1. Introduction
Fibrosis is defined as excessive accumulation of extracellular matrix (ECM) proteins, mainly consisting of collagen whose thickened fibers are highly crosslinked. The massive accumulation of matrix components disrupts the physiological architecture leading to dysfunction and finally loss of function of the affected organ. Fibrosis may occur in single organs such as idiopathic pulmonary fibrosis (IPF) or affect multiple organs as in systemic sclerosis (SSc). Systemic sclerosis, a systemic subform of scleroderma, is a rare, chronic, immune-mediated rheumatic disease characterized by vascular abnormalities, immunological alterations and fibrosis of the skin and visceral organs. SSc is associated with high morbidity and mortality and classified into two major subsets: limited cutaneous SSc (lcSSc) with restricted fibrotic lesions at the limbs distal to elbows and knees, and diffuse cutaneous SSc (dcSSc) with fibrotic lesions also affecting the proximal part of the limbs and the trunk [
1,
2]. The most prominent feature of SSc is progressive fibrosis, resulting from excessive ECM protein accumulation [
1]. The exact cause of SSc is unknown, but it is likely to involve environmental factors in genetically susceptible individuals [
3]. The pathophysiology is incompletely understood so far, but it is generally accepted that vasculopathy and autoimmunity precede fibroblast activation and fibrosis [
4].
In addition, aberrant fibrotic tissue remodeling contributes to morbidity and mortality in many other diseases such as liver cirrhosis, atherosclerosis, chronic obstructive pulmonary disease (COPD), and inflammatory bowel disease. Furthermore, it has been linked to tumor invasion and metastasis, and chronic graft rejection. Consequently, fibrosis has been recognized as major health care burden in industrial societies with an estimated contribution to deaths of up to 45 % in the developed world [
5].
Myofibroblasts are the key cellular mediators of wound healing and fibrosis in all tissues [
6]. They are a heterogeneous cell subset, commonly defined by the co-expression of fibroblast specific markers and the contractile protein α-smooth muscle actin (αSMA) [
6], and with the capacity to release ECM proteins [
7]. According to literature, myofibroblast differentiation is initially dependent on external stimuli such as cytokines. However, upon prolonged exposure to these stimuli, myofibroblasts remain persistently activated and escape regulatory mechanisms [
5,
8]. Under normal conditions, fibrogenesis is a tightly regulated process during wound healing and tissue repair. During tissue injury and granulation tissue formation, local fibroblasts as well as other cells migrate toward the wound centre. In the same time, myofibroblasts appear, secrete ECM proteins and facilitate wound contraction to prompt wound repair [
9]. When the wound is closed and the damage repaired, myofibroblasts disappear through different mechanisms [
10]. In fibrotic diseases, the regulatory mechanisms are disrupted leading to an uncontrolled and subsequent self-amplifying activation of fibroblasts and myofibroblasts. Of note, myofibroblasts accumulate in all fibrotic-related diseases, highlighting the cardinal role of myofibroblasts in fibrotic diseases [
11]. Different cell types can differentiate into myofibroblasts including resident fibroblasts, pericytes, endothelial cells, epithelial cells, and smooth muscle cells amongst others [
12].
This review article summarizes selected pathways and mechanisms of fibroblast activation with subsequent myofibroblast differentiation with an emphasis on SSc.
2. Core Pathways of Fibrotic Tissue Remodelling
Defined by Mehal et al, core pathways are pathways, which are crucial to turn initial stimuli into the development of fibrosis and are shared across different organs and fibrotic diseases [
13]. The permanent signaling activity via these core pathways keeps fibroblasts active and drives fibroblast-to-myofibroblast transition. In the past years, several signaling cascades were identified to be core pathways. Some of them will be presented in this section.
2.1. Transforming Growth Factor Beta (TGFβ) Pathway
TGFβ signaling is a generally accepted key regulator of fibrotic tissue remodeling. The pluripotent growth factor acts in many physiological and pathological tissue responses. The latent form of TGFβ can be released from different cell types such as platelets, T cells, and fibroblasts amongst others. Upon secretion, TGFβ is stored in the ECM by binding to latency-associated peptide (LAP) and latent TGFβ-binding protein (LTBP). Increased release of latent TGFβ from this reservoir by integrins is a major mechanism of aberrant TGFβ signaling during fibrogenesis [
14,
15]. Upon activation, TGFβ binds to TGFβ receptor type II (TGFBR2), which then dimerizes with TGFBR1 to induce signaling. The main intracellular mediators of “canonical” TGFβ signaling are SMAD proteins, which activate the transcription of a plethora of pro-fibrotic genes [
16]. In addition to SMAD signaling, TGFβ can activate several other “non-canonical” pathways signaling via mitogen-activated protein kinases (MAPKs), RhoA-ROCK or cAbl as examples [
16]. The activation of this number of different intracellular signaling cascades enables also cross-activation and regulation at different levels leading to a vicious cycle of permanent pro-fibrotic pathway activation.
2.2. Developmental Pathways
Developmental pathways, or so called morphogen or stem cell pathways, are tightly regulated pathways due to their importance in embryonic organ development and adult tissue homeostasis. The central or “classical” stem cell pathways include the WNT pathway, Hedgehog signaling, and the Notch signaling cascade. Loss of the tight control of those pathways leads to aberrant signaling, which is contributing to the pathogenesis of several diseases. These three mentioned pathways can cross-regulate each other and are also interlinked with TGFβ signaling [
17].
