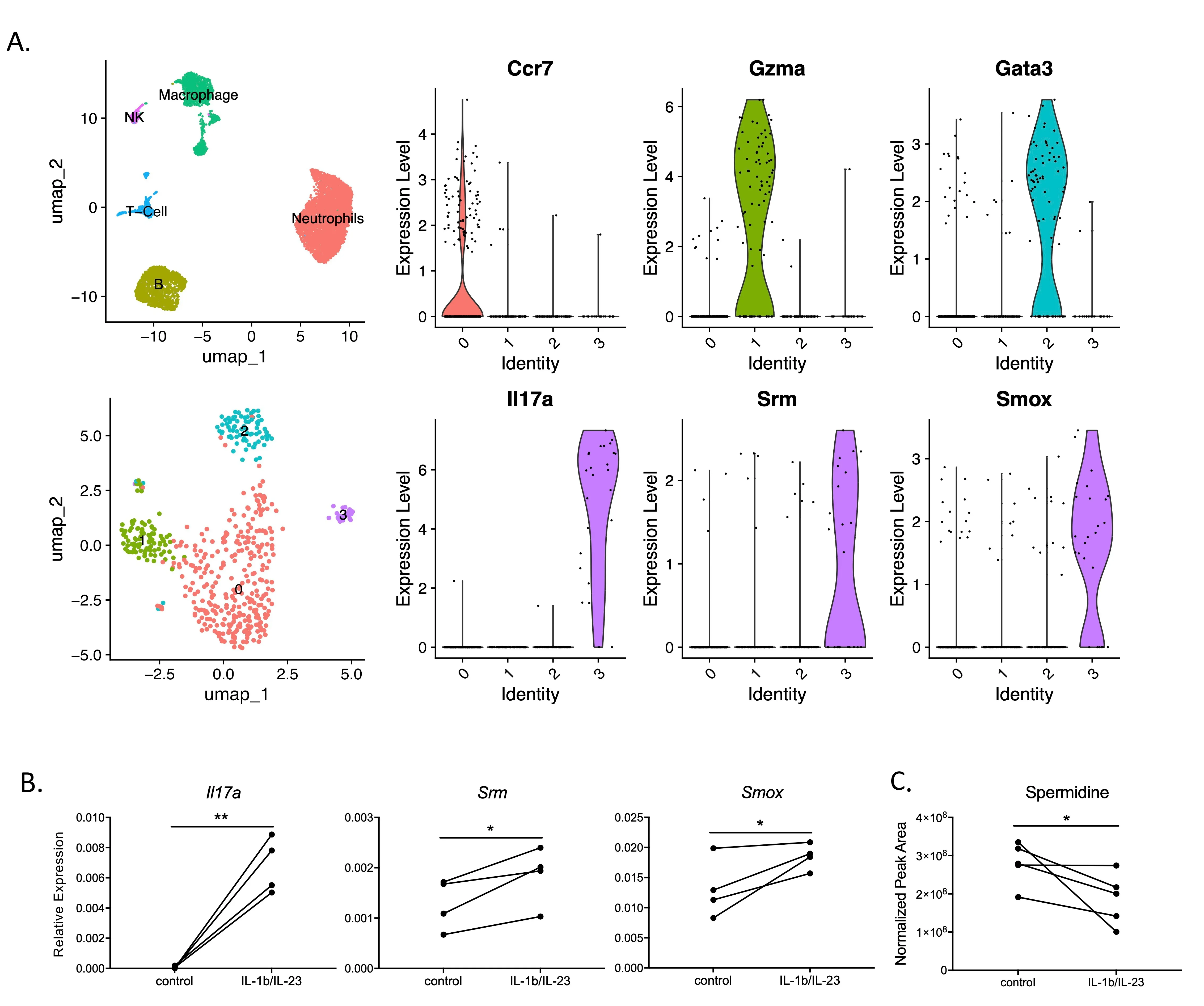
The activation of IL-17 signaling has been linked to the pathogenesis of many chronic, inflammatory lung diseases including cystic fibrosis. Through unbiased single-cell RNAseq screening, we found that IL-17+ T cells highly express Srm and Smox, which encode two key enzymes involved in spermidine synthesis, spermidine synthase and spermine oxidase respectively. Spermidine has been shown to reduce inflammation by regulating macrophage activation and balancing Th17/Treg differentiation; however its direct effects on Th17 cytokine production have not been carefully investigated. Here, using already differentiated Th17 cells from cultured mouse splenocytes, we found that exogenous spermidine directly inhibits IL-1β/IL-23-induced IL-17 production. Blockade of endogenous spermidine synthesis enhanced IL-17 production above native levels, further supporting the notion that spermidine is a direct regulator of cytokine secretion independent of differentiation. In vivo, spermidine alleviates lung inflammation in both Pseudomonas aeruginosa (PA) and LPS induced acute lung injury models. Further