Comparative Transcriptome Analyses Highlight Distinct Pathogenetic Mechanisms for Pleuropulmonary Blastoma and Congenital Pulmonary Airway Malformations

Comparative Transcriptome Analyses Highlight Distinct Pathogenetic Mechanisms for Pleuropulmonary Blastoma and Congenital Pulmonary Airway Malformations
Josselin Guéno
1
Nicolas Houde
2
Kim Landry-Truchon
2
Béatrice Frenette
1
William D. Foulkes
3
Christophe Delacourt
4,5
Lucie Jeannotte
1,2,*

Received: 30 October 2025 Revised: 03 December 2025 Accepted: 18 December 2025 Published: 31 December 2025

© 2025 The authors. This is an open access article under the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/).
Graphical Abstract
1. Introduction
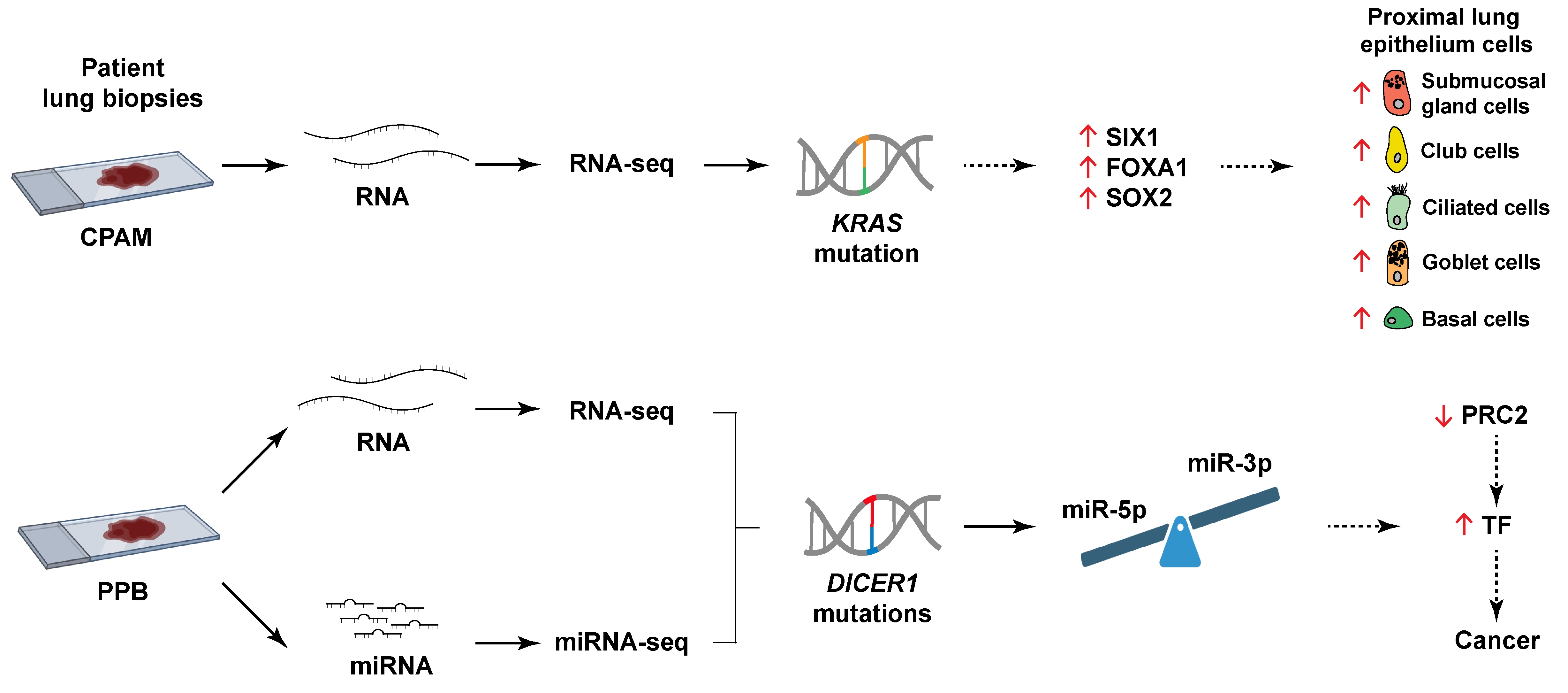
Congenital cystic lung malformations are uncommon. They encompass several types of rare pulmonary disorders, including congenital pulmonary airway malformation (CPAM), one of the most frequent pediatric lung lesions with an incidence of about 1:3000 births, and pleuropulmonary blastoma (PPB), a very rare lung neoplasm often confounded with CPAM [1,2]. In addition to sharing cystic morphological features, PPB and CPAM are thought to originate from developmental defects that impair the lung morphogenesis program. However, the molecular mechanisms underlying the pathogenesis of these clinically significant conditions remain largely elusive.
PPB is the most common pediatric primary lung neoplasm [3]. It manifests in early childhood and progresses through three distinct clinicopathological subtypes. Type I PPB consists of low-grade epithelial cystic lesions that are described to contain rhabdomyoblasts and immature cartilage cells within the cyst walls [4,5]. Overgrowth of these primitive mesenchymal cells may result in a mixed cystic and solid neoplasm (Type II), which can further evolve into a purely solid high-grade sarcoma with poor prognosis (Type III) [4]. Early diagnosis of Type I PPB is associated with improved outcomes, whereas delayed detection or failure to resect cystic PPB worsens prognosis, as Type I lesions may progress into aggressive Type III tumors within a few years [6,7].
PPB is the hallmark tumor of the DICER1 syndrome (also known as DICER1-related tumor predisposition; MONDO: 0100216), an autosomal dominant condition predisposing individuals to multiple cancers [8]. The DICER1 gene encodes a ribonuclease (RNase) III essential for processing pre-microRNAs (miRNAs) into mature, functional miRNAs [9]. DICER1 syndrome typically involves a germline loss-of-function mutation in one allele of the DICER1 gene, coupled with an acquired somatic missense mutation in the second allele [10]. These tumor-specific mutations usually occur in five hotspots, codons E1705, D1709, G1809, D1810, and E1813, affecting critical amino acids within the metal-ion binding catalytic cleft of the RNase IIIb domain of the DICER1 protein [11,12]. The resulting neomorphic DICER1 function disturbs miRNA biogenesis, selectively impairing miRNA-5p processing and favoring miRNA-3p production from the double-stranded precursors [13,14]. These quantitative and qualitative changes in the miRNA expression panel ensuing from DICER1 mutations may thus perturb target gene expression and contribute to PPB pathogenesis.
Diagnosing PPB remains challenging due to its overlap of clinical and histopathological features with other cystic lung malformations. Type I PPB is particularly prone to misidentification as CPAM, which generally has a more favorable outcome [2]. Accurate recognition of PPB at the Type I stage is therefore critical, as it represents the most curable phase of the disease.
A complex set of classification systems and terminologies has emerged over the years to describe CPAM. Historically, CPAM was divided into five subtypes based on cyst location along the tracheobronchial tree, cyst size, and histological characteristics [1,15]. However, cystic Type I PPB and Type IV CPAM are now considered to be the same pathological entity [16]. The widespread use of prenatal ultrasound has also prompted to revisit CPAM and to group them into two broad categories: Type 1, characterized by larger cysts and frequent mucous cell clusters, and Type 2 featuring smaller cysts [17,18]. However, this classification remains debated, as several CPAM cases exhibit overlapping features of both types [2,18,19].
Despite morphologic similarities, notable distinctions between PPB and CPAM have been documented. PPB cases frequently present an alveolar-type epithelial lining, whereas CPAM cysts are predominantly bordered by a ciliated respiratory bronchial-type epithelium [1,15,17]. Moreover, CPAM lesions very rarely contain primitive mesenchymal cells or cartilage nodules within their septa [4]. Finally, proliferation in CPAM samples is comparable to that observed in controls, in contrast to the increased mesenchymal proliferation commonly seen in PPB lung tissues, which likely reflects neoplastic expansion and disease progression [20,21]. While PPB is associated with DICER1 pathogenic variants, the molecular etiology of CPAM remains unresolved. Nevertheless, mutations in genes such as KRAS, FGFR2, and TP53 have been identified in CPAM cases [19,22,23].
Comparative expression analysis of candidate genes in PPB and CPAM lung biopsies has revealed differences in gene expression in PPB that were not observed in CPAM, indicating distinct molecular signatures [21,24]. Among the genes differentially expressed in PPB, several encode transcription factors (TF) and signaling molecules important for lung development [21,25]. Furthermore, misexpression of genes associated with epithelial-mesenchymal transition (EMT) suggests that this process may contribute to PPB pathogenesis. Altered gene expression is expected in PPB due to biallelic compound mutations in the DICER1 gene and the resulting dysregulation of miRNA biogenesis. For instance, miR-125a-3p negatively regulates lung branching as well as the expression of the ubiquitous TF YY1. It is also abnormally overexpressed in PPB lung tissues and in patient sera, a possible explanation for the decreased expression of the YY1 protein in PPB biopsies [21,24,26]. However, despite the identification of candidate genes, the sequence of molecular events driving PPB development remains to be elucidated.
To gain a comprehensive view of the molecular impact occurring in PPB and CPAM, we performed a transcriptome analysis of lesions at the genome-wide level, leveraging valuable patient lung specimens for these two rare lung diseases. RNA-seq data revealed that each pathology exhibits a unique expression profile, indicating distinctive molecular mechanisms. Analysis of the miRNA repertoire in PPB lung samples confirmed the predominance of miRNA-3p over miRNA-5p. It also unveiled the potential role of specific dysregulated miRNAs in the misexpression of components of the Polycomb Repressive Complex 2 (PRC2), with repercussions on developmental TF genes and cancer.
2. Materials and Methods
2.1. Human Tissues
This study was conducted using anonymized specimens from control, CPAM, and PPB patients from the Department of Anatomo-Pathology of Hôpital Necker-Enfants Malades (Paris, France) and from McGill University/Segal Cancer Centre (Montréal, QC, Canada). CCAM and PPB tissue samples were obtained from surgical resections of the respective malformations. Decisions regarding whether to perform surgery and its timing were made at the discretion of the participating centers. All procedures were conducted according to the standard protocols of these centers, each of which is a certified or well-established pediatric surgery facility. Tissue samples for the malformation site were preserved for research purposes, either as frozen or formalin-fixed paraffin-embedded (FFPE) specimens. Informed consent was obtained from all subjects, and all clinical investigations were conducted in accordance with the principles expressed in the Declaration of Helsinki. The procedures were approved by the respective institutional ethics committee and by the CHU de Québec-Université Laval research ethics committee (project MP-20-2020-4809). When possible, the sex of specimens was determined by RNA-seq or RT-qPCR analyses of the Y-specific lung-expressed gene, UTY [27]. Characteristics of patients are summarized in Table S1.
2.2. Histology, Immunohistochemistry (IHC), and Immunofluorescence (IF) Analyses
Alcian Blue staining, for the detection of acid mucus-producing cells, and immunostaining experiments were performed as described [28]. IHC staining was revealed with DAB+ Substrate Chromogen kit (3,3′-diaminobenzidine; Dako K3468, Agilent Technologies, Santa Clara, CA, USA). IF labelling was performed with Vectastain ABC kit (Vector Laboratories, Newark, NJ, USA) and HRP activity was detected with the Tyramide Signal Amplification (TSA) plus Cyanine 5 (1/150; NEL745001KT Akoya Biosciences, Marlborough, MA, USA). Slides were counterstained with hematoxylin or nuclear Fast Red. For IF studies, nuclei were visualized with DAPI staining. Primary and secondary antibodies are listed in Table S2. IHC sections were imaged with a Leica DMR microscope (Leica Camera, Wetzlar, Germany) and a HP-PL-Fluostar 20× 0.50, while IF sections were imaged with a Zeiss Axio Observer 7 microscope (Jena, Germany) inverted with a Zeiss Axiocam 705 color camera (Jena, Germany) and a Plan-Apochromat 20× 0.80NA or with a Zeiss microscope LSM 900 AiryScan II (Jena, Germany) inverted with a Plan-Apochromat 20× 0.80NA.
