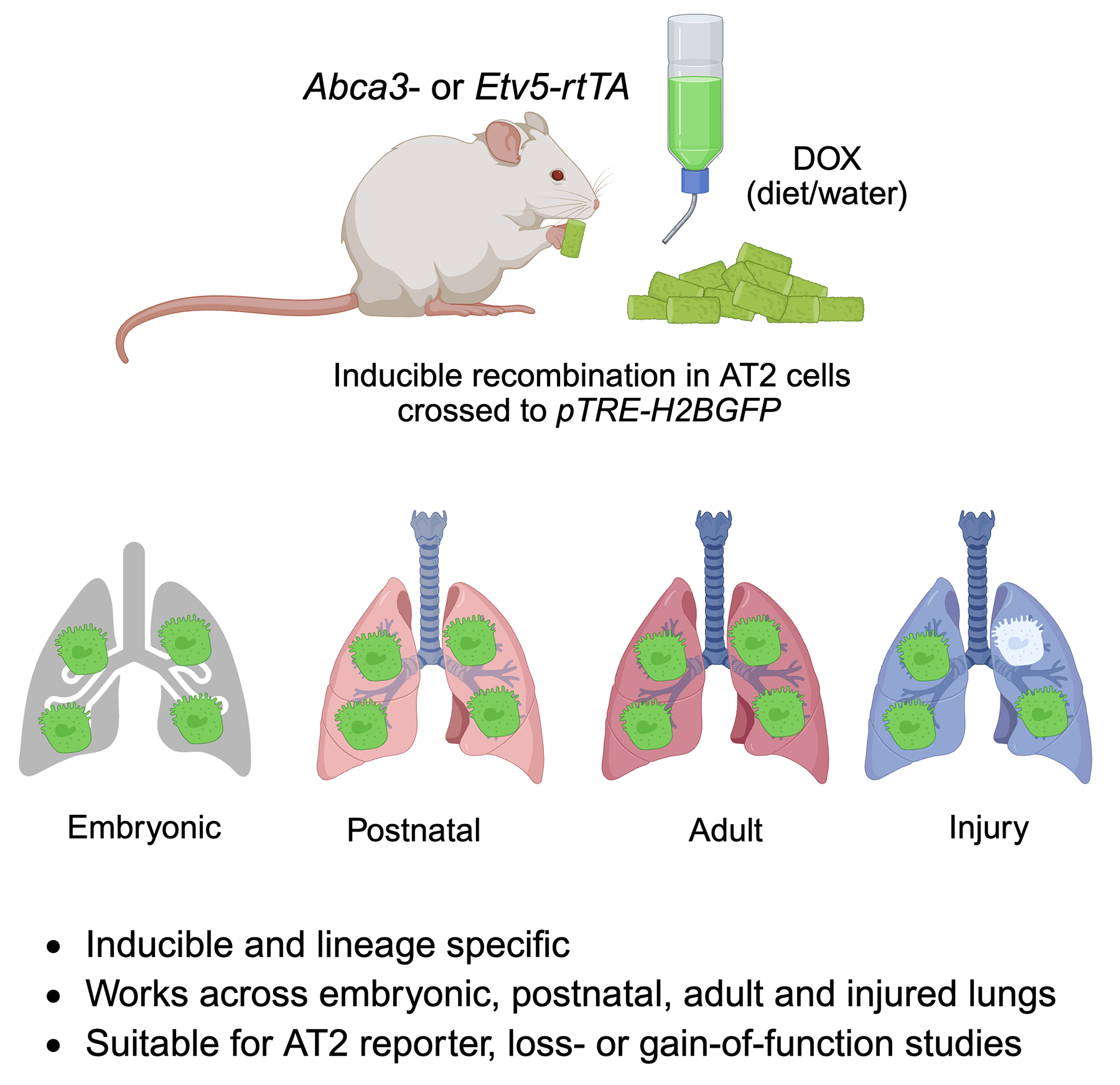
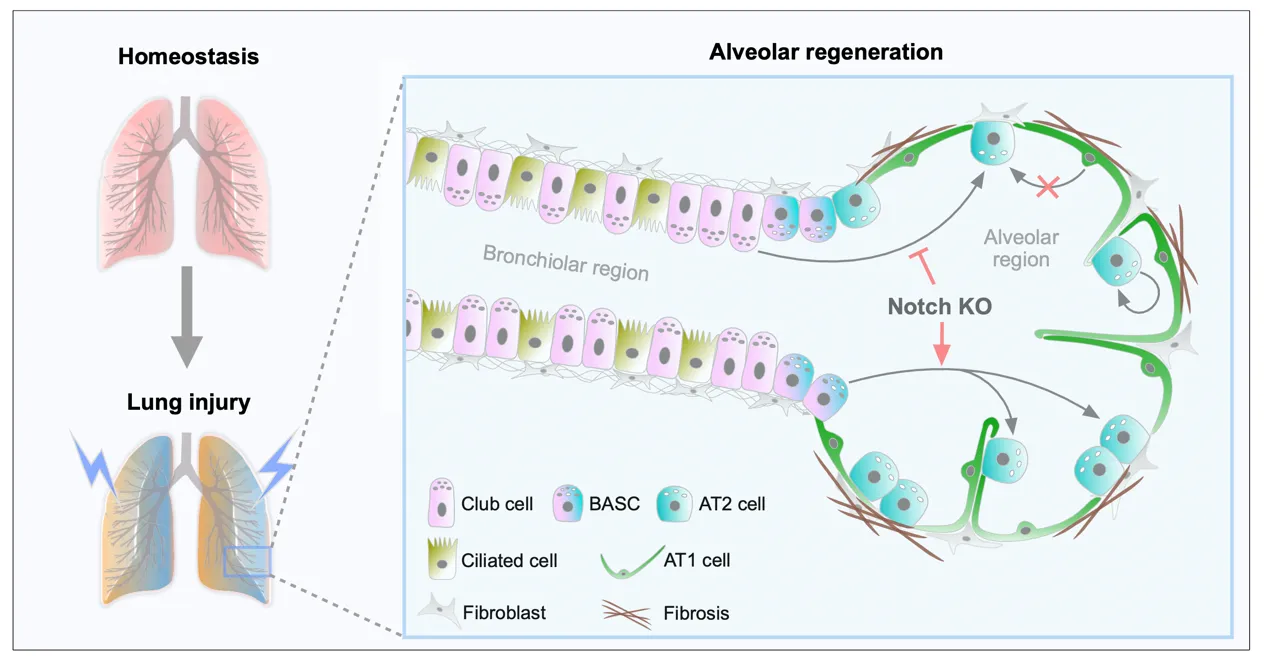
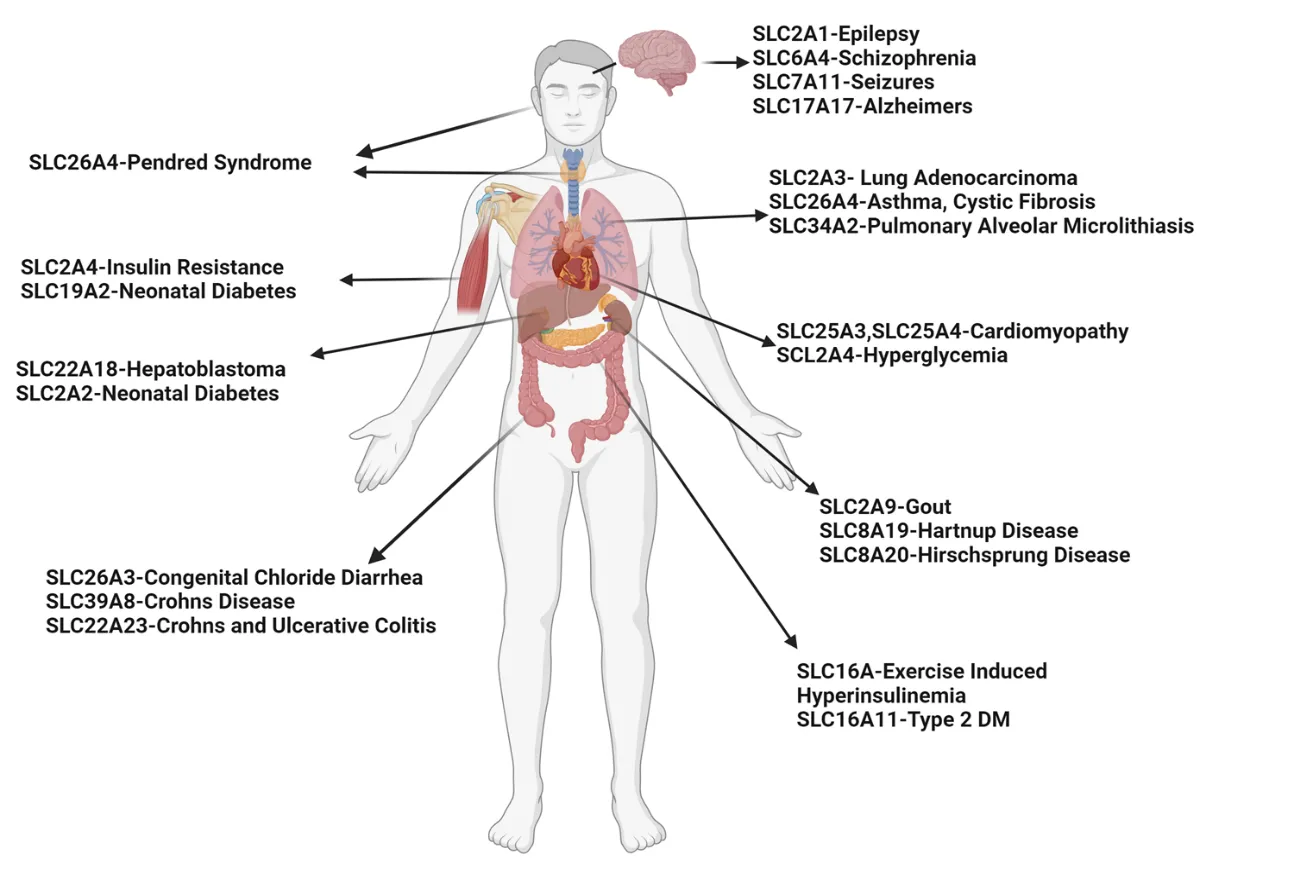
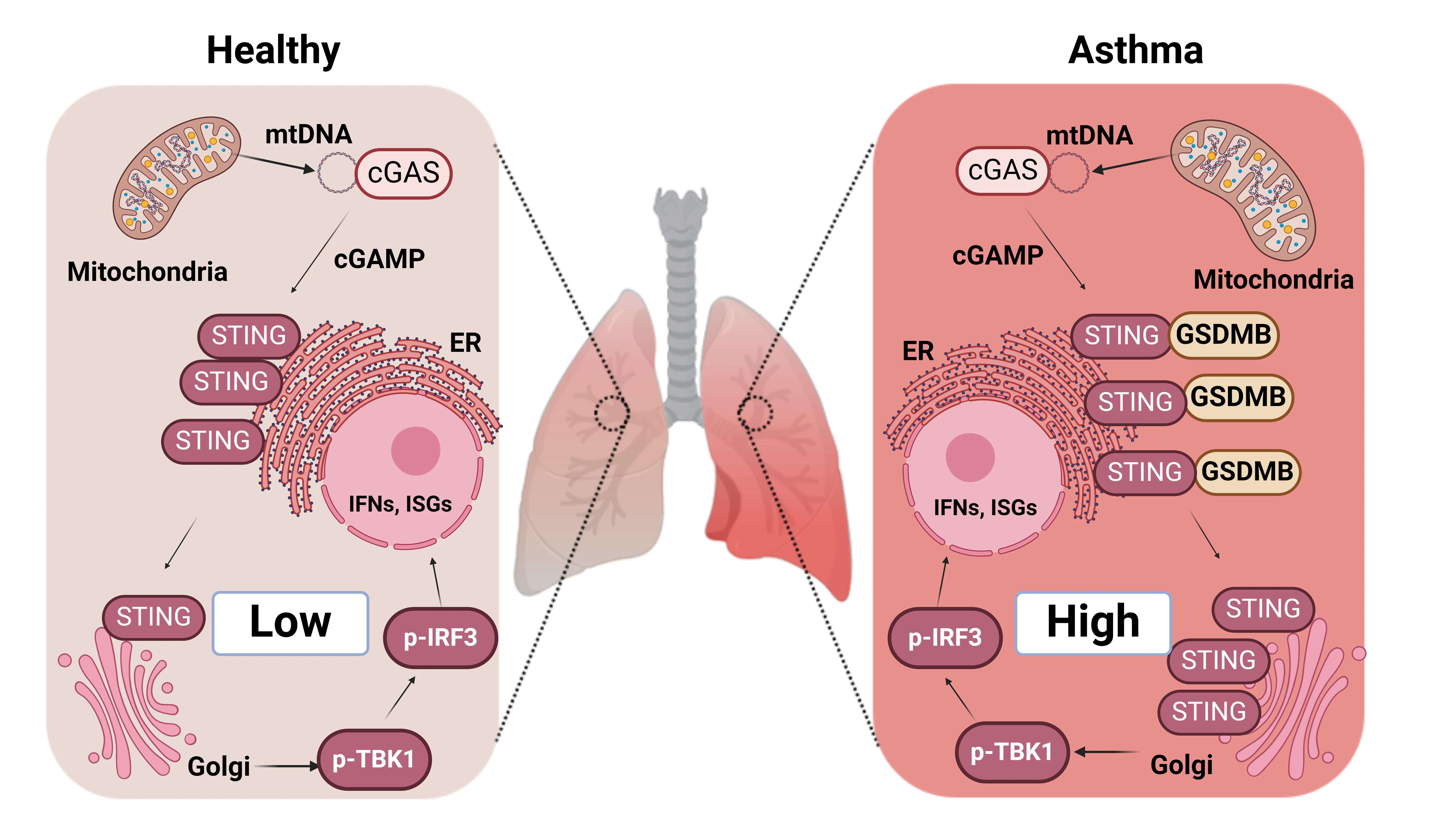
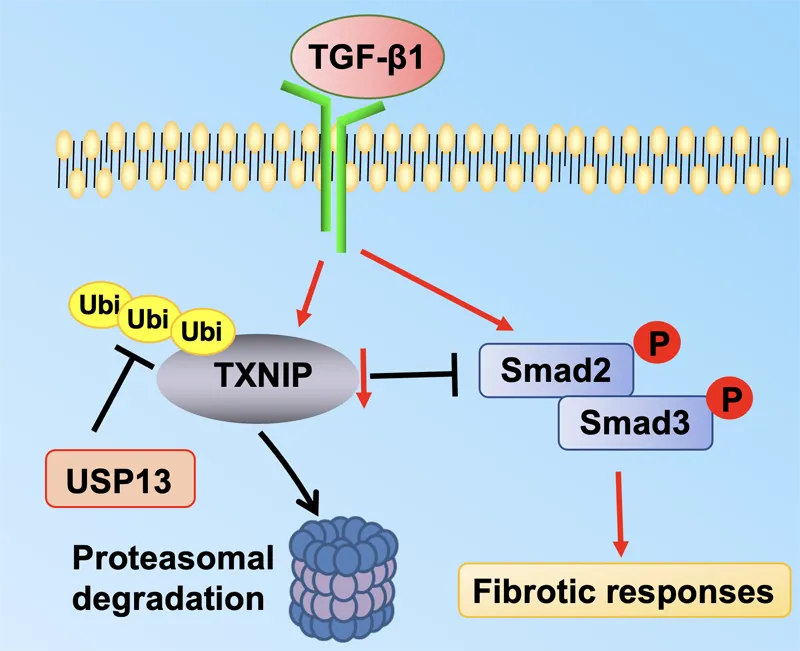
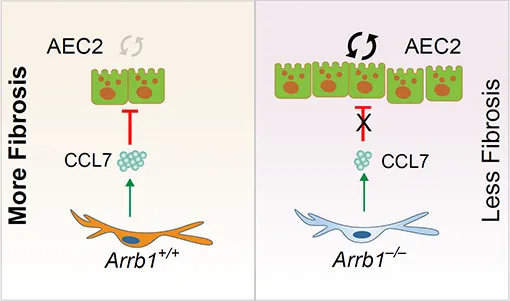
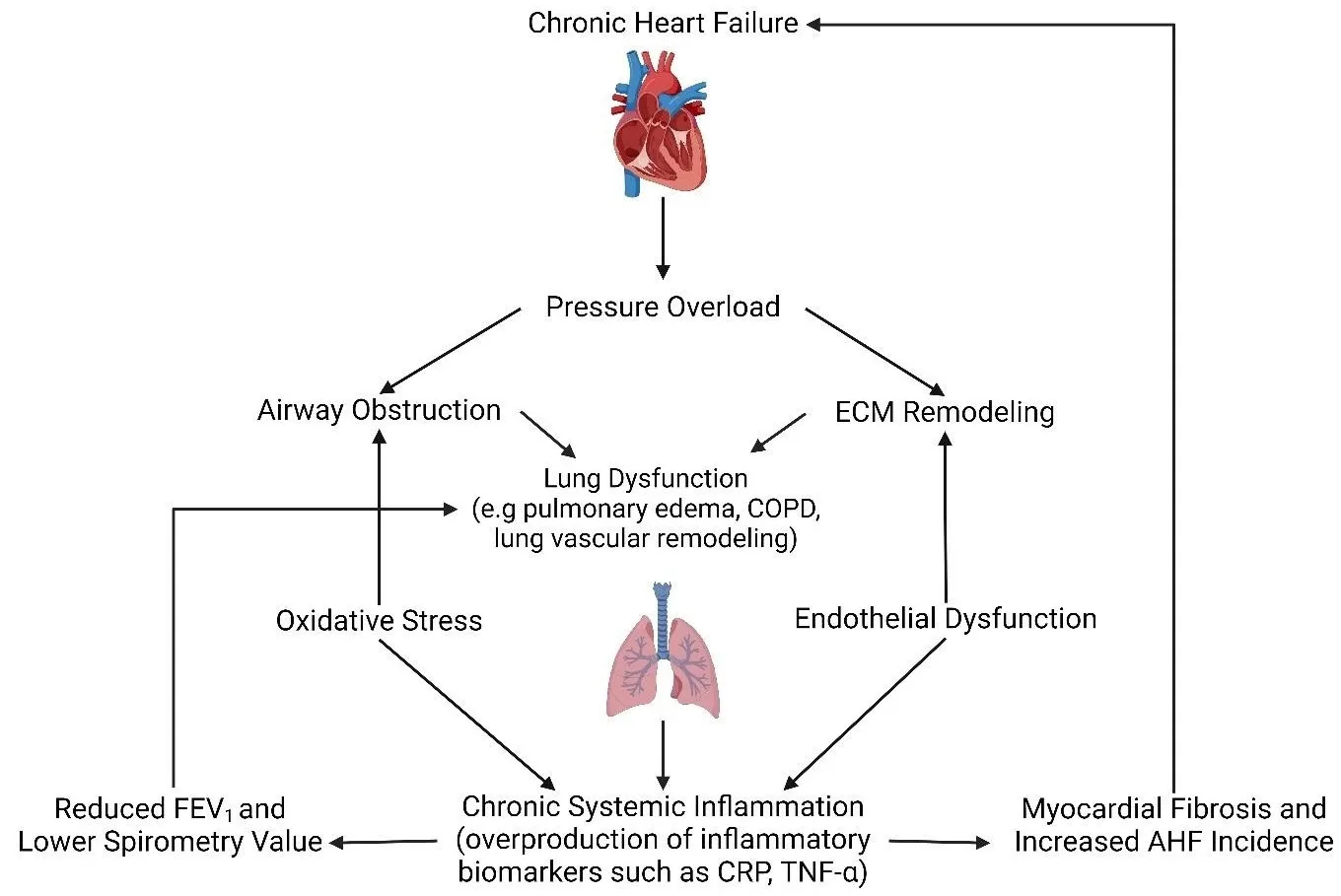
