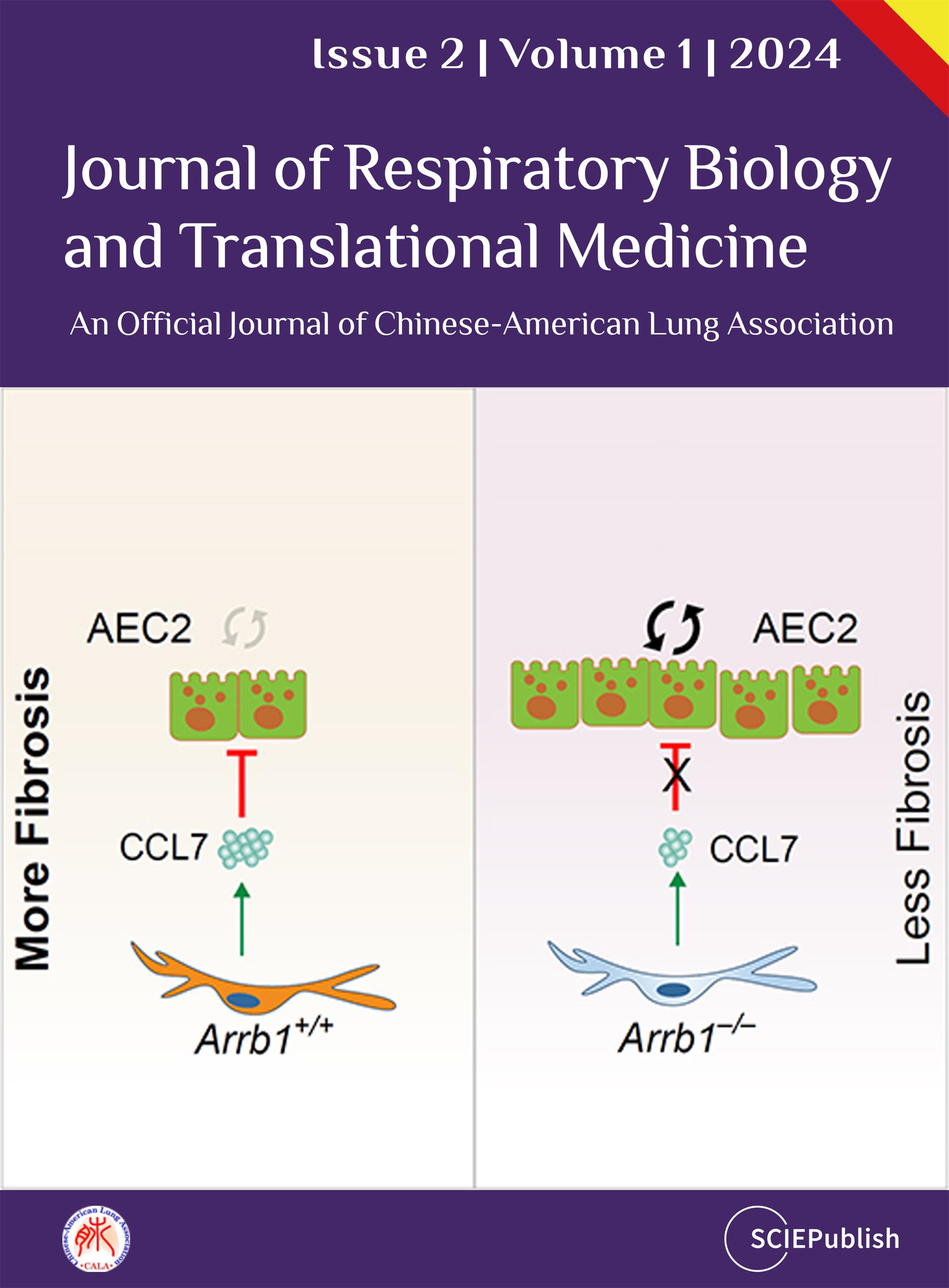
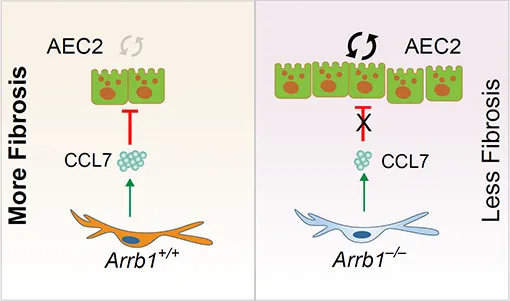
The molecular mechanisms that regulate progressive pulmonary fibrosis remain poorly understood. Type 2 alveolar epithelial cells (AEC2s) function as adult stem cells in the lung. We previously showed that there is a loss of AEC2s and a failure of AEC2 renewal in the lungs of idiopathic pulmonary fibrosis (IPF) patients. We also reported that beta-arrestins are the key regulators of fibroblast invasion, and beta-arrestin 1 and 2 deficient mice exhibit decreased mortality, decreased matrix deposition, and increased lung function in bleomycin-induced lung fibrosis. However, the role of beta-arrestins in AEC2 regeneration is unclear. In this study, we investigated the role and mechanism of Arrestin beta 1 (ARRB1) in AEC2 renewal and in lung fibrosis. We used conventional deletion as well as cell type-specific deletion of ARRB1 in mice and found that Arrb1 deficiency in fibroblasts protects mice from lung fibrosis, and the knockout mice