Quantification of SOX2 IF staining was performed using the QuPath open-source software for digital pathology image analysis (v0.6.0; https://qupath.github.io, accessed on 27 June 2025) [29]. Epithelial nuclei were identified by applying the Cell Detection command to the Blue Channel (DAPI) and using QuPath’s integrated segmentation algorithms to ensure accurate nuclear detection. For each detected nucleus, the total SOX2 signal intensity from the RED channel was calculated. To normalize the nuclear size, a ratio of SOX2 signal per nuclear area (mm2) was computed. The basal epithelial length was measured using the Freehand Polyline annotation tool within QuPath. For each image, values were obtained from 55 to 567 epithelial cells, and 3 to 7 images were analyzed per specimen.
2.3. Bulk RNA-Seq Analysis
Total RNA from four PPB (PPB1-4), four CPAM (CPAM1-4), and four control (CTL1-4) lung biopsies was extracted as previously described (Table S1) [21]. cDNA libraries were constructed using SMART-Seq v4 Ultra-low input RNA Kit (Takara Bio USA, San Jose, CA, USA), and paired-end sequenced at 100 bp reads on Illumina Novaseq 6000 (San Diego, CA, USA) at the Next-Generation Sequencing (NGS) Platform, Genomics Center, Centre de recherche du CHU de Québec-Université Laval (Québec, QC, Canada). The quality of the sequencing reads was assessed using FastQC software (v0.12.1; http://www.bioinformatics.babraham.ac.uk/projects/fastqc, accessed on 1 March 2023). Reads were trimmed with Trim Galore (v0.6.5; https://github.com/FelixKrueger/TrimGalore, accessed on 19 November 2019) and mapped with STAR (v2.7.9a) [30] to the human reference genome from ENSEMBL GRCh38.p14 (release 3 February 2022) [31]. Between 77% and 87% of the reads corresponded successfully to the human genome (Table S3A). The mapped sequencing data were then processed with the software featureCounts to obtain the number of reads associated with genes (v2.0.3) [32]. Among the aligned reads, an average of 44% mapped to exon sequences that were further considered for analysis (Table S3A). Gene expression levels were represented as transcripts per million (TPM). Genes with a mean of TPM < 1 in both control and pathological conditions were filtered out before performing DESeq2 package (Bioconductor) differential expression analysis with an alpha parameter of 0.05 (v1.42.0) [33]. Genes were considered differentially expressed (DE) when they exhibited at least a 2-fold difference (fold change; FC) in expression between control and pathological conditions, with a false discovery rate (FDR) adjusted p value (padj) < 0.05. The value of a |Log2FC| ≥ 1 was chosen considering the small number of replicates and the important biological variation between specimens. Lists of the differentially expressed genes (DEG) are provided in Table S4.
Gene Set Enrichment Analysis (GSEA) was performed on TPM data from CPAM samples versus control samples and from PPB samples versus control samples with the GSEA software (v4.2.3) using the MSigDB database [34]. FDR-adjusted Q value < 0.05 and |Normalized Enrichment Score (NES)| > 1 were used as thresholds to define statistic gene set enrichment.
2.4. Bulk miRNA-Seq Analysis
Total RNA from three PPB (PPB1-3) and four control (CTL1-4) lung biopsies was used for miRNA-seq assay (Table S1). After a purification step with GenElute PCR clean-up kit (Sigma-Aldrich, St. Louis, MI, USA), an appropriate range of cDNA fragments ~150 bp was extracted using a Pippin Prep instrument (Sage Science, Beverly, MA, USA). miRNA libraries were constructed and paired-end sequenced for 100 bp reads on the Illumina Novaseq 6000 of the NGS platform mentioned above. Data quality was evaluated using FastQC software, and reads were trimmed with Trim Galore. Only R1 reads were mapped with the software Bowtie (v1.3.1) [35] to the human reference genome from ENSEMB GRCh38.p14. More than 99% of the reads mapped successfully to the human genome (Table S3B). The mapped sequencing data were processed with the software featureCounts to obtain the number of reads associated with genes (v2.0.3) [32]. Among the aligned reads, an average of 84% mapped to miRNA sequences that were further analyzed (Table S3B). miRNA expression levels were represented as reads per million (RPMs). miRNAs with a mean of RPM < 1 in both control and PPB conditions were filtered out before performing DESeq2 package (Bioconductor) differential expression analysis with an alpha parameter of 0.05 (v1.42.0) [33]. miRNAs were considered to be differentially enriched when they exhibited at least a 2-fold change in expression between PPB and control conditions with a false discovery rate (FDR) padj < 0.05. Differentially expressed miRNAs are listed in Table S5.
2.5. Whole Exome Sequencing
Genomic DNA from the four PPB lung specimens (PPB1-4) was prepared using the Agilent SureSelectXT Low Input Human All Exon V7 (Agilent Technologies). 200 ng of DNA was fragmented on a Covaris instrument (Covaris, Woburn, MA, USA), and fragments of 150–200 bp were purified and used for library preparation. Following hybridization to the target-specific capture library and purification using streptavidin-coated beads (Dynabeads MyOne Streptavidin T1, Thermo Fisher Scientific, Waltham, MA, USA), libraries were amplified for 10 cycles with indexing primers. Exome libraries were paired-end sequenced for 150 bp reads on the Illumina Novaseq 6000 of the NGS platform previously mentioned. Data quality was evaluated using FastQC software, and reads were trimmed with Trim Galore before being mapped with the software Bowtie (v2.5.2) [35] to the human reference genome from ENSEMB GRCh38.p14. More than 99% of the reads mapped successfully to the human genome, and an average of 78% aligned to exon sequences (Table S3C). Variant detection was done using the variant calling software bcftools (v1.19) [36]. We found 17,202 variants, Single Nucleotide Polymorphism (SNP) and Insertion-Deletion (INDEL), for PPB1, 15,265 for PPB2, 16,996 for PPB3, and 17,469 for PPB4. Using this variant detection approach, we were able to identify DICER1 mutations in lung biopsy samples from PPB patients (Figure 1).
2.6. PCR Assay and KRAS Status
Genomic DNA from CPAM specimens was recovered from paraffin sections using QIAamp DNA FFPE Advanced Kit (Qiagen, Venlo, The Nethernands). DNA fragments encompassing the KRAS G12 protein position were amplified by PCR with Taq DNA Polymerase (Bio Basic, Markham, ON, Canada) with three different primer sets: #1: Forward: 5′-TGTTGGATCATATTCGTCCACAA-3′, Reverse: 5′-GTACTGGTGGAGTATTTGATAGTGT-3′; #2: Forward: 5′-CGATACACGTCTGCAGTCAAC-3′, Reverse: 5′-TCATGAAAATGGTCAGAGAAACC-3′; and #3: Forward: 5′-GTGTGACATGTTCTAATATAGTCAC-3′, Reverse: 5′-AAAACAAGATTTACCTCTATTGTTGG-3′. DNA fragments were sequenced by Sanger technology, and the KRAS G12 status was established by analysis of chromatograms.
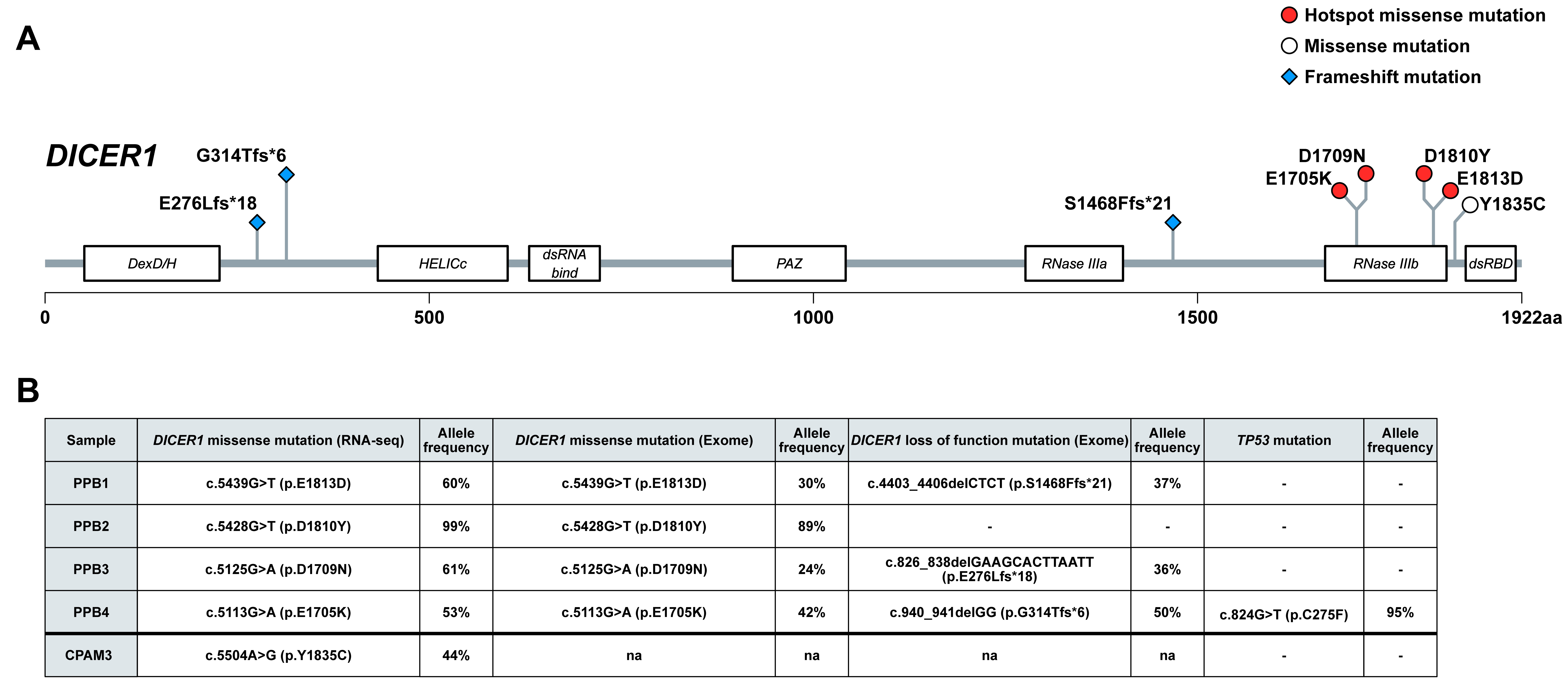
Figure 1. RNA-seq and exome analyses revealed DICER1 and TP53 mutations in PPB and CPAM specimens. (A) Lollipop diagram of the missense and frameshift DICER1 mutations found by RNA-seq and exome sequencing analyses in PPB and CPAM lung specimens. The annotated functional domains of the DICER1 protein are indicated. Red circles represent Hotspot missense mutations found in PPB lesions, while the missense mutation present in CPAM3 is shown in white. Blue diamonds correspond to loss-of-function frameshift mutations of PPB specimens. (B) Table listing the DICER1 and TP53 mutations and their allelic frequency in PPB and CPAM specimens.
2.7. Chromatin States and Statistical Analyses
To estimate the chromatin state of the 92 genes coding for TF and presenting a gain in expression in PPB samples, we used ChIP-seq peak called data for human lung from 16-, 30- and 40-years old individuals obtained from ENCODE for the three following histone post-translational modifications: H3K27ac (ENCODE:ENCSR510RPC/GEO:GSE175318; ENCODE:ENCSR540ADS/GEO:GSM906395; ENCODE:ENCSR494WCX/GEO:GSE167619), H3K27me3 (ENCODE: ENCSR800MYV/GEO:GSE175189; ENCODE:ENCSR204NFO/GEO:GSM1220283; ENCODE:ENCSR975GDL/GEO: GSE167807), and H3K36me3 (ENCODE:ENCSR793LDG/GEO:GSE175165; ENCODE:ENCSR671NXL/GEO:GSM956014; ENCODE:ENCSR906YQO/GEO:GSE167769). Only peaks present in the three data sets with an overlap of at least 50% were kept using the software BEDTools option intersect (v2.31.0) [37]. The genome-wide distribution of the three histone marks was analyzed. The combination of histone marks at the gene level enabled to define a 3-state model. The sole presence of H3K27me3 corresponds to heterochromatin, while H3K27ac and H3K36me3 combined or alone are associated with permissive chromatin. Finally, the absence of all three marks, or the presence of the repressive H3K27me3 mark with the activating H3K36me3 and H3K27ac marks, was considered an undetermined state.
