Found 13 results
Open Access
Review
12 January 2026The Anti-Fibrotic Potential of GLP-1 and GIP Receptor Agonists in Chronic Inflammatory Disorders: Mechanisms and Therapeutic Horizons
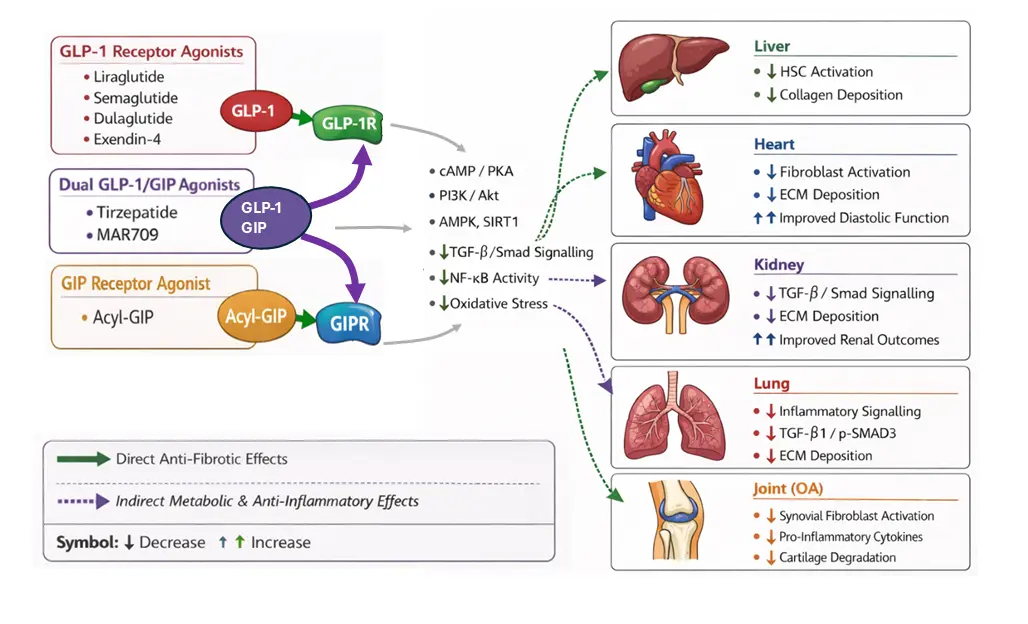
Fibrosis, characterised by the excessive deposition of extracellular matrix via activated fibroblasts, is a pathological feature of several chronic inflammatory disorders, which collectively contribute significantly to global morbidity and mortality. Despite this, current anti-fibrotic therapies are of limited efficacy. However, incretin-based therapies, primarily glucagon-like peptide-1 (GLP-1) receptor agonists, are now emerging as candidate drugs for modulating fibrotic signalling pathways. This review synthesises the growing body of preclinical and clinical evidence that incretin receptor agonists exert direct and indirect anti-fibrotic effects. We detail the molecular mechanisms and survey the promising data across hepatic, cardiac, renal, lung, and joint tissues, which underscore the potential for repurposing of this drug class as a therapeutic strategy for fibro-inflammatory conditions.
Open Access
Commentary
20 October 2025Sulfatide Inhibits Growth of Fibroblasts and Is a Potential Treatment against Fibrosis
Fibrosis of vital organs such as the lungs, liver, and kidneys is a serious condition without effective causal treatment. Here, we suggest the use of the sphingolipid sulfatide and its isoform C16, which we have found to inhibit the growth of fibroblasts. In the lungs, sulfatide can be easily administered via an inhalation spray. Alternatively, fenofibrate, an anti-cholesterol drug with no major side effects, may be used, as it enhances the body’s own production of sulfatide.

