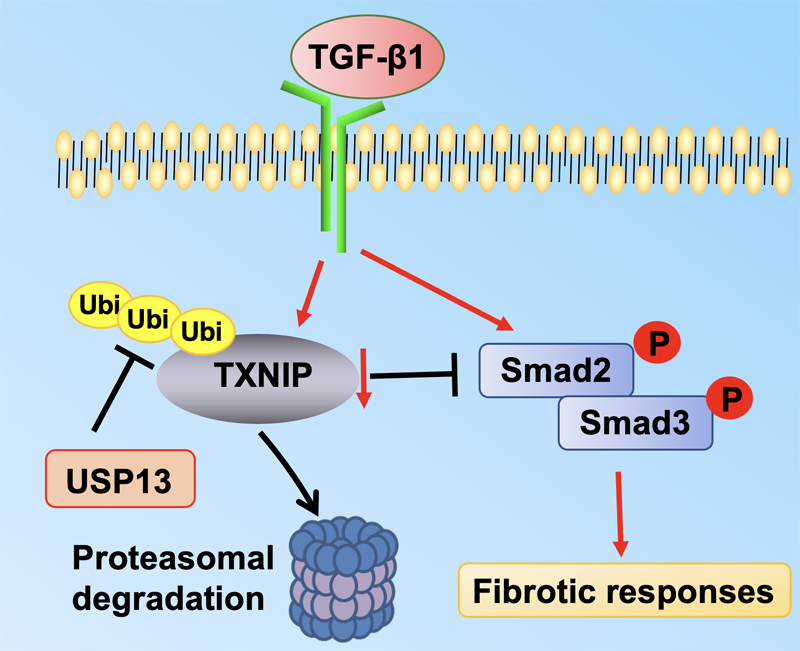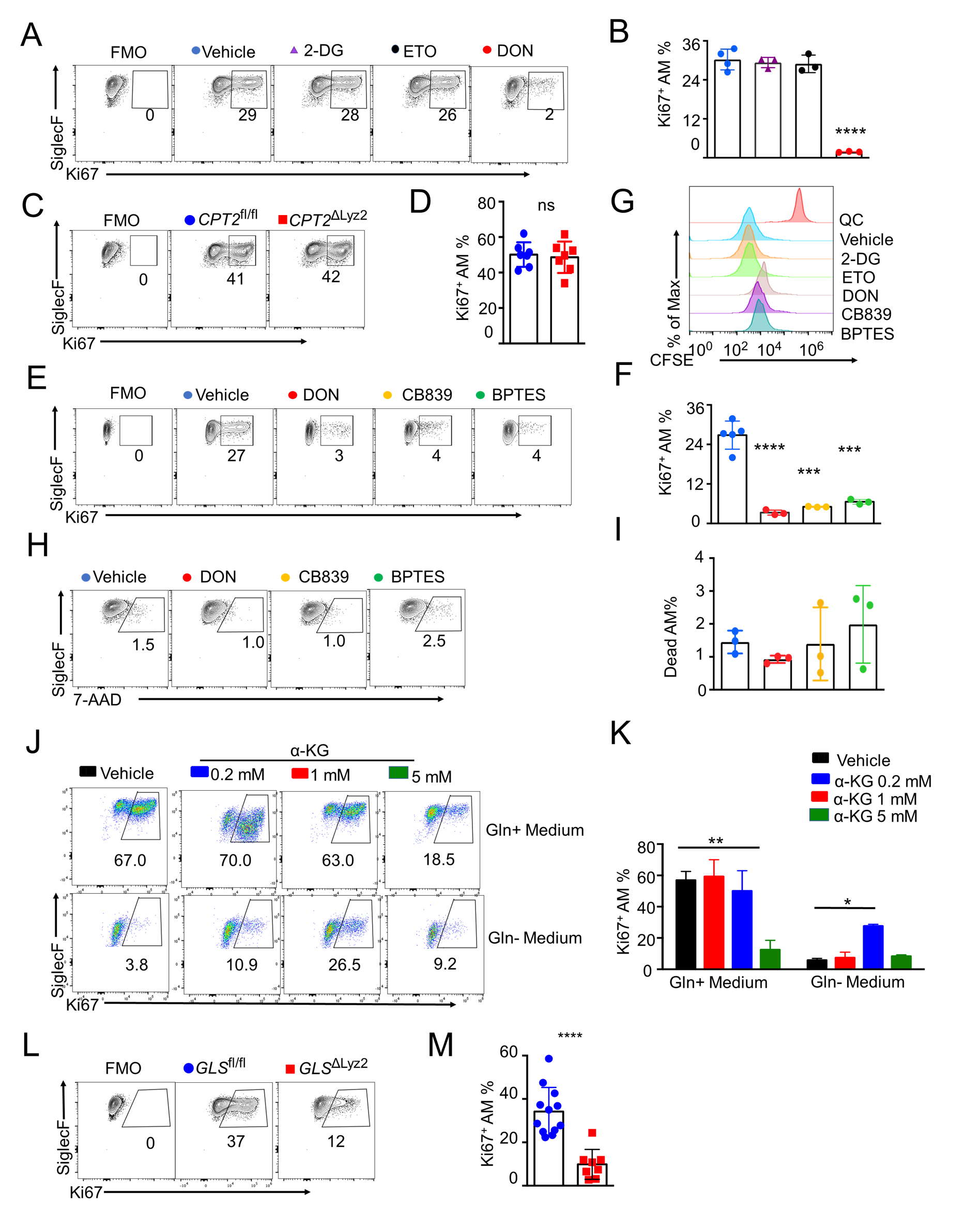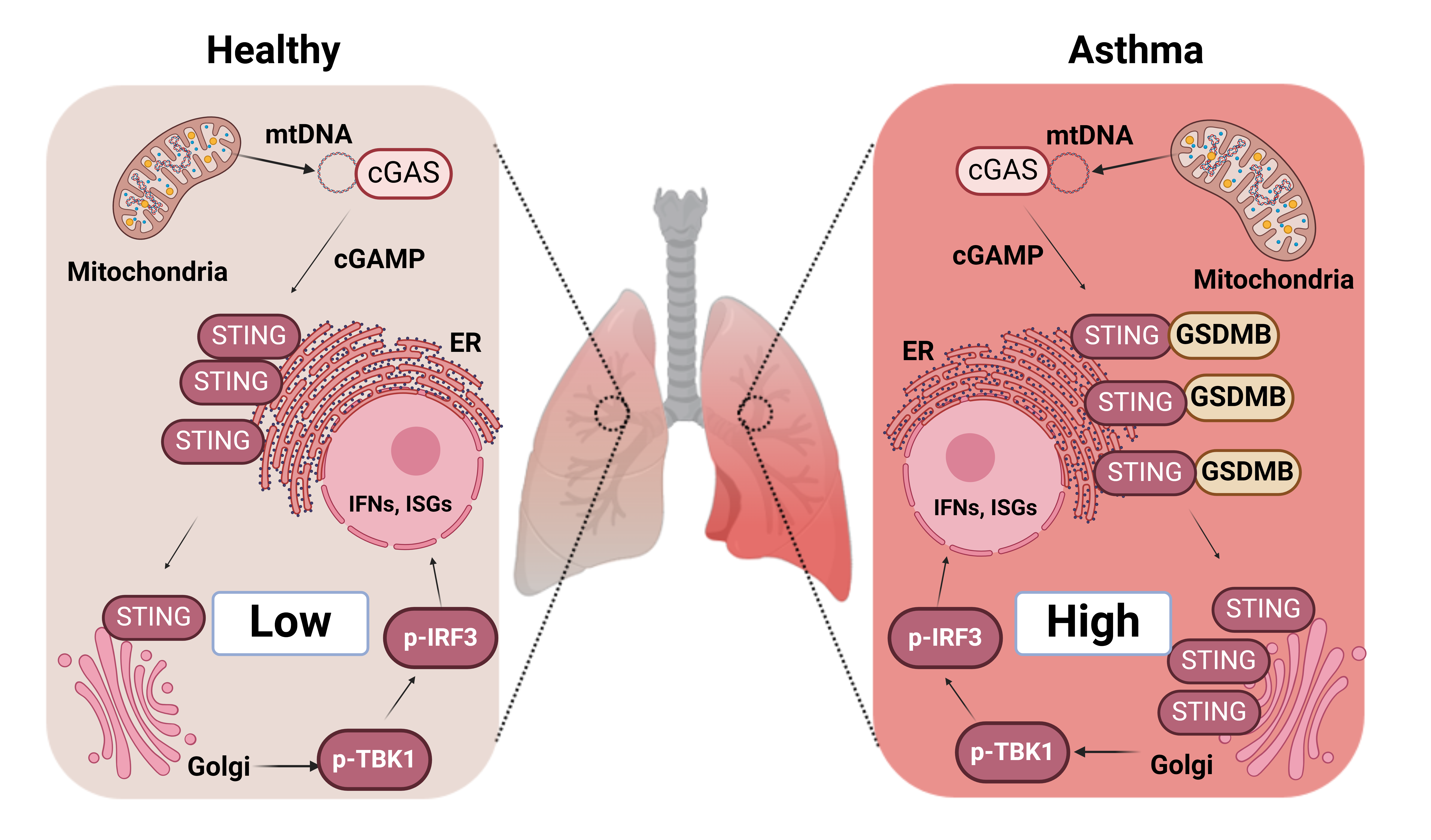
Issue 1, Volume 1 – 5 articles
Cover Story (View full-size image):
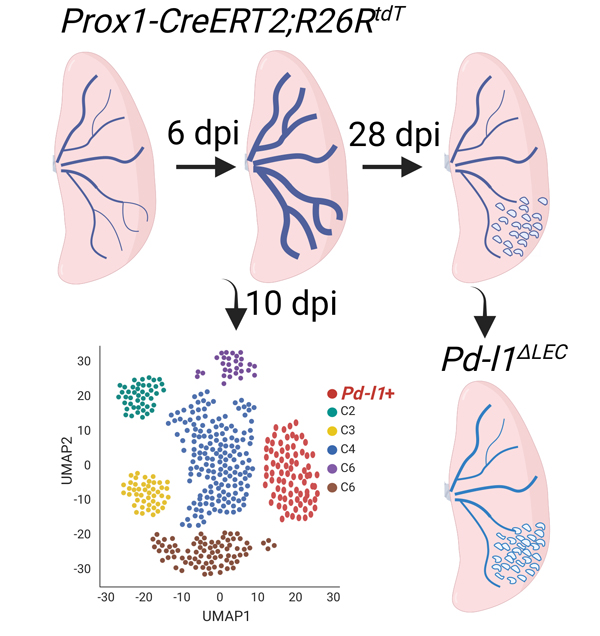
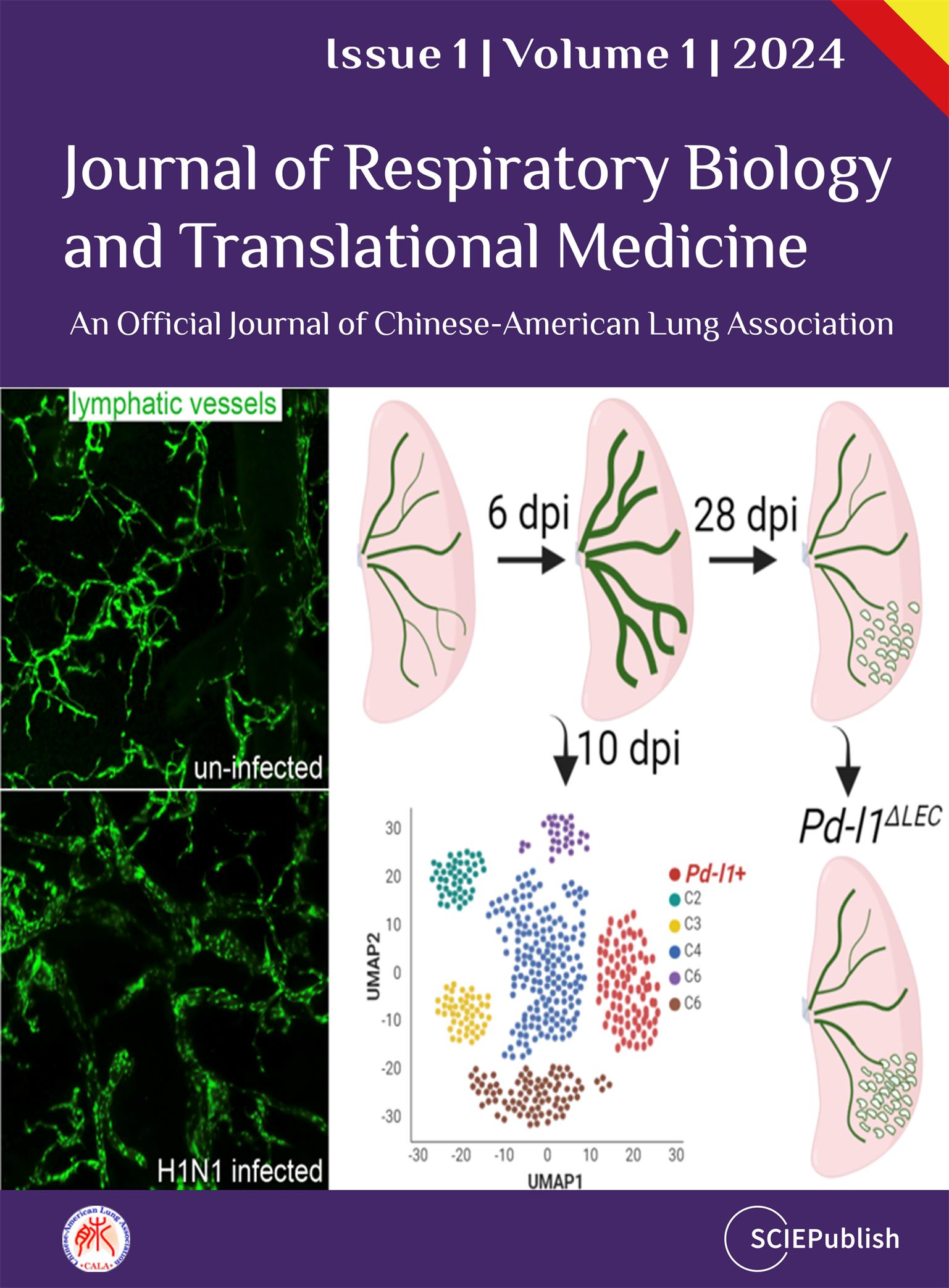
Tissue lymphatic vessels network plays critical roles in immune surveillance and tissue homeostasis in response to pathogen invasion, but how lymphatic system per se is remolded during infection is less understood. Here, we observed that influenza infection induces a significant increase of lymphatic vessel numbers in the lung, accompanied with extensive proliferation of lymphatic endothelial cells (LECs). Single-cell RNA sequencing illustrated the heterogeneity of LECs, identifying a novel PD-L1+ subpopulation that is present during viral infection but not at steady state. Specific deletion of Pd-l1 in LECs elevated the expansion of lymphatic vessel numbers during viral infection. Together these findings elucidate a dramatic expansion of lung lymphatic network in response to viral infection, and reveal a PD-L1+ LEC subpopulation that potentially modulates lymphatic vessel remolding.
View this paper