Statistical analyses were performed in R (v4.3.1). Random sampling was used to study the variations in proportions of chromatin states in seven different groups of genes: (i) ‘all genes’, (ii) ‘all TF’, (iii) ‘DEG down in PPB’, (iv) ‘DE TF down in PPB’, (v) ‘DEG up in PPB’, (vi) ‘DE TF up in PPB’, and (vii) ‘DE TF with a gain in expression in PPB’. Gain in expression was defined as expression < 1 TPM in controls, but ≥1 TPM in PPB specimens. To determine if the gain in expression of the 92 DE TF genes in PPB was random or significant, a proportion test based on the chromatin states was performed. To do so, 92 genes from the ‘all genes’, ‘all TF genes’, and ‘DEG up in PPB’ categories were randomly sub-sampled from the genome 10,000 times [38]. Observed and simulated Chi-square statistics were then compared to assess the observed differences in chromatin state proportions of the 92 DE TF genes with a gain in expression in PPB. The difference was statistically significant with a p value < 2.2 × 10−16 for all three comparisons, indicating that the variation in proportions was not due to chance. Significant differences in gene expression (Log2(mean of TPM + 1)) between PPB and control specimens for the seven different groups of genes mentioned above were assessed using the pairwise Wilcoxon rank sum test.
3. Results
3.1. DICER1 Biallelic Mutations in PPB Lung Lesions
To characterize transcriptome-wide changes occurring in PPB and CPAM, we performed a comparative RNA-seq analysis of lung lesions from four PPB patients and four CPAM patients, as well as of normal lung tissues from four controls (Table S1). We first examined the DICER1 sequences to determine whether mutations were present in PPB patients. All four PPB specimens carried a missense mutation in the RNase IIIb catalytic domain, corresponding to one of the previously reported mutational hotspots (Figure 1) [12,14]. For patients PPB1, PPB3, and PPB4, the variant frequency ranged from 53% to 61% indicating a heterozygous mutation. In contrast, PPB2 exhibited an allelic frequency of 99%, suggesting that the mutation was either homozygous or hemizygous with a loss of heterozygosity. The availability of DNA from the four PPB lung tissues also enabled whole exome sequencing, which confirmed the presence of these missense mutations. Again, the variant frequency of the mutation was higher in PPB2 (89%) than in the other three specimens (24% to 42%). Moreover, whole exome sequencing data from PPB1, PPB3, and PPB4 revealed a loss-of-function mutation caused by nucleotide deletion, resulting in a frameshift and premature termination of the DICER1 protein. No loss-of-function mutation was found in the PPB2 specimen. Since we did not have access to DNA from paired normal samples, we could not determine whether the DICER1 mutations identified in the lung lesions were germline predisposition variants or tumor-specific alterations.
RNA-seq data did not reveal DICER1 mutations in CPAM specimens or in controls, with the exception of CPAM3 lung tissue, which carried the DICER1 missense mutation Y1835C. This mutation lies in a region with no known functional annotation, located between the RNase IIIb and the double-stranded RNA binding domain (Figure 1). Previous studies have shown that carriers of non-hotspot DICER1 mutations do not present biases in miRNA biogenesis, suggesting that the functional impact of the Y1835C mutation on miRNA processing is probably limited [39].
Further analysis of the whole exome sequence for cancer gene mutations using the Catalogue of Somatic Mutations in Cancer (COSMIC) and the IntOGen databases revealed that the PPB4 lesion carried a missense mutation in the TP53 gene (Figure 1) [40,41]. This C275F mutation, located in the DNA-binding domain of the TP53 protein, is considered a hotspot in several carcinomas, including lung adenocarcinoma and lung squamous cell carcinoma (cBioPortal.org, accessed on April 2018). The high allelic frequency (95%) of this TP53 mutation suggested it was either homozygous or hemizygous with a loss of heterozygosity. Biallelic TP53 mutations are a frequent event in PPB [14].
3.2. Bulk RNA-Seq Reveals Distinct Gene Expression Profiles for PPB and CPAM
To determine whether PPB and CPAM have similar transcriptomic impacts in the lung, RNA-seq assays were carried out on PPB, CPAM, and control specimens. Principal Component Analysis (PCA) was conducted on normalized gene counts to assess the variability within the RNA-seq dataset. This analysis revealed a clear clustering of specimens according to their pathological status (Figure 2A). Despite the limited sample size, PC1 captured a well-defined separation of PPB from CPAM and controls, while PC2 showed some overlap among CPAM1, CPAM2, and control specimens. Clustering was further supported by the comparison of the differentially expressed genes (DEG) exhibiting at least a 2-fold difference in expression with a padj < 0.05. A Venn diagram showed 2416 DEG between PPB and control, 2301 DEG between PPB and CPAM, and only 250 DEG between CPAM and control specimens. Notably, the DEG profiles of PPB versus control and PPB versus CPAM superimposed by more than 70% (1725 DEG; Figure 2B). This was reflected in the comparable numbers of downregulated and upregulated DEG in PPB: 890 and 876 DEG were downregulated in PPB versus control and PPB versus CPAM comparisons, respectively, while 1526 and 1425 DEG were upregulated (Figure 2C,D). In contrast, only seven DEG were downregulated in CPAM versus control, while 243 DEG were upregulated, a pattern similar to that reported by Zhang et al., 2024 (Figure 2E) [42].
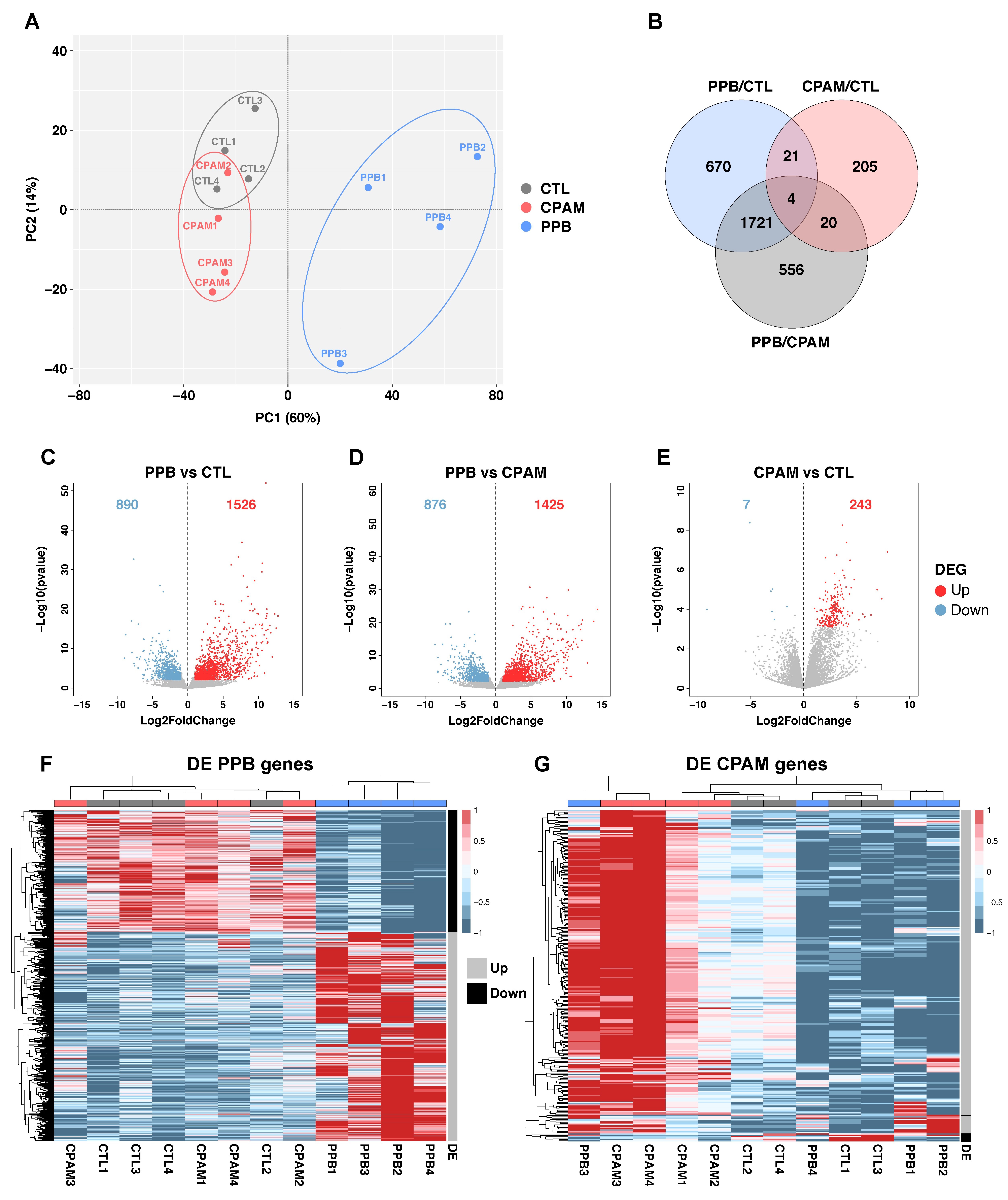
Figure 2. Transcriptomic analysis of PPB and CPAM specimens reveals distinct gene expression profiles. (A) Principal Component Analysis (PCA) of controls (CTL, grey), CPAM (red), and PPB (blue) specimens. (B) Venn diagrams comparing differentially expressed genes (DEG) between PPB, CPAM, and CTL. Differential expression analysis was performed with DESeq2 with an alpha parameter of 0.05. Statistically significant differential expression was defined as |Log2FC| ≥ 1 and padj < 0.05, and a value of TPM ≥ 1 in at least one condition. (C–E) Volcano plots representing DEG between PPB and CTL (C), PPB and CPAM (D), and CPAM versus CTL (E) specimens. Numbers of upregulated (red) and downregulated (blue) DEG with a cutoff of |Log2FC| ≥ 1 and padj < 0.05 are indicated. (F) Heatmap and hierarchical clustering based on comparison of DEG in PPB versus CTL. CTL, CPAM, and PPB specimens are indicated in grey, red, and blue, respectively. Data are represented in the z-score of the Log2(mean of TPM + 1). Genes with a mean of TPM < 1 in all conditions were excluded from analysis. (G) Heatmap and hierarchical clustering based on comparison of DEGs in CPAM versus CTL using the same parameters as in (F). Upregulated and downregulated DE genes are indicated on the right side of panels F and G by grey and black bars, respectively.
Heatmap analysis of DEG expression across all 12 samples confirmed the hierarchical clustering by pathology. For the 2416 PPB DEG, PPB specimens were grouped together while CPAM and control samples clustered jointly (Figure 2F). Similarly, for the 250 CPAM DEG, CPAM specimens were sorted together (Figure 2G). However, as shown by the PCA analysis, heterogeneity between CPAM cases was observed. CPAM1 and CPAM2 formed a subgroup that clustered with the control samples, which may explain the moderate number of DEG between CPAM and controls. In contrast, CPAM3 and CPAM4 were part of another subgroup with markedly increased DEG expression (Figure 2G). This subgroup also included PPB3, which had been identified as a Type I cystic form. This may justify why its transcriptome shares molecular characteristics with CPAM, while retaining a distinct PPB profile (Figure 2F,G; Table S1). The dual behavior of PPB3 may offer molecular clues on the intermediate phenotypes that arise during PPB tumor progression. In summary, transcriptome-wide data indicate that CPAM and PPB are globally different pathological entities with a unique molecular signature, supporting our previous findings [21]. Furthermore, transcriptional heterogeneity among CPAM specimens highlights the complexity of this disease.