exhibit enhanced AEC2 regeneration in vivo, suggesting a role of fibroblast-derived ARRB1 in AEC2 renewal. We further found that Arrb1-deficient fibroblasts promotes AEC2 renewal in 3D organoid assays. Mechanistically, we found that CCL7 is among the top downregulated cytokines in Arrb1 deficient fibroblasts and CCL7 inhibits AEC2 regeneration in 3D organoid experiments. Therefore, fibroblast ARRB1 mediates AEC2 renewal, possibly by releasing chemokine CCL7, leading to fibrosis in the lung.
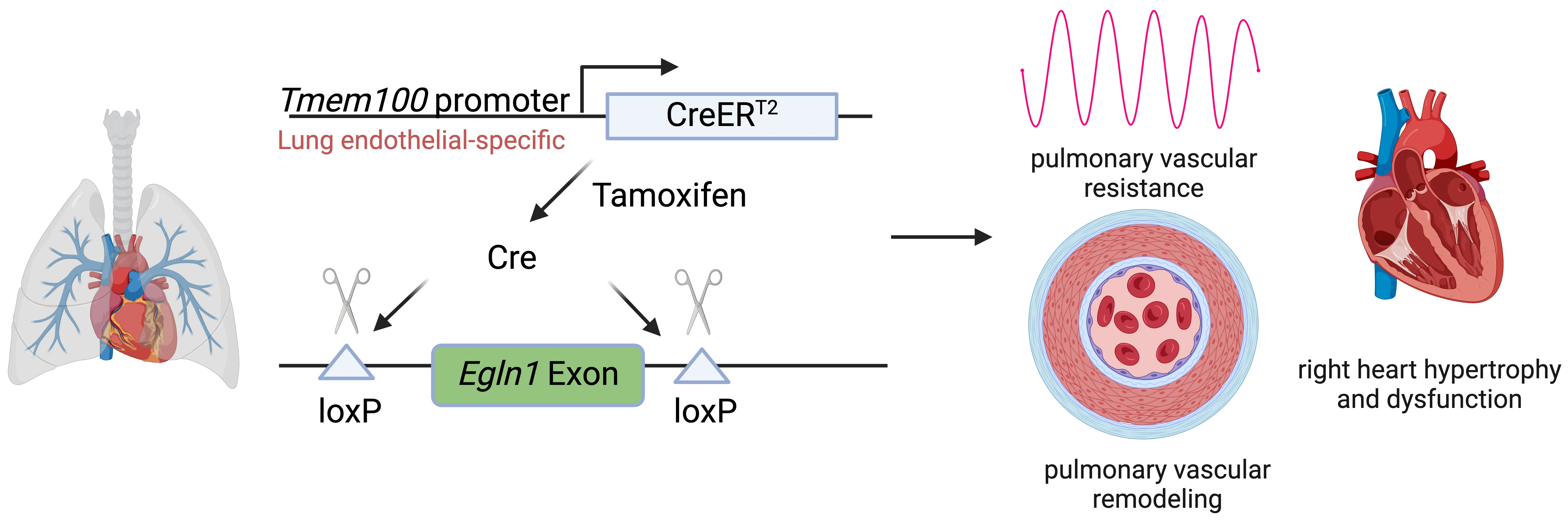
Pulmonary arterial hypertension (PAH) is a devastating disease characterized by high blood pressure in the pulmonary arteries, which can potentially lead to heart failure over time. Previously, our lab found that endothelia-specific knockout of Egln1, encoding prolyl 4-hydroxylase-2 (PHD2), induced spontaneous pulmonary hypertension (PH). Recently, we elucidated that Tmem100 is a lung-specific endothelial gene using Tmem100-CreERT2 mice. We hypothesize that lung endothelial-specific deletion