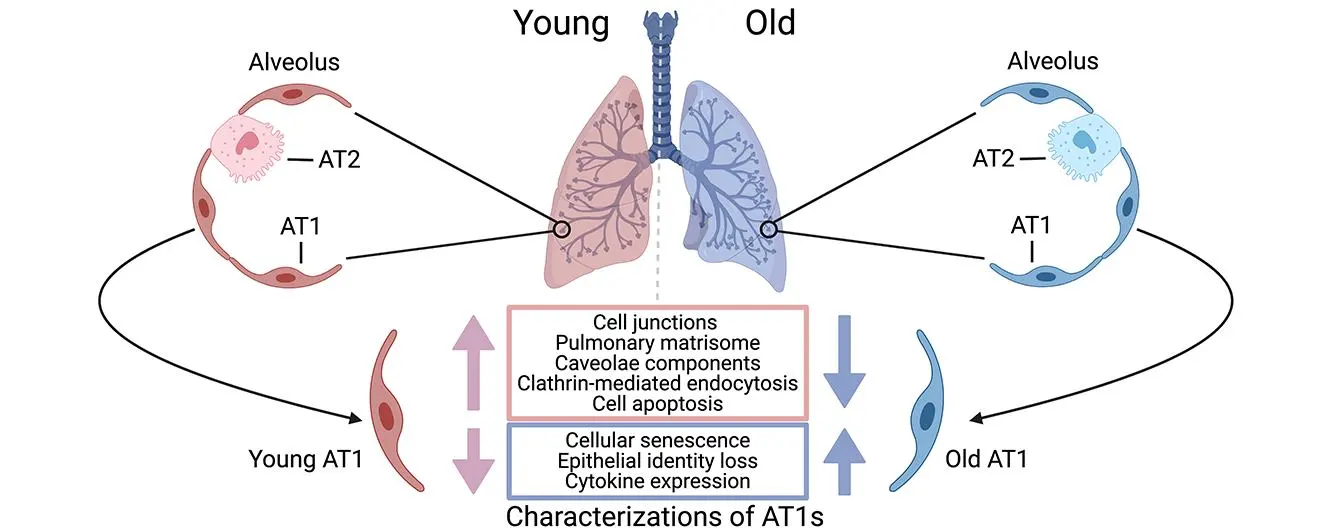
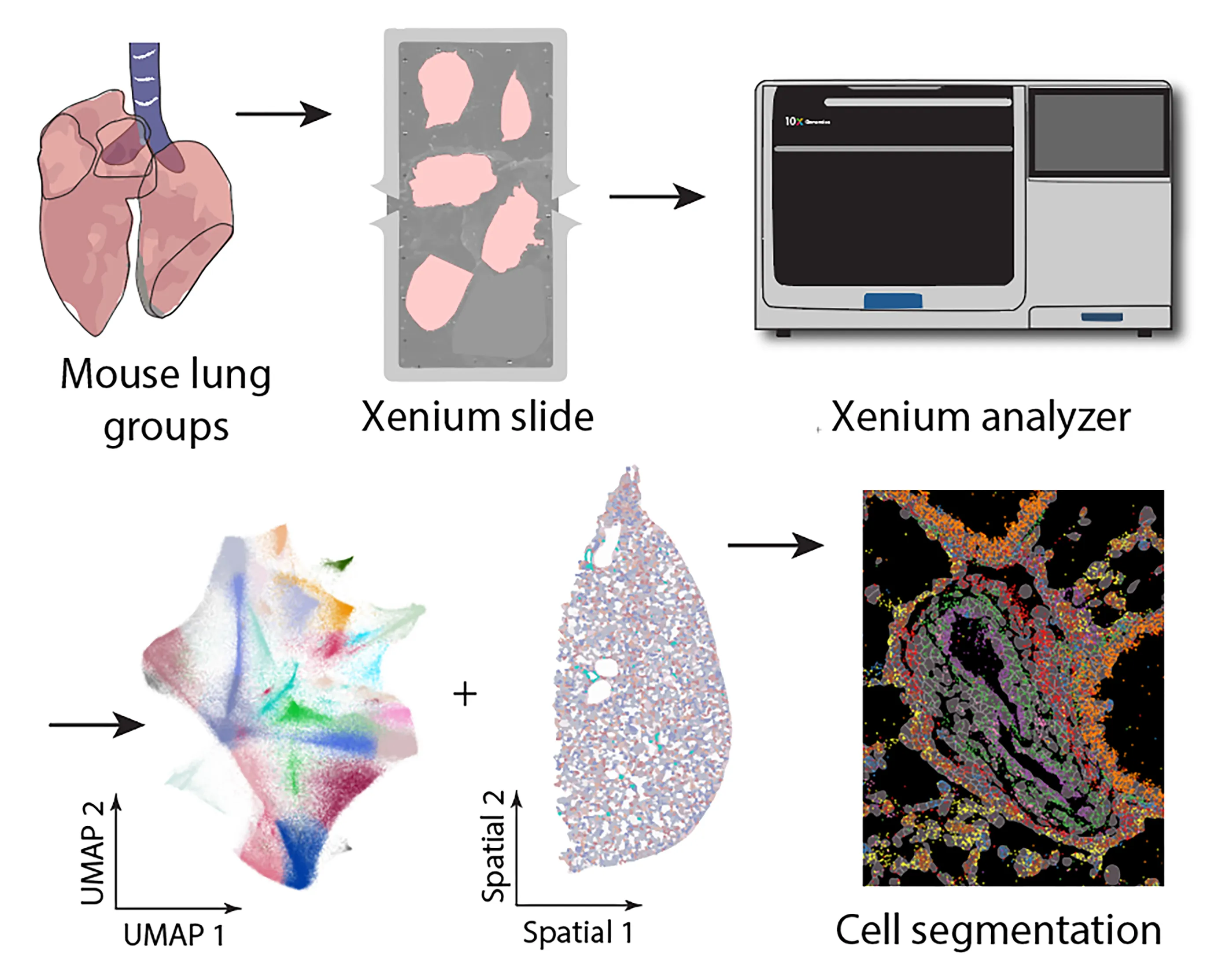
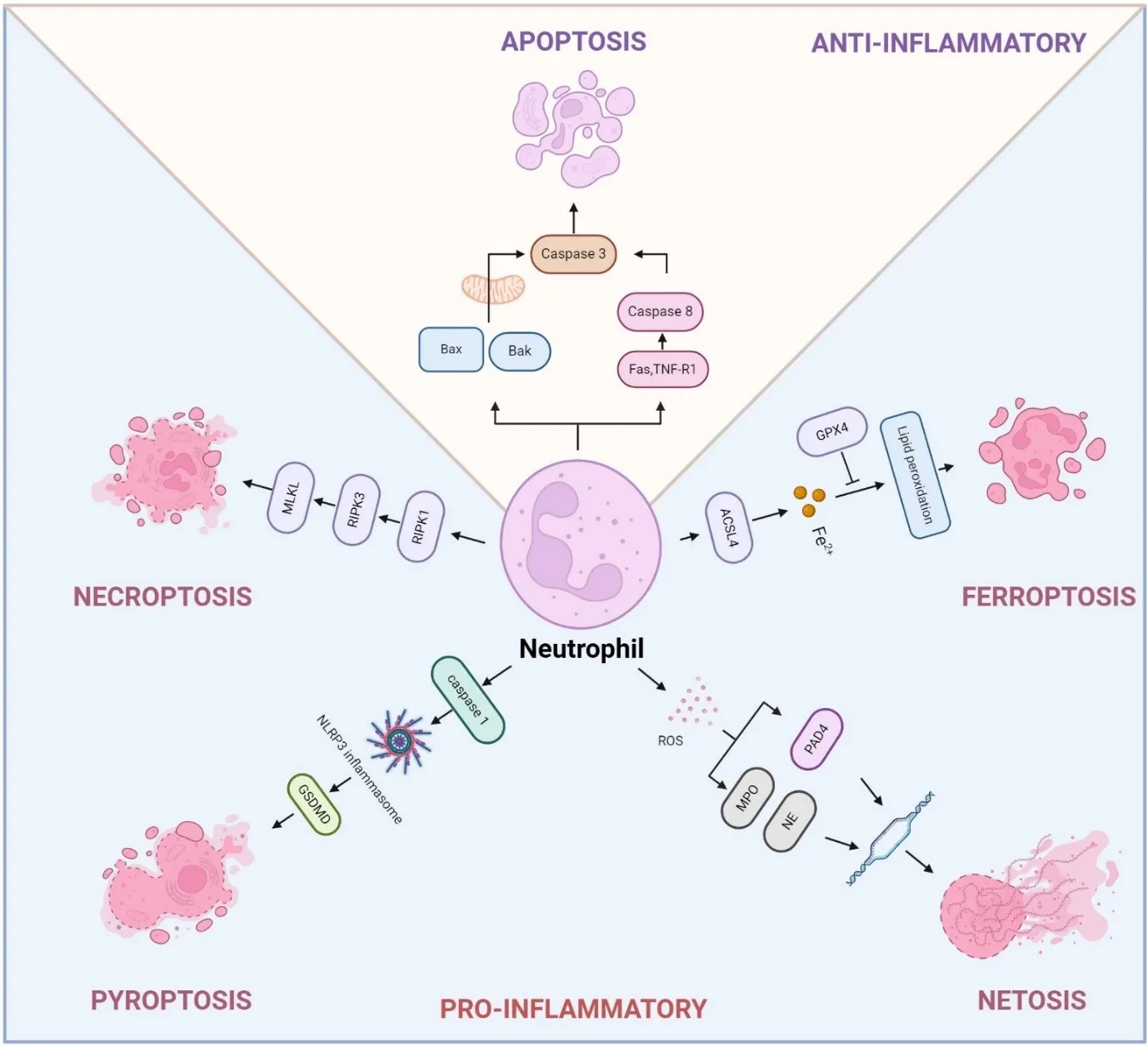
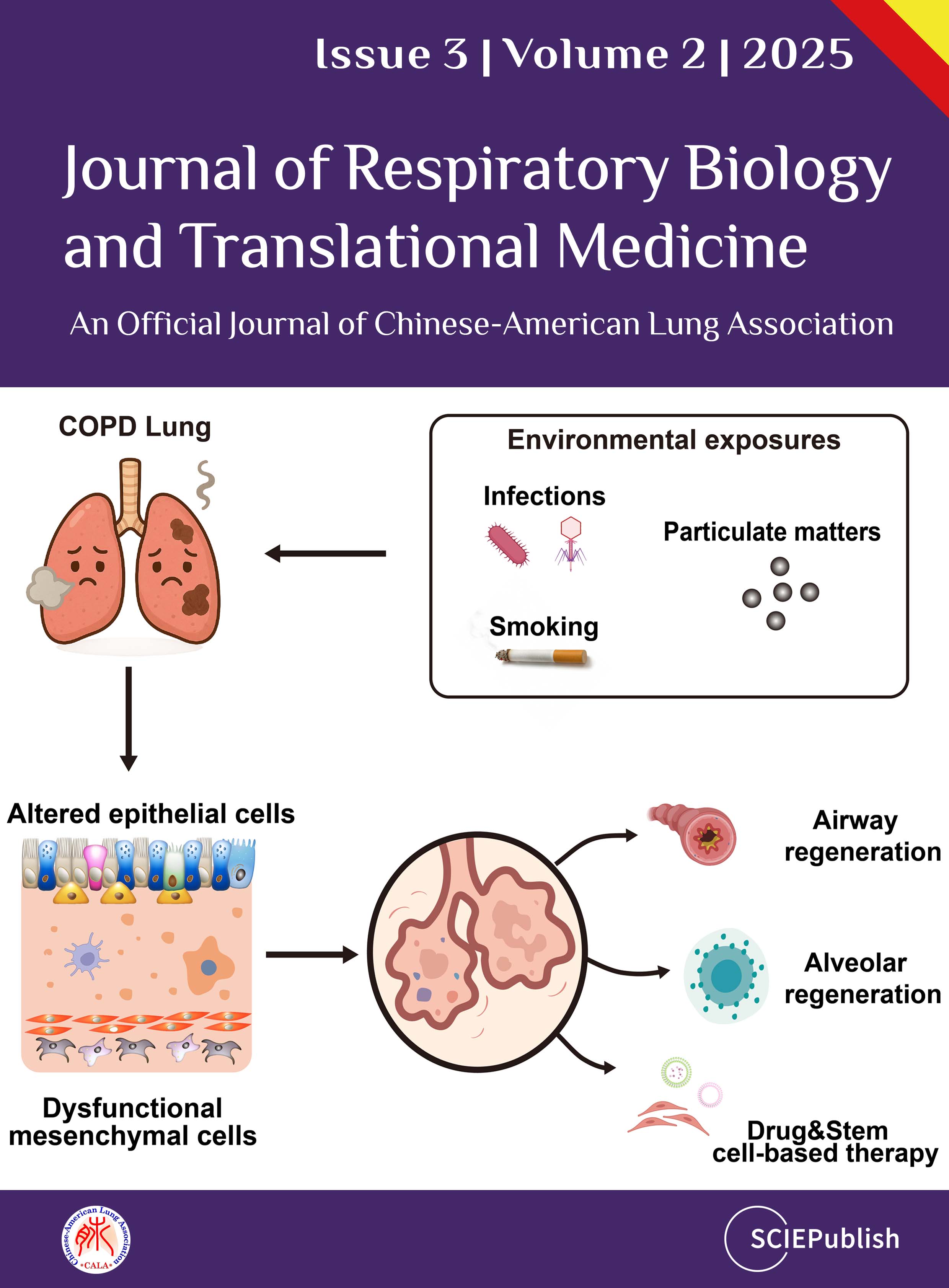
Chronic obstructive pulmonary disease (COPD) is
a leading cause of global morbidity and mortality, characterized by progressive