Open Access
Article
09 October 2025Identification of Pathways That Drive Myofibroblast Transformation in Hypertrophic Scars
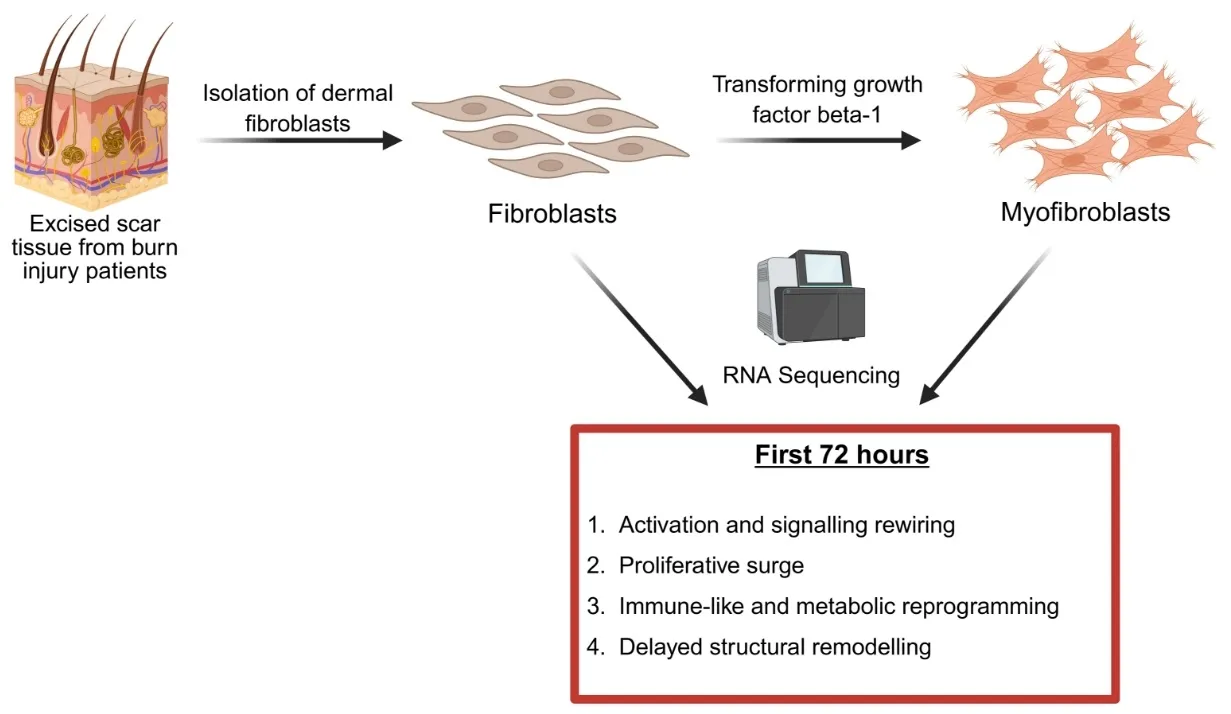
Hypertrophic scars (HTS) are a common complication of burn injuries and are characterized by excessive dermal fibrosis driven by the transformation of resident dermal fibroblasts to profibrotic myofibroblasts. Although single cell and bulk RNA transcriptomics analysis of HTS and normal skin tissue samples were performed previously, transcriptomics of the transformation of fibroblasts to myofibroblasts has not been studied. Here, we report the data obtained from RNA sequencing of fibroblasts before and after exposure to transforming growth factor beta 1 (TGF-β1) and highlight the pathways that are up- and down-regulated during myofibroblast transformation. Our results suggest increased cellular signalling and rewiring, proliferative surge, immune-like and metabolic reprogramming, and delayed structural remodelling as four groups of events during the transformation of human primary dermal fibroblasts to myofibroblasts.