of Egln1 could lead to the development of PH without affecting Egln1 gene expression in other organs. Tmem100-CreERT2 mice were crossed with Egln1flox/flox mice to generate Egln1f/f;Tmem100-CreERT2 (LiCKO) mice. Western blot and immunofluorescent staining were performed to verify the knockout efficacy of Egln1 in multiple organs of LiCKO mice. PH phenotypes, including hemodynamics, right heart size and function, pulmonary vascular remodeling, were evaluated by right heart catheterization and echocardiography measurements. Tamoxifen treatment induced Egln1 deletion in the lung endothelial cells (ECs) but not in other organs of adult LiCKO mice. LiCKO mice exhibited an increase in right ventricular systolic pressure (RVSP, ~35 mmHg) and right heart hypertrophy. Echocardiography measurements showed right heart hypertrophy, as well as cardiac and pulmonary arterial dysfunction. Pulmonary vascular remodeling, including increased pulmonary wall thickness and muscularization of distal pulmonary arterials, was enhanced in LiCKO mice compared to wild-type mice. Tmem100 promoter-mediated lung endothelial knockout of Egln1 in mice leads to development of spontaneous PH. LiCKO mice could serve as a novel mouse model for PH to study lung and other organ crosstalk.
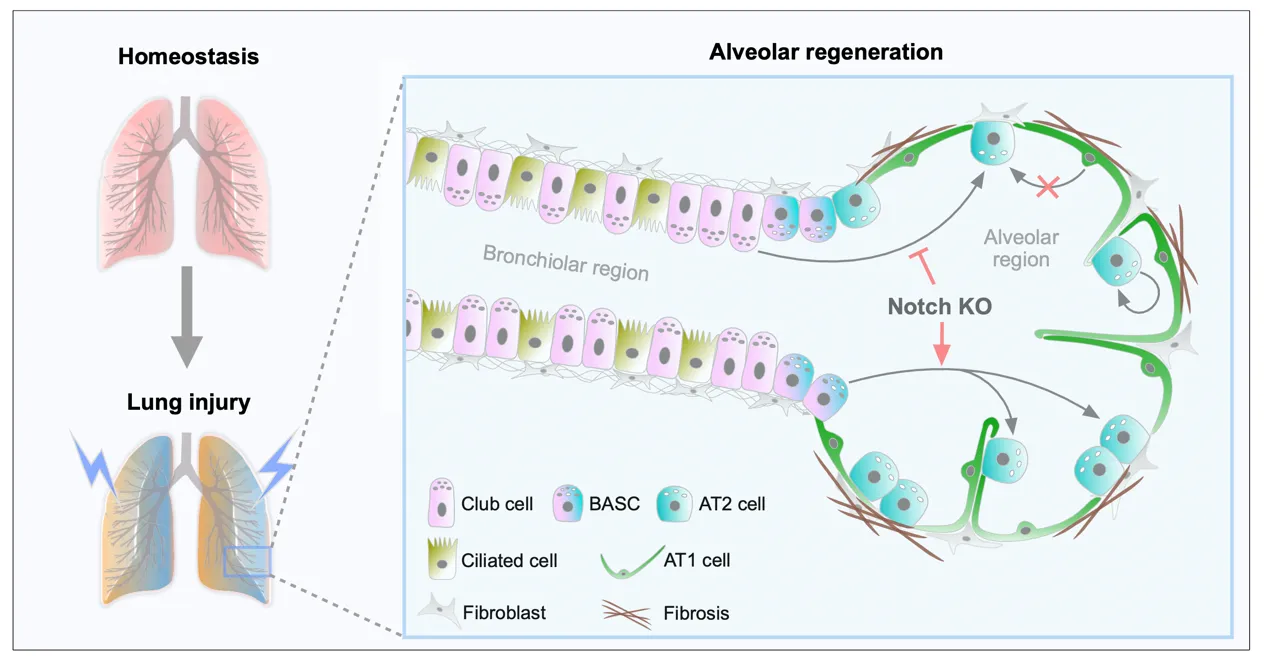
As alveolar epithelial stem cells, alveolar type II (AT2) cells play a pivotal role in sustaining alveolar homeostasis and facilitating repair processes. However, the sources of AT2 cell regeneration have remained contentious due to the non-specific labeling limitations of traditional single recombinase-based lineage tracing techniques. To address this issue, we employed dual recombination systems to develop more precise lineage tracing methodologies, effectively bypassing the shortcomings of conventional approaches and enabling specific labeling of lung epithelial cells. Our findings demonstrate that, following lung injury, regenerated AT2 cells do not originate from alveolar type I (AT1) cells, but instead derive from bronchiolar