Found 26 results
Open Access
Article
06 February 2026Genetic Strategies for Labeling AT2 Cells in Murine Lung via Abca3 and Etv5-Driven Reporters
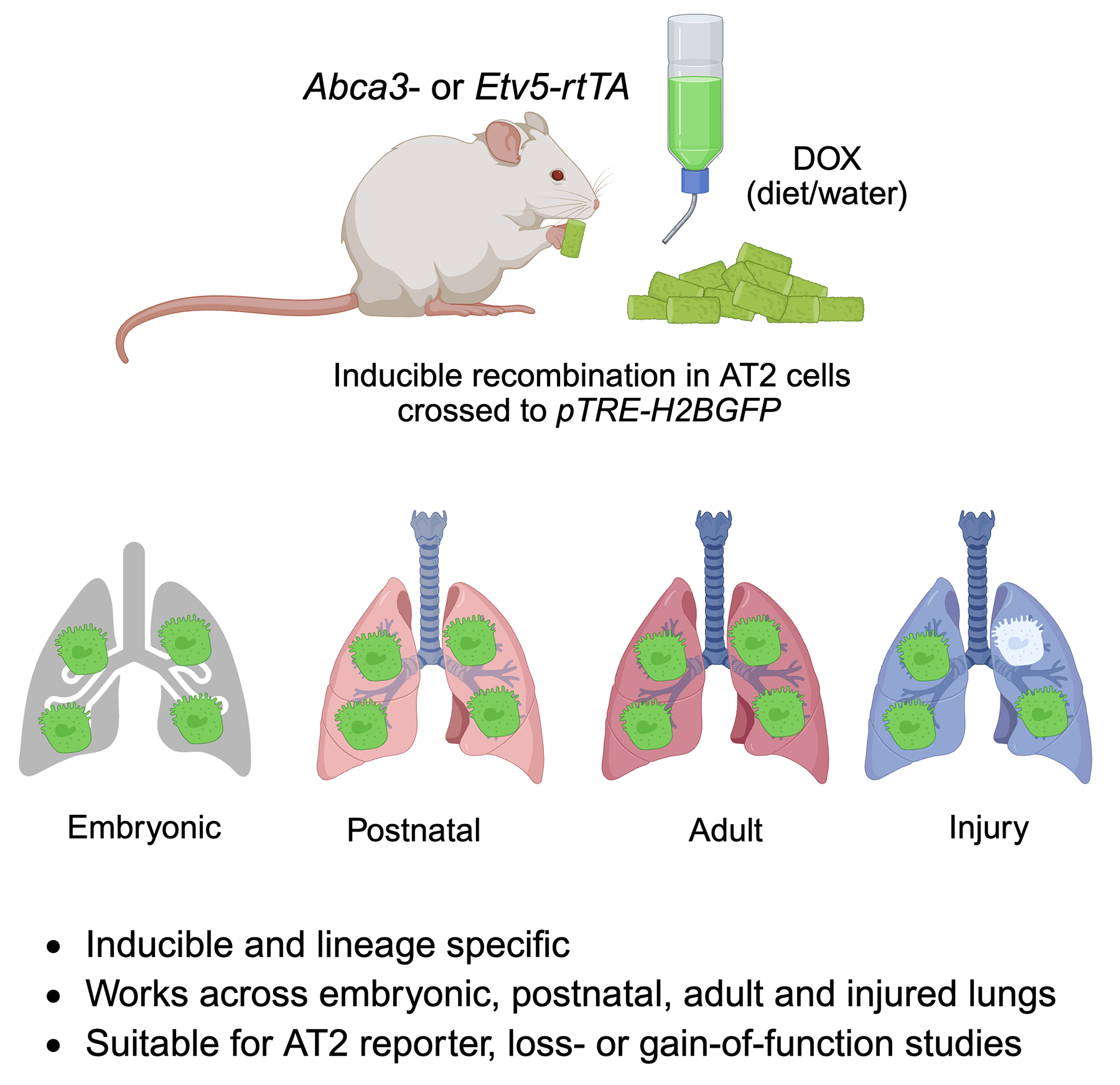
Precise labeling of alveolar type 2 (AT2) cells is essential for elucidating lung development and injury responses. In this study, we evaluated Abca3 and Etv5-based genetic strategies for labeling AT2 cells in murine models. Using targeted genetic approaches, we generated Abca3-rtTA and Etv5-rtTA knock-in mouse lines and crossed them with pTRE-H2BGFP to create inducible reporter models driven by Abca3 or Etv5. Labeling specificity and efficiency were assessed by flow cytometry and co-immunostaining. Our results show that both Abca3 and Etv5 strategies faithfully label AT2 cells across developmental stages and following lung injury. Comprehensive analyses confirmed the high specificity and efficiency of labeling. These Abca3- and Etv5-driven systems offer robust tools for investigating AT2 cell biology and pathology and may serve as effective drivers for tetO-mediated gene knockout or overexpression studies specifically in AT2 cells in mouse models.
