1. Introduction
Idiopathic pulmonary fibrosis (IPF) remains one of the most challenging chronic lung diseases, with limited treatment options and a poor prognosis [
1,
2]. The incidence of IPF increases markedly with age, and its prevalence is rising globally [
3,
4], largely driven by the rapidly expanding elderly population [
5]. Although the antifibrotic agents pirfenidone and nintedanib have been approved by the FDA and shown to slow the rate of lung function decline modestly, neither therapy reverses fibrosis or improves overall survival [
2]. Despite advances in understanding its pathophysiology, lung transplantation remains the only definitive treatment for end-stage IPF. However, this option is severely limited by a shortage of suitable donors, along with considerable surgical risks and long-term complications following transplantation [
6,
7].
IPF is widely believed to result from repetitive alveolar epithelial injury coupled with inadequate repair [
8]. While the precise role of the immune system, particularly macrophages, in IPF pathogenesis remains incompletely understood, macrophages are among the earliest responders to tissue damage and play pivotal roles in repair processes [
9,
10]. Over the past decade, macrophages have emerged as central players in pulmonary fibrosis, exhibiting dual functions that either promote resolution or drive fibrotic progression [
11]. Although early immunomodulatory therapies were unsuccessful [
12,
13], recent studies have shown that different macrophage subtypes can either drive or resolve fibrosis through specific mechanisms, including the expression of surface receptors, changes in cellular metabolism, and activation of immune signaling pathways. Their functional heterogeneity positions them as promising therapeutic targets in IPF [
14]. Within the alveolar macrophage subtypes, tissue-resident alveolar macrophages (TR-AMs) support lung homeostasis through self-renewal and are protective during early life; however, they are depleted following severe injury [
15,
16]. In contrast, monocyte-derived alveolar macrophages (Mo-AMs) are recruited in response to injury, initially contributing to acute defense, but can perpetuate chronic fibrosis by producing profibrotic factors [
17,
18]—especially in aged individuals, where myeloid-biased hematopoiesis and systemic age related-chronic inflammation (inflammaging) enhance their pathological impact.
Bone marrow transplantation (BMT) offers a promising and accessible approach to treating end-stage diseases, especially with the increased availability of living donors compared to organ transplantation [
19]. While traditionally used for hematologic malignancies, BMT is now being explored for its potential to modulate immune function and promote tissue regeneration in non-malignant diseases. Recent research highlights the crucial roles of bone marrow–derived monocytes in tissue repair and immune reprogramming, particularly through the replenishment of macrophage populations, including TR-AMs and Mo-AMs, which influence the inflammatory environment of injured organs [
20,
21,
22]. Aging disrupts hematopoietic function by promoting myeloid bias and systemic inflammaging, impairing repair mechanisms, and driving maladaptive immune responses. Replacing aged hematopoietic stem cells with healthy donor cells can rejuvenate immune function, enhance regenerative capacity, and potentially restore the balance of macrophage populations [
23,
24]. Although BMT shows promise, its therapeutic potential in non-hematologic, degenerative conditions such as IPF, where TR-AMs and Mo-AMs are key players in fibrosis progression, remains underexplored and warrants further investigation.
2. Hematopoietic Age Exacerbates Fibrosis Independent of Initial Lung Injury
The practical and biological rationale for exploring BMT in IPF therapy lies in both logistics and mechanistic insights. Lung transplantation, while curative, suffers from a severe shortage of suitable donor lungs and requires lifelong immunosuppression. In contrast, bone marrow donors are far more accessible and primarily come from living individuals, making the procedure logistically more feasible. Furthermore, BMT has a long-established safety record and is routinely used in hematologic disorders. This contrast highlights the clinical potential of BMT as a more scalable intervention for IPF, particularly when informed by mechanistic studies that link hematopoietic age to fibrotic outcomes.
A compelling mechanistic basis for BMT in IPF emerges from recent work by Farhat et al. [
25], who used a heterochronic BMT model in which young adult recipient mice received bone marrow from either aged or young donors. This design allowed the researchers to isolate the influence of hematopoietic aging independent of lung tissue age. Following bleomycin-induced injury, young mice reconstituted with aged bone marrow exhibited significantly worsened fibrotic outcomes—including increased collagen deposition, α-SMA expression, and Ashcroft scores—despite equivalent levels of early lung injury. This dissociation between injury and fibrosis highlights that immune aging, rather than epithelial vulnerability, plays a pivotal role in fibrotic progression. Using heterochronic BMT models, researchers successfully decoupled the effects of tissue aging from hematopoietic aging, demonstrating that bone marrow-derived signals dominate the fibrotic response, irrespective of the age of the lung parenchyma.