WNT signaling is divided into “canonical” (β-catenin-dependent) and “non-canonical” (β-catenin-independent) signaling. Both parts are implicated in fibroblast activation, however, most studies so far investigated canonical WNT signaling. The aberrant signaling is mainly driven by increased expression and release of WNT proteins, repression of endogenous inhibitors and stabilization and nuclear accumulation of β-catenin in fibroblasts and other cell types [
18,
19,
20,
21]. The WNT proteins WNT1, WNT3a, and WNT10b are capable of inducing fibroblast activation and transdifferentiation to myofibroblasts
in vitro [
18,
21,
22,
23], whereas WNT10b is sufficient to induce fibroblast activation and fibrosis
in vivo [
23]. Also WNT8b has recently been found to be overexpressed in resident pulmonary mesenchymal cells driving them to differentiation into myofibroblasts [
24]. In addition, downregulation of endogenous WNT inhibitors such as dickkopf WNT signaling pathway inhibitor 1 (DKK1), secreted frizzled related protein 1 (SFRP1), and WNT inhibitory factor 1 (WIF1) contributes to the accumulation of β-catenin in the nucleus of fibroblasts [
18,
19,
20,
21]. WNT signaling can be cross-activated by the TGFβ pathway directly by repressing the expression of endogenous inhibitors [
18,
19] or indirectly by regulating the expression of signaling mediators that control the signaling activity of β-catenin [
22]. One such mediator is X-linked inhibitor of apoptosis protein (XIAP), which is upregulated by TGFβ. XIAP then binds transducin-like enhancer of split 3 (TLE3), and thus preventing it from blocking the β-catenin-TCF/LEF transcription factor assembly [
22]. Non-canonical WNT signaling is less studied, however, the ligand WNT5A has been shown to be dysregulated in different cell types in IPF [
25,
26].
In parallel to the WNT pathway, the hedgehog signaling cascade is also divided into “canonical” and “non-canonical” signaling. The canonical cascade is activated by binding of hedgehog ligands SHH (sonic hedgehog), IHH (indian hedgehog), and DHH (desert hedgehog) to the receptor patched (PTCH) releasing smoothend (SMO) from its inhibition leading to subsequent activation of GLI transcription factors [
27]. The activation of GLI proteins independent of ligands and SMO is considered as non-canonical signaling. In dermal and lung fibroblasts, SHH induces accumulation of active GLI2, myofibroblast differentiation, and collagen release [
28,
29]. However, also TGFβ can induce GLI2 expression and activation during fibroblast activation [
29,
30].
Notch signaling mainly relies on direct cell-cell contact with one cell expressing the single-pass transmembrane receptor and the neighboring cell expressing the ligand. Four Notch receptors and five ligands have been described in mammals: Notch1-4 and Jagged-1 (JAG1), JAG2, and Delta-like 1 (DLL1), DLL3, and DLL4 [
31]. In addition to the membrane-bound ligands, also soluble “non-canonical” ligands have been described [
32]. Aberrant Notch signaling has been implicated in SSc and IPF amongst other fibrotic diseases. Increased Notch1 signaling with enhanced expression of receptor and ligands has been demonstrated in fibroblasts in skin of SSc patients [
33,
34,
35], and in patients with IPF [
36]. Consistently, fibroblast-specific targeting of Notch1 signaling has been shown to ameliorate experimental pulmonary fibrosis [
37]. Additionally, active Notch1 signaling is inducing the differentiation of lung pericytes into myofibroblasts [
38].
2.3. Nuclear Receptors
Nuclear receptors are transcriptional regulators and are summarized in a superfamily comprising 48 members in humans [
39]. Several nuclear receptors have been implicated to be dysregulated in fibroblasts during fibrogenesis. One of the most studied in this context seems to be peroxisome proliferator activated receptor gamma (PPARγ, also known as NR1C3). The expression of PPARγ is severely reduced in pulmonary fibrosis and in fibrotic tissue and in cultured dermal fibroblasts explanted from SSc patients [
40,
41,
42,
43]. The downregulation of PPARγ contributes to the amplification of TGFβ signaling, which in turn is responsible for the repression of PPARγ [
41,
43,
44]. Activation of PPARγ blocks the pro-fibrotic effects of TGFβ-SMAD signaling, but treatment with selective PPARγ agonists is accompanied by severe side effects, such as bone fractures and an increased risk for cardiovascular events. Nevertheless, the use of panPPAR agonists showed promising effects in pre-clinical studies. Lanifibranor ameliorated both bleomycin-induced lung and dermal fibrosis, and was also effective in lung fibrosis in mice with fibroblast-specific transgenic expression of kinase-deficient TGFBR2 [
40]. Derret-Smith et al demonstrate in their study that panPPAR activation not only ameliorated pulmonary fibrosis, but also pulmonary hypertension in these mice [
40].
Another nuclear receptor being investigated in fibrogenesis is the vitamin D receptor (VDR or NR1I1). Deficiency of vitamin D is shared across different fibrotic diseases and VDR has been shown to be downregulated in skin and cultured fibroblasts from SSc patients [
45]. Treatment with vitamin D not only reduces the outcome of experimental pulmonary and dermal fibrosis, but also restores TGFβ-induced downregulation of VDR expression [
45,
46].
Systemic sclerosis is more prevalent in women than in men, with men having a higher risk of a severe phenotype, suggesting that female hormones play a role during pathogenesis [
47,
48]. A study by Avouac et al. demonstrates that mice deficient for the estrogen receptor alpha (ERα) are more susceptible to bleomycin-induced dermal fibrosis via increased SMAD signaling. Also hypodermal thickening in Tsk1 mice is exacerbated upon inhibition of ER by tamoxifen. Furthermore, incubation of human dermal fibroblasts with estrogens abrogated the pro-fibrotic effects of TGFβ in cultured human dermal fibroblasts. However, analysis of the expression of ERα and ERβ in cultured fibroblasts from control subjects and SSc patients showed only a trend towards decreased expression of ERβ, whereas the expression of ERα was not altered in SSc dermal fibroblasts [
47]. However, other studies showed an increased pro-fibrotic response of fibroblasts on incubation with estrogen with higher ECM production [
49,
50].