Open Access
Commentary
06 February 2026Novel Therapeutic Targets of Endothelial Inflammation in Acute Lung Injury and Acute Respiratory Distress Syndrome
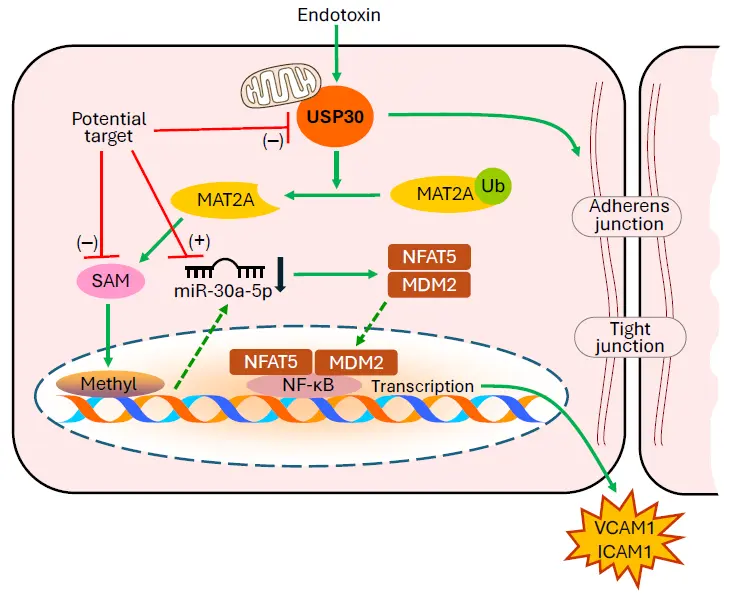
Lung microvascular endothelial inflammation and barrier dysfunction play critical roles in the pathogenesis of acute lung injury (ALI)/acute respiratory distress syndrome (ARDS). Despite recent scientific advances, the mortality of ALI/ARDS is still extremely high because the molecular mechanisms involved in ALI/ARDS remain unclear. In a recent issue of the journal Advanced Science, Baoyinna and colleagues reported that deubiquitinase USP30 induces lung microvascular inflammation and endothelial barrier disruption through the S-adenosylmethionine (SAM) cycle, DNA methylation, and miR-30a-5p down-regulation in ALI/ARDS. Their findings provide a strong rationale for targeting microRNAs, S-adenosylmethionine, DNA methylation, and deubiquitinating enzymes as potential therapeutic strategies for the treatment of ALI/ARDS.
Open Access
Perspective
28 January 2026The Double Face of Exosomes Derived from Mesenchymal Stromal Cells in Fibrotic Lung Diseases: Pathology Contribution or Treatment?
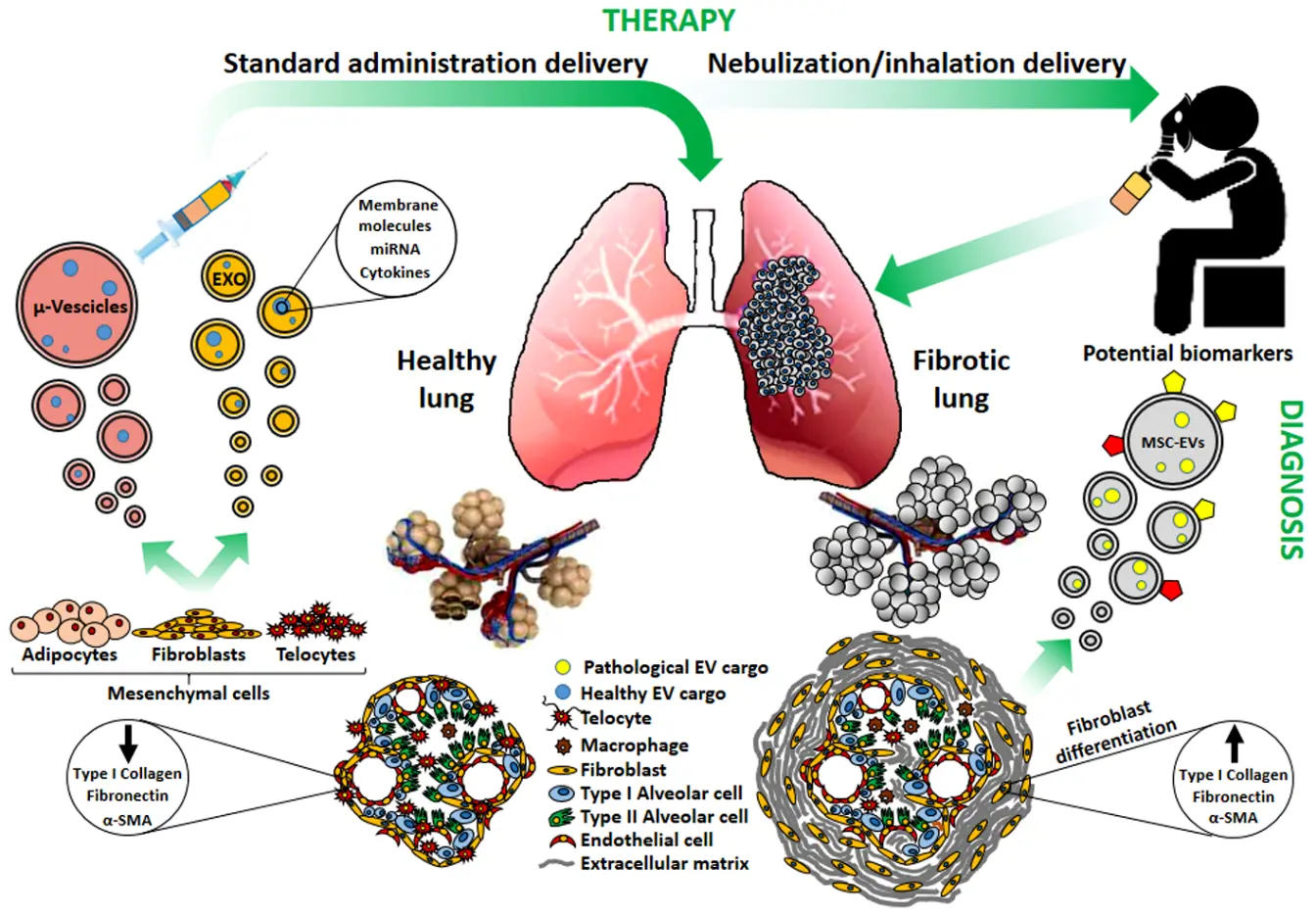
Several studies have attempted to clarify the role of exosomes and/or microvesicles derived from mesenchymal stromal cells (MSCs) (collectively indicated as extracellular vesicles: MSCs-EVs) in pulmonary fibrosis. Depending on their origin and on the micro-environmental context, MSCs-EVs may support or attenuate the fibrotic invasion of the lung, a hallmark of all Interstitial Lung Diseases (ILDs). Indeed, EVs have emerged as pivotal intercellular mediators and their potential diagnostic and therapeutic applications have been suggested. We aim here to elucidate the dual role of MSCs-derived exosomes and microvesicles: the contribution to pulmonary fibrosis progression, exerted by the MSCs-EVs originated from resident MSCs, or the potential therapeutic activity of those generated from healthy MSCs. Actually, MCSs-EVs appear as the frontiers of cell-free therapy and nano-medicine research in a great number of pre-clinical studies, but developments are