club cells and bronchioalveolar stem cells (BASCs), alongside the self-renewal of resident AT2 cells. Furthermore, we discovered that the transition of club cells and BASCs into AT2 cells is distinctly modulated by the Notch signaling pathway. This study not only provides novel insights into lung regeneration, but the innovative lineage tracing technology developed herein also holds promise as a technical support for research in diverse fields.
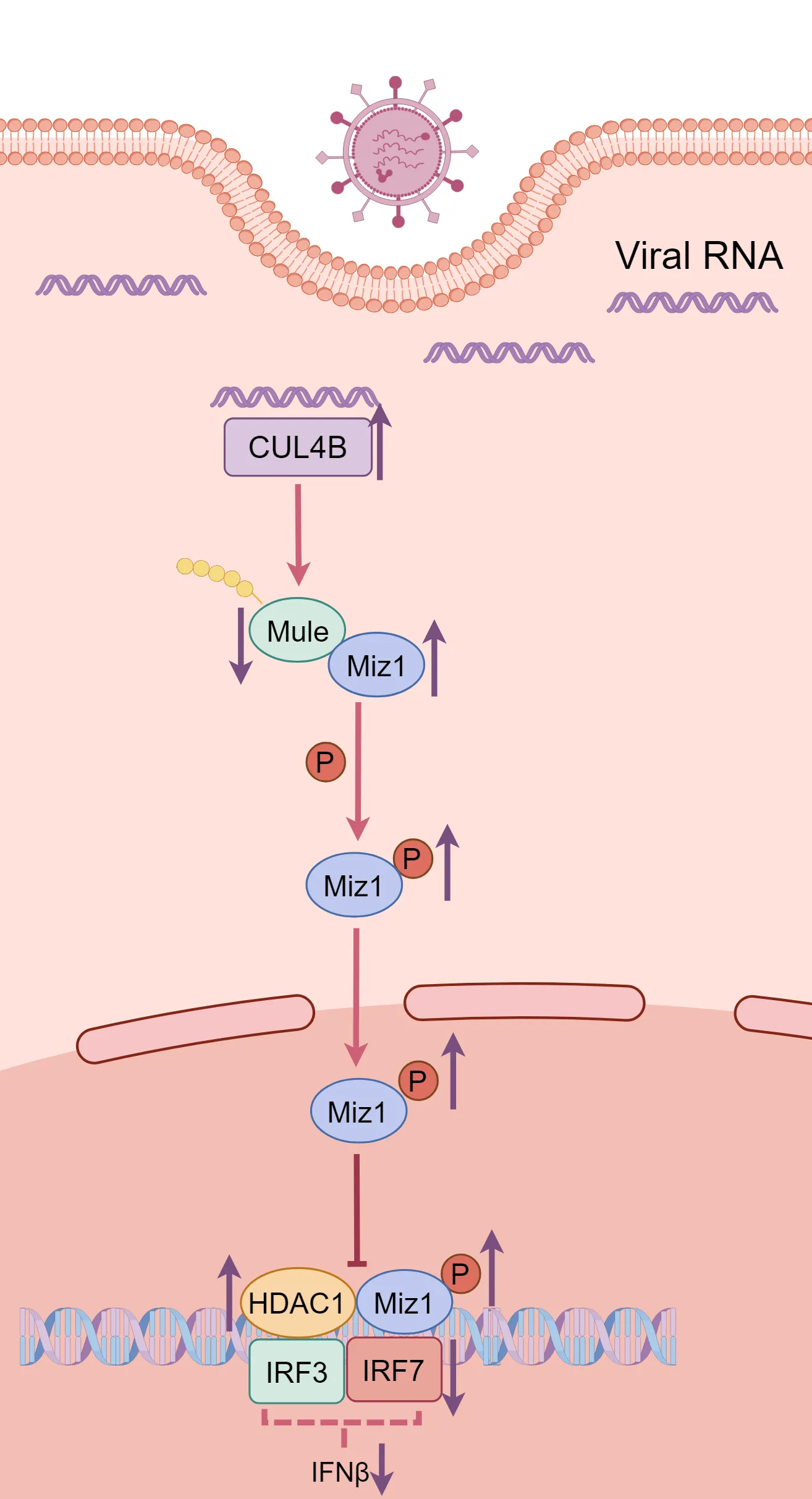
The ubiquitin system has been shown to play an important role in regulation of immune responses during viral infection. In a recent article published in Science Signaling, Wu and colleagues revealed that transcriptional factor Miz1 plays a pro-viral role in influenza A virus (IAV) infection by suppressing type I interferons (IFNs) production through recruiting HDAC1 to ifnb1 promoter. They show that a series of E3 ligases combinatorially regulates Miz1 ubiquitination and degradation and modulates IFNs production and viral replication.