Open Access
Review
27 June 2025Fibroblast Migration in Fibrosis
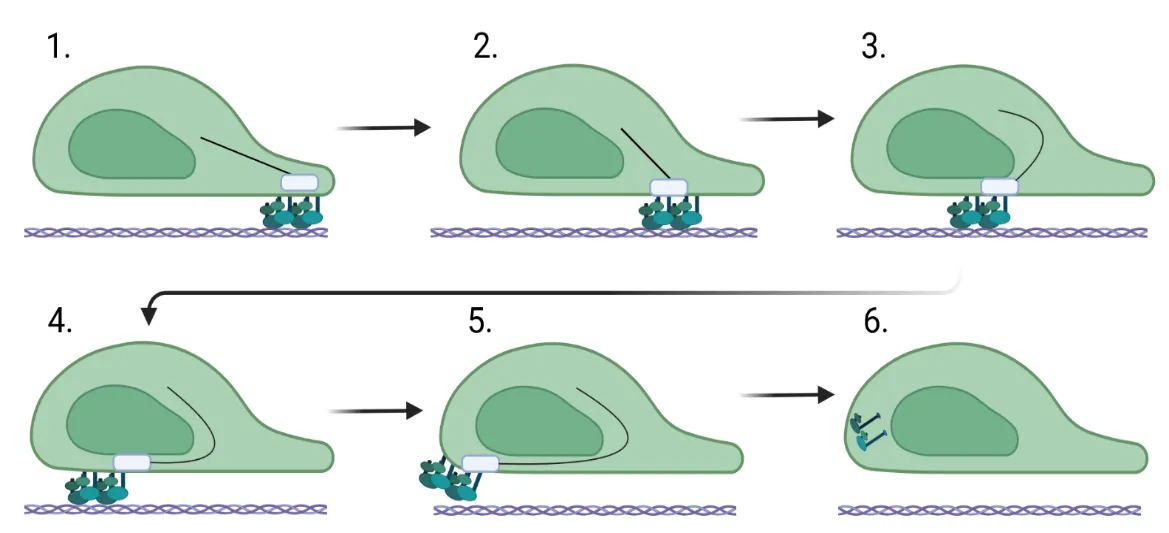
Fibroblast migration is a critical factor in wound healing, but also plays a fundamental role in fibrosis. For a fibroblast to migrate, the cell must be able to assemble factors that help it crawl across the extracellular matrix. Most of this movement is facilitated through the assembly and stability of the cytoskeleton that connects focal adhesion engagement with the extracellular matrix to intracellular stress fibers that wrap around the nucleus. These intracellular stress fibers help to polarize the fibroblast and orient the nucleus in the direction it is traveling. Changes in intracellular signaling for the fibroblast to move are also required, and this is necessitated by downstream signaling mediated by sonic hedgehog, WNT/β-catenin, ROCK/Rho, and PI3K/AKT. These changes regulate the stability of the cytoskeleton and, in addition, increase the expression of genes involved in cell migration. This review assimilates what is known about the function of the cytoskeleton in migration and the role of intracellular signaling pathways in fibrosis.
Open Access
Review
22 May 2025NLRP3 Inflammasome and IL-11 in Systemic Sclerosis Pulmonary Fibroblasts
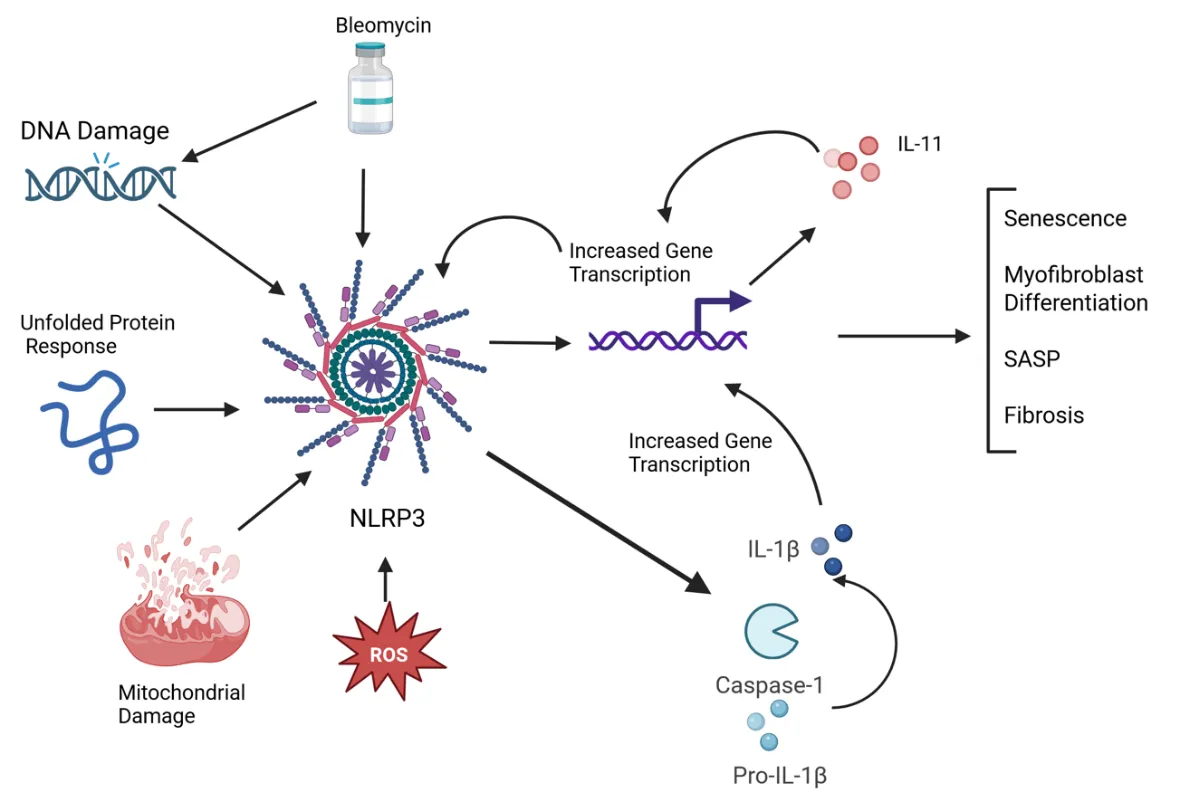
Systemic sclerosis (SSc) is an autoimmune disease characterized by widespread fibrosis affecting multiple organ systems. There is clinical heterogeneity among patients with SSc in terms of the organs affected. However, the pathophysiology of the disease remains elusive. The NLRP3 inflammasome is upregulated in SSc and exerts its fibrotic effects through activation of caspase-1, which in turn activates a fibrotic signaling cascade, resulting in increased collagen deposition and myofibroblast transition. Recently, IL-11 has been shown to be elevated in disease and has been shown to participate in downstream signaling via the NLRP3 inflammasome. A significant number of patients with SSc will develop pulmonary involvement, termed interstitial lung disease (SSc-ILD). Though this type of pulmonary involvement is distinct from other types of