airway and alveolar remodeling. The disease pathogenesis is commonly driven by
chronic environmental insults, leading to airway obstruction, emphysema, and
chronic bronchitis. This review synthesizes emerging evidence that altered
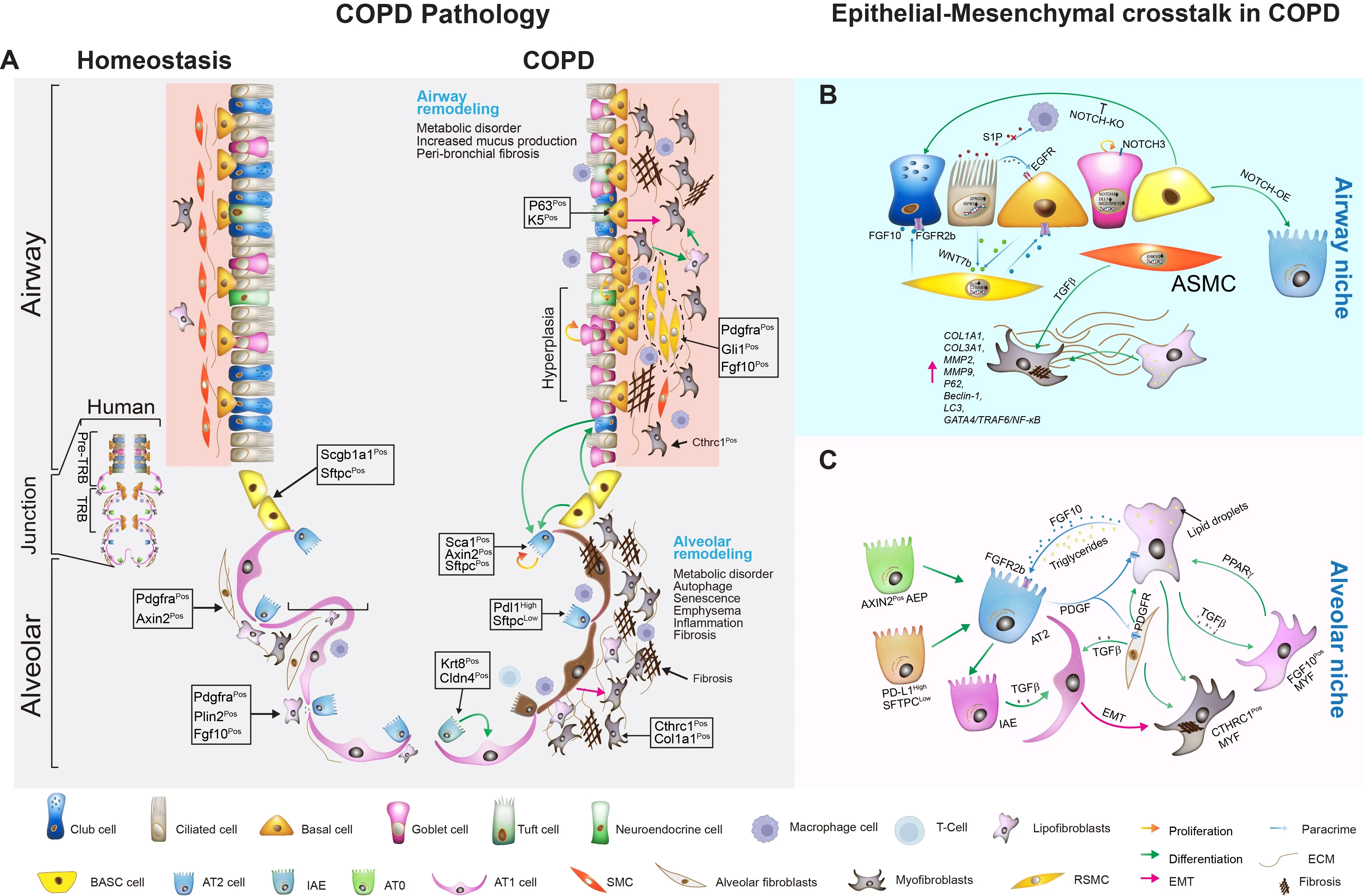
epithelial cell behavior and dysfunctional epithelial-mesenchymal