needed to optimize and standardize their isolation, production and delivery. Interestingly, since the respiratory system directly communicates with the external environment, lung treatment could be approached by MSCs-EVs nebulization as a preferential administration route, integrating targeted pulmonary delivery with an enhanced patient’s compliance. Hence MSCs-EVs may contribute to ILD pathogenesis, display a potential as biomarkers, and still hold promise as therapeutic agents to reduce lung fibrosis. However further researches are needed to validate their clinical application.
Open Access
Article
22 January 2026Therapies Targeting Metabolic Pathways in Lung Fibrosis: Advances and Future Perspectives
Pulmonary fibrosis is a progressive lung disease associated with high morbidity and mortality. Increasing evidence indicates that metabolic reprogramming is a central driver of fibrogenesis. Multiple cell types in the fibrotic lung, including fibroblasts, alveolar epithelial type II (AEC2) cells, and macrophages, exhibit enhanced glycolysis, dysregulated lipid turnover, and altered amino acid utilization. These metabolic changes promote fibroblast activation, sustain ECM production, and impair epithelial repair. Recent studies have identified key regulatory pathways—such as hypoxia-inducible factor-1α(HIF-1α)-mediated glycolysis, aberrant fatty acid and cholesterol metabolism, and glutamine-dependent anabolic processes—that collectively shape the profibrotic microenvironment. Targeting these metabolic vulnerabilities has shown promising antifibrotic effects in preclinical studies, supporting glycolysis inhibitors, lipid-modulating agents, and amino acid metabolism blockers as potential therapeutic approaches. This review summarizes recent advances in glucose, lipid, and amino acid metabolic reprogramming in pulmonary fibrosis, with IPF discussed as a representative and well-studied subtype, and highlights emerging metabolic-targeted therapeutic strategies. Understanding cell-specific metabolic adaptations may provide new opportunities to develop effective interventions for pulmonary fibrosis, whereas most metabolic mechanisms are shared across fibrotic lung diseases.
