Novel Therapeutic Targets of Endothelial Inflammation in Acute Lung Injury and Acute Respiratory Distress Syndrome

Novel Therapeutic Targets of Endothelial Inflammation in Acute Lung Injury and Acute Respiratory Distress Syndrome
Yunchao Su
1,2,3,4,*

Received: 13 December 2025 Accepted: 14 January 2026 Published: 06 February 2026

© 2026 The authors. This is an open access article under the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/).
Acute lung injury (ALI)/acute respiratory distress syndrome (ARDS), characterized by diffuse lung microvascular inflammation, endothelial dysfunction and pulmonary edema, is a clinical syndrome caused by severe organ injury, infection or sepsis [1,2]. Despite recent scientific advances, the mortality of ALI/ARDS is still extremely high because molecular mechanisms involved in ALI/ARDS remain obscure. Therefore, there is an urgent need to identify potential therapeutic targets for the treatment of ALI/ARDS.
In a recent issue of the journal Advanced Science, Baoyinna et al. [3] reported that deubiquitinase USP30 is a critical regulator of lung microvascular inflammation and endothelial barrier integrity in ALI/ARDS (Figure 1). They found that endothelial-specific USP30-deficient mice exhibit reduced lung endothelial inflammation and microvascular barrier dysfunction in models of endotoxin-induced and ischemia-reperfusion lung injury. They discovered a novel mechanism by which endotoxin induces inflammatory responses in ALI/ARDS. The data revealed that USP30 deubiquitinates and stabilizes methionine adenosyltransferase 2A (MAT2A), leading to increased S-adenosylmethionine (SAM) and DNA methylation, and reduced miR-30a-5p expression in ALI/ARDS. Their results further indicate that miR-30a-5p suppresses the expression of mouse double minute 2 homolog (MDM2) and nuclear factor of activated T cells 5 (NFAT5), thereby alleviating endothelial inflammation and protecting endothelial barrier function. Their findings provide a strong rationale for targeting microRNAs, S-adenosylmethionine, DNA methylation and deubiquitinating enzymes for potential therapeutic strategies in ALI/ARDS (Figure 1).
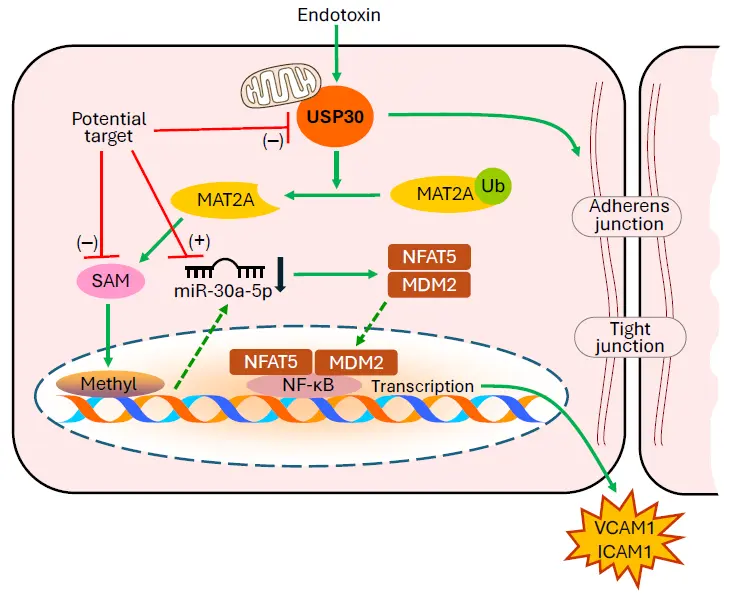
Figure 1. MicroRNAs, S-adenosylmethionine, DNA methylation, and deubiquitinating enzymes are potential targets of endothelial inflammation for therapeutic strategies in the treatment of ALI/ARDS. USP30, ubiquitin-specific peptidase 30; MAT2A, methionine adenosyltransferase 2A; SAM, S-adenosylmethionine; NFAT5, nuclear factor of activated T cells 5; MDM2, mouse double minute 2 homolog; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; VCAM1, vascular cell adhesion molecule 1; ICAM1, intercellular adhesion molecule 1.