3. Activation by Immune Cells
Different cell types of the immune system have been implicated in the pathogenesis of fibrotic diseases in general and SSc in particular [
51,
52]. By releasing cytokines, they contribute to the activation of fibroblasts. Particularly, interleukin-4 (IL4), IL6 and TGFβ have been demonstrated to directly activate fibroblasts and are considered as key mediators in fibroblast activation. Interleukin-4 can stimulate expression of collagen and other ECM components in fibroblasts [
53,
54,
55,
56]. Both IL6 and TGFβ can activate the JAK2-STAT3 cascade, which has been shown to be activated in SSc skin and fibroblasts [
57,
58,
59]. Leukocytes might not only activate fibroblasts by releasing cytokines, but also by direct cell-cell contact. Co-culture of fibroblasts with macrophages results in the reciprocal amplification of fibroblast-monocyte adhesion and chemokine release [
60]. Also Notch signaling, as explained above, can be activated by direct contact of fibroblasts with immune cells with the ligand expressed by the leukocytes and the receptor expressed by the fibroblast [
33,
35].
4. Activation by ECM Components
It is known for quite a while that fibroblasts are activated by stiff substrates [
61]. Fibroblasts are adherent cells and thus interact with the ECM. Increased tissue stiffness due to accumulating collagen fibers and loss of the normal ECM structure leads to increased mechanical force and stress for the fibroblasts [
62]. The mechanisms behind this mechanotranduction in fibroblasts are extensively reviewed elsewhere [
61,
62,
63]. We will give here some examples of recent publications.
4.1. Mechanosensing and Transduction
Several studies identified Yes1 associated transcriptional regulator (YAP) and WW domain containing transcription regulator 1 (WWTR1, also known and widely referred to as TAZ) as mediators of mechanotransduction in fibroblasts [
64]. In kidney fibrosis, pharmacological inhibition of YAP or specific deletion of YAP and TAZ in GLI1+ cells ameliorated experimental fibrosis and significantly reduced the number of αSMA-positive myofibroblasts [
65]. In vitro, inhibition of YAP/TAZ signaling blocked both stiffness- and TGFβ-induced myofibroblast differentiation [
65]. A younger study showed that blockade of tank binding protein kinase 1 (TBK1), either pharmacologically or genetically, reduced αSMA levels, ECM deposition and traction force in TGFβ-stimulated normal and IPF lung fibroblasts [
66]. Of note, inhibition of TBK1 also reduced the total and nuclear levels of YAP/TAZ [
66]. Inhibition of YAP by different pharmacological approaches also reduced collagen expression, stress fiber formation and contraction forces in human normal dermal and SSc fibroblasts at baseline or upon TGFβ stimulation [
67,
68]. One of these substances – celastrol – was also effective in bleomycin-induced skin fibrosis with reductions in dermal thickness, myofibroblast differentiation and in the spatial expression of pro-fibrotic markers [
68]. Another study also demonstrated that knockdown of either YAP or TAZ or both combined ameliorated bleomycin-induced dermal as well as lung fibrosis [
69].
A very recent study introduced the calcium binding protein S100A4 as another player in the mechanosensitive activation of fibroblasts [
70]. S100A4 is induced by increased ECM stiffness in primary lung fibroblasts and mediates transdifferentiation to myofibroblasts in response to stiffness. The study also elegantly shows that upon knockout of S100A4 in fibroblasts, peripheral actin is locked in filamentous bundles and can no longer assemble to mechanically active stress fibers [
70]. Also the mechanical sensor PIEZO1 has been linked to fibroblast activation during tissue remodelling [
71].
Integrins are cell surface receptors connecting the cell to the ECM and thus enabling adhesion, mechanosensing, and mechanotransductive signaling. In pulmonary fibroblasts, α6 integrin is induced by stiff matrices and is mediating MMP2-dependent pericellular proteolysis of collagen IV in the basement membrane and thus promotes myofibroblast invasion. Increased expression can be found in lung fibroblasts from IPF patients, which remains in
in vitro culture [
72]. Blockade of α6 integrin expression or signaling ameliorated experimental lung fibrosis [
72]. Recently, a study involving triple knockout mice for α1β1, α2β1 and α11β1 integrins demonstrates increased expression of the collagen receptor discoidin domain receptor tyrosine kinase 2 (DDR2) in fibroblasts with triple integrin knockout, whereas the levels of activated and total YAP decreased [
73]. Triple knockout mice also had a better outcome of bleomycin-induced dermal fibrosis [
73].
4.2. Matrix Remodelling Enzymes
Extracellular matrix proteins such as collagens are mainly released from fibroblasts. The ECM undergoes permanent remodeling with deposition, degradation and modification of its components [
74]. Crosslinking of collagen fibers after the release from fibroblasts is mediated by the family of lysyl oxidases (LOX), degrading enzymes are matrix metalloproteinases (MMPs), a disintegrin and metalloproteases (ADAMs), and ADAM with thrombospondin motifs (ADAMTS) amongst others [
74]. The degrading enzymes are counteracted by the tissue inhibitors of MMPs (TIMPs) and other inhibitors [
74]. Many of these enzymes are also released by fibroblasts. Dysregulation of these mechanisms or dysbalance in the expression of these enzymes also contribute to fibroblast activation and fibrosis.