Open Access
Article
31 December 2025Comparative Transcriptome Analyses Highlight Distinct Pathogenetic Mechanisms for Pleuropulmonary Blastoma and Congenital Pulmonary Airway Malformations
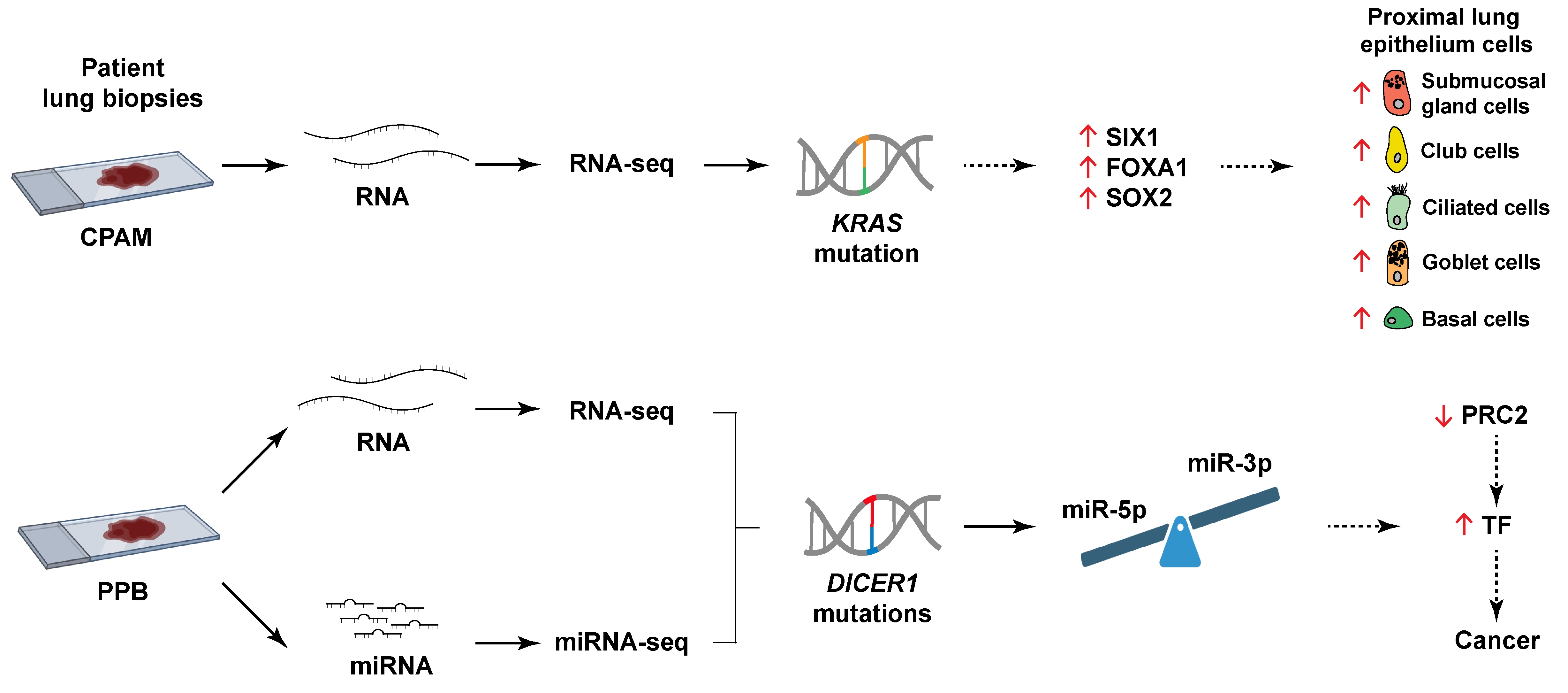
Pleuropulmonary blastoma (PPB) and congenital pulmonary airway malformations (CPAM) are two rare cystic lung diseases occurring in childhood. PPB can evolve from a low-grade epithelial cyst lesion to a high-grade sarcoma with a poor prognosis, whereas CPAM usually has a favorable non-tumorous outcome. Clinical similarities complicate diagnosis and may delay appropriate care. PPB is associated with DICER1 mutations that disturb miRNA biogenesis, altering the miRNA repertoire. Conversely, KRAS mutations are detected in CPAM, but their implication remains unclear. To decipher the mechanisms underlying these diseases, we undertook a comprehensive analysis of molecular variations in CPAM and PPB lung lesions using genome-wide RNA-seq and miRNA-seq assays. Each pathology displayed a distinct expression profile revealing a unique etiology. CPAM presented misexpression of bronchial epithelial markers correlating with KRAS mutation, while changes in expression of distal lung epithelial and mesenchymal markers were PPB-specific. PPB also exhibited abnormal gain of expression of developmental transcription factors, likely due to perturbed Polycomb Repressive Complex 2 (PRC2) activity. Overexpression of miR-323a-3p, which targets the PRC2 subunit EED, correlated with decreased EED expression. Together, these observations propose a PPB pathogenetic mechanism connecting DICER1 mutations and altered miRNA profile to defective PRC2 activity, misexpression of developmental transcription factors, and cancer.