1. MicroRNAs
MicroRNAs target specific mRNAs to regulate gene expression, either by blocking translation or promoting mRNA degradation in cells, thereby influencing endothelial cell homeostasis by modulating processes such as cell monolayer permeability, cell apoptosis, and the inflammatory response in the pathogenesis of ALI/ARDS [4]. miRNAs are significantly upregulated or downregulated in lung endothelial cells or other lung cells and play injurious or protective roles in lung endothelial inflammation [5]. For example, miR-34a-5p, miR-1246 and miR-92a are upregulated in endotoxin-treated pulmonary microvascular endothelial cells and promote endothelial damage and inflammation in ALI [6,7,8]. miR-34a-5p has been found to target histone deacetylase 1 (Sirt1) gene [6], miR-1246 to target the angiotensin-converting enzyme 2 (Ace2) gene [7], and miR-92a to target integrin a5 (Itga5) gene [8]. These microRNAs could be potential therapeutic targets and diagnostic biomarkers of ALI/ARDS [9,10,11]. miR-339-3p, miR-539-5p and miR-33 are down-regulated in endotoxin-stimulated lung microvascular endothelial cells and play a protective role in endothelial inflammation in ALI [12]. miR-339-3p was confirmed to target annexin A3 gene [12,13], miR-539-5p to target Rho kinase (Rock) gene [13], miR-33 to target receptor interacting protein 140 (Rip140) gene [14]. Over-expressions of miR-339-3p, miR-539-5p and miR-33 alleviates lung endothelial dysfunction, inflammation, and vascular injury in ALI/ARDS. In the study of Baoyinna et al. [3], they reported that miR-30a-5p overexpression downregulated the expression of co-transcriptional factor NFAT5 and MDM2 and inhibited endotoxin-induced expression of vascular cell adhesion molecule 1 (VCAM1) and intercellular adhesion molecule 1 (ICAM1) in pulmonary microvascular endothelial cells, suggesting that miR-30a-5p is a protective microRNA that alleviates endothelial inflammation and barrier dysfunction. Together with miR-339-3p, miR-539-5p and miR-33, these protective microRNAs could be used as novel pharmacologic agents in ALI/ARDS.
2. S-Adenosylmethionine and DNA Methylation
DNA methylation is an epigenetic regulatory mechanism that involves the transfer of a methyl group to the C5 position of cytosine at CpG dinucleotide sites, forming 5-methylcytosine in the mammalian genome. Genomic DNA methylation is a dynamic process of methylation and demethylation through DNA methyltransferases (DNMTs) and demethylases. The primary methyl donor for DNA methylation is S-adenosylmethionine (SAM), a species generated in the methionine cycle of one-carbon metabolism [15]. In ALI/ARDS, DNA methylation suppresses gene expression, resulting in pathophysiologic changes in endothelial inflammation and permeability [16,17]. Alterations or manipulations of DNMTs and demethylases would influence inflammatory response and lung vascular barrier function [18,19,20]. Another mechanism for the regulation of DNA methylation is through the modification of the level of methyl donor SAM [21]. Baoyinna and colleagues found that the reduction of intracellular SAM level caused by MAT2A degradation (inhibition) mitigates DNA methylation of the gene for miR-30a-5p, which leads to down-regulation of inflammation and endothelial permeability [3]. Targeting the SAM cycle and DNA methylation could be a novel therapeutic strategy for acute inflammatory diseases. Interestingly, SAM is also the methyl donor for histone methyltransferase [22], and the SAM level has an impact on histone methylation and gene expression in lung inflammation [23]. Baoyinna and colleagues did not study whether the reduction of intracellular SAM level caused by MAT2A degradation (inhibition) mitigates histone methylation in ALI/ARDS. Further studies are needed to disclose more epigenetic mechanisms of lung inflammation and endothelial barrier regulation.