A study with two 3D skin-like models with either SSc or normal dermal fibroblasts demonstrates increased expression of lysyl oxidase-like 4 (LOXL4) in SSc skin biopsies as well as in SSc dermal fibroblasts [
75]. Knockdown of LOXL4 reduced αSMA expression, stromal stiffness, and TGFβ-induced collagen accumulation [
75]. In contrast, a recent study analyzing LOXL2 in IPF lung samples and fibroblasts also registered increased expression of LOXL2 in IPF lungs, however, inhibition by the monoclonal antibody simtuzumab promoted myofibroblast differentiation and invasion, and enhanced experimental pulmonary fibrosis in mice, which is in line with a failed phase 2 clinical trial in primary sclerosing cholangitis [
76,
77]. Other enzymes of matrix maturation are transglutaminases whereof transglutaminase-2 (TGM2) is the most widely expressed member. TGM2 has been shown to be upregulated in renal and pulmonary fibrotic diseases [
78,
79,
80]. Recently, it has been also demonstrated to be upregulated in skin and serum samples from SSc patients with particularly higher expression in patients with diffuse cutaneous SSc (dcSSc) and in patients with SSc-related interstial lung disease (SSc-ILD) [
81]. This increased expression and a higher activity of TGM2 could be also found in cultured SSc dermal fibroblasts. Inhibition of TGM2 did not show efficacy in 2D culture models, but reduced dermal thickness and type I collagen deposition in a 3D skin culture model with SSc dermal fibroblasts [
81].
Another player in matrix remodeling is extracellular matrix metalloproteinase inducer (EMMPRIN, also known as CD147, encoded by the basigin gene
BSG). EMMPRIN is a transmembrane protein that induces the expression of MMPs in neighbouring stromal cells. Overexpression of EMMPRIN in lung fibroblasts leads to upregulation of WNT/β-catenin signaling and increased proliferation and resistance to apoptosis [
82]. Blockade of EMMPRIN by an antibody inhibited TGFβ-induced αSMA expression, activation of MMP2 and induced apoptosis in normal human lung fibroblasts [
82]. Another more recent study also demonstrated that blocking of EMMPRIN
in vivo prevented bleomycin-induced pulmonary fibrosis [
83]. Increased serum levels of soluble CD147/EMMPRIN have been also found in SSc patients and have been associated with renal crisis [
84].
MMP3 is downregulated in SSc dermal fibroblasts and incubation of SSc dermal fibroblasts with recombinant MMP3 is reversing the activated phenotype of SSc fibroblasts with lower expression of αSMA, type I collagen, and α2-antiplasmin – a circulating inhibitor of plasmin and a key regulator of fibrinolysis [
85]. Targeting TIMP1 with miR-29a also decreased the levels of α2-antiplasmin [
85]. In the lungs, MMP expression and activity is in part regulated by glykogen synthase kinase 3 (GSK3). Inhibition of GSK3 in bleomycin-induced pulmonary fibrosis reduced the activity of MMP2 and MMP9 in bronchoalveolar lavage fluid (BALF), and also reduced bleomycin-induced expression of MMP2, MMP9, TIMP1 and TIMP2 in inflammatory cells isolated from BALFs and in different cell types in the lung tissue of bleomycin-challenged mice [
86]. Mass spectrometry analysis revealed increased levels of active MMP1, ADAM9, ADAM10 and ADAM17 in lung tissue of IPF patients, whereas the levels of MMP8 and MMP14 were decreased [
87]. Pharmacological inhibition of MMP1 and ADAM10 reduced αSMA expression in cultured lung fibroblasts [
87].
5. Epigenetic Changes as Cell Endogenous Self-amplifying Activating Mechanisms
When fibroblasts are explanted from fibrotic tissue and kept in culture, they remain activated for several passages. This persistent profibrotic phenotype can be explained by intrinsic changes, namely epigenetic modifications. Epigenetics describe changes in gene expression, which are not encoded in the nucleic sequence. The classical epigenetic mechanisms comprise methylation of the DNA, different histone modifications (e.g. methylation and acetylation), and non-coding RNAs like microRNAs (miRNA) and long non-coding RNAs (lncRNA). All of these mentioned mechanisms have been implicated in the process of fibrogenesis. It is assumed that prolonged exposure to external stimuli induce these epigenetic changes, which stabilize the activated phenotype of fibroblasts rendering them at least in part independent of external stimulation.
5.1. DNA Methylation
DNA can be methylated at the fifth position of the pyrimidine ring of cytosine residues. This methylation mainly occurs in CG rich areas, so called CpG islands. Three DNA methyltransferases (DNMTs) are responsible for methylating DNA: DNMT1 has a preference for hemimethylated sites and is responsible for the maintenance of DNA methylation during the cell cycle, whereas DNMT3A and DNMT3B are both de novo methyltransferases [
88,
89,
90].