Open Access
Editorial
30 December 2025Immune Escape Mechanisms in Non-Small Cell Lung Cancer: From Biological Complexity to Actionable Targets
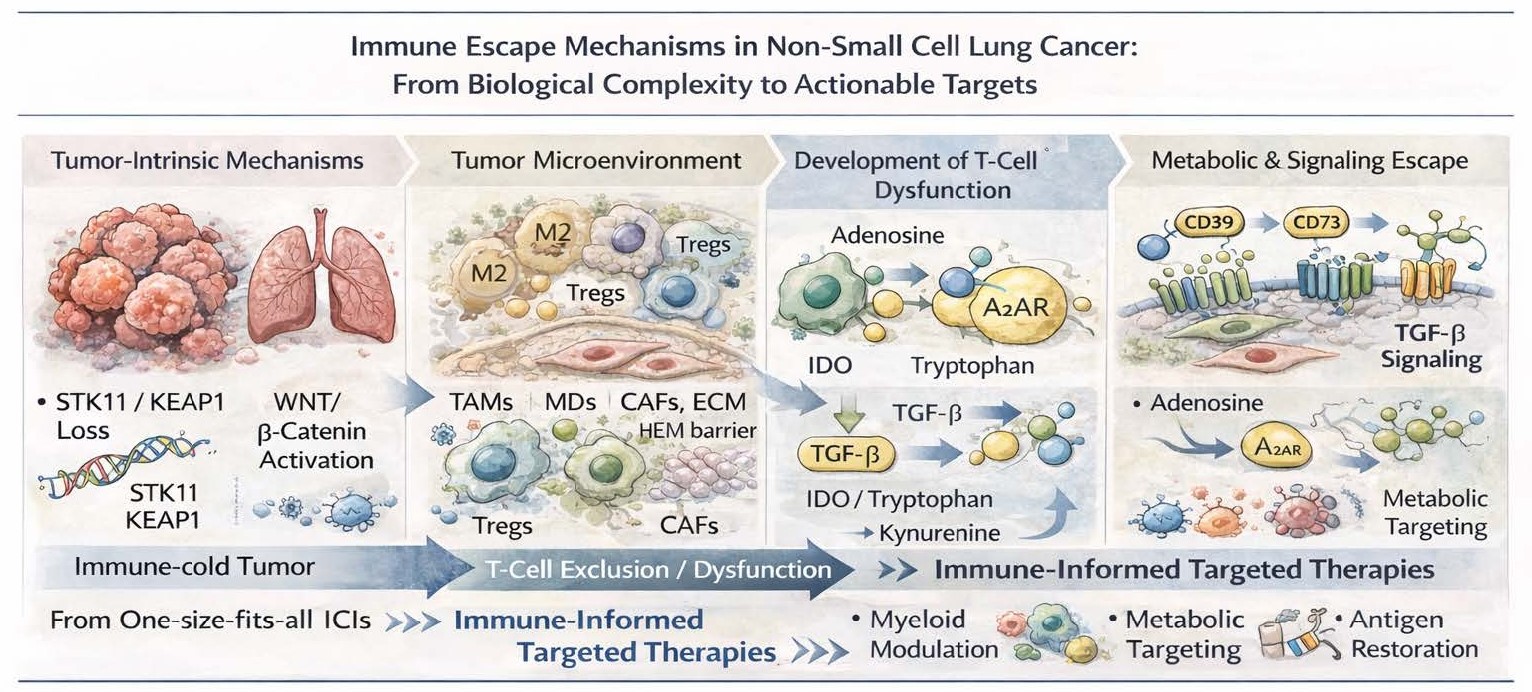
Despite significant progress in immune checkpoint inhibitors (ICIs) and targeted therapies, non-small cell lung cancer (NSCLC) continues to be associated with high rates of primary and acquired resistance. Although PD-1/PD-L1 blockade has revolutionized treatment, its clinical development has largely followed a one-size-fits-all approach, relying on limited biomarkers such as PD-L1 expression or tumor mutational burden. It is now increasingly clear that immune escape in NSCLC is orchestrated by a multifaceted, multilayered network of both tumor-intrinsic alterations and TME (tumor microenvironment)–driven mechanisms. The challenge has been to understand and to therapeutically exploit these immune escape pathways and this knowledge is now needed so that rather than embark on empirical combinations we can advance to rational, immune-informed targeted therapies.