3.3. Misexpression of Bronchial Epithelium Markers in CPAM Lesions
Gene Set Enrichment Analysis (GSEA) was applied to CPAM and PPB datasets. For CPAM, upregulated transcripts were significantly enriched for genes specific to basal, ciliated, secretory, goblet, and club cell types from the bronchial epithelium, known to line CPAM cysts (Figure 3A–C) [1]. GSEA also revealed several terms related to cilia, such as cilia development and movement, axoneme assembly, microtubule motor activity, and dynein complex (Figure 3A). This analysis agreed with previous studies that have suggested the importance of cilia in CPAM disease [42,43,44]. Heatmap representation of upper airway cell type-specific gene expression data confirmed the higher expression of proximal cell markers for basal, ciliated, goblet, club, and submucosal gland (SMG) cells (Figure 3D). However, the four CPAM specimens behaved differently. Increased expression of bronchial cell markers was more important in CPAM3 and CPAM4 than in CPAM1 and CPAM2, which presented similarities with controls (Figure 3D). Elevated expression of proximal cell markers was also detected in PPB3, consistent with the clustering of PPB3 with CPAM3 and CPAM4. Moreover, the high expression of specific bronchial cell genes in CPAM3, CPAM4, and PPB3 correlated with the increased expression of the TF sex-determining region Y (SRY)-box 2 (SOX2) gene, a lineage commitment marker of proximal endodermal lung progenitor cells (Figure 3D) [45]. Increased SOX2 expression in CPAM was previously reported [20,43,46]. In addition, gain of Sox2 expression in mice hampers lung branching and causes cystic formation, supportive of a role in CPAM pathogenesis [46,47]. Interestingly, expression of FOXA1 and SIX1 genes, which encode the winged-helix TF FOXA1 and the sine oculis homeobox transcriptional regulator SIX1, respectively, was also augmented in CPAM3, CPAM4, and PPB3. FOXA1 and SIX1 proteins were both shown to act positively on SOX2 expression by direct binding to SOX2 specific enhancers [48,49]. Moreover, the loss of function of Six1 or Foxa1 in mice affects lung morphogenesis [50,51]. Finally, SOX2 was shown to directly activate the expression of the basal cell specific Trp63 gene [46]. Together, these correlations suggest that the concomitant increased expression of FOXA1 and SIX1 can trigger SOX2 upregulation and consequently perturb bronchial epithelial cell differentiation in CPAM lung tissues.

Figure 3. CPAM transcriptomic analyses. (A) Gene Set Enrichment Analysis (GSEA) of CPAM upregulated genes compared to CTL data. The x-axis corresponds to the Normalized Enrichment Score (NES), the dot size represents the percentage of upregulated genes in each gene set, and the color of dots illustrates the gene set category. (B) A subset of statistically significant enrichment plots of terms associated with proximal pulmonary epithelium with a NES > 1 among CPAM upregulated genes is shown. Green curves correspond to the running enrichment score. (C) Schematic representation of the proximal pulmonary epithelium. (D) Heatmap based on the expression of different cell type markers of proximal lung epithelium in CTL, CPAM, and PPB specimens. Data are represented in the z-score of the Log2(mean of TPM + 1). Genes indicated in bold correspond to those further studied by immunostaining.
3.4. KRAS Mutation in CPAM Affects the Bronchial Epithelium
To validate the changes in expression of proximal cell genes revealed by RNA-seq, we performed immunostaining assays. For these analyses, we considered the KRAS status of the CPAM specimens. Somatic gain-of-function mutations in the KRAS (Kirsten rat sarcoma viral oncogene homolog) gene are established driver mutations for the development of several cancers, including lung mucinous adenocarcinoma, a condition that can arise from CPAM [52,53,54]. KRAS mutations at codon 12 were also reported in CPAM cases containing mucinous cell clusters, raising questions on the cancer risk associated with the cystic disease [18,19,22,55,56,57]. Using Sanger sequencing of PCR-amplified KRAS specific DNA fragments, we determined the KRAS status of CPAM lung biopsies. Out of 25 CPAM lesions, 12 (48%) carried either a p.G12D (83%) or a p.G12V (17%) constitutive active mutation (KRASmut), while no KRAS mutation (KRASwt) was detected in 13 specimens (52%) (Table S1). The prevalence of KRAS p.G12D in CPAM cases was previously reported [57]. Foci of goblet cell hyperplasia and regions of pseudostratified epithelium and papillary architecture were frequent in KRASmut specimens but rarely seen in KRASwt ones (Figure 4) [22].

Figure 4. Perturbed expression of proximal lung epithelium cell-specific markers in CPAM. (A–X) Comparative immunostaining for SOX2 (A–C), SIX1 (D–F), FOXA1 (G–I), Ki-67 (J–L), TP63 (M–O), FOXJ1 (P–R), CC10 (S–U), and for Alcian Blue histological staining (V–X) between controls (A,D,G,J,M,P,S,V), CPAM lesions with no KRAS mutation at position G12 (KRASwt; B,E,H,K,N,Q,T,W) and CPAM samples carrying a KRAS mutation at position G12 (KRASmut; C,F,I,L,O,R,U,X). Increased signal for SOX2, SIX1, FOXA1, TP63, FOXJ1, and CC10 proteins, as well as more Alcian Blue positive cells, were detected in CPAM KRASmut specimens. No change in proliferation was observed by Ki-67 staining. Representative specimens are shown. Mucinous cell clusters are indicated by arrowheads. Scale bars: 50 mm (A–X).
Variations between specimens were detected, but an overall trend of augmented SOX2 protein expression in KRASmut specimens was observed, as shown by IF staining and SOX2 intensity measurement using QuPath (Figure 4A–C and Figure S1). SOX2 staining in CPAM KRASwt tissues was comparable to that of controls (Figure 4A,B). As well, expression of SIX1, FOXA1, and TP63 was also increased in KRASmut specimens versus CPAM KRASwt and controls (Figure 4D–I,M–O). A similar result was obtained for FOXJ1, a marker of ciliated cells and CC10, which labels club secretory cells (Figure 4P–U). Mucus-producing cells detected by Alcian Blue staining were also more frequent in CPAM KRASmut specimens (Figure 4V–X). Ki-67 staining was similar in the three conditions analyzed, indicating that the increased protein staining detected in CPAM KRASmut tissues was not due to enhanced proliferation (Figure 4J–L).
Thus, gene expression changes revealed by RNA-seq assay were confirmed by differential protein expression detection in a larger CPAM cohort. It also showed a correlation between the presence of a KRAS constitutive active mutation, altered epithelial architecture, and variations in expression of bronchial cell specific markers.
3.5. Changes in Expression of Distal Lung Epithelial and Mesenchymal Cell Markers in PPB Lesions
While CPAM DEG were almost exclusively upregulated, PPB DEG included both downregulated and upregulated genes (Figure 2). GSEA for PPB downregulated transcripts showed significant enrichment for genes associated with cell types from the alveolar epithelium, such as alveolar epithelial Type 1 cells that permit gas exchanges with the underlying vascular endothelium, and Type 2 cells that secrete surfactant proteins (Figure 5A,B and Figure S2) [58]. Moreover, a significant enrichment for downregulated genes specific to capillary endothelial cells, alveolar fibroblasts, aerocytes, pericytes, and the Gene Ontology (GO) term “lung alveolus development” was observed, suggesting a loss of cell identity in the distal lung. Heatmap analysis clearly demonstrated the decreased expression of markers for alveolar epithelial and mesenchymal cells (Figure 5C,D). No significant variation was detected in SOX9 and ID2 expression, both of which encode lineage markers of distal epithelial lung progenitors, suggesting that cellular commitment was not affected (Figure 5C) [58]. In contrast, expression of GATA6, NKX2-1, and ICAM1, known to be important for distal airway cell differentiation, was diminished in PPB [59,60,61]. Thus, impaired alveolar cell differentiation likely contributes to PPB pathogenesis.

Figure 5. PPB transcriptomic analyses. (A) GSEA of PPB downregulated and upregulated genes compared to CTL data. The x-axis corresponds to NES, the dot size represents the percentage of misregulated genes in each gene set, and the color of dots illustrates the gene set category. (B) Schematic representation of cell types in the distal lung. (C,D) Heatmaps based on the expression of cell type markers of distal lung epithelium (C) and mesenchyme (D) in CTL, CPAM, and PPB specimens. Data are represented in the z-score of the Log2(mean of TPM + 1). (E) Heatmap based on the expression of epithelium and mesenchyme markers associated with EMT in CTL, CPAM, and PPB specimens.
GSEA also revealed decreased expression of genes associated with epithelial cell characteristics, including cell adhesion, cell junctions, epithelial cell development, and tube branching. Interestingly, epithelial genes known to be suppressed in EMT were found to be downregulated in PPB (Figure 5A,E). In parallel, upregulated transcripts were enriched in genes involved in mesenchyme-derived cell types and in mesenchyme development, as well as in genes highly expressed in EMT, supporting the notion that EMT contributes to the progression of PPB (Figure 5A,E) [21]. Finally, upregulated transcripts were enriched in genes involved in pediatric cancer markers, cell cycle, translation, ribosome assembly, and chromatin remodeling, suggestive of an enhanced proliferative status (Figure S2).
3.6. Abnormal Gain of Expression of Transcription Factors Involved in Embryonic Development in PPB
GSEA analysis of PPB data also unveiled enrichment in upregulated genes associated with embryonic organ morphogenesis, proximal-distal pattern formation, and limb development, categories that were not observed in CPAM upregulated DEG. Among these PPB upregulated genes, a significant number corresponded to TF included in the GO term “DNA-binding transcription factor binding activity” (Figure 5A).
We questioned what proportion of TF was abnormally expressed in PPB. Among the PPB DEG, 7.5% corresponded to upregulated TF and 2% to downregulated TF (Figure 6A). Interestingly, in the top 10, 50, and 100 upregulated DEG, TF represented respectively 80%, 56% and 39%. Thus, despite TF constituting only a small proportion of upregulated DEG, they were among the most strongly upregulated ones. This pattern was not observed for downregulated DEG, where TF corresponded to only 2% of the top 50 DEG (Figure 6A,B). Most upregulated TF genes (51%) were associated with a gain of expression, i.e., their expression was <1 TPM in controls, but ≥1 TPM in PPB specimens (Figure 6C). Thus, ectopic expression of developmental TF not normally found in the lung occurs in PPB.

Figure 6. Abnormal gain of expression of transcription factors in PPB. (A) Proportion of differentially expressed (DE) transcription factor (TF) genes in all PPB DE genes (DEG), Top 10 DEG up, Top 50 DEG up, Top 100 DEG up, and Top 50 DEG down in PPB versus CTL. TF DEG up and down are shown in red and blue, respectively. (B) Diagram representing all PPB DEG sorted from the most DEG up (red) to the most DEG down (blue) in Log2FC. Red lines correspond to TF DEG up, blue lines to TF DEG down. (C) Comparison of gene transcript levels (Log2(mean of TPM + 1)) in PPB and CTL. PPB TF DEG up and down are represented by red and blue dots, respectively. Dotted lines surround TF genes with a gain in expression in PPB. (D) ChIP-seq profiles for H3K36me3 and H3K27ac positive histone marks (in dark and light green, respectively), and H3K27me3 repressive mark (red) covering the HOXA10, HOXA11, and HOXA13 region in human lung. Data were obtained from ENCODE. (E) Chromatin signatures assigned to genes based on a combination of H3K36me3, H3K27ac, and H3K27me3 histone marks in human lung. Three broad categories were defined: (i) permissive (green) corresponding to open chromatin, (ii) repressive (red) corresponding to closed chromatin, and (iii) indetermined (grey). The proportion of each chromatin signature for the seven groups is indicated in percentage. A Chi-square statistics test was performed to determine whether the proportion of chromatin states observed for ‘DE TF gain PPB’ was specific. *** p value < 2.2 × 10−16. (F) Comparison of transcript expression levels for the different groups in PPB (blue) and CTL (white) specimens. *** p value < 0.001, NS = not significant (Wilcoxon test).