This paradigm-shifting observation underscores the need to reevaluate fibrosis not merely as a consequence of failed epithelial repair but also as a dysfunction of the immune system’s resolution capacity—one that worsens with age. The ability of aged bone marrow to potentiate fibrosis in an otherwise youthful lung environment reveals novel therapeutic targets in hematopoietic regulation.
3. Delayed Mo-AM Transition Drives Persistent Fibrosis
Monocyte-derived alveolar macrophages (Mo-AMs) have emerged as central drivers of lung fibrosis [
17,
18]. These cells are recruited to the lung following injury, where they are expected to transition into TR-AMs, helping to restore homeostasis. In this study, a longitudinal analysis of the Mo-AM compartment from the shielded BMT chimeras model using flow cytometry, bone marrow chimera tracking, and transcriptomics revealed that aged bone marrow–derived Mo-AMs exhibit a delayed transition into TR-AMs. Instead of resolving inflammation, they persist in a pro-inflammatory and profibrotic state, contributing to sustained fibroblast activation and the deposition of extracellular matrix. This stalled differentiation correlates with impaired fibrosis resolution and delayed recovery of lung homeostasis. Notably, this effect is intrinsic to the age of the hematopoietic progenitors, as evidenced by competitive bone marrow chimera experiments. Aged Mo-AMs outcompete their younger counterparts, indicating that aged hematopoietic progenitors may exhibit enhanced proliferative capacity or increased migratory efficiency rather than being favored by expanded niche availability.
These findings significantly advance our understanding of macrophage dynamics in fibrosis, positioning Mo-AM differentiation as a key checkpoint in disease progression. Therapeutic strategies that enhance the transition of Mo-AMs into a TR-AM phenotype may offer a new avenue for halting or even reversing fibrosis.
4. Treg-Derived IL-10 as a Master Regulator of Macrophage Resolution
The study further identifies a critical immunoregulatory axis involving regulatory T cells (Tregs) and their secretion of interleukin-10 (IL-10). In young bone marrow recipients, IL-10 production facilitates the differentiation of Mo-AMs into homeostatic cells, tempering the inflammatory response and promoting epithelial repair. In contrast, aged bone marrow recipients exhibit reduced Treg-derived IL-10, leading to the persistence of inflammatory macrophage phenotypes and enhanced fibrogenesis.
This reduction in IL-10 is not simply due to lower Treg numbers but rather reflects intrinsic dysfunction in aged Tregs, including TH1 skewing and transcriptional alterations that impair their reparative capacity. This Treg defect represents a crucial bottleneck in the resolution of lung inflammation, suggesting that rejuvenating Treg function or boosting IL-10 production could represent targeted interventions to accelerate fibrosis resolution. Importantly, the fibrogenic influence of Treg dysfunction is phase-specific. Loss of IL-10 or Tregs during the fibrotic development phase (day 7–21 post-injury) drives aberrant Mo-AM differentiation, whereas early depletion during the injury phase (day 0–7) affects inflammation but not long-term fibrosis. This temporal distinction opens a therapeutic window where immune modulation can be optimally timed for maximal antifibrotic benefit.
5. Microenvironmental Cues and Crosstalk with Other Immune Cells
Beyond Tregs, the inflammatory microenvironment shaped by aged hematopoietic cells exhibits elevated IFN-γ and IL-6, and reduced IL-10, setting the stage for the persistence of profibrotic Mo-AM. While this study focused on Treg-derived IL-10, other cell types—including group 2 innate lymphoid cells (ILC2s) [
25] and basophils—may also modulate macrophage differentiation and fibrotic outcomes.
Moreover, the lung’s ability to repair itself through alveolar epithelial cell differentiation (AT2 to AT1) is heavily influenced by early immune signals [
26]. Prior research shows that excessive or prolonged activation of monocyte-derived macrophages disrupts epithelial integrity, promoting the formation of aberrant intermediate cell states that resist resolution [
27,
28]. In this context, the persistent activation state of aged Mo-AMs likely impairs alveolar repair and promotes chronic fibrosis.