Open Access
Review
10 December 2025Intracellular Chloride Channels: A Rising Target in Lung Disease Research
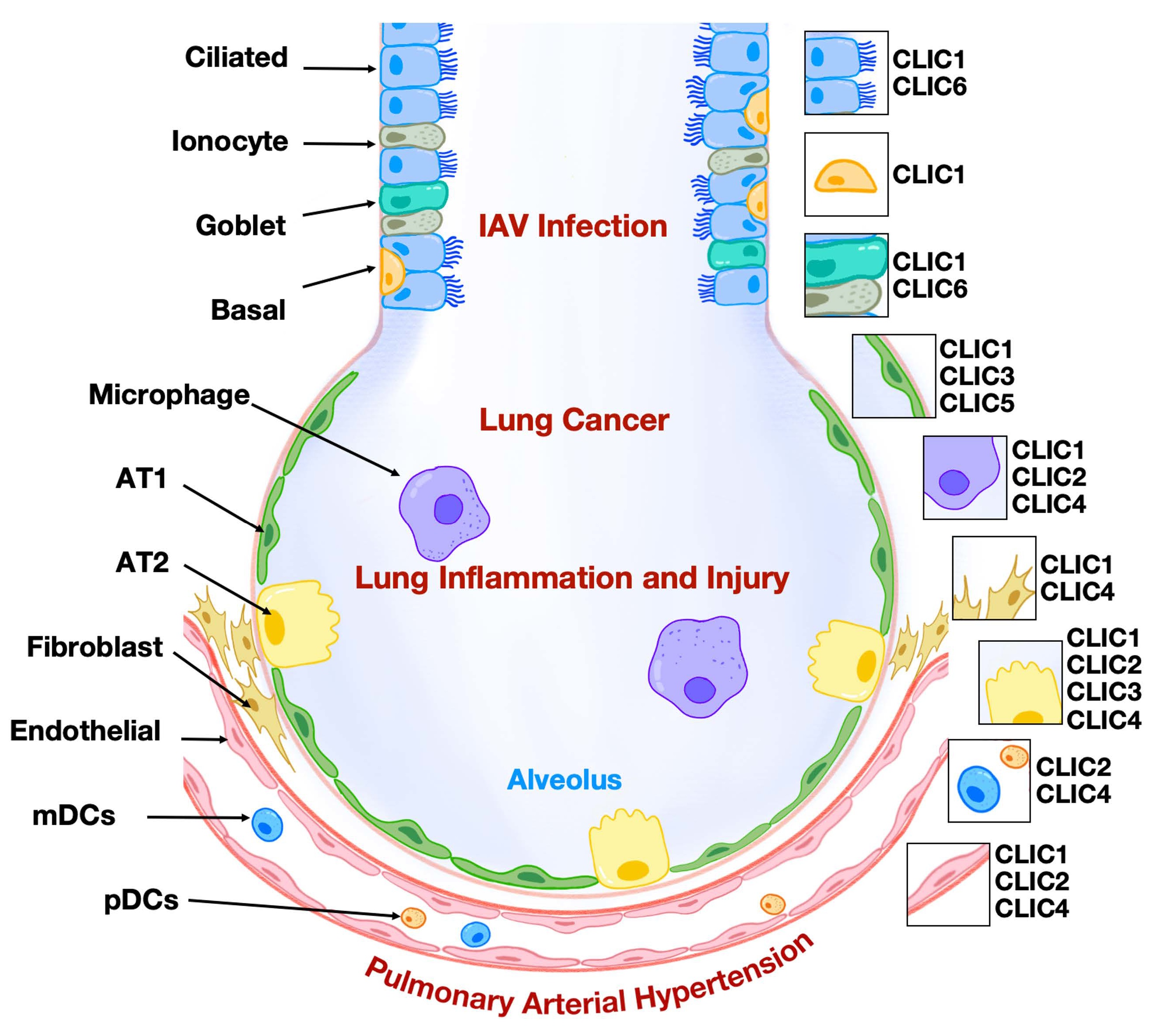
Chloride intracellular ion channels (CLICs) represent a relatively underexplored class of chloride channels and are included in a research initiative that focuses on druggable genes that have not been well studied yet. As a unique family, CLICs exist in membrane and soluble forms and play a role in regulating chloride flux and modulating various aspects of cellular biology. To date, six mammalian CLICs have been cloned and characterized at molecular and physiological levels. The respiratory system, responsible for gas exchange between the atmosphere and the human body, has recently been shown to express CLICs with functional relevance in lung pathophysiology, including lung carcinoma, inflammation, and endothelial dysfunction. Notably, the expression patterns of CLIC isoforms in lung cell types are distinct. Among them, CLIC1, CLIC3, and CLIC4 have been investigated more extensively, particularly in the context of lung cancer, inflammatory diseases, and pulmonary arterial hypertension. A deeper understanding of the role of CLICs in regulating lung cellular function may pave the way for developing novel therapeutic strategies to treat pulmonary disorders. In this review, we summarize the expression and functional roles of CLICs in lung pathophysiology, with particular emphasis on CLIC1, CLIC3, and CLIC4.