RNA-seq analysis suggests that spermidine suppression of Th17 cytokine production is mediated through its PRDX1-dependent antioxidant activity. Our data suggests that spermidine is a direct regulator of Type-17 T cell cytokine production and has potent anti-inflammatory effects against lung inflammation.
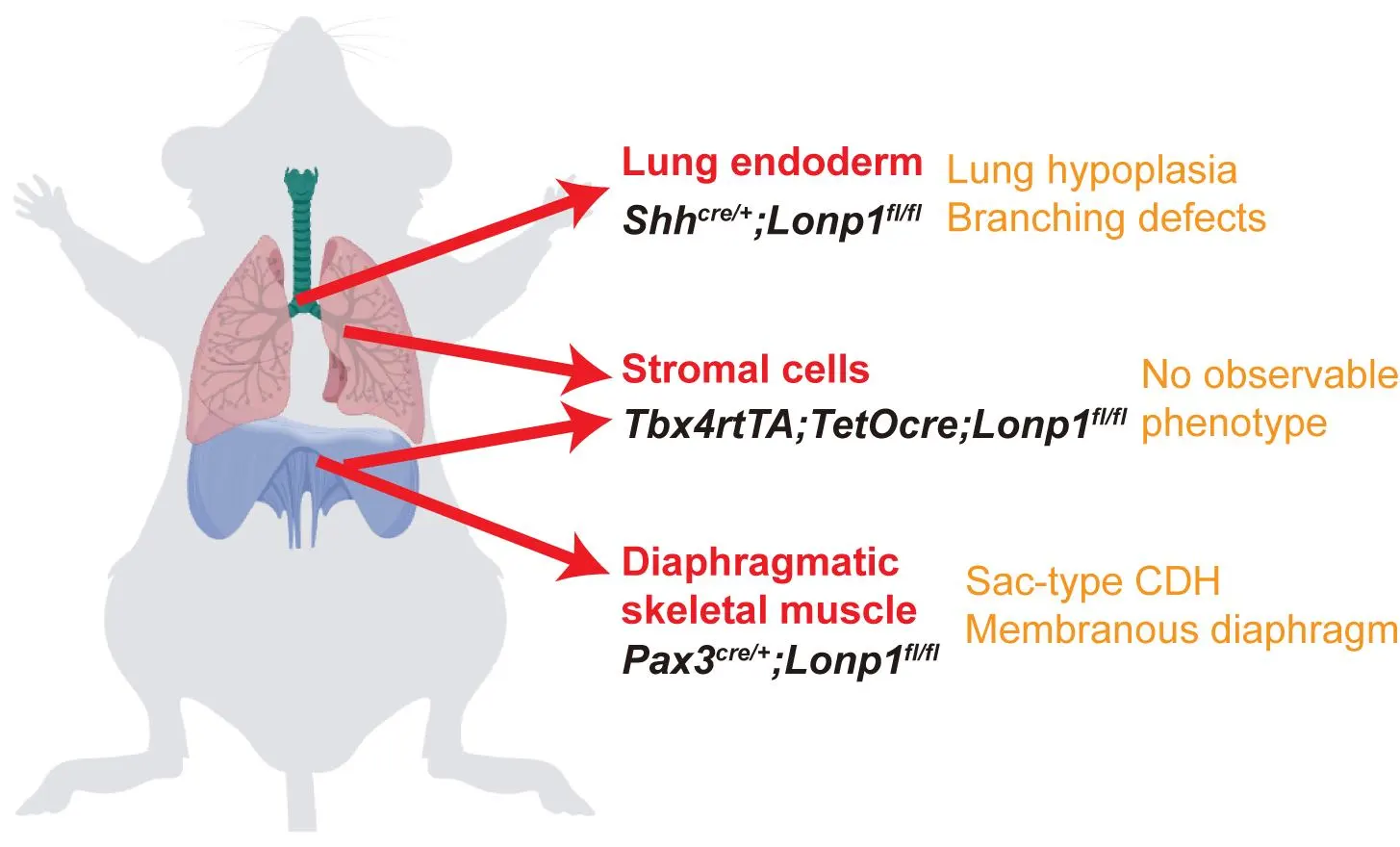
Congenital Diaphragmatic Hernia (CDH) is a rare neonatal disorder causing diaphragmatic defects and cardiopulmonary hypoplasia, traditionally attributed to mechanical compression from organ herniation. However, emerging evidence suggests genetic mutations may independently impair lung development, prompting debate over CDH etiology. Here, we investigated the requirement of mitochondrial function guarded by LON peptidase 1 (Lonp1), a CDH risk gene, in either diaphragm or lung development. Lonp1 loss in skeletal muscles of the diaphragm led to its thinning and membranization, recapitulating the pathology of sac-type CDH. On the other hand, lung-specific inactivation caused severe hypoplasia with defective branching morphogenesis, independent of diaphragm anomalies. Molecularly, Lonp1 