pulmonary fibrosis (such as idiopathic pulmonary fibrosis), it may be a valuable model to study mechanisms of fibrosis that could apply to other fibrotic diseases. Here, we discuss recent advances in understanding the mechanisms of the NLRP3 inflammasome and IL-11 in SSc pulmonary fibroblasts. We tie together some of the recent findings, such as senescence, the unfolded protein response, and reactive oxygen species, that contribute to fibrotic pathology via modulating NLRP3 activation, possibly leading to IL-11 expression.
Open Access
Review
07 March 2025Mechanisms and Therapeutic Potential of Myofibroblast Transformation in Pulmonary Fibrosis
Idiopathic pulmonary fibrosis (IPF) is a progressive, irreversible, and fatal disease with an increasing incidence and limited therapeutic options. It is characterized by the formation and deposition of excess extracellular matrix proteins resulting in the gradual replacement of normal lung architecture by fibrous tissue. The cellular and molecular mechanism of IPF has not been fully understood. A hallmark in IPF is pulmonary fibroblast to myofibroblast transformation (FMT). During excessive lung repair upon exposure to harmful stimuli, lung fibroblasts transform into myofibroblasts under stimulation of cytokines, chemokines, and vesicles from various cells. These mediators interact with lung fibroblasts, initiating multiple signaling cascades, such as TGFβ1, MAPK, Wnt/β-catenin, NF-κB, AMPK, endoplasmic reticulum stress, and autophagy, contributing to lung FMT. Furthermore, single-cell transcriptomic analysis has revealed significant heterogeneity among lung myofibroblasts, which arise from various cell types and are adapted to the altered microenvironment during pathological lung repair. This review provides an overview of recent research on the origins of lung myofibroblasts and the molecular pathways driving their formation, with a focus on the interactions between lung fibroblasts and epithelial cells, endothelial cells, and macrophages in the context of lung fibrosis. Based on these molecular insights, targeting the lung FMT could offer promising avenues for the treatment of IPF.