Open Access
Article
27 November 2025Lyz1-Expressing Alveolar Type II Cells Contribute to Lung Regeneration
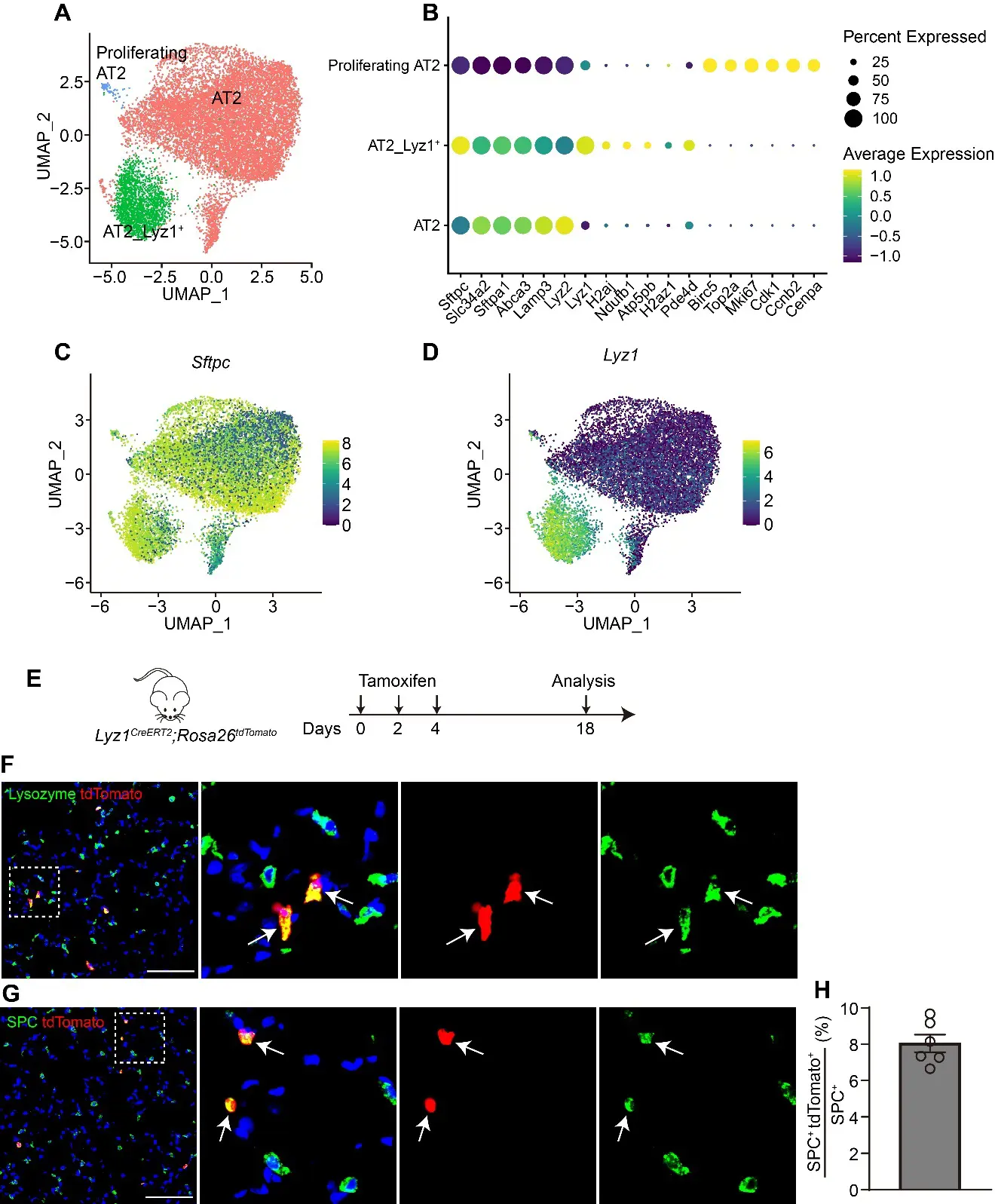
The alveolar units, composed of alveolar epithelial type II cells (AT2) and type I cells (AT1), are essential for efficient gas exchange. While AT2 cells are known to play critical roles in alveolar homeostasis and regeneration, the contribution of heterogeneous AT2 cells to lung repair remains poorly understood. Here, we identified a distinct AT2 subpopulation that exclusively expressed Lysozyme 1 (Lyz1) through single-cell RNA sequencing (scRNA-seq) analyses. Cell fate mapping revealed that the Lyz1CreERT2 mouse strain specifically labeled Lyz1-expressing AT2 cells in vivo at homeostasis. Following lung injury, Lyz1+ AT2 cells expanded and contributed to alveolar regeneration by generating both self-renewing AT2 cells and differentiating AT1 cells. We further observed the emergence of de novo Lyz1-expressing cells in the airways after lung injury. Additionally, Lyz1+ AT2 cells displayed significantly enhanced proliferative capacity compared with general bulk AT2 cells in 3D organoid cultures. These findings define Lyz1+ AT2 cells as a previously unrecognized progenitor population, expanding the paradigm of alveolar regeneration and providing insight into how epithelial diversity supports lung regeneration.