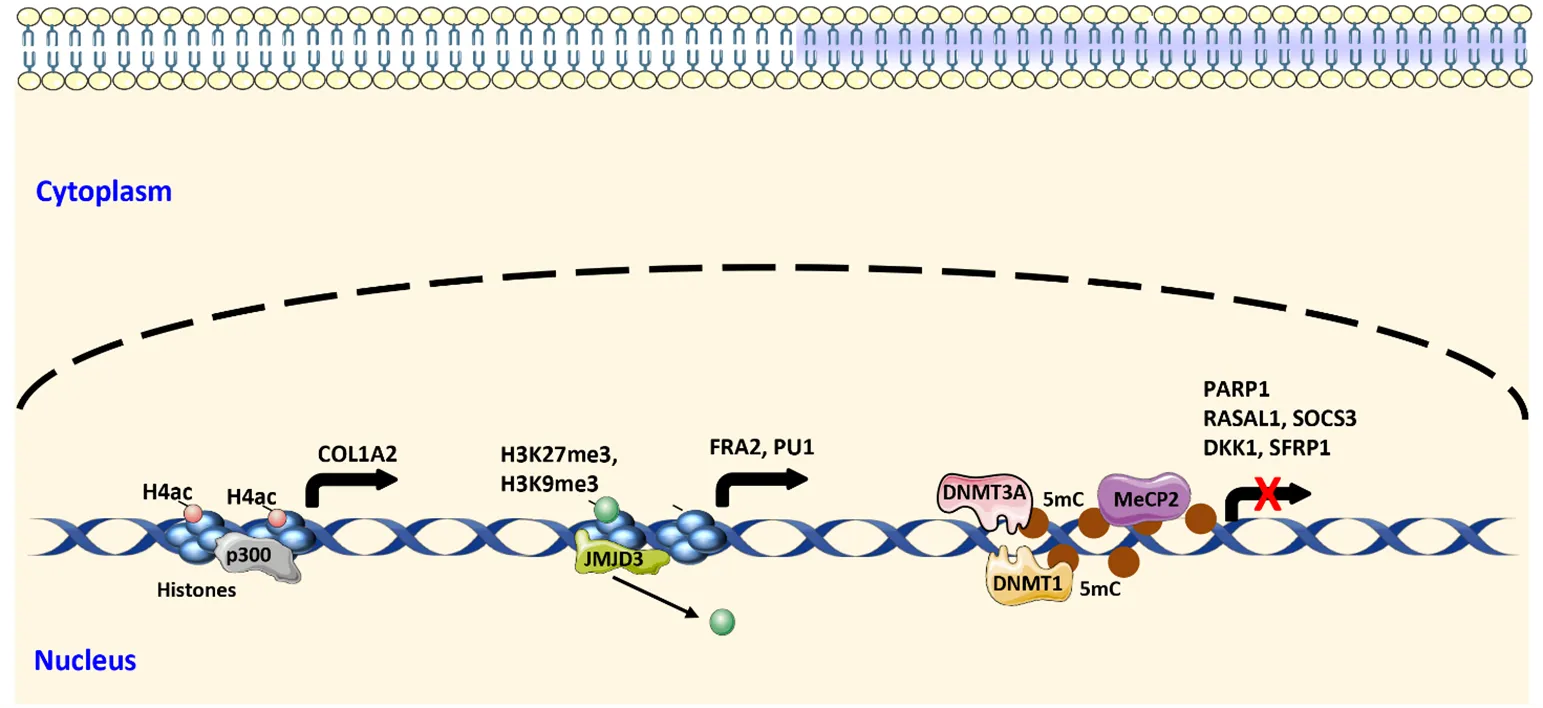
First evidence for the concept of induction of DNA methylation by pro-fibrotic stimuli was provided in experimental models of renal fibrosis, in which long-term stimulation with TGFβ induced hypermethylation of the
RASAL1 promoter, thereby causing aberrant RAS signaling () [
91]. A more recent study extended those findings to fibroblasts from SSc patients. In this study, chronic exposure of dermal fibroblasts to TGFβ induced the expression of DNMT1 and DNMT3A resulting in the repression of suppressor of cytokine signaling 3 (SOCS3) by DNA hypermethylation within the promoter region and increased STAT3 signaling () [
92]. Repressor complexes are recruited to methylated DNA regions upon binding of methyl-binding domain (MBD) proteins. One of these proteins is methyl cap binding protein 2 (MECP2), which has been implicated in repression of genes during fibrogenesis. MECP2 has been demonstrated to bind to the hypermethylated promoter of the WNT antagonist SFRP1 leading to aberrant Wnt signaling () [
93]. This finding highlights the interaction of the different pro-fibrotic mechanisms, and that also core pathways are affected by differential DNA methylation. The two endogenous WNT antagonists SFRP1 and DKK1 have both been shown to be repressed by increased DNA methylation in dermal fibroblasts from SSc patients [
19]. In addition, the TGFβ pathway is partly enhanced by DNA methylation induced silencing of regulatory proteins. One of these proteins is poly(ADP-ribose) polymerase 1 (PARP1), which, under normal conditions, PARylates SMAD3. This PARylation induces the dissociation of SMAD3 from DNA and thus limits its transcriptional activity. PARP1 is downregulated by promoter hypermethylation in fibroblasts from SSc patients and can be reactivated by incubation with the inhibitor of DNA methyltransferases 5-aza-2’-deoxycytidine (5-aza, Decitabine) (). Of note, TGFβ also induces this hypermethylation, suggesting a self-amplifying loop [
94]. A twin study published in 2022 analyzing genome-wide DNA methylation in dermal fibroblasts from twin pairs discordant to SSc, revealed 55 hypomethylated and 16 hypermethylated CpG sites. On the gene level, these data showed 13 genes with increased methylation and 22 genes with decreased methylation including several homeobox domain (HOX) genes [
95]. A previous genome-wide DNA methylation study with primary dermal fibroblasts from 15 SSc patients and 15 healthy individuals of African Americans also revealed 17 and 11 differentially methylated genes and promoters, respectively [
96]. Several non-coding RNAs can be found within these differentially methylated genes [
96].
5.2. Histone Modifications
In contrast to DNA methylation, histone modifications are more divers and complex. The tails of histones can be acetylated at lysine residues, mono-, di-, and trimethylated at lysine (K) and arginine (R) residues, phosphorylated at serine (S), tyrosine (Y) and threonine (T) residues, ubiquitinylated, sumoylated, and biotinylated. The most studied modifications regarding fibrosis are acetylation and methylation of histones.
Acetylation of lysine residues results in the repulsion from the negatively charged DNA opening the chromatin structure, which leads to facilitated transcriptional activity [
97]. Histone acetylation is regulated by histone acetyl transferases (HAT) and histone deacetylases (HDAC). One of these histone acetyltransferases is p300, which has been shown to amplify TGFβ signaling [
98,
99]. Activation of the nuclear receptor PPARγ either by small molecules or forced overexpression, resulted in the disruption of TGFβ-SMAD-dependent collagen expression by preventing hyperacetylation of histone H4 at the COL1A2 locus mediated by p300 () [
44]. Another histone acetyltransferase MYST1 (lysine acetyltransferase 8; KAT8) is downregulated in fibroblasts of SSc patients [
100]. This downregulation leads to lower levels of H4K16 acetylation resulting in increased levels of autophagy related 7 (ATG7) and BECLIN1, and enhanced autophagy [
100].