3. E3 Ubiquitin Ligases and Deubiquitinating Enzymes
Protein ubiquitination links ubiquitin to target proteins via E3 ubiquitin ligases, leading to protein degradation via the ubiquitin-proteasome system. Deubiquitinating enzymes (DUBs) can reverse the ubiquitination process by removing the ubiquitin chain from the target protein. The equilibrium between ubiquitination and deubiquitination is essential for maintaining intracellular protein homeostasis and signaling [24]. Ubiquitination and deubiquitination can directly modulate the lung endothelial barrier by controlling the stability and expression of proteins in adherens junction (AJ) and tight junction (TJ), as well as small Rho GTPases that adjust the actin cytoskeleton in lung microvascular endothelium in ALI/ARDS [25,26,27]. Hakai, an E3 ubiquitin ligase, mediates E-cadherin ubiquitination and degradation [28]. β-Catenin is ubiquitinated by the E3 ubiquitin ligase β-transducin repeat-containing protein (β-TrCP) and then degraded by the proteasome [29]. Itch, a HECT domain-containing E3 ubiquitin ligase, interacts and ubiquitinates occludin in the tight junction, resulting in occludin degradation by the proteasome [30]. Several E3 ubiquitin ligases, such as Smurf1, Cullin3/BACURD, Fbxl19, and Fbxw7, have been identified as regulators of RhoA ubiquitination and stability [31]. Two DUBs, OTUB1 and USP17, have been shown to regulate RhoA ubiquitination and degradation [25].
Ubiquitination and deubiquitination regulate the NLRP3 inflammasome and NF-κB (nuclear factor kappa-light-chain-enhancer of activated B cells) activation in lung inflammation in ALI/ARDS [32,33]. E3 ubiquitin ligases target NLRP3 proteins or their constituents to degrade NLRP3 inflammasomes. In endotoxin-induced ALI model, the expression of E3 ubiquitin ligases Pellino2, WW domain-containing E3 ubiquitin ligase protein 1 (WWP1) and E3 ubiquitin ligase adaptor BTB/POZ domain-containing protein 2 (BPOZ-2) was found to be downregulated [34,35,36]. Pellino2, WWP1 and BPOZ-2 promote ubiquitination of NLRP3 inflammasomes, thereby downregulating the expression of NLRP3 inflammasomes, IL-1β and TNF-α proteins [34,35,36]. In ALI/ARDS, YAP, a transcriptional co-activator, interacts with the E3 ubiquitin ligase TRAF6 (tumor necrosis factor receptor-associated factor 6) to promote its degradation via K48-linked ubiquitination and to inhibit K63-linked autoubiquitination in ECs, thereby inhibiting NF-κB activation [37].
Different DUBs act on their specific target proteins and exert different roles in lung inflammation [33]. Several DUBs such as BRCC3, ABRO1, USP7, and USP47 have been shown to be implicated in NLRP3 inflammasome activation [38]. USP9X was found to be upregulated and participate in the pathogenesis of ALI by promoting NLRP3 inflammasome deubiquitination and activation [39]. USP14 protein is upregulated, which reduces I-κB protein levels and thus increases cytokine release in lung inflammation [40]. On the other hand, USP7 acts as a negative regulator of the NF-κB pathway by mediating the deubiquitination of NEMO, TRAF6, and IKK, thereby retaining NF-κB in the cytosol and suppressing its activity and the expression of inflammatory cytokines [41]. In the study by Baoyinna et al. [3], USP30 was inhibited using siRNA in vitro and endothelial-specific gene knockout in vivo. Inhibition of USP30 reduced MAT2A ubiquitination and degradation, DNA methylation, and increased miR-30a-5p expression, thereby ameliorating vascular leakage, cytokine production, and lung inflammation in ALI/ARDS. USP30 may represent a potential therapeutic target warranting further preclinical and clinical trials in ALI/ARDS.
Ethics Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Funding
This work was supported, in whole or in part, by NIH grants HL134934 and HL158909 to Y.S., and by the Department of Veterans Affairs BX005350 to Y.S.