As listed in Figure 6B, most of the upregulated TF were members of gene families having an established role in embryo development. One of the main representatives was the HOX gene family, which is involved in embryo patterning, axis specification, and organogenesis [62]. In PPB, 22 out of the 39 HOX genes presented changes in their expression, 15 showing a gain in expression (HOXA1, HOXA9, HOXA10, HOXA11, HOXA13, HOXC5, HOXC8, HOXC9, HOXC10, HOXC11, HOXC12, HOXC13, HOXD10, HOXD11 and HOXD13). These 15 HOX genes are not normally expressed in the lung [63]. Repression of developmental TF is known to be associated with heterochromatin, and silencing is mediated by the histone H3K27me3, an epigenetic negative mark (Figure 6D) [64]. Therefore, ectopic gain in TF expression could be explained by defective chromatin regulation due to the loss of H3K27me3. This hypothesis was further supported by the GSEA data showing an enrichment in upregulated genes for the “Polycomb Repressive Complex 2 (PRC2) targets” category (Figure 5A). PRC2 is the chromatin-modifying multi-protein complex that adds methyl groups to lysine 27 on histone 3 causing gene silencing [65].
To further determine if PPB TF DEG are mainly associated with a repressive chromatin state compared to other genes in the lung, we looked at the histone mark distribution over all genes of the entire human genome. To do so, we exploited ChIP-seq data for lungs of healthy human adults obtained from ENCODE for three histone post-translational modifications: H3K27ac, H3K36me3, and H3K27me3. H3K27ac is enriched in promoters and enhancers of transcribed genes, H3K36me3 is present in coding sequences of actively transcribed genes, while, as mentioned, H3K27me3 is a repressive mark [64,66,67]. Combinations of these three marks define three broad chromatin states: permissive, repressive, and undetermined (Figure 6E). A similar distribution of chromatin states was observed for the ‘all genes’, ‘all TF’, ‘DEG down in PPB’, ‘DE TF down in PPB’ and ‘DEG up in PPB’ groups, which were mainly associated with a permissive chromatin state (from 53% to 71%) with a low % corresponding to the repressive state (from 5% to 21%; Figure 6E). In contrast, ‘DE TF up in PPB’ and ‘DE TF with a gain in expression in PPB’ were found predominantly in a repressive chromatin state (50% and 78.5%, respectively) with a small % in the permissive state (28% and 4%; Figure 6E). To confirm that the pattern of chromatin states for the 92 DE TF showing a gain in expression in PPB was not due to arbitrary events, we randomly sub-sampled 92 genes from the ‘all genes’, ‘all TF genes’, and ‘DEG up in PPB’ categories from the genome 10,000 times as described in the Materials & Methods section. This analysis demonstrated that the enrichment in H3K27me3 observed for the ‘DE TF with a gain in expression in PPB’ group was significant, indicating that the chromatin state distribution was not due to chance (Figure 6E).
We also looked at the changes in transcript levels between PPB and control specimens for the seven different groups. No significant variation was detected for the ‘all genes’ and ‘all TF’ categories, whereas a significant difference was measured for the other five categories (Wilcoxon test, p value < 0.001; Figure 6F). Moreover, the expression levels observed in control specimens corroborated with the chromatin state deduced from the histone mark distribution. For the ‘DE TF with a gain in expression in PPB’ group, transcript levels were close to 0 in controls, supporting the repressive chromatin state, while significant expression levels were detected in PPB specimens (Figure 6F). Together, these analyses highlighted a gain of expression for a high proportion of TF associated with embryonic development but normally repressed in the developing lung. Correlation with the presence of the H3K27me3 repressive histone post-translational modification on these TF genes in normal lung tissue suggests a connection between their upregulation in PPB and impaired PRC2 function. This is further supported by the loss of H3K27me3 signal reported in PPB specimens [68].
3.7. Deregulation of miRNA Processing in PPB Could Participate in Decreased Activity of the PRC2 Complex
DICER1 mutations in PPB favor the production of miRNA-3p over miRNA-5p [13,14]. miRNA-seq assay was therefore carried out on PPB and control specimens to identify differentially expressed miRNA-5p and -3p. Data obtained agreed with previous reports, as 98% of upregulated miRNAs were miRNA-3p, while miRNA-5p constituted 83% of the downregulated DE miRNAs in PPB specimens (Figure 7A,B). The majority (52%) of the remaining 17% downregulated DE miRNAs were miRNA-3p, for which miRNA-5p expression was also decreased, suggesting impaired synthesis of the pre-miRNA. Heatmap analysis revealed heterogeneity between the three PPB specimens studied. PPB1 and PPB3 presented similar miRNA expression profiles, while PPB2 showed a more pronounced deregulation of miRNA-3p expression. The latter might be explained by the sole presence of the missense mutation (Figure 1 and Figure 7A).
Despite differences in their miRNA patterns, the impact on gene expression was largely comparable between PPB specimens (Figure 2F). We thus hypothesized that miRNAs showing similar expression changes in the three PPB specimens should be involved in the disease. We separately examined upregulated miRNA-3p and downregulated miRNA-5p (Figure 7 and Figure S3). To identify functional categories statistically over-represented among the misregulated miRNA, a microRNA Enrichment Analysis and Annotation (miEAA; v2.1) with the Over-Representation Analysis (ORA) option was performed for each population [69]. For the upregulated miRNA-3p, miEAA revealed significant enrichment in lung development, lung disease, and lung cancer, including EMT, TP53, and cell cycle. Categories associated with gene regulation, transcription factors, and chromatin were also significantly enriched (Figure 7D). For the downregulated miRNA-5p, significant enrichment was obtained for categories associated with embryonic development, FGF pathway, cancer, miRNA processing, chromatin and translation (Figure S3B).

Figure 7. Impact of miRNA deregulation on expression of PRC2 component EED in PPB. (A) Heatmap and hierarchical clustering of miRNA-5p and miRNA-3p in PPB and CTL specimens. Data are represented in z-score of the Log2(mean of RPM + 1). miRNAs with a mean of RPM < 1 in all conditions were excluded from the analysis. (B) Comparison of miRNA-5p (triangle) and miRNA-3p (circle) expression levels (Log2(mean of RPM + 1)) in PPB and CTL. DE upregulated and downregulated miRNAs in PPB are represented in red and blue, respectively. Misregulated miRNAs known to target PRC2 components are labelled. Expression data are shown in the table. (C) Heatmap based on the expression of upregulated miRNA-3p common to the three PPB specimens analyzed. They were selected according to a cutoff of the z-score value > 0.1 in all PPB samples. Statistically significant DE miRNA-3p are shown by red squares on the left. Data are represented in z-score of the Log2(mean of RPM + 1). Upregulated miRNAs known to target PRC2 components are written in red on the right. (D) A functional enrichment analysis of upregulated miRNA-3p was performed using microRNA Enrichment Analysis and Annotation (miEAA) with the Over-Representation Analysis (ORA) option surveying across pathway (yellow), disease (green), and GO (violet) databases. The x-axis corresponds to the −Log10(padj). (E) Comparative immunofluorescence staining for EED between control (a), PPB Type I (b), PPB Type II (c), and PPB Type III (d). Decreased and mosaic expression of the EED protein was detected in all PPB lesions. Representative specimens are shown. EED labelling and DAPI staining are detected in red and blue, respectively. Scale bars: 100 mm (a–d), 250 mm (a’–d’).
Attention was given to upregulated miRNA-3p. Among the DE upregulated miRNA-3p common to all specimens, some were known to target protein subunits of the PRC2 complex. Furthermore, loss of expression of SUZ12 or EED protein subunits was shown to cause an increased expression of transcription factors associated with embryonic development [70,71]. We thus decided to look closely at components of the PRC2 complex (Figure 7B,C). Interestingly, 68% of the DE TF showing a gain in expression in PPB, including the 15 HOX genes listed above, were found upregulated in non-small-cell lung murine cancer cells carrying an Eed deletion [72]. In PPB, we observed that miR199a-3p, miR-199b-3p, and miR-148b-3p, as well as miR-128-3p and miR148a-3p, all known to target SUZ12, were either upregulated or presented a trend for overexpression [73,74,75]. Moreover, miR-323a-3p, which was shown to target Eed in mice directly, was upregulated in PPB specimens [76]. To determine if miR-323a-3p overexpression correlates with changes in EED protein expression, we performed immunofluorescence staining on sections from control lung tissues and Type I, Type II, and Type III PPB lung lesions. In controls, EED protein was detected throughout the tissue as previously reported (https://www.proteinatlas.org/ENSG00000074266-EED/tissue/bronchus#, release 19 June 2023) [77]. In contrast, the EED signal appeared mosaic with regions devoid of EED expression in the PPB specimens tested (Figure 7E). Thus, the increased expression of specific miRNA-3p due to the DICER1 mutations in PPB may negatively affect PRC2 activity with repercussions on chromatin epigenetic landscape and expression of developmentally involved transcription factors not normally expressed in the lung.
4. Discussion
The morphologic overlap between PPB and CPAM poses a significant challenge to accurate diagnosis. Improving the ability to distinguish PPB from CPAM would represent a significant advance in diagnostic precision and clinical management. To address this, we investigated the molecular differences between the two diseases by profiling their transcriptional landscapes at a genome-wide scale using comprehensive bulk RNA-seq and miRNA-seq analyses. The transcriptome-wide data support our previous gene expression analysis, indicating that PPB and CPAM are different pathologies possessing distinct molecular signatures [21]. Expression of proximal lung epithelium markers was specifically increased in CPAM (Figure 3). In contrast, distal lung epithelium and mesenchyme markers were less expressed in PPB, with the exceptions of mesenchyme markers associated with EMT and selected developmental transcription factors that showed increased and even gain of expression (Figure 5 and Figure 6). Apart from specimen PPB3, diagnosed as a Type I cystic form and presenting an RNA-seq profile sharing both PPB and CPAM characteristics, a clear difference was thus seen between PPB and CPAM specimens (Figure 2F,G).
We questioned if DEG common to PPB and CPAM transcriptomes, as compared to CTL, may participate in cyst formation for both pathologies. Among the 25 common DEG, EYA1 appears as a candidate that could play such a role (Table S4). EYA1 is a transcriptional co-activator of SIX1 [78]. In the lung, EYA1 and SIX1 were shown to interact synergistically in the control of epithelial branching and mesenchymal development by inhibiting SHH signaling activity, coherent with a role in cyst formation [79]. EYA1 and SIX1 expression levels were increased in PPB and CPAM specimens, with a more pronounced effect in PPB. This may contribute to the reduced SHH expression observed in PPB (Figure 3D; Table S4). However, in CPAM, there is no evidence for altered expression of SHH pathway members, suggesting that either the modest upregulation of EYA1 and SIX1 expression has minimal impact on SHH signaling, or that EYA1/SIX1 acts through a distinct cascade that remains to be identified (Table S4) [21,80].
A point to consider is that, given the limited number of CPAM samples analyzed, we disregarded their CPAM classification in the RNA-seq data analysis. This could explain the heterogeneity between transcriptomic data and the low number of DEGs retrieved compared to other studies with larger cohorts or that considered CPAM classification based on cyst size [42,43,44]. This raises the possibility that other genes not detected by our transcriptomic analysis may be shared between PPB and CPAM during cyst formation.