6. Toward a Novel Therapeutic Framework
This work illuminates the far-reaching influence of hematopoietic aging on pulmonary fibrosis and builds a compelling case for bone marrow transplantation as a novel therapeutic strategy in IPF. By restoring youthful hematopoietic output, particularly in the context of end-stage disease with preserved lung niche capacity, BMT may offer a means to reprogram the immune landscape of the fibrotic lung. Importantly, this approach circumvents some of the limitations of current therapies by addressing a root cause of fibrosis persistence—dysregulated immune resolution driven by aged hematopoietic progenitors [
29]. Moreover, the identification of key immune checkpoints, such as the Treg–IL-10–Mo-AM-TR-AM axis (), provides actionable targets for adjunct therapies that could enhance the efficacy of bone marrow replacement.
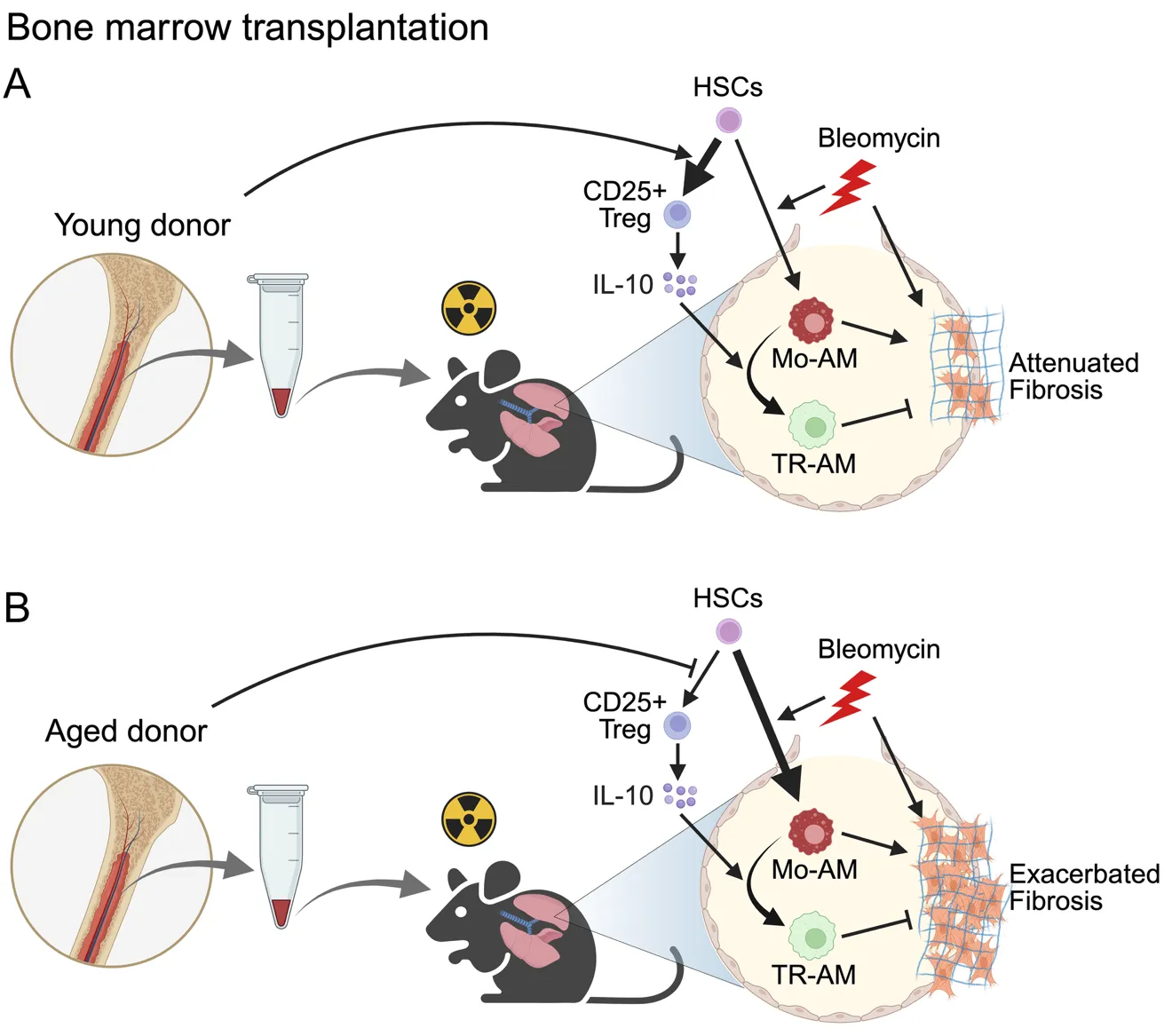
. Experimental overview of bone marrow transplantation in a mouse model of pulmonary fibrosis created with BioRender. C57BL/6J WT recipient mice were irradiated and transplanted with bone marrow from young (<strong>A</strong>) or aged (<strong>B</strong>) donor mice followed by intratracheal bleomycin administration two months after transplantation. (<strong>A</strong>) Mice receiving young bone marrow exhibited reduced differentiation of Mo-Ams and, increased CD25<sup>+</sup> T<sub>regs</sub> as well as elevated IL-10 production. This immunoregulatory environment supported the transition of Mo-AMs toward a homeostatic phenotype, leading to the attenuation of fibrosis. (<strong>B</strong>) In contrast, recipients of aged bone marrow showed enhanced Mo-AM differentiation, diminished CD25<sup>+</sup> T<sub>regs</sub> development, and lower IL-10 levels, creating a profibrotic environment that exacerbated lung fibrosis. HSC, Hematopoietic stem cell; Mo-AM, Monocyte-derived alveolar macrophage; TR-AM, Tissue-resident alveolar macrophages marrow.
7. Limitations and Future Directions
This study significantly extends prior studies on macrophage heterogeneity in IPF by mechanistically linking hematopoietic aging to aberrant Mo-AM fate decisions. While previous research has recognized the dual roles of TR-AMs and Mo-AMs in lung fibrosis, this study identifies a critical age-dependent regulatory axis—Treg-derived IL-10—as a central modulator of Mo-AM resolution. This discovery not only clarifies why Mo-AMs remain pathologically activated in aged hosts but also reframes the fibrotic process as a failure of immune reprogramming. By integrating aging biology with immune regulation, the study opens new avenues for age-tailored immunotherapies targeting macrophage plasticity in fibrotic lung disease.