Declaration of Competing Interest
The author declares that he has no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
-
Zhou K, Qin Q, Lu J. Pathophysiological mechanisms of ARDS: A narrative review from molecular to organ-level perspectives. Respir. Res. 2025, 26, 54. DOI:10.1186/s12931-025-03137-5 [Google Scholar]
-
Su Y, Lucas R, Fulton DJR, Verin AD. Mechanisms of pulmonary endothelial barrier dysfunction in acute lung injury and acute respiratory distress syndrome. Chin. Med. J. Pulm. Crit. Care Med. 2024, 2, 80–87. DOI:10.1016/j.pccm.2024.04.002 [Google Scholar]
-
Baoyinna B, He J, Miao J, Shaheen N, Xia B, Wang C, et al. Activation of USP30 Disrupts Endothelial Cell Function and Aggravates Acute Lung Injury Through Regulating the S-Adenosylmethionine Cycle. Adv. Sci. (Weinh. Baden-Wurtt. Ger.) 2025, 13, e12807. DOI:10.1002/advs.202512807 [Google Scholar]
-
Wang P, Lai D, Jin L, Xue Y. Roles of microRNAs in acute lung injury and acute respiratory distress syndrome: Mechanisms and clinical potential. Front. Immunol. 2025, 16, 1570128. DOI:10.3389/fimmu.2025.1570128 [Google Scholar]
-
Lu Q, Yu S, Meng X, Shi M, Huang S, Li J, et al. MicroRNAs: Important Regulatory Molecules in Acute Lung Injury/Acute Respiratory Distress Syndrome. Int. J. Mol. Sci. 2022, 23, 5545. DOI:10.3390/ijms23105545 [Google Scholar]
-
Shah D, Das P, Alam MA, Mahajan N, Romero F, Shahid M, et al. MicroRNA-34a Promotes Endothelial Dysfunction and Mitochondrial-mediated Apoptosis in Murine Models of Acute Lung Injury. Am. J. Respir. Cell Mol. Biol. 2019, 60, 465–477. DOI:10.1165/rcmb.2018-0194OC [Google Scholar]
-
Fang Y, Gao F, Hao J, Liu Z. microRNA-1246 mediates lipopolysaccharide-induced pulmonary endothelial cell apoptosis and acute lung injury by targeting angiotensin-converting enzyme 2. Am. J. Transl. Res. 2017, 9, 1287–1296. [Google Scholar]
-
Xu F, Zhou F. Inhibition of microRNA-92a ameliorates lipopolysaccharide-induced endothelial barrier dysfunction by targeting ITGA5 through the PI3K/Akt signaling pathway in human pulmonary microvascular endothelial cells. Int. Immunopharmacol. 2020, 78, 106060. DOI:10.1016/j.intimp.2019.106060 [Google Scholar]
-
Jiang ZF, Zhang L, Shen J. MicroRNA: Potential biomarker and target of therapy in acute lung injury. Hum. Exp. Toxicol. 2020, 39, 1429–1442. DOI:10.1177/0960327120926254 [Google Scholar]
-
Lee LK, Medzikovic L, Eghbali M, Eltzschig HK, Yuan X. The Role of MicroRNAs in Acute Respiratory Distress Syndrome and Sepsis, From Targets to Therapies: A Narrative Review. Anesth. Analg. 2020, 131, 1471–1484. DOI:10.1213/ANE.0000000000005146 [Google Scholar]
-
Wang Q, Huang Y, Fu Z. Bone mesenchymal stem cell-derived exosomal miR-26a-3p promotes autophagy to attenuate LPS-induced apoptosis and inflammation in pulmonary microvascular endothelial cells. Cell. Mol. Biol. 2024, 70, 104–112. DOI:10.14715/cmb/2024.70.2.15 [Google Scholar]
-
Wu XM, Ji KQ, Wang HY, Zhao Y, Jia J, Gao XP, et al. MicroRNA-339-3p alleviates inflammation and edema and suppresses pulmonary microvascular endothelial cell apoptosis in mice with severe acute pancreatitis-associated acute lung injury by regulating Anxa3 via the Akt/mTOR signaling pathway. J. Cell Biochem. 2018, 119, 6704–6714. DOI:10.1002/jcb.26859 [Google Scholar]