Open Access
Review
22 November 2024Dermal Fibrosis and the Current Scope of Hydrogel Strategies for Scarless Wound Healing
Dermal fibrosis poses a significant challenge in wound healing, affecting both the appearance and functionality of the scarred skin tissue. Beyond aesthetic implications, fibrosis can lead to pruritus, chronic pain, loss of mechanical flexibility, and impeded restoration of skin appendages, blood vessels, and nerves. Therefore, scar prevention remains a priority in wound management, and hydrogels, with their hydrophilic three-dimensional network and extracellular matrix-mimicking properties, have emerged as promising biomaterials for achieving scarless wound regeneration. In this review, we explore advancements in various hydrogel strategies designed to regulate myofibroblast differentiation, control the wound microenvironment, and mitigate dermal fibrosis. We provide an overview of dermal fibrosis, the scar-forming cells involved, and the various types of dermal scars. We then summarise advancements made in antifibrotic hydrogel formulations, emphasizing their practical applications in scarless skin wound healing. By reviewing the current research landscape and highlighting key hydrogel-based biomaterial strategies employed in this field, we aim to offer insights into design principles and underlying mechanisms of action. We intend for this review to serve as a valuable resource for researchers and clinicians interested in entering this field or exploring the potential of hydrogels to promote scarless wound healing.
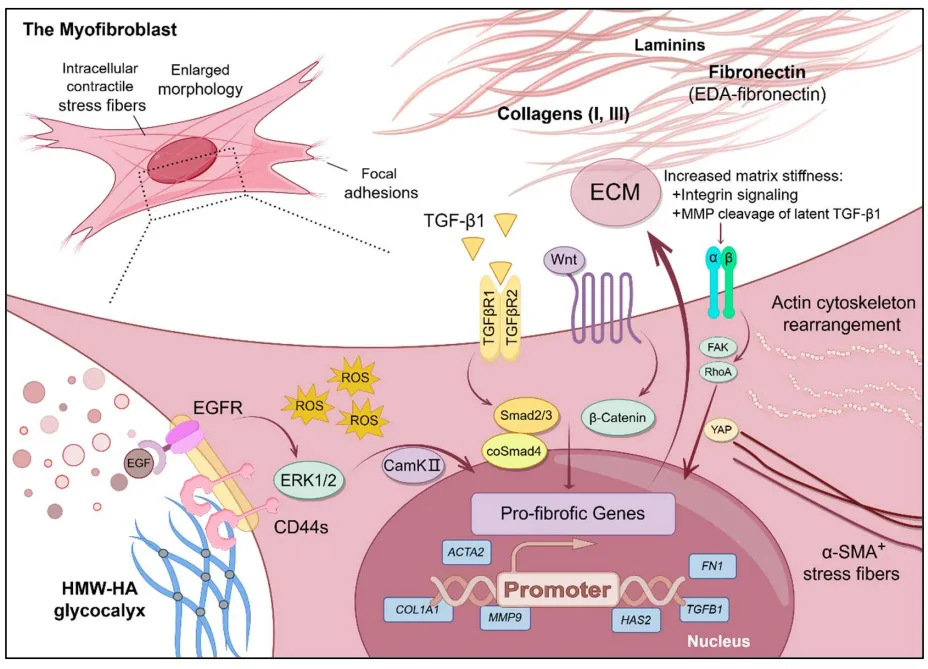
Open Access
Review
23 October 2024The Notch3 Pathway in Organ Fibrosis
Fibrosis occurs in many organs, including the lung, heart, skin, liver or kidney, and is characterized by progressive tissue scarring in response to repetitive or chronic non-resolving injury, ultimately leading to organ failure and death. It is, in fact, a major cause of morbidity and mortality worldwide, being estimated to account for 45% of deaths in the world. Despite this fact, little progress has been made therapeutically, and fibrosis remains a major clinical and therapeutic challenge. Although significant advances in our understanding of cellular and molecular mechanisms driving tissue fibrosis have been made, the lack of an efficient treatment reflects the limited insight into the pathophysiological mechanisms underlying the initiation and progression of the fibrotic process. Thus, there is an urgent need for better understanding of tissue fibrosis and repair mechanisms that later lead to the development of new therapeutic approaches to fight fibrosis. The Notch pathway is a highly conserved signaling pathway that has been linked to tissue fibrosis in many organs and promises to open new therapeutic opportunities. This manuscript reviews the relevance of Notch signaling in the development and progression of tissue fibrosis in several organs with a special focus on the Notch3 pathway due to the unique features of this receptor.