Open Access
Review
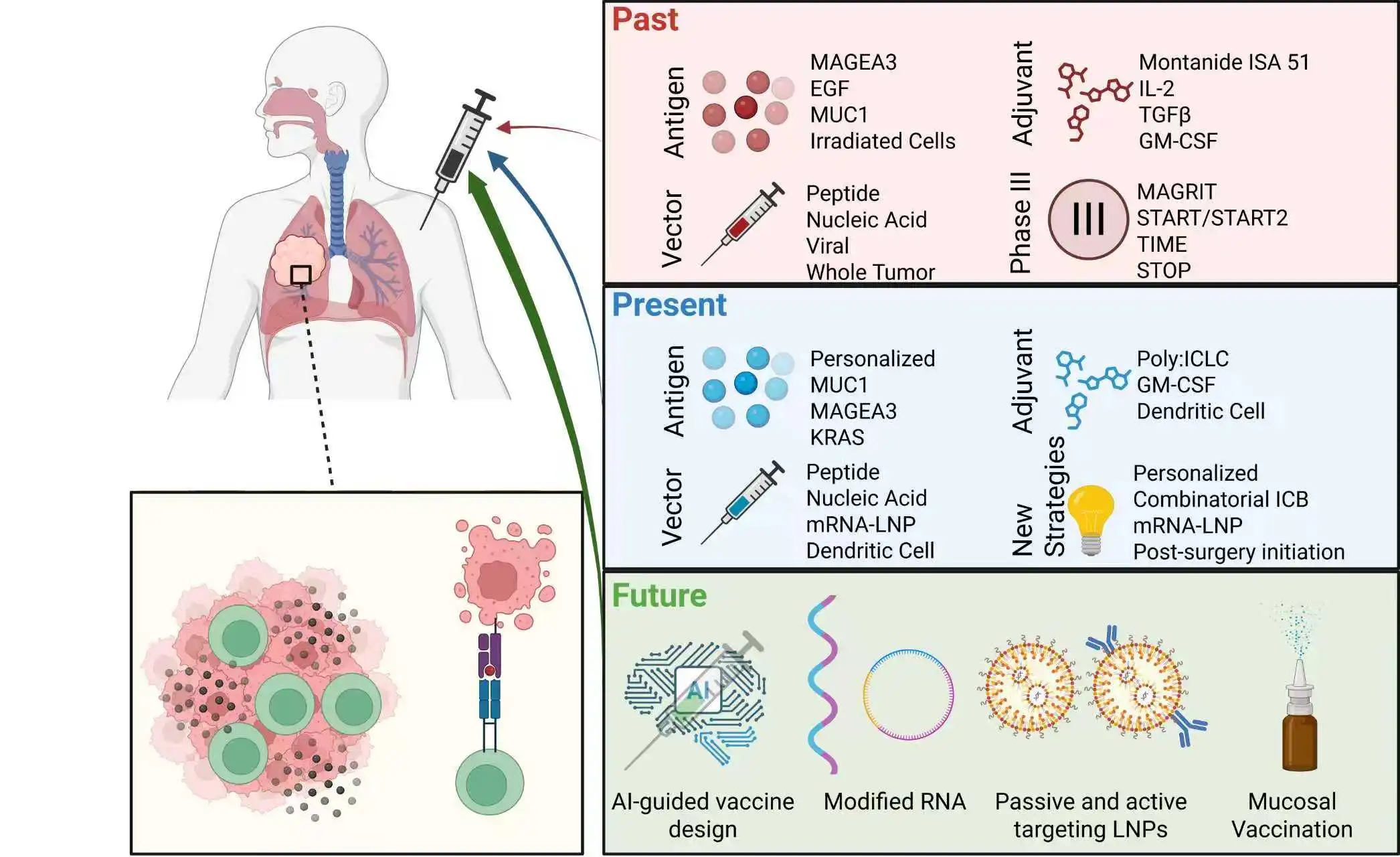
04 November 2025Therapeutic Vaccination in Lung Cancer: Past Attempts, Current Approaches and Future Promises
Lung cancer represents a significant burden on global health, necessitating the need for new and effective treatment strategies that expand our current therapeutic repertoire. Immunotherapy, namely immune checkpoint blockade (ICB), has revolutionized lung cancer therapy over the last decade by invigorating anti-tumor T cell responses to prolong survival and quality of life. However, not all patients benefit from ICB, emphasizing the need for novel immunotherapeutic strategies that engage other immune functionalities to offer synergy with already available therapies. There has been a longstanding interest in deploying lung cancer vaccines to generate or enhance tumor antigen-specific T cell responses for greater tumor control. Thus far, success has been limited to early-stage clinical trials, where safety, generation of antigen-specific T cell responses in blood sampling, and some patient benefits have been established. Moving forward, the establishment of widespread clinical success in large-scale trials is a necessity to bring lung cancer vaccines into the therapeutic arsenal. In this review, we examine the logic and mechanisms behind therapeutic lung cancer vaccines, before critically and iteratively examining past and current attempts in lung cancer vaccinology. We also look at early pre-clinical studies and outline the future for therapeutic lung cancer vaccines.
Open Access
Commentary
20 October 2025Sulfatide Inhibits Growth of Fibroblasts and Is a Potential Treatment against Fibrosis
Fibrosis of vital organs such as the lungs, liver, and kidneys is a serious condition without effective causal treatment. Here, we suggest the use of the sphingolipid sulfatide and its isoform C16, which we have found to inhibit the growth of fibroblasts. In the lungs, sulfatide can be easily administered via an inhalation spray. Alternatively, fenofibrate, an anti-cholesterol drug with no major side effects, may be used, as it enhances the body’s own production of sulfatide.