-
Meng L, Cao H, Wan C, Jiang L. MiR-539-5p alleviates sepsis-induced acute lung injury by targeting ROCK1. Folia Histochem. Cytobiol. 2019, 57, 168–178. DOI:10.5603/FHC.a2019.0019 [Google Scholar]
-
Li H, Hou H, Liu S, Feng Y, Zhong W, Hu X, et al. miR-33 and RIP140 participate in LPS-induced acute lung injury. Turk. J. Med. Sci. 2019, 49, 422–428. DOI:10.3906/sag-1804-173 [Google Scholar]
-
Zhang N. Role of methionine on epigenetic modification of DNA methylation and gene expression in animals. Anim. Nutr. 2018, 4, 11–16. DOI:10.1016/j.aninu.2017.08.009 [Google Scholar]
-
Bossardi Ramos R, Adam AP. Molecular Mechanisms of Vascular Damage During Lung Injury. Adv. Exp. Med. Biol. 2021, 1304, 95–107. DOI:10.1007/978-3-030-68748-9_6 [Google Scholar]
-
Zhang XQ, Lv CJ, Liu XY, Hao D, Qin J, Tian HH, et al. Genome‑wide analysis of DNA methylation in rat lungs with lipopolysaccharide‑induced acute lung injury. Mol. Med. Rep. 2013, 7, 1417–1424. DOI:10.3892/mmr.2013.1405 [Google Scholar]
-
Samanta S, Zhou Z, Rajasingh S, Panda A, Sampath V, Rajasingh J. DNMT and HDAC inhibitors together abrogate endotoxemia mediated macrophage death by STAT3-JMJD3 signaling. Int. J. Biochem. Cell Biol. 2018, 102, 117–127. DOI:10.1016/j.biocel.2018.07.002 [Google Scholar]
-
Lu CH, Chen CM, Ma J, Wu CJ, Chen LC, Kuo ML. DNA methyltransferase inhibitor alleviates bleomycin-induced pulmonary inflammation. Int. Immunopharmacol. 2020, 84, 106542. DOI:10.1016/j.intimp.2020.106542 [Google Scholar]
-
Shih CC, Hii HP, Tsao CM, Chen SJ, Ka SM, Liao MH, et al. Therapeutic Effects of Procainamide on Endotoxin-Induced Rhabdomyolysis in Rats. PLoS ONE 2016, 11, e0150319. DOI:10.1371/journal.pone.0150319 [Google Scholar]
-
Glier MB, Green TJ, Devlin AM. Methyl nutrients, DNA methylation, and cardiovascular disease. Mol. Nutr. Food Res. 2014, 58, 172–182. DOI:10.1002/mnfr.201200636 [Google Scholar]
-
Serefidou M, Venkatasubramani AV, Imhof A. The Impact of One Carbon Metabolism on Histone Methylation. Front. Genet. 2019, 10, 764. DOI:10.3389/fgene.2019.00764 [Google Scholar]
-
Dai Y, Chen J, Duan Q. Epigenetic mechanism of EZH2-mediated histone methylation modification in regulating ferroptosis of alveolar epithelial cells in sepsis-induced acute lung injury. Drug Dev. Res. 2024, 85, e22263. DOI:10.1002/ddr.22263 [Google Scholar]
-
Suresh B, Lee J, Kim KS, Ramakrishna S. The Importance of Ubiquitination and Deubiquitination in Cellular Reprogramming. Stem Cells Int. 2016, 2016, 6705927. DOI:10.1155/2016/6705927 [Google Scholar]
-
Cai J, Culley MK, Zhao Y, Zhao J. The role of ubiquitination and deubiquitination in the regulation of cell junctions. Protein Cell 2018, 9, 754–769. DOI:10.1007/s13238-017-0486-3 [Google Scholar]
-
Wang Y, Zhan Y, Wang L, Huang X, Xin HB, Fu M, et al. E3 Ubiquitin Ligases in Endothelial Dysfunction and Vascular Diseases: Roles and Potential Therapies. J. Cardiovasc. Pharmacol. 2023, 82, 93–103. DOI:10.1097/FJC.0000000000001441 [Google Scholar]
-
Magnani ND, Dada LA, Sznajder JI. Ubiquitin-proteasome signaling in lung injury. Transl. Res. 2018, 198, 29–39. DOI:10.1016/j.trsl.2018.04.003 [Google Scholar]
-