Analysis of CPAM RNA-seq data allowed us to distinguish two subgroups with distinct molecular signatures. CPAM3 and CPAM4 specimens showed strong expression of proximal pulmonary epithelial cell markers, a feature associated with Type 1 CPAM. In contrast, gene expression changes were less pronounced in CPAM1 and CPAM2 (Figure 2G) [17]. Type 1 CPAM is generally characterized by large cyst lesions lined with a pseudostratified epithelium and papillary structures, mucus cell hyperplasia, and the presence of a KRAS oncogenic mutation at codon 12 [17,18,57]. Although CPAM3 and CPAM4 displayed transcriptomic profiles consistent with Type 1 CPAM, they were clinically diagnosed as Type 2, and they lacked a KRAS mutation. Conversely, CPAM2, which had a transcriptomic profile resembling that of controls, was diagnosed as a Type 1 and carried a KRASG12V mutant allele (Table S1). These inconsistencies highlight the challenges of CPAM classification. Discrepancies between KRAS mutation status and clinical diagnosis may also reflect intralesional heterogeneity of the mutation, as previously reported [57]. Moreover, limited dissection of cystic tissue may hinder KRAS mutation detection by the Sanger sequencing method. Finally, the possibility that Type 1 CPAM cases lack KRAS mutations cannot be excluded.
For the validation by immunostaining of DEG encoding proximal lung epithelium markers, we considered the KRAS status of CPAM samples from a larger cohort. A clear correlation was established between the KRASmut status of CPAM lesions, increased expression of bronchial epithelial cell markers, presence of pseudostratified epithelium with papillary projections and abrupt transitions, and the occurrence of mucus islands. This agrees with previously established links between KRAS activating mutations and increased differentiation of secretory and ciliated cells, epithelial cell hyperplasia and mucinous cell clusters [57,81,82]. However, we did not observe that the thickening of the cyst epithelium in CPAM KRASmut specimens was associated with changes in cell proliferation, as observed in human airway basal cells overexpressing a constitutively active KRAS protein (Figure 4) [82]. Our findings concurred with the lack of change in lung epithelial proliferation seen in mice carrying a KrasG12D mutation and presenting a defective lung branching phenotype that causes the formation of large, fluid-filled sacs reminiscent of lung cysts [83]. While these data further support the developmental origin of CPAM, they also suggest that the presence of KRAS oncogenic mutation may cause an intrinsic epithelial anomaly that leads to a disordered epithelial growth and differentiation of proximal airway structures and participates in cyst development.
The increased expression of SOX2 observed in CPAM KRASmut specimens may constitute an additional and accurate molecular marker to classify CPAM cystic lesions. Evidence from genetically modified mice has established a role for SOX2 in cyst formation [46]. Moreover, transcription of SOX2 is under the control of SIX1 and FOXA1, two TF with augmented expression in CPAM KRASmut specimens. It is therefore possible that the KRAS activating mutation may affect the expression of several molecules, including SOX2, SIX1, and FOXA1, and consequently impact bronchial epithelial cell differentiation in CPAM lesions.
On the other hand, SOX2, SIX1, and FOXA1 were also highly expressed in PPB3, a specimen positive for both DICER1 loss-of-function and missense mutations but negative for the KRAS mutation. This observation raises the possibility that misregulation of the SOX2, SIX1, and FOXA1 transcription factors occurs independently of the KRAS mutation status.
In PPB, genomic heterogeneity was observed between specimens. Notably, PPB2 only exhibited a DICER1 missense mutation, rather than the typical combination of a loss-of-DICER1 function mutation on one allele and a missense hotspot mutation on the other. Due to limited material, we were unable to determine whether the mutation present in PPB2 was homozygous or hemizygous with a loss of heterozygosity. Cases of tumor-specific, biallelic DICER1 pathogenic variants were reported to give rise to sporadic, non-syndromic PPB [8,10]. Furthermore, we cannot exclude the possibility that the DICER1 missense mutation in PPB2 is heterozygous and exerts a dominant-negative effect over the wild-type allele, as recently reported [84].
Despite its specific genomic status, PPB2 presented an RNA transcriptome profile similar to that of the three other specimens analyzed, each with a significant decrease in alveolar epithelial cell markers that distinguishes this pathology from CPAM (Figure 5) [43]. The reduced expression of alveolar epithelial cell markers did not result from fewer progenitor cells, since the alveolar epithelial progenitor cell markers SOX9 and ID2 did not present significant changes in their expression levels. Moreover, diminished expression of markers of specialized alveolar mesenchymal cells, such as capillaries, alveolar fibroblasts, aerocytes, and pericytes, suggested a widespread de-differentiation process occurring in the distal lung that might be indicative of tumoral progression. Tumor development was also supported by changes in the expression of genes promoting EMT as observed in the PPB specimens except for PPB3, coherent with the Type I classification of this sample [85].
One striking feature observed in all four PPB specimens was the abnormal upregulation of genes encoding developmental transcription factors (Figure 6). These TF are not normally involved in lung development and are primarily associated with heterochromatin-enriched regions, where their expression is repressed in healthy lung tissue. Genomic regions encompassing these genes are characterized by the presence of the histone H3K27me3 mark, product of the PRC2 complex catalysis, and essential for the transcriptional repression of PRC2 target genes [64]. In ES cells, it was shown that the PRC2 complex occupies numerous genes, most coding for transcription factors playing key roles in development, such as the HOX genes that are canonical PRC targets [65].
PRC2 complex comprises numerous protein subunits, including EED (for Embryonic Ectoderm Development). EED deficiency was shown to cause de-repression of TF genes due to the loss of H3K27me3 repressive mark [70,86]. Furthermore, inactivation of the PRC2 complex through EED or SUZ12 protein loss has been identified in human cancers, leading to abnormal expression of transcription factors linked to embryonic development and morphogenesis, including HOX TF [71,72]. Defective deposition of the repressive chromatin mark H3K27me3 has previously been reported in PPB, consistent with the aberrant gain of expression of developmental TF and the reduced expression of EED detected by immunostaining in PPB specimens (Figure 6 and Figure 7) [68]. The decreased EED expression in PPB may be explained by the increased expression of miRNA-323-3p, which is known to target EED. This suggests a mechanistic link between altered miRNA production, resulting from DICER1 mutations, and the abnormal expression of developmental transcription factors [76].
A close relationship exists between development and cancer. It is well established that misregulation of developmental TF genes can perturb downstream effector expression, leading to inappropriate activation of embryonic developmental cascades. This, in turn, can interfere with normal growth and differentiation programs, contributing to neoplastic transformation [87]. Aberrant activation of HOX genes has been reported in numerous cancers [88]. For instance, HOXC10 expression is abnormally upregulated in non-small cell lung cancer due to functional suppression of the PRC2 complex [89].
In conclusion, for the first time, and despite the limited sample size, integration of multi-omics analyses has enabled the identification of a potential pathogenetic mechanism in PPB that connects DICER1 mutations and altered miRNA profiles to impaired PRC2 activity, misexpression of developmental transcription factor genes, and tumorigenesis. To validate the significance of this pathway in PPB pathogenesis, additional RNA- and miRNA-seq experiments on larger cohorts will be required, along with functional assays to assess the impact of DICER1 mutations on other PRC2 complex components and to identify the transcription factors driving the oncogenic process. scRNA-seq assays should also be performed to dissect the dynamic cellular states involved. Similarly, further studies involving more patients are needed to clarify the relationship between KRAS status and SOX2 expression in CPAM pathology, and to disentangle the underlying molecular mechanisms.
Supplementary Materials
The following supporting information can be found at: https://www.sciepublish.com/article/pii/822, Table S1: General characteristics of patients; Table S2: List of primary and secondary antibodies; Table S3: RNA-seq, miRNA-seq and whole exome statistics; Table S4: RNA-seq data—Gene expression PPB-CPAM-CTL; Table S5: miRNA-seq data—miRNA expression PPB-CTL; Figure S1: Box-and-whisker plot representation of SOX2 fluorescence signal along lung epithelium in control, CPAM KRASwt and CPAM KRASmut specimens; Figure S2: Statistically significant enrichment plots of terms associated to GSEA analysis of PPB transcriptome with an FDR-adjusted Q < 0.05 and a NES > 1; Figure S3: (A) Heatmap and hierarchical clustering of downregulated miRNA-5p common to the three PPB specimens analyzed. (B) A functional enrichment analysis of downregulated miRNA-5p was performed using microRNA Enrichment Analysis and Annotation (miEAA) with the Over-Representation Analysis (ORA) option surveying across GO (violet) and pathway (yellow) databases.
Acknowledgments
We thank Olivier Boucherat, Jean Charron and Tom Moss for comments on the manuscript and sharing reagents. We also thank Venkata Manem for guidance in bioinformatic analyses, Stéphane Dubois from the Next-Generation Sequencing Platform, Genomics Center, CRCHUQ-ULaval for help in RNA-seq assays, and Carl St-Pierre from the Cellular Imaging Unit CRCHUQ-ULaval for advice.
Author Contributions
Conceptualization: J.G., N.H., K.L.-T. and L.J.; Methodology: J.G., N.H., K.L.-T. and B.F.; Validation: J.G., N.H. and L.J.; Formal analysis: J.G., N.H. and L.J.; Resources: C.D., W.D.F. and L.J.; Data curation: J.G., N.H. and L.J.; Writing—original draft preparation: J.G. and L.J.; Writing—review and editing: J.G., N.H., K.L.-T., B.F., C.D., W.D.F. and L.J.; Supervision: L.J.; Project administration: L.J.; Funding acquisition: L.J. All the authors read and approved the manuscript before submission.
Ethics Statement
The study was conducted according to the guidelines of the Declaration of Helsinki and approved by the respective institutional ethics committees and by the CHU de Québec-Université Laval research ethics committee (project MP-20-2020-4809, approved: 5 August 2019).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
RNA-seq and miRNA-seq data are available through NCBI GEO (accession numbers GSE311205 and GSE311209) and exome data are available through SRA (accession number PRJNA1356925).
Funding
This work was supported by the Canadian Institutes of Health Research (FRN169158 to L.J.) and the Cancer Research Society (24217 to L.J.). B.F. holds studentships from NSERC (BESC M) and Fonds de Recherche du Québec en Santé (BF1-325367).