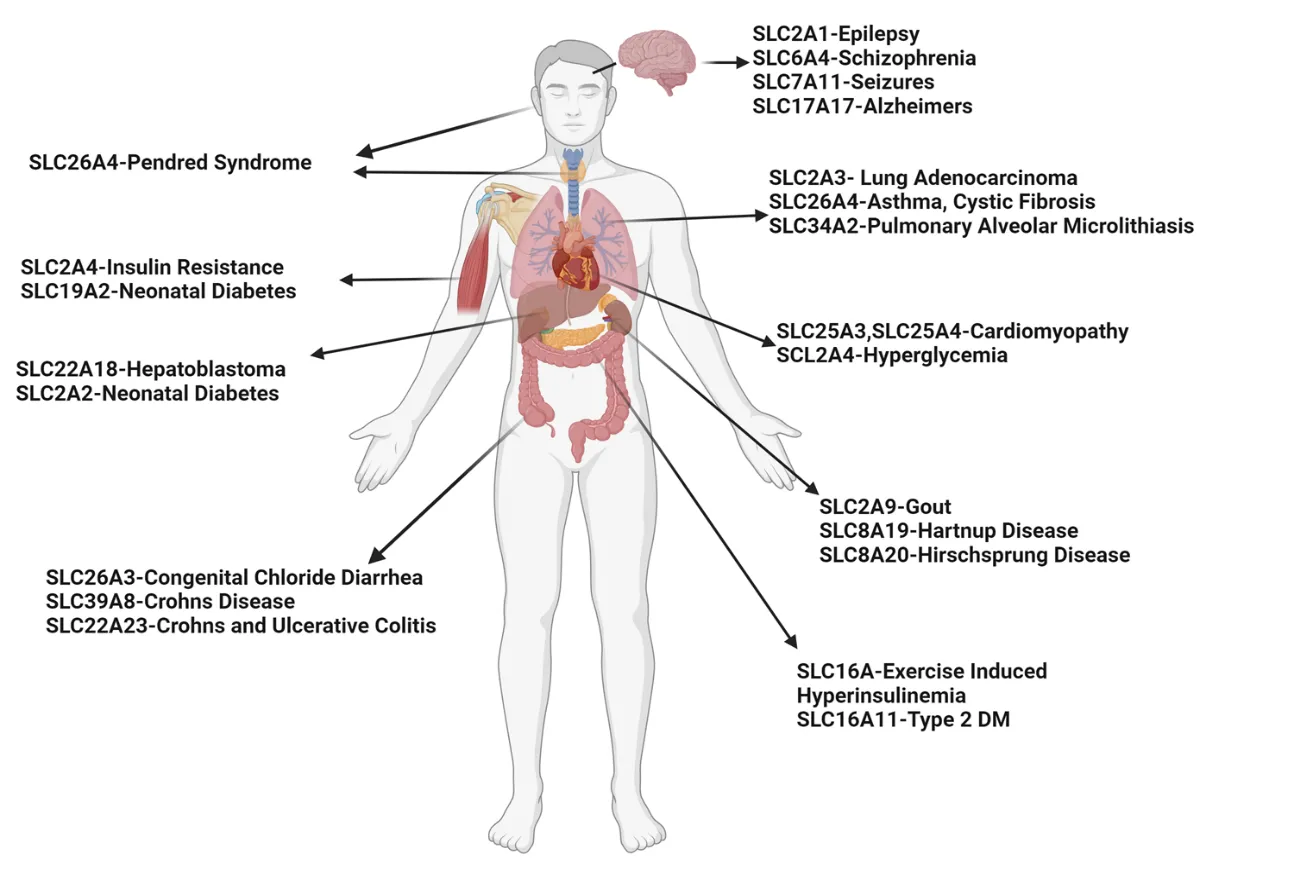
Asthma is a prevalent respiratory condition with multifaceted pathomechanisms, presenting challenges for therapeutic development. The SLC (Solute Carrier) gene family, encompassing diverse membrane transport proteins, plays pivotal roles in various human diseases by facilitating solute movement across biological membranes. These solutes include ions, sugars, amino acids, neurotransmitters, and drugs. Mutations in these ion channels have been associated with numerous disorders, underscoring the significance of SLC gene families in physiological processes. Among these, the SLC26A4 gene encodes pendrin, an anion exchange protein involved in transmembrane transport of chloride, iodide, and bicarbonate. Mutations in SLC26A4 are associated with Pendred syndrome. Elevated SLC26A4 expression has been linked to airway inflammation, hyperreactivity, and mucus production in asthma. Here, we review novel insights from SLC gene family members into the mechanisms of substrate transport and disease associations, with specific emphasis on SLC26A4. We explore triggers inducing SLC26A4 expression and its contributions to the pathogenesis of pulmonary diseases, particularly asthma. We summarize the inhibitors of SLC26A4 that have shown promise in the treatment of different phenotypes of diseases. While SLC26A4 inhibitors present potential treatments for asthma, further research is imperative to delineate their precise role in asthma pathogenesis and develop efficacious therapeutic strategies targeting this protein.