While this study highlights the promising potential of BMT to reverse immune dysfunction and promote fibrosis resolution in IPF, several limitations must be acknowledged when considering clinical translation. BMT, although logistically more feasible than lung transplantation, is not without significant risks. One of the most critical concerns is graft-versus-host disease (GVHD), a life-threatening complication that can arise from immune incompatibility between donor and recipient [
30]. Even with autologous or matched sibling transplants, the risks of infection, immunosuppression-related complications, and prolonged recovery remain substantial. These factors necessitate a thorough risk-benefit analysis before considering BMT as a therapeutic strategy for patients with chronic lung disease, particularly those with already compromised respiratory function. Moreover, the use of preclinical mouse models, while instrumental in dissecting the role of hematopoietic aging and macrophage dysfunction, does not fully replicate the complexity of human IPF pathogenesis. Differences in immune responses, lung architecture, and comorbidities in aged human populations may influence the effectiveness and safety of BMT-based interventions.
Future research should focus on refining BMT approaches to reduce associated risks. This may include the use of non-myeloablative conditioning regimens, selective immune cell replacement strategies, or ex vivo gene editing of hematopoietic stem cells to enhance reparative functions without full transplantation. Additionally, adjunctive therapies such as IL-10 agonists, Treg-enhancing biologics, or agents targeting monocyte-to-macrophage differentiation pathways could synergize with BMT to improve outcomes and minimize adverse effects. Importantly, the precise mechanisms by which the aged bone marrow microenvironment impairs Treg function remain incompletely understood. Whether this dysfunction arises from myeloid-biased hematopoiesis, altered Treg progenitor niches or epigenetic reprogramming within aged hematopoietic systems is an open question that warrants further investigation. Addressing these gaps will be essential for developing more targeted and effective interventions. Furthermore, pharmacologic targeting of immune pathways provides alternative strategies for patients ineligible for transplantation [
10]. Inhibitors of GM-CSF, CCR2, galectin-3, and JAK/STAT pathways, as well as modulation of PD-1 and IL-33/ILC2 signaling, have demonstrated antifibrotic potential. Notably, Src family kinase inhibition, implicated in pulmonary inflammation [
31], has shown efficacy in preclinical IPF models [
32], suggesting possible combinatorial benefits with BMT. Ultimately, while BMT represents a novel and promising avenue to address the immune component of IPF, clinical application will require careful consideration of safety, feasibility, and regulatory challenges. Expanding preclinical models to better reflect human disease and developing combinatorial therapeutic strategies are critical next steps for translating these findings into clinical benefit.