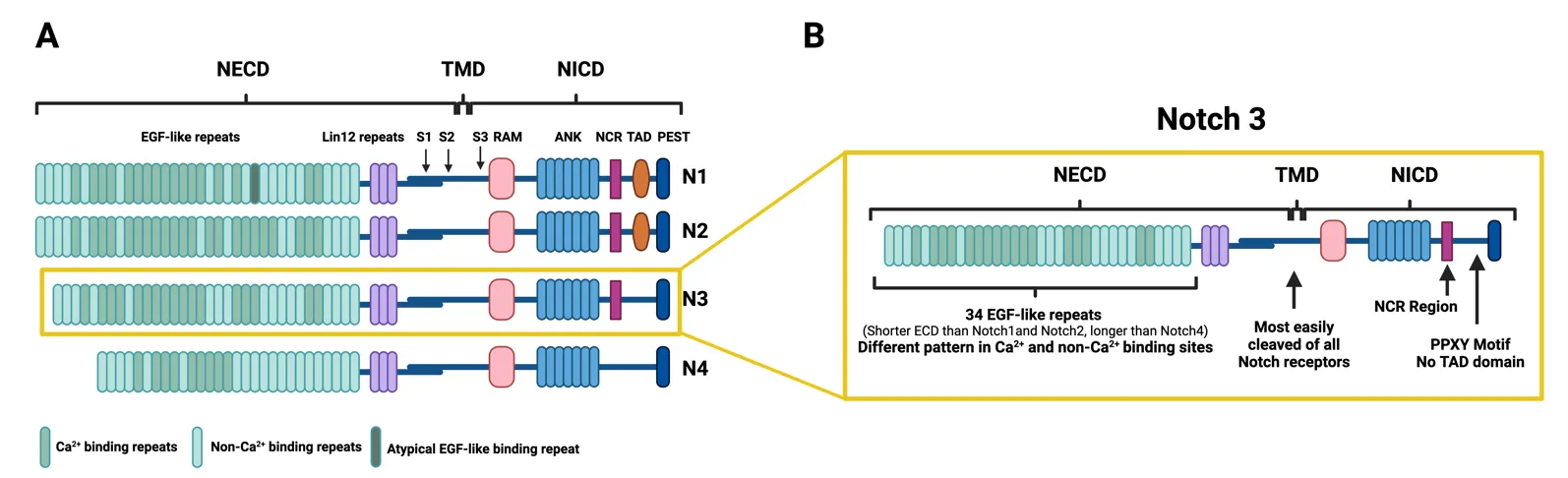
Open Access
Review
26 February 2024Mechanisms of Fibroblast Activation during Fibrotic Tissue Remodeling
Fibrosis can occur in almost every organ system. It can occur in single organs, such as in idiopathic pulmonary fibrosis (IPF), or affect multiple organs as in systemic sclerosis (SSc). Fibrotic diseases are recognized as major cause of morbidity and mortality in modern societies due to the dysfunction or loss of function of the affected organs. This dysfunction is caused by progressive deposition of extracellular matrix proteins released by activated fibroblasts. Activation of fibroblasts and differentiation into myofibroblasts is required for physiological tissue remodeling, e.g, during wound healing. Disruption of regulatory mechanisms, however, results in chronic and uncontrolled activity of fibroblasts and myofibroblasts. Intensive research during the past years identified several core pathways of pathophysiological relevance, and described different fibroblast subsets based on their expression profile in fibrotic tissue. Herein, we discuss the molecular changes in fibroblasts leading to persistent activation during fibrotic tissue remodeling with a focus on lung fibrosis and SSc.