disruption dysregulated key transcription factors and signaling pathways known to be critical for early lung development. Our findings here revealed that mitochondrial defects contribute to the pathogenesis of CDH in an organ and cell type specific manner, opening new avenues for drug and therapeutic development.
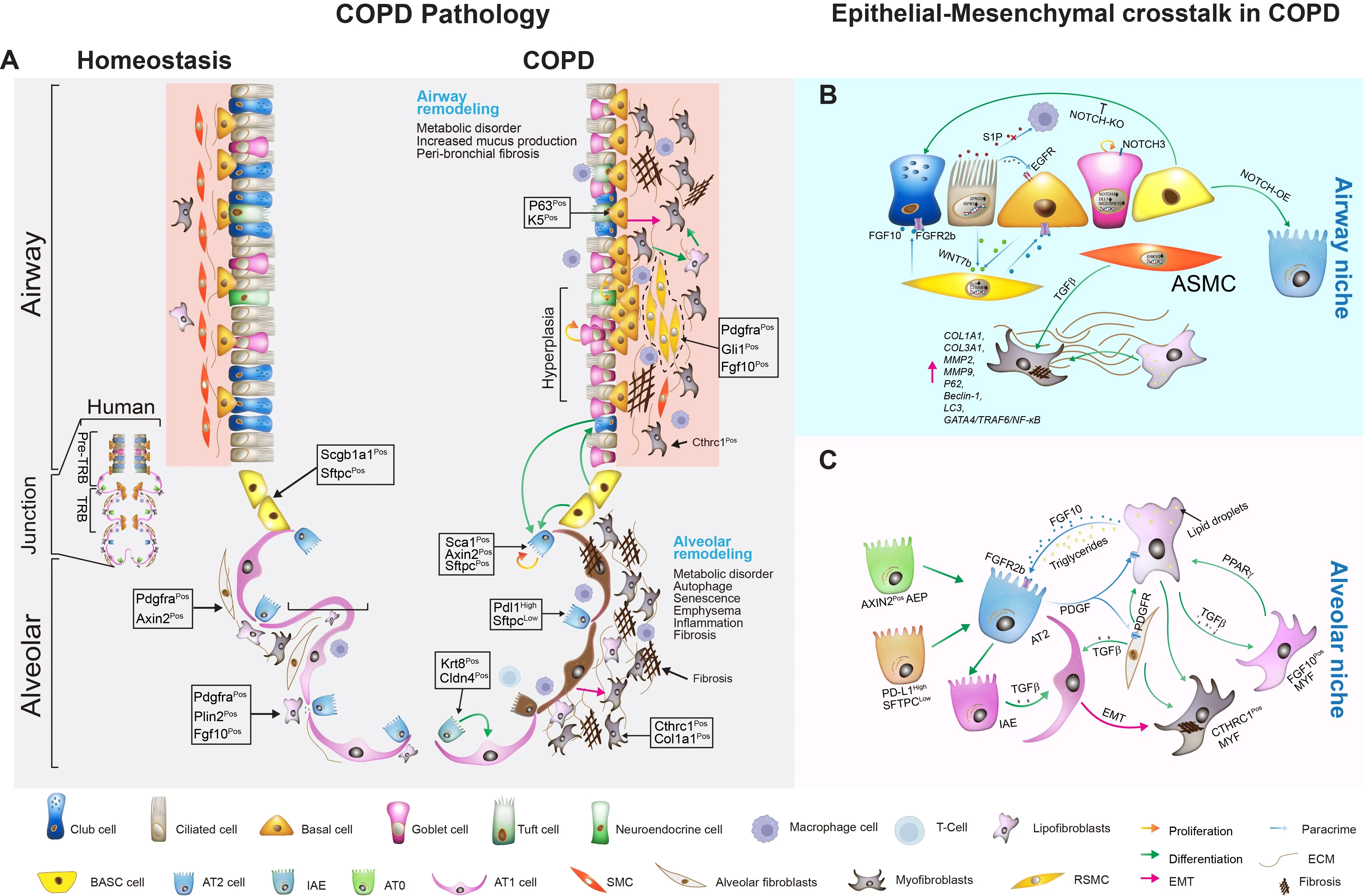
Chronic obstructive pulmonary disease (COPD) is a leading cause of global morbidity and mortality, characterized by progressive airway and alveolar remodeling. The disease pathogenesis is commonly driven by chronic environmental insults, leading to airway obstruction, emphysema, and chronic bronchitis. This review synthesizes emerging evidence that altered epithelial cell behavior and dysfunctional epithelial-mesenchymal interactions serve as pivotal drivers of COPD pathogenesis, orchestrating failed repair and structural degeneration. We detail how altered responses of airway (ciliated, club, basal, goblet) and alveolar (AT1 and AT2) epithelial cells lead to cellular senescence, metaplasia, defective regeneration, and barrier disruption, acting as primary instigators of pathogenesis. We also summarize current knowledge on the mechanisms of activation and pathogenic role of mesenchymal cells, which drive peribronchiolar fibrosis, alveolar destruction, and metabolic reprogramming, alongside the compromised reparative function of mesenchymal stem cells (MSCs). We emphasize how distinct mesenchymal niches (e.g., PDGFRαPos MANCs, FGF10Pos lipofibroblasts, SFRP1Pos fibroblasts) and distinct epithelial stem/progenitor subpopulations critically contribute to pathogenesis. Key signaling pathways—including FGF10/FGFR2b, WNT, Hippo, NOTCH, and TGF-β—mediate epithelial-mesenchymal transition (EMT), stem cell niche function, and structural remodeling. By dissecting how epithelial injury responses and mesenchymal niche failure collaboratively drive COPD progression, we identify actionable targets to disrupt pathogenesis and restore endogenous repair. We propose targeting EMT, including inhibiting EMT/fibrosis, promoting alveolar regeneration, MSC-based therapies, exosome-delivered biomolecules, and precision cell transplantation strategies, as promising future therapeutic strategies.