Young patients with IPF are relatively uncommon and tend to have lower mortality rates and reduced need for lung transplantation [
33,
34]. Since hematopoietic aging contributes to maladaptive immune responses—such as impaired Mo-AM differentiation and diminished Treg-derived IL-10—BMT is most likely to confer therapeutic benefits in older patients, where these age-associated defects are more pronounced. In contrast, younger individuals, who may exhibit fewer hematopoietic abnormalities, could derive limited benefit from this approach. However, key fibrotic mechanisms, including persistent Mo-AM activation and impaired immune resolution, may still be present, albeit to a lesser extent. Future studies comparing outcomes across age groups will be essential to identify the most responsive patient populations and refine clinical strategies.
In summary, the transplantation of young bone marrow into aged hosts demonstrates striking antifibrotic effects by modulating monocyte-to-macrophage differentiation and restoring immune resolution through IL-10–producing Tregs. These findings mark a major advance in our understanding of how hematopoietic aging influences pulmonary fibrosis and opens the door to innovative therapies based on bone marrow rejuvenation. As IPF continues to impose a high burden with limited treatment options [
2], this line of research brings hope for more accessible and effective interventions. Bone marrow transplantation, either alone or in combination with targeted immune modulators [
30], may soon complement or even replace lung transplantation as a viable strategy for reversing the course of fibrotic lung disease.
Acknowledgments
We thank lab members for their insightful and helpful discussions.
Author Contributions
X.L. wrote the original manuscript, prepared the figure, reviewed and edited the manuscript.
Ethics Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Funding
This research was funded by the American Heart Association award 24CDA1268568 and the Pulmonary Fibrosis Foundation Scholars Program 1272558.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
-
1.
Noble PW, Barkauskas CE, Jiang D. Pulmonary fibrosis: Patterns and perpetrators.
J. Clin. Investig. 2012,
122, 2756–2762. doi:10.1172/JCI60323.
[Google Scholar]
-
2.
Richeldi L, Collard HR, Jones MG. Idiopathic pulmonary fibrosis.
Lancet 2017,
389, 1941–1952. doi:10.1016/S0140-6736(17)30866-8.
[Google Scholar]
-
3.
Martinez FJ, Collard HR, Pardo A, Raghu G, Richeldi L, Selman M, et al. Idiopathic pulmonary fibrosis.
Nat. Rev. Dis. Primers 2017,
3, 17074. doi:10.1038/nrdp.2017.74.
[Google Scholar]
-
4.
Rojas M, Mora AL, Kapetanaki M, Weathington N, Gladwin M, Eickelberg O. Aging and Lung Disease. Clinical Impact and Cellular and Molecular Pathways.
Ann. Am. Thorac. Soc. 2015,
12, S222–S227. doi:10.1513/AnnalsATS.201508-484PL.
[Google Scholar]
-
5.
Hauer ME. Population projections for U.S. counties by age, sex, and race controlled to shared socioeconomic pathway.
Sci. Data 2019,
6, 190005. doi:10.1038/sdata.2019.5.
[Google Scholar]
-
6.
Swaminathan AC, Hellkamp AS, Neely ML, Bender S, Paoletti L, White ES, et al. Disparities in Lung Transplant among Patients with Idiopathic Pulmonary Fibrosis.
Ann. Am. Thorac. Soc. 2022,
19, 981–990. doi:10.1513/AnnalsATS.202105-589OC.
[Google Scholar]
-
7.
Mason DP, Brizzio ME, Alster JM, McNeill AM, Murthy SC, Budev MM, et al. Lung transplantation for idiopathic pulmonary fibrosis.
Ann. Thorac. Surg. 2007,
84, 1121–1128. doi:10.1016/j.athoracsur.2007.04.096.
[Google Scholar]
-
8.