Histone methylation, mainly histone trimethylation, has also been implicated in fibrosis. The best studied locus is trimethylation at histone 3 lysine 27 (H3K27me3), which is a repressive histone mark. A study by Krämer et al. demonstrated that inhibition of the H3K27 methyltransferases and polycomb repressor complex 2 (PRC2) components enhancer of zeste 2 (EZH2) and suppressor of zeste 12 (SUZ12) with 3-Deazaneplanocin A (DZNep) promoted fibroblast activation and induced dermal fibrosis [
101]. These effects might result from stimulatory effects on the expression of the pro-fibrotic transcription factor fos-related antigen 2 (FRA2), a member of the AP1 transcription factor complex, whose transgenic, non-conditional overexpression in mice drives SSc-like skin and lung fibrosis [
101,
102]. The H3K27me3 mark can be actively removed by the demethylases ubiquitously transcribed tetratricopeptide repeat on chromosome X (UTX, also known as KDM6A) and jumonji domain-containing protein 3 (JMJD3, also known as KDM6B). Another study by Bergmann et al. provides evidence that the expression of JMJD3 is upregulated in SSc dermal fibroblasts as well as in experimental fibrosis [
103]. Inactivation of JMJD3 reversed the activated phenotype of SSc fibroblasts, and pharmacological inhibition prevented bleomycin-induced skin fibrosis as well as dermal and lung fibrosis induced by injection of recombinant human topoisomerase I. The provided mechanism in this study also includes FRA2, whose expression was reduced upon inhibition of JMJD3 due to an increase in H3K27 trimethylation () [
103]. Nevertheless, not only so called anti-fibrotic genes are affected by epigenetic changes, also pro-fibrotic genes silenced during physiological homeostasis can be reactivated during fibroblast-to-myofibroblast transition. Such a pro-fibrotic gene is the ETS transcription factor PU.1, which has been shown to be silenced by the repressive histone marks H3K27me3 and H3K9me3 in resting fibroblasts (). Upon fibroblast activation and myofibroblast transdifferentiation, these histone marks get lost and PU.1 can activate the expression of several pro-fibrotic mediators and collagen [
104].
. Selected epigenetic changes during fibroblast activation. Increased histone acetylation promotes COL1A2 expression, removing repressive histone methylation marks activates the expression of the pro-fibrotic mediators FRA2 and PU.1. Hypermethylation of the promoter regions suppresses the expression of different anti-fibrotic genes.
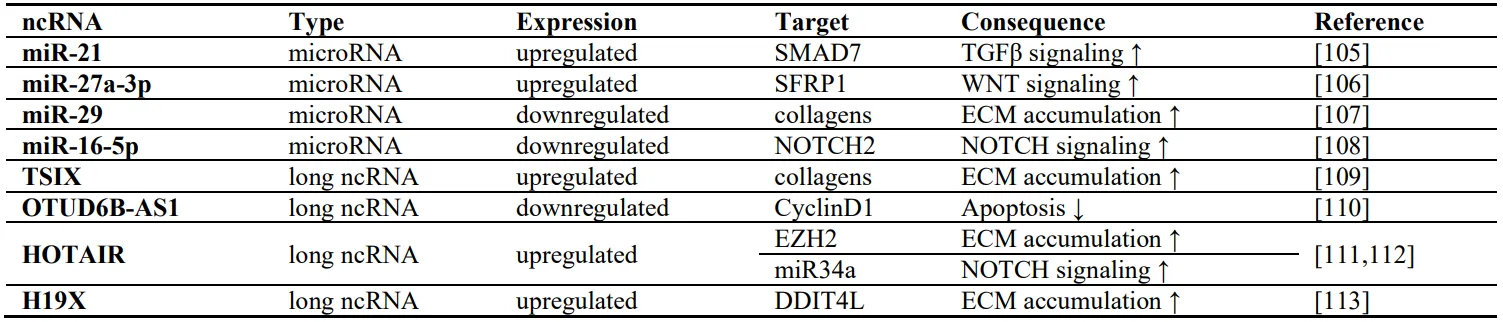
Dysregulated expression of non-coding RNAs in fibroblasts is also driving fibrogenesis. Micro RNAs (miRNAs) are defined as short RNA molecules of 20-25 nucleotides, whereas long non-coding RNAs (lncRNAs) have a length of over 200 nucleotides. MicroRNAs miR-21, miR-29, miR-16-5p, and miR-27a-3p are examples of such dysregulated non-coding RNAs (). miR-21 has been demonstrated to be upregulated in lungs of IPF patients as well as in mice with bleomycin-induced lung fibrosis [
105]. Stimulation of primary lung fibroblasts with TGFβ induced the expression of miR-21, whereas the knockdown interrupted TGFβ signaling. Mechanistically, it was proposed that miR-21 targets the mRNA of the inhibitory SMAD7 leading to its degradation and enhanced TGFβ signaling [
105]. Another miRNA overexpressed in activated fibroblasts from SSc patients is miR-27a-3p [
106]. The overexpression of miR-27a-3p in cultured fibroblasts led to reduced levels of SFRP1 and increased release of collagens [
106]. In contrast to the pro-fibrotic miR-21 and miR-27a-3p, miR-29 exerts anti-fibrotic effects and is downregulated in fibroblasts obtained from SSc patients [
107]. The repression could be induced by stimulation of fibroblasts with TGFβ, IL4 or PDGF-B. miR-29 targets mRNAs of several collagens, whose translation is no longer blocked upon repression of miR-29 expression [
107]. Another anti-fibrotic miRNA is miR-16-5p, which is downregulated in SSc patients [
108]. miR-16-5p targets and downregulates the NOTCH2 receptor, thereby blocking myofibroblast differentiation [
108].