interactions serve as pivotal drivers of COPD pathogenesis, orchestrating
failed repair and structural degeneration. We detail how altered responses of
airway (ciliated, club, basal, goblet) and alveolar (AT1 and AT2) epithelial
cells lead to cellular senescence, metaplasia, defective regeneration, and
barrier disruption, acting as primary instigators of pathogenesis. We also
summarize current knowledge on the mechanisms of activation and pathogenic role
of mesenchymal cells, which drive peribronchiolar fibrosis, alveolar
destruction, and metabolic reprogramming, alongside the compromised reparative
function of mesenchymal stem cells (MSCs). We emphasize how distinct
mesenchymal niches (e.g., PDGFRαPos MANCs, FGF10Pos lipofibroblasts, SFRP1Pos fibroblasts) and distinct epithelial
stem/progenitor subpopulations critically contribute to pathogenesis. Key
signaling pathways—including FGF10/FGFR2b, WNT, Hippo, NOTCH, and TGF-β—mediate
epithelial-mesenchymal transition (EMT), stem cell niche function, and
structural remodeling. By dissecting how epithelial injury responses and
mesenchymal niche failure collaboratively drive COPD progression, we identify
actionable targets to disrupt pathogenesis and restore endogenous repair. We
propose targeting EMT, including inhibiting EMT/fibrosis, promoting alveolar
regeneration, MSC-based therapies, exosome-delivered biomolecules, and
precision cell transplantation strategies, as promising future therapeutic
strategies.

 Open Access
Open Access