Fujita Y, Krause G, Scheffner M, Zechner D, Leddy HE, Behrens J, et al. Hakai, a c-Cbl-like protein, ubiquitinates and induces endocytosis of the E-cadherin complex. Nat. Cell Biol. 2002, 4, 222–231. DOI:10.1038/ncb758 [Google Scholar]
-
Zhang L, Zhou F, Li Y, Drabsch Y, Zhang J, van Dam H, et al. Fas-associated factor 1 is a scaffold protein that promotes beta-transducin repeat-containing protein (beta-TrCP)-mediated beta-catenin ubiquitination and degradation. J. Biol. Chem. 2012, 287, 30701–30710. DOI:10.1074/jbc.M112.353524 [Google Scholar]
-
Murakami T, Felinski EA, Antonetti DA. Occludin phosphorylation and ubiquitination regulate tight junction trafficking and vascular endothelial growth factor-induced permeability. J. Biol. Chem. 2009, 284, 21036–21046. DOI:10.1074/jbc.M109.016766 [Google Scholar]
-
Zhao J, Mialki RK, Wei J, Coon TA, Zou C, Chen BB, et al. SCF E3 ligase F-box protein complex SCF(FBXL19) regulates cell migration by mediating Rac1 ubiquitination and degradation. FASEB J. 2013, 27, 2611–2619. DOI:10.1096/fj.12-223099 [Google Scholar]
-
Tang S, Geng Y, Wang Y, Lin Q, Yu Y, Li H. The roles of ubiquitination and deubiquitination of NLRP3 inflammasome in inflammation-related diseases: A review. Biomol. Biomed. 2024, 24, 708–721. DOI:10.17305/bb.2023.9997 [Google Scholar]
-
Li T, Zou C. The Role of Deubiquitinating Enzymes in Acute Lung Injury and Acute Respiratory Distress Syndrome. Int. J. Mol. Sci. 2020, 21, 4842. DOI:10.3390/ijms21144842 [Google Scholar]
-
Liu X, Lin Z, Yin X. Pellino2 accelerate inflammation and pyroptosis via the ubiquitination and activation of NLRP3 inflammation in model of pediatric pneumonia. Int. Immunopharmacol. 2022, 110, 108993. DOI:10.1016/j.intimp.2022.108993 [Google Scholar]
-
Zhang S, Guan X, Liu W, Zhu Z, Jin H, Zhu Y, et al. YTHDF1 alleviates sepsis by upregulating WWP1 to induce NLRP3 ubiquitination and inhibit caspase-1-dependent pyroptosis. Cell Death Discov. 2022, 8, 244. DOI:10.1038/s41420-022-00872-2 [Google Scholar]
-
Li J, Lin H, Fan T, Huang L, Zhang X, Tai Y, et al. BPOZ-2 is a negative regulator of the NLPR3 inflammasome contributing to SARS-CoV-2-induced hyperinflammation. Front. Cell Infect. Microbiol. 2023, 13, 1134511. DOI:10.3389/fcimb.2023.1134511 [Google Scholar]
-
Lv Y, Kim K, Sheng Y, Cho J, Qian Z, Zhao YY, et al. YAP Controls Endothelial Activation and Vascular Inflammation Through TRAF6. Circ. Res. 2018, 123, 43–56. DOI:10.1161/CIRCRESAHA.118.313143 [Google Scholar]
-
Lopez-Castejon G. Control of the inflammasome by the ubiquitin system. FEBS J. 2020, 287, 11–26. DOI:10.1111/febs.15118 [Google Scholar]
-
Xiang Y, Li X, Cai M, Cai D. USP9X promotes lipopolysaccharide-stimulated acute lung injury by deubiquitination of NLRP3. Cell Biol. Int. 2023, 47, 394–405. DOI:10.1002/cbin.11932 [Google Scholar]
-
Mialki RK, Zhao J, Wei J, Mallampalli DF, Zhao Y. Overexpression of USP14 protease reduces I-kappaB protein levels and increases cytokine release in lung epithelial cells. J. Biol. Chem. 2013, 288, 15437–15441. DOI:10.1074/jbc.C112.446682 [Google Scholar]
-
Li T, Guan J, Li S, Zhang X, Zheng X. HSCARG downregulates NF-kappaB signaling by interacting with USP7 and inhibiting NEMO ubiquitination. Cell Death Dis. 2014, 5, e1229. DOI:10.1038/cddis.2014.197 [Google Scholar]