Barkauskas CE, Cronce MJ, Rackley CR, Bowie EJ, Keene DR, Stripp BR, et al. Type 2 alveolar cells are stem cells in adult lung.
J. Clin. Investig. 2013,
123, 3025–3036. doi:10.1172/JCI68782.
[Google Scholar]
-
9.
Heukels P, Moor CC, von der Thusen JH, Wijsenbeek MS, Kool M. Inflammation and immunity in IPF pathogenesis and treatment.
Respir. Med. 2019,
147, 79–91. doi:10.1016/j.rmed.2018.12.015.
[Google Scholar]
-
10.
Shenderov K, Collins SL, Powell JD, Horton MR. Immune dysregulation as a driver of idiopathic pulmonary fibrosis.
J. Clin. Investig. 2021,
131, 2. doi:10.1172/JCI143226.
[Google Scholar]
-
11.
Pokhreal D, Crestani B, Helou DG. Macrophage Implication in IPF: Updates on Immune, Epigenetic, and Metabolic Pathways.
Cells 2023,
12, 2193. doi:10.3390/cells12172193.
[Google Scholar]
-
12.
Raghu G, Brown KK, Costabel U, Cottin V, du Bois RM, Lasky JA, et al. Treatment of Idiopathic Pulmonary Fibrosis with Etanercept An Exploratory, Placebo-controlled Trial.
Am. J. Resp. Crit. Care 2008,
178, 948–955. doi:10.1164/rccm.200709-1446OC.
[Google Scholar]
-
13.
Raghu G, Anstrom KJ, King TE, Lasky JA, Martinez FJ, Clinical IPF. Prednisone, Azathioprine, and N-Acetylcysteine for Pulmonary Fibrosis.
N. Engl. J. Med. 2012,
366, 1968–1977. doi:10.1056/NEJMoa1113354.
[Google Scholar]
-
14.
Byrne AJ, Maher TM, Lloyd CM. Pulmonary Macrophages: A New Therapeutic Pathway in Fibrosing Lung Disease?
Trends. Mol. Med. 2016,
22, 303–316. doi:10.1016/j.molmed.2016.02.004.
[Google Scholar]
-
15.
Misharin AV, Morales-Nebreda L, Reyfman PA, Cuda CM, Walter JM, McQuattie-Pimentel AC, et al. Monocyte-derived alveolar macrophages drive lung fibrosis and persist in the lung over the life span.
J. Exp. Med. 2017,
214, 2387–2404. doi:10.1084/jem.20162152.
[Google Scholar]
-
16.
Perrot CY, Karampitsakos T, Herazo-Maya JD. Monocytes and macrophages: Emerging mechanisms and novel therapeutic targets in pulmonary fibrosis.
Am. J. Physiol. Cell. Physiol. 2023,
325, C1046–C1057. doi:10.1152/ajpcell.00302.2023.
[Google Scholar]
-
17.
McQuattie-Pimentel AC, Budinger GRS, Ballinger MN. Monocyte-derived Alveolar Macrophages: The Dark Side of Lung Repair?
Am J Respir Cell Mol Biol 2018,
58, 5–6. doi:10.1165/rcmb.2017-0328ED.
[Google Scholar]
-
18.
Joshi N, Watanabe S, Verma R, Jablonski RP, Chen CI, Cheresh P, et al. A spatially restricted fibrotic niche in pulmonary fibrosis is sustained by M-CSF/M-CSFR signalling in monocyte-derived alveolar macrophages.
Eur. Respir. J. 2020,
55, 1. doi:10.1183/13993003.00646-2019.
[Google Scholar]
-
19.
Kean LS, Blazar BR. Major breakthroughs in hematopoietic stem cell transplantation and future challenges in clinical implementation.
J. Clin. Investig. 2024,
134, e179944. doi:10.1172/JCI179944.
[Google Scholar]
-
20.
Kingsley PD, Rust ET, Kingsley AJ, Pinney JJ, Rivera-Escalera F, Bennett CP, et al. Tissue-Resident Macrophages in the Bone Marrow Comprise a Diverse Cellular Population Derived from Hematopoietic Stem Cells.
Blood 2023,
142, 3910. doi:10.1182/blood-2023-188067.
[Google Scholar]
-
21.
Nasser H, Adhikary P, Abdel-Daim A, Noyori O, Panaampon J, Kariya R, et al. Establishment of bone marrow-derived M-CSF receptor-dependent self-renewing macrophages.