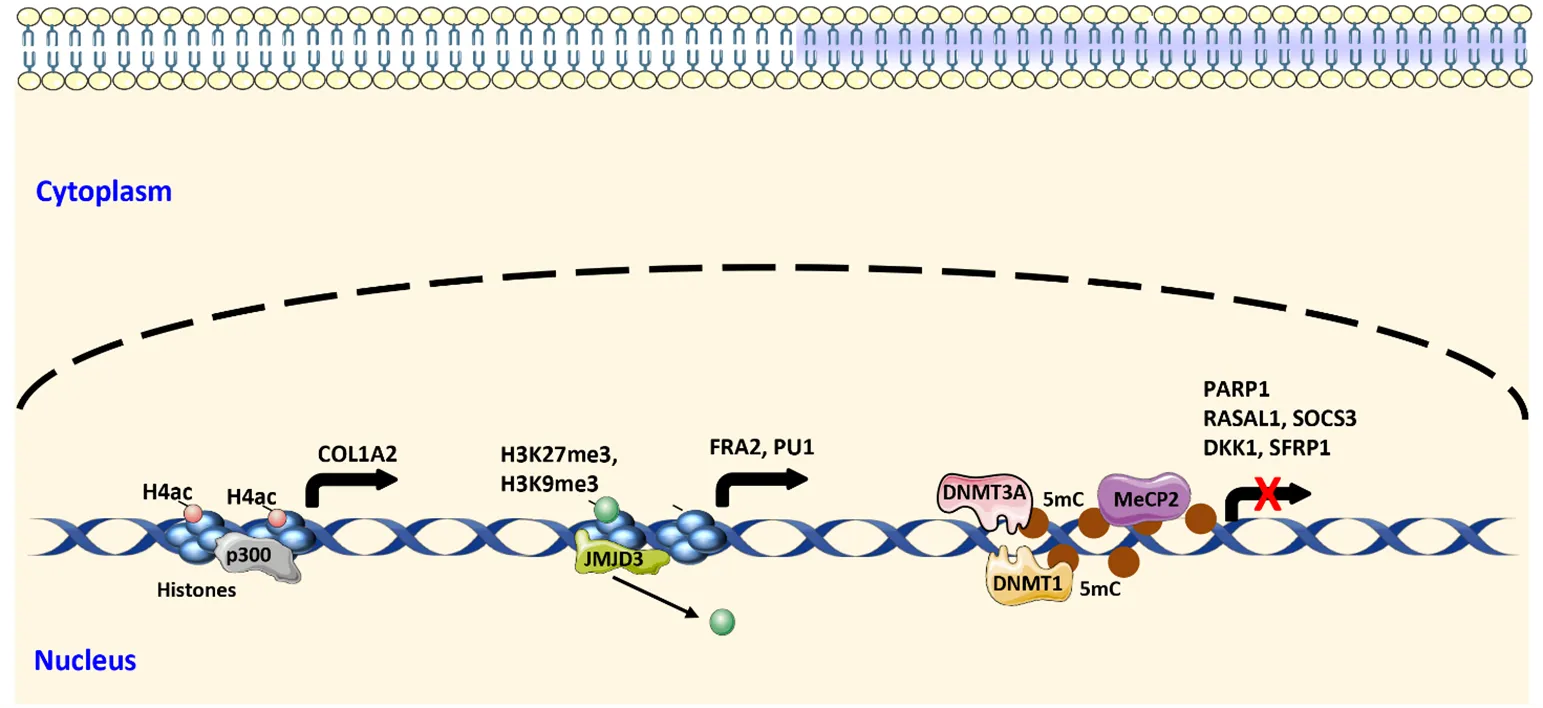
Open Access
Meeting Report
20 February 2024The Cellular and Metabolic Bases of Organ Fibrosis: UNIA Workshop 2023 in Baeza, Spain
Fibrosis is defined by scarring and tissue hardening caused by excess deposition of extracellular matrix components, mainly collagens. A fibrotic response can occur in any tissue of the body and is the final outcome of an unbalanced reaction to inflammation and wound healing induced by a variety of insults, including persistent infections, autoimmune reactions, allergic responses, chemical exposure, radiation, and tissue injury. The accumulation of extracellular matrix proteins replaces the living tissue and disrupts the architecture leading to organ malfunction. Fibrosis remains a major clinical and therapeutic challenge and has been estimated to account for 45% of deaths in the developed world. While major advances regarding mechanistic knowledge on the underlying cell biology alterations in fibrosis have helped to characterize the main phases and mediators involved, this knowledge has not yielded significant progress in treatment. Only recently, the metabolic features associated to fibrosis have begun to emerge. This information, likely representing only the tip of the iceberg, suggests that metabolic derangement is a key culprit in the pathophysiology of fibrogenesis. The Workshop on The Cellular and Metabolic Bases of Organ Fibrosis, International University of Andalusia, Baeza, Spain, October 8–11, 2023 aimed to discuss the current knowledge and novel perspectives on the mechanisms contributing to the development of fibrosis in different organs and tissues, with particular focus on new methodological developments in metabolomics and therapeutic strategies.