Long non-coding RNAs have been implicated into fibrogenesis later than the miRNAs. In 2016, the lncRNA TSIX was shown to be upregulated in dermal fibroblasts of fibrotic tissue. TSIX stabilized collagen mRNA and its knockdown by siRNA reduced the levels of type I collagen [
109]. Some years later in 2019, the lncRNA OTUD6B-AS1 was implicated in the regulation of apoptosis in dermal fibroblasts from SSc patients and healthy controls by controlling Cyclin D1 expression [
110]. Further on, the lncRNA HOTAIR was found to be upregulated in αSMA-positive SSc dermal fibroblasts [
111]. The overexpression of HOTAIR increased EZH2-dependent collagen expression and decreased the expression of miR34a, thereby inducing Notch pathway activation [
111]. A follow-up study of the same group further revealed that the HOTAIR-dependent activation of Notch signaling induced the expression of the hedgehog pathway transcription factor GLI2, once more highlighting the trans-activation of the different pro-fibrotic pathways [
112]. The X-chromosome encoded lncRNA H19X is also upregulated in SSc dermal fibroblasts and in other fibrotic diseases as well as in physiological wounds [
113]. This upregulation can be induced by TGFβ. Mechanistically, the authors show that H19X negatively regulates the expression of DDIT4L, whose knockdown strongly induces collagen expression in dermal fibroblasts [
113]. Silencing of H19X, on the contrary, blocked TGFβ-induced collagen expression and myofibroblast differentiation, and triggered fibroblast apoptosis [
113].
. Summary of selected non-coding RNAs and their role in fibrotic tissue remodelling.
6. Alterations in Cell Metabolism
Mitochondria are intracellular organelles, which are thought to originate from bacteria taken up by eukaryotic cells, and thus forming an endosymbiotic relationship. They are the main producers of cellular energy by converting glucose, free fatty acids and glutamine. Under aerobic conditions, mitochondria produce ATP and acetyl-coenzyme A (acetyl-CoA) via respiration and the citric acid cycle. Oxidation of products of the citric acid cycle sustains a proton gradient through the mitochondrial inner membrane, which is driving ATP synthesis. This process is called oxidative phosphorylation (OXPHOS). Dysfunctional mitochondria can cause the production of reactive oxygen species (ROS) by releasing free electrons. These ROS further damage mitochondria resulting in a vicious cycle of ROS release and mitochondrial damage and dysfunction [
114]. The extracellular environment in the pathogenesis of fibrotic diseases such as SSc with hypoxia, ROS and chronic inflammation can drive metabolic reprogramming in fibroblasts. Mitochondrial damage has been reported in different fibrotic diseases such as kidney fibrosis [
115], IPF, connective tissue disease-associated lung fibrosis [
116,
117], and SSc [
118,
119]. Bueno and coworkers investigated the role of PINK1 (PTEN-induced putative kinase 1), a protein implicated in the process of mitophagy, in IPF. Under physiological conditions, PINK1 can enter mitochondria where it is degraded. Upon mitochondrial damage, PINK1 proteins accumulate at the outer mitochondrial membrane and induce mitophagy by recruiting the ubiquitin ligase Parkin [
120]. The work of Bueno et al. not only shows abnormal changes of mitochondria in lungs, particularly in alveolar type II cells of IPF patients, but also that these changes are associated with upregulation of markers of endoplasmatic reticulum (ER) stress and with increasing age of the individual. Induction of ER stress by tunicamycin in mice leads to the downregulation of PINK1 expression. In addition, knockdown or knockout of PINK1 in lung epithelial cells or mice resulted in the upregulation of profibrotic markers [
116]. Another study adds to these findings by demonstrating that TGFβ contributes to epithelial cell death by disrupting mitochondrial membrane integrity. However, Patel et al. also show that TGFβ upregulates the expression of PINK1, but confirm the study of Bueno et al. that mice with knockout of PINK1 are more susceptible to bleomycin-induced lung fibrosis [
121]. No explanation exists so far for the difference between these two studies with, on the one hand, downregulation of PINK1 upon ER stress and, on the other hand, upregulation of PINK1 by TGFβ. Another two studies analyzing whole tissue extracts of experimental bleomycin-induced lung fibrosis and of patients with connective tissue disease-related interstitial lung disease (CTD-ILD) show that the development of fibrosis is associated with ROS formation, mitochondrial dysfunction, impairment of the respiratory chain, and deletions in mitochondrial DNA (mtDNA), which might in turn sustain ROS formation with subsequent additional mitochondrial damage and fibrotic tissue remodeling [
117,
122]. Another protein of interest regarding mitochondrial function during fibrogenesis is SIRT3. Sirtuins form the class III of the superfamily of HDACs. Within the sirtuin family, SIRT3 belongs to the class I proteins with NAD
+-dependent deacetylating activity and is exclusively expressed in mitochondria [
123]. Regarding mitochondria in fibroblasts, Sosulski and coworkers revealed a massive downregulation of SIRT3 in lungs of bleomycin-injected mice as well as in human lung fibroblasts treated with TGFβ. The TGFβ-induced suppression of SIRT3 expression was paralleled by an accumulation of the acetylated forms of IDH2
K413 and SOD2
K68 in mitochondria suggesting a deficient antioxidant response in mitochondrial homeostasis upon TGFβ-induced SIRT3 repression [
124]. Also in fibroblasts of SSc patients, many mitochondria are damaged with deletions of mtDNA and increased release of mtDNA into the extracellular space [
125]. The same study shows that the respiratory capacity is profoundly decreased in SSc fibroblasts whereas the non-mitochondrial oxygen consumption increases as potential compensatory mechanism. Analysis of microarray data from a North American patient cohort revealed that 39.5% of mitochondrial genes are deregulated in skin samples of SSc patients, one of which is the mitochondrial transcription factor A (TFAM). This transcription factor controls the transcription of core proteins required for mitochondrial homeostasis. Zhou et al. found TFAM severely downregulated in skin and particularly in fibroblasts from SSc patients. Experimentally, fibroblast-specific knockout of TFAM in mice exacerbated dermal as well as lung fibrosis. Of note, the mitochondrial damages and the downregulation of TFAM observed in SSc samples could be mimicked by prolonged stimulation with TGFβ in normal fibroblasts [
125]. In contrast to the previous mentioned study, Cantanhede and coworkers found increased ATP-producing mitochondrial respiration in SSc fibroblasts which could be further increased upon stimulation with TGFβ [
118]. The authors explain this increase with the hyperfusion of mitochondria in stressed cells [
118].