Cell Death Discov. 2020,
6, 63. doi:10.1038/s41420-020-00300-3.
[Google Scholar]
-
22.
Scott CL, Zheng F, De Baetselier P, Martens L, Saeys Y, De Prijck S, et al. Bone marrow-derived monocytes give rise to self-renewing and fully differentiated Kupffer cells.
Nat. Commun. 2016,
7, 10321. doi:10.1038/ncomms10321.
[Google Scholar]
-
23.
Alibhai FJ, Li RK. Targeting aged bone marrow for systemic rejuvenation.
Aging 2020,
12, 2024–2025. doi:10.18632/aging.102838.
[Google Scholar]
-
24.
Hou J, Chen KX, He C, Li XX, Huang M, Jiang YZ, et al. Aged bone marrow macrophages drive systemic aging and age-related dysfunction via extracellular vesicle-mediated induction of paracrine senescence.
Nat. Aging 2024,
4, 1562–1581. doi:10.1038/s43587-024-00694-0.
[Google Scholar]
-
25.
Farhat A, Radhouani M, Deckert F, Zahalka S, Pimenov L, Fokina A, et al. An aging bone marrow exacerbates lung fibrosis by fueling profibrotic macrophage persistence.
Sci. Immunol. 2025,
10,
eadk5041. doi:10.1126/sciimmunol.adk5041.
[Google Scholar]
-
26.
Guenin-Mace L, Konieczny P, Naik S. Immune-Epithelial Cross Talk in Regeneration and Repair.
Annu. Rev. Immunol. 2023,
41, 207–228. doi:10.1146/annurev-immunol-101721-062818.
[Google Scholar]
-
27.
Choi J, Park JE, Tsagkogeorga G, Yanagita M, Koo BK, Han N, et al. Inflammatory Signals Induce AT2 Cell-Derived Damage-Associated Transient Progenitors that Mediate Alveolar Regeneration.
Cell. Stem. Cell. 2020,
27, 366. doi:10.1016/j.stem.2020.06.020.
[Google Scholar]
-
28.
McQuattie-Pimentel AC, Ren Z, Joshi N, Watanabe S, Stoeger T, Chi M, et al. The lung microenvironment shapes a dysfunctional response of alveolar macrophages in aging.
J. Clin. Investig. 2021,
131, 4. doi:10.1172/JCI140299.
[Google Scholar]
-
29.
Lv JJ, Zhang C, Liu XX, Gu CY, Liu YD, Gao YH, et al. An aging-related immune landscape in the hematopoietic immune system.
Immun. Aging. 2024,
21, 3. doi:10.1186/s12979-023-00403-2.
[Google Scholar]
-
30.
Yanik G, Cooke KR. The lung as a target organ of graft-versus-host disease.
Semin. Hematol. 2006,
43, 42–52. doi:10.1053/j.seminhematol.2005.09.004.
[Google Scholar]
-
31.
Ernst M, Inglese M, Scholz GM, Harder KW, Clay FJ, Bozinovski S, et al. Constitutive activation of the Src family kinase Hck results in spontaneous pulmonary inflammation and an enhanced innate immune response.
J. Exp. Med. 2002,
196, 589–604. doi:10.1084/jem.20020873.
[Google Scholar]
-
32.
Hu M, Che P, Han X, Cai GQ, Liu G, Antony V, et al. Therapeutic targeting of SRC kinase in myofibroblast differentiation and pulmonary fibrosis.
J. Pharmacol. Exp. Ther. 2014,
351, 87–95. doi:10.1124/jpet.114.216044.
[Google Scholar]
-
33.
Leuschner G, Reiter F, Stocker F, Crispin A, Kneidinger N, Veit T, et al. Idiopathic Pulmonary Fibrosis Among Young Patients: Challenges in Diagnosis and Management.
Lung 2018,
196, 401–408. doi:10.1007/s00408-018-0123-9.
[Google Scholar]
-
34.
Raghu G, Chen SY, Hou Q, Yeh WS, Collard HR. Incidence and prevalence of idiopathic pulmonary fibrosis in US adults 18-64 years old.
Eur. Respir. J. 2016,
48, 179–186. doi:10.1183/13993003.01653-2015.
[Google Scholar]