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
-
Stocker JT. Cystic lung disease in infants and children. Fetal Pediatr. Pathol. 2009, 28, 155–184. doi:10.1080/15513810902984095. [Google Scholar]
-
Dehner LP, Schultz KAP, Hill DA. Congenital pulmonary airway malformations with a reconsideration and current perspective on the Stocker Classification. Pediatr. Dev. Pathol. 2023, 26, 241–249. doi:10.1177/10935266221146823. [Google Scholar]
-
Dehner LP, Messinger YH, Schultz KA, Williams GM, Wikenheiser-Brokamp K, Hill DA. Pleuropulmonary blastoma: Evolution of an entity as an entry into a familial tumor predisposition syndrome. Pediatr. Dev. Pathol. 2015, 18, 504–511. doi:10.2350/15-10-1732-OA.1. [Google Scholar]
-
Hill DA, Jarzembowski JA, Priest JR, Williams G, Schoettler P Dehner LP. Type I pleuropulmonary blastoma: Pathology and biology study of 51 cases from the international pleuropulmonary blastoma registry. Am. J. Surg. Pathol. 2008, 32, 282–295. doi:10.1097/PAS.0b013e3181484165. [Google Scholar]
-
Kommoss FKF, Chaong AS, Chong AL, Pfaff E, Jones DTW, Hiemcke-Jiwa LS, et al. Genomic characterization of DICER1-associated neoplasms uncovers molecular classes. Nat. Commun. 2023, 14, 1677. doi:10.1038/s41467-023-37092-w. [Google Scholar]
-
Priest JR, Hill DA, Williams GM, Moertel CL, Messinger Y, Finkelstein MJ, et al. International Pleuropulmonary Blastoma Registry. Type I pleuropulmonary blastoma: A report from the International Pleuropulmonary Blastoma Registry. J. Clin. Oncol. 2006, 24, 4492–4498. doi:10.1200/JCO.2005.05.3595. [Google Scholar]
-
Messinger YH, Stewart D, Priest JR, Williams GM, Harris AK, Schultz KAP, et al. Pleuropulmonary blastoma: A report on 350 central pathology-confirmed pleuropulmonary blastoma cases by the International pleuropulmonary blastoma registry. Cancer 2015, 121, 276–285. doi:10.1002/cncr.29032. [Google Scholar]
-
Schultz KAP, Nelson AT, Mallinger PHR, Harris AK, Kamihara J, Baldinger S, et al. DICER1-related tumor predisposition: Identification of at-risk individuals and recommended surveillance strategies. Clin. Cancer Res. 2024, 30, 5681–5692. doi:10.1158/1078-0432.CCR-24-1532. [Google Scholar]
-
Bartel DP. Metazoan microRNAs. Cell 2018, 173, 20–51. doi:10.1016/j.cell.2018.03.006. [Google Scholar]
-
Brenneman M, Field A, Yang J, Williams G, Doros L, Rossi C, et al. Temporal order of RNaseIIIb and loss-of-function mutations during development determines phenotype in pleuropulmonary blastoma/DICER1 syndrome: A unique variant of the two-hit tumor suppression model (version 2). F1000Research 2022, 4, 214. doi:10.12688/f1000research.6746.2. [Google Scholar]
-
Blaszczyk J, Gan J, Tropea JE, Court DL, Waugh DS, Ji X. Noncatalytic assembly of ribonuclease III with double-stranded RNA. Structure 2004, 12, 457–466. doi:10.1016/j.str.2004.02.004. [Google Scholar]
-
Foulkes WD, Priest JR, Duchaine TF. DICER1: Mutations, microRNAs and mechanisms. Nat. Rev. Cancer 2014, 14, 662–672. doi:10.1038/nrc3802. [Google Scholar]
-
Anglesio MS, Wang Y, Yang W, Senz J, Wan A, Heravi-Moussavi A, et al. Cancer-associated somatic DICER1 hotspot mutations cause defective miRNA processing and reverse-strand expression bias to predominantly mature 3p strands through loss of 5p strand cleavage. J. Pathol. 2013, 229, 400–409. doi:10.1002/path.4135. [Google Scholar]
-
Pugh TJ, Yu W, Yang J, Field AL, Ambrogio L, Carter SL, et al. Exome sequencing of pleuropulmonary blastoma reveals frequent biallelic loss of TP53 and two hits in DICER1 resulting in retention of 5p-derived miRNA hairpin loop sequences. Oncogene 2014, 33, 5295–5302. doi:10.1038/onc.2014.150. [Google Scholar]
-
Stocker JT, Madewell JE, Drake RM. Congenital cystic adenomatoid malformation of the lung. Classification adn morphologic spectrum. Hum. Pathol. 1977, 8, 155–171. doi:10.1016/s0046-8177(77)80078-6. [Google Scholar]
-
Dehner LP, Messinger YH, Williams GM, Stewart DR, Harney LA, Schultz KA, et al. Type I pleuropulmonary blastoma versus congenital pulmonary airway malformation Type IV. Neonatology 2017, 111, 76. doi:10.1159/000447992. [Google Scholar]
-
Langston C. New concepts in the pathology of congenital lung malformations. Semin. Pediatr. Surg. 2003, 12, 17–37. doi:10.1053/spsu.2003.00001. [Google Scholar]
-
Pogoriler J, Swarr D, Kreiger P, Adzick NS, Peranteau W. Congenital cystic lung lesions: Redefining the natural distribtuion of subtypes and assessing the risk of malignancy. Am. J. Surg. Pathol. 2019, 43, 47–55. doi:10.1097/PAS.0000000000000992. [Google Scholar]
-
Garinet S, Rahshenas M, Galmiche-Rolland L, Abbo O, Bonnard A, Hameury F, et al. Cancer-associated mutations in congenital pulmonary malformations: A prospective cohort. Am. J. Respir. Crit. Care Med. 2023, 207, 615–619. doi:10.1164/rccm.202208-1573LE. [Google Scholar]
-
Barazzone-Argiroffo C, Lascano Maillard J, Vidal I, Bochaton-Piallat ML, Blaskovic S, Donati Y, et al. New insights on congenital pulmonary airways malformations revealed by proteomic analyses. Orphanet J. Rare Dis. 2019, 14, 272. doi:10.1186/s13023-019-1192-4. [Google Scholar]
-
Landry-Truchon K, Houde N, Lhuillier M, Charron L, Hadchouel A, Delacourt C, et al. Deletion of Yy1 in mouse lung epithelium unveils molecular mechanisms governing pleuropulmonary blastoma pathogenesis. Dis. Model. Mech. 2020, 13, dmm045989. doi:10.1242/dmm.045989. [Google Scholar]
-
Lantuejoul S, Nicholson AG, Sartori G, Piolat C, Danel C, Brabencova E, et al. Mucinous cells in type 1 pulmonary congenital cystic adenomatoid malformation as mucinous bronchioalveolar carcinoma precursors. Am. J. Surg. Pathol. 2007, 31, 961–969. doi:10.1097/01.pas.0000249444.90594.27. [Google Scholar]
-
Schweitzer S, Stiller M, Lacher M, Aubert O, Krâmer S, Hirsch FW, et al. Comprehensive morphological, immunohistochemical, and molecular characterization of congenital pulmonary airway malformation (CPAM). Virchows Arch. 2025. doi:10.1007/s00428-025-04131-4. [Google Scholar]
-
Boucherat O, Landry-Truchon K, Bérubé-Simard FA, Houde N, Beuret L, Lezmi G, et al. Epithelial inactivation of Yy1 abrogates lung branching morphogenesis. Development 2015, 142, 2981–2995. doi:10.1242/dev.120469. [Google Scholar]
-
Yin Y, Castro AM, Hoekstra M, Yan TJ, Kanakamedala C, Dehner LP, et al. Fibroblat growth factor 9 regulation by microRNAs controls lung development and links DICER1 loss to the pathogenesis of pleuropulmonary blastoma. PLoS Genet. 2015, 11, e1005242. doi:10.1371/journal.pgen.1005242. [Google Scholar]
-
Murray MJ, Bailey S, Raby KL, Saini HK, de Kock L, Burke GA, et al. Serum levels of mature microRNAs in DICER1-mutated pleuropulmonary blastoma. Oncogenesis 2014, 3, e87. doi:10.1038/oncsis.2014.1. [Google Scholar]
-
Greenfield A, Scott D, Pennisi D, Ehrmann I, Ellis P, Cooper L, et al. An H-YDb epitope is encoded by a novel mouse Y chromosome gene. Nat. Genet. 1996, 14, 474–478. doi:10.1038/ng1296-474. [Google Scholar]
-
Boucherat O, Nadeau V, Charron J, Jeannotte L. Crucial requirement of ERK/MAPK signaling in respiratory tract development. Development 2014, 141, 3197–3211. doi:10.1242/dev.110254. [Google Scholar]
-
Bankhead P, Loughrey MB, Fernandez JA, Dombrowski Y, McArt DG, Dunne PD, et al. QuPath: Open source software for digital pathology image analysis. Sci. Rep. 2017, 7, 16878. doi:10.1038/s41598-017-17204-5. [Google Scholar]
-
Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. doi:10.1093/bioinformatics/bts635. [Google Scholar]
-
Martin FJ, Amode MR, Aneja A, Austine-Orimoloye O, Azov AG, Barnes I, et al. Ensembl 2023. Nucleic Acids Res. 2023, 51, D933–D941. doi:10.1093/nar/gkac958. [Google Scholar]
-
Liao Y, Smyth GK, Shi W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. doi:10.1093/bioinformatics/btt656. [Google Scholar]
-
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. doi:10.1186/s13059-014-0550-8. [Google Scholar]
-
Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. doi:10.1073/pnas.0506580102. [Google Scholar]
-
Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. doi:10.1038/nmeth.1923. [Google Scholar]
-
Danecek P, Bonfield JK, Liddle J, Marshall J, Ohan V, Pollard MO, et al. Twelve years of SAMtools and BCFtools. GigaScience 2021, 10, giab008. doi:10.1093/gigascience/giab008. [Google Scholar]
-
Quinlan AR, Hall IM. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. doi:10.1093/bioinformatics/btq033. [Google Scholar]
-
Guéno J, Borg M, Bourdareau S, Cossard G, Godfroy O, Lipinska A, et al. Chromatin landscape associated with sexual differentiation in a UV sex determination system. Nucleic Acids Res. 2022, 50, 3307–3322. doi:10.1093/nar/gkac145. [Google Scholar]
-
Vedanayagam J, Chatila WK, Aksoy BA, Majumbar S, Skanderup AJ, Demir E, et al. Cancer-associated mutations in DICER1 RNase IIIa and IIIb domains exert similar effects on miRNA biogenesis. Nat. Commun. 2019, 10, 3682. doi:10.1038/s41467-019-11610-1. [Google Scholar]
-
Tate JG, Bamford S, Jubb HC, Sondka Z, Beare DM, Bindal N, et al. COSMIC: The catalogue of somatic mutations in cancer. Nucleic Acids Res. 2019, 47, D941–D947. doi:10.1093/nar/gky1015. [Google Scholar]
-
Martínez-Jiménez F, Muiños F, Sentís I, Deu-Pons J, Reyes-Salazar I, Arnedo-Pac C, et al. A compendium of mutational cancer driver genes. Nat. Rev. Cancer 2020, 20, 555–572. doi:10.1038/s41568-020-0290-x. [Google Scholar]
-
Zhang G, Lou L, Shen L, Zeng H, Cai C, Wu R, et al. The underlying molecular mechanisms of ciliated epithelium dysfunction and TGF-ß signaling in children with congenital pulmonary airway malformations. Sci. Rep. 2024, 14, 4430. doi:10.1038/s41598-024-54924-x. [Google Scholar]
-
Swarr DT, Peranteau WH, Pogoriler J, Frank DB, Adzick NS, Hedrick HL, et al. Novel molecular and phenotypic insights into congenital lung malformations. Am. J. Respir. Crit. Care Med. 2018, 197, 1328–1339. doi:10.1164/rccm.201706-1243OC. [Google Scholar]
-
Zhang G, Cai C, Li X, Lou L, Zhou B, Zeng H, et al. Application of second-generation sequencing in congenital pulmonary airway malformations. Sci. Rep. 2022, 12, 20459. doi:10.1038/s41598-022-24858-3. [Google Scholar]
-
Que J, Okubo T, Goldenring JR, Nam KT, Kurotani R, Morrisey EE, et al. Multiple dose-dependent roles for Sox2 in the patterning and differentiation of anterior foregut endoderm. Development 2007, 134, 2521–2531. doi:10.1242/dev.003855. [Google Scholar]
-
Ochieng JK, Schilders K, Kool H, Boerema-De Munck A, Buscop-Van Kempen M, Gontan C, et al. Sox2 regulates the emergence of lung basal cells by directly activating the transcription of Trp63. Am. J. Respir. Cell Mol. Biol. 2014, 51, 311–322. doi:10.1165/rcmb.2013-0419OC. [Google Scholar]
-
Gontan C, de Munck A, Vermeij M, Grosveld F, Tibboel D, Rottier R. Sox2 is important for two crucial processes in lung development: Branching morphogenesis and epithelial cell differentiation. Dev. Biol. 2008, 317, 296–309. doi:10.1016/j.ydbio.2008.02.035. [Google Scholar]
-
Abatti LE, Lado-Fernández P, Huynh L, Collado M, Hoffman MM, Mitchell JA. Epigenetic reprogramming of a distal developmental enhancer cluster drives SOX2 overexpression in breast and lung carcinoma. Nucleic Acids Res. 2023, 52, 10109–10131. doi:10.1093/nar/gkad734. [Google Scholar]
-
De Lope C, Martín-Alonso S, Auzmendi-Iriarte J, Escudero C, Mulet I, Larrasa-Alonso J, et al. SIX1 represses senescence and promotes SOX2-mediated cellular plasticity during tumorigenesis. Sci. Rep. 2019, 9, 1412. doi:10.1038/s41598-018-38176-0. [Google Scholar]
-
Besnard V, Wert SE, Kaestner KH, Whitsett JA. Stage-specific regulation of respiratory epithelial cell differentiation by Foxa1. Am. J. Physiol. Lung Cell Mol. Physiol. 2005, 289, L750–L759. doi:10.1152/ajplung.00151.2005. [Google Scholar]
-
El-Hashash AH, Al Alam D, Turcatel G, Rogers O, Li X, Bellusci S, et al. Six1 transcription factor is critical for coordination of epithelial, mesenchymal and vascular morphogenesis in the mammalian lung. Dev. Biol. 2011, 353, 242–258. doi:10.1016/j.ydbio.2011.02.031. [Google Scholar]
-
Chang WC, Zhang YZ, Wolf JL, Hermelijn SM, Schnater JM, von der Thüsen JH, et al. Mucinous adenocarcinoma arising in congenital pulmonary airway malformation: Clinicopathological analysis of 37 cases. Histopathology 2021, 78, 434–444. doi:10.1111/his.14239. [Google Scholar]
-
Han G, Sinjab A, Rahal Z, Lynch AM, Treekitkarnmongkol W, Liu Y, et al. An atlas of epithelial cell states and plasticity in lung adenocarcinoma. Nature 2024, 627, 656–663. doi:10.1038/s41586-024-07113-9. [Google Scholar]
-
Kadota K, Yeh YC, D’Angelo SP, Moreira AL, Kuk D, Sima CS, et al. Associations between mutations and histologic patterns of mucin in lung adenocarcinoma: Invasive mucinous pattern and extracellular mucin are associated with KRAS mutation. Am. J. Surg. Pathol. 2014, 38, 1118–1127. doi:10.1097/PAS.0000000000000246. [Google Scholar]
-
Muntean A, Banias LE, Ade-Ajayi N, Patel SB, McKinney O, Davenport M. Neonatal congenital pulmonary airway malformation associated with mucinous adenocarcinoma and KRAS mutations. J. Pediatr. Surg. 2022, 57, 520–526. doi:10.1016/j.jpedsurg.2021.12.018. [Google Scholar]
-
Singh G, Coffey A, Neely R, Lambert D, Sonett J, Borczuk AC, et al. Pulmonary Kirsten Rat Sarcoma virus mutation positive mucinous adenocarcinoma arising in a congenital pulmonary airway malformation, mixed type 1 and 2. Ann. Thorac. Surg. 2016, 102, e335–e337. doi:10.1016/j.athoracsur.2016.01.104. [Google Scholar]
-
Nelson ND, Xu F, Chandrasekaran P, Litzky LA, Peranteau WH, Frank DB, et al. Defining the spatial landscape of KRAS mutated congenital pulmonary airway malformations: A distinct entity with a spectrum of histopathologic features. Mod. Pathol. 2022, 35, 1870–1881. doi:10.1038/s41379-022-01129-0. [Google Scholar]
-
Morrisey EE, Hogan BL. Preparing for the first breath: Genetic and cellular mechanisms in lung development. Dev. Cell 2010, 18, 8–23. doi:10.1016/j.devcel.2009.12.010. [Google Scholar]
-
Minoo P, Su G, Drum H, Bringas P, Kimura S. Defects in tracheoesophageal and lung morphogenesis in Nkx2.1(−/−) mouse embryos. Dev. Biol. 1999, 209, 60–71. doi:10.1006/dbio.1999.9234. [Google Scholar]
-
Keijzer R, van Tuyl M, Meijers C, Post M, Tibboel D, Grosveld F, et al. The transcription factor GATA6 is essential for branching morphogenesis and epithelial cell differentiation during fetal pulmonary development. Development 2001, 128, 503–511. doi:10.1242/dev.128.4.503. [Google Scholar]
-
Ke X, van Soldt B, Vlahos L, Zhou Y, Qian J, Laise P, et al. Morphogenesis and regeneration share a conserved core transition cell state program that controls lung epithelial cell fate. Dev. Cell 2025, 60, 2533. doi:10.1016/j.devcel.2025.08.014. [Google Scholar]
-
Krumlauf R. Hox genes in vertebrate development. Cell 1994, 78, 191–201. doi:10.1016/0092-8674(94)90290-9. [Google Scholar]
-
Frenette B, Guéno J, Houde N, Landry-Truchon K, Giguère A, Ashok T, et al. Loss of Hoxa5 function affects Hox gene expression in different biological contexts. Sci. Rep. 2024, 14, 30903. doi:10.1038/s41598-024-81867-0. [Google Scholar]
-
Cao R, Wang L, Wang H, Xia L, Erdjument-Bromage H, Tempst P, et al. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science 2002, 298, 1039–1043. doi:10.1126/science.1076997. [Google Scholar]
-
Schuettengruber B, Bourbon HM, Di Croce L, Cavalli G. Genome regulation by Polycomb and Trithorax: 70 years and counting. Cell 2017, 171, 34–57. doi:10.1016/j.cell.2017.08.002. [Google Scholar]
-
Bannister AJ, Schneider R, Myers FA, Thorne AW, Crane-Robinson C, Kouzarides T. Spatial distribution of di- and tri-methyl lysine 36 of histone H3 at active genes. J. Biol. Chem. 2005, 280, 17732–17736. doi:10.1074/jbc.M500796200. [Google Scholar]
-
Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. USA 2010, 107, 21931–21936. doi:10.1073/pnas.1016071107. [Google Scholar]
-
Alturkustani M, Yang B, Bockoven C, Mahabir R, Shillingford N, Schmidt RJ, et al. Histone H3 trimethylation on lysine 27 immunostaining pattern in DICER1-assocaited tumors. Trans. Pediatr. 2024, 13, 624–633. doi:10.21037/tp-24-61. [Google Scholar]
-
Aparicio-Puerta E, Hirsch P, Schmartz GP, Kern F, Fehlmann T, Keller A. miEAA 2023: Updates, new functional microRNA sets and improved enrichment visualizations. Nucleic Acids Res. 2023, 51, W319–W325. doi:10.1093/nar/gkad392. [Google Scholar]
-
Boyer LA, Plath K, Zeitlinger J, Brambrink T, Medeiros LA, Lee TI, et al. Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature 2006, 441, 349–353. doi:10.1038/nature04733. [Google Scholar]
-
Lee W, Teckie S, Wiesner T, Ran L, Prieto Granada CN, Lin M, et al. PRC2 is recurrently inactivated through EED or SUZ12 loss in malignant peripheral nerve sheath tumors. Nat. Genet. 2014, 46, 1227–1232. doi:10.1038/ng.3095. [Google Scholar]
-
Serresi M, Gargiulo G, Proost N, Siteur B, Cesaroni M, Koppens M, et al. Polycomb repressive complex 2 is a barrier to KRAS-driven inflammation and epithelial-mesenchymal transition in non-small-cell lung cancer. Cancer Cell 2016, 29, 17–31. doi:10.1016/j.ccell.2015.12.006. [Google Scholar]
-
Peruzzi P, Bronisz A, Nowicki MO, Wang Y, Ogawa D, Price R, et al. MicroRNA-128 coordinately targets Polycomb Repressor Complexes in glioma stem cells. Neuro-oncology 2013, 15, 1212–1224. doi:10.1093/neuonc/not055. [Google Scholar]
-
Xu Q, Wang K, Ahsan S, Rodriguez F, Eberhart C, Jiang Y, et al. miR-148-3p regulation of OTX2 and PRC2 in group 3 and 4 medulloblastoma. Cancer Res. 2017, 77, 1471. doi:10.1158/1538-7445.AM2017-1471. [Google Scholar]
-
Fukuoka M, Fujita H, Numao K, Nakamura Y, Shimizu H, Sekiguchi M, et al. MiR-199-3p enhances muscle regeneration and ameliorates aged muscle and muscular dystrophy. Commun. Biol. 2021, 4, 421. doi:10.1038/s42003-021-01952-2. [Google Scholar]
-
Zhang Y, Teng F, Luo GZ, Wang M, Tong M, Zhao X, et al. MicroRNA-323-3p regulates the activity of Polycomb repressice complex 2 (PRC2) via targeting the mRNA of embryonic ectoderm development (Eed) gene in mouse embryonic stem cells. J. Biol. Chem. 2013, 288, 23659–23665. doi:10.1074/jbc.M113.475608. [Google Scholar]
-
Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, et al. Tissue-based map of the human proteome. Science 2015, 347, 1260419. doi:10.1126/science.1260419. [Google Scholar]
-
Li X, Oghi KA, Zhang J, Krones A, Bush KT, Glass CK, et al. Eya protein phosphatase activity regulates Six1-Dach-Eya transcriptional effects in mammalian organogenesis. Nature 2003, 426, 247–254. doi:10.1038/nature02083. [Google Scholar]
-
Lu K, Reddy R, Berika M, Warburton D, El-Hashash AHK. Abrogation of Eya1/Six1 disrupts the saccular phase of lung morphogenesis and causes remodeling. Dev. Biol. 2013, 382, 110–123. doi:10.1016/j.ydbio.2013.07.019. [Google Scholar]
-
Lezmi G, Verkarre V, Khen-Dunlop N, Vibhushan S, Hadchouel A, Rambaud C, et al. FGF10 signaling differences between type I pleuropulmonary blastoma and congenital cystic adenomatoid malformation. Orphanet J. Rare Dis. 2013, 8, 130. doi:10.1186/1750-1172-8-130. [Google Scholar]
-
Xu X, Rock JR, Lu Y, Futtner C, Schwab B, Guinney J, et al. Evidence for type II cells as cells of origin of K-Ras-induced distal lung adenocarcinoma. Proc. Natl. Acad. Sci. USA 2012, 109, 4910–4915. doi:10.1073/pnas.1112499109. [Google Scholar]
-
Ogawa F, Walters MS, Shafquat A, O’Beirne SL, Kaner RJ, Mezey JG, et al. Role of KRAS in regulating normal human airway basal cell differentiation. Respir. Res. 2019, 20, 181. doi:10.1186/s12931-019-1129-4. [Google Scholar]
-
Shaw AT, Meissner A, Dowdle JA, Crowley D, Magendantz M, Ouyang C, et al. Sprouty-2 regulates oncogenic K-ras in lung development and tumorigenesis. Genes Dev. 2007, 21, 694–707. doi:10.1101/gad.1526207. [Google Scholar]
-
Pelletier D, Sabbaghian N, Chong AL, Priest JR, Elsheikh Ahmed Y, Fox GP, et al. Extraskeletal chondroma of the toe in a child with DICER1 tumor predisposition syndrome: Support for a dominant negative mechanism. Virchows Arch. 2024, 484, 1023–1027. doi:10.1007/s00428-024-03759-y. [Google Scholar]
-
Zhang CX, Huang RY, Sheng G, Thiery JP. Epithelial-mesenchymal transition. Cell 2025, 188, 5436–5486. doi:10.1016/j.cell.2025.08.033. [Google Scholar]
-
Montgomery ND, Yee D, Chen A, Kalantry S, Chamberlain SJ, Otte AP, et al. The murine polycomb group protein Eed is required for global histone H3 lysine-27 methylation. Curr. Biol. 2005, 15, 942–947. doi:10.1016/j.cub.2005.04.051. [Google Scholar]
-
Abate-Shen C. Deregulated homeobox gene expression in cancer: Cause or consequence? Nat. Rev. Cancer 2002, 2, 777–785. doi:10.1038/nrc907. [Google Scholar]
-
Feng Y, Zhang T, Wang Y, Xie M, Ji X, Luo X, et al. Homeobox genes in cancers: From carcinogenesis to recent therapeutic intervention. Front. Oncol. 2021, 11, 770428. doi:10.3389/fonc.2021.770428. [Google Scholar]
-
Guerra SL, Maertens O, Kuzmickas R, De Raedt T, Adeyemi RO, Guild CJ, et al. A deregulated HOX gene axis confers an epigenetic vulnerability in KRAS-mutant lung cancers. Cancer Cell 2020, 37, 705–719. doi:10.1016/j.ccell.2020.03.004. [Google Scholar]