6.1. Fibroblast Subsets
In the past, fibroblasts were considered as passive cells with the only function of construction and remodeling of the ECM [
126]. In addition, this cell type was considered to form a stable and homogenous cell population. During recent years, however, different fibroblast populations in part with additional subpopulations have been identified in fibrotic diseases, which also are thought to have different functions both in physiological (“healthy”) and pathological conditions () [
127].
Regarding SSc, Franks and coworkers of the Whitfield group analyzed different expression datasets including independent patient cohorts and defined four subsets of fibroblasts, namely inflammatory, fibroproliferative, normal-like, and limited. However, these subsets are intrinsic to individual patients rather than to different organs [
128].
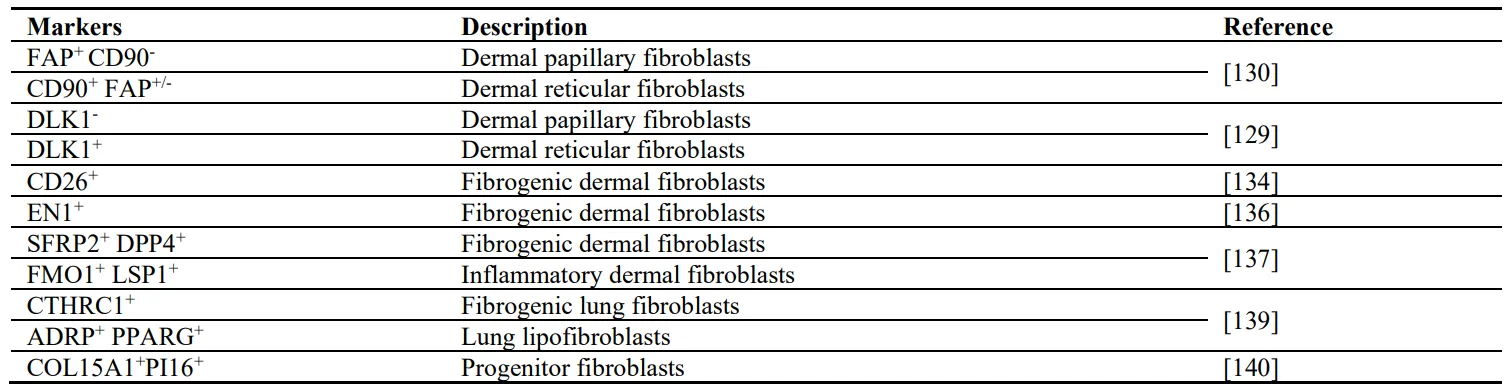
The dermal layer of the skin is divided into two parts: the upper papillary dermis and the lower reticular dermis. Different studies characterized and divided dermal fibroblasts according to their localization [
129,
130,
131]. Papillary fibroblasts are considered to be positive for fibroblast activation protein (FAP) and negative for Thy1 cell surface antigen (THY1, also known as CD90; FAP
+ CD90
-), and to have a high proliferative capacity [
130]. Reticular fibroblasts on the contrary have been identified as CD90
+ with an adipogenic potiential [
130]. Interestingly, THY1 has been implicated in both skin and lung fibrosis [
132,
133]. A study investigating dermal fibrosis in SSc patients demonstrated that THY1 is markedly overexpressed in the deeper dermis of SSc patients. Knockdown in human dermal fibroblasts or ubiquitous knockout of THY1 in mice, reduced the expression of fibrotic genes and ameliorated experimental dermal fibrosis, which is contrary to findings in experimental lung fibrosis where deficiency for THY1 exacerbated the fibrotic tissue response [
132,
133]. These differences may be explained by organ-specific differences and the potentially different origins of fibroblasts in the two organs. Another study characterized two different fibroblast lineages in the skin of mice. One of these lineages can be localized in the upper dermis, whereas the other one is found in the lower dermis including reticular fibroblasts, but also preadipocytes and adipocytes of the hypodermis [
129]. According to Driskell et al, the papillary fibroblasts can be identified as negative for delta like non-canonical Notch ligand 1 (DLK1
−), the reticular fibroblasts are defined as DLK1
+. The study also identified different function of these lineages: the papillary subtype is required for new hair follicle formation, whereas the reticular fibroblasts mediate wound repair and synthesize the majority of fibrillary ECM [
129].
With an elegant study published eight years ago, Rinkevich and coworkers identified and isolated a dermal fibroblast lineage with fibrogenic potential. This lineage is derived from embryonic precursor cells expressing engrailed-1 (EN1) and could be identified by the presence of the surface protein dipeptidyl peptidase 4 (DPP4, also known as CD26) [
134]. This lineage of fibroblasts has been shown to be responsible for scarring and fibrosis [
134,
135]. Of note, another recent study highlighted the importance of EN1 for the progression of fibrotic tissue remodeling [
136]. The Lafyatis group in Pittsburgh later separated different fibroblast lineages by single cell RNA sequencing (scRNAseq) of human skin biopsies [
137]. They describe two major lineages distinguished by either expressing secreted frizzled-related protein 2 (SFRP2) and DPP4 (SFRP2
+ DPP4
+) or flavin containing dimethylaniline monoxygenase 1 (FMO1) and lymphocyte specific protein 1 (LSP1) (FMO1
+ LSP1
+). Both of these lineages have several sub-lineages identified by the expression of different genes [
137].
Another study performing scRNAseq from all collagen-producing cells in the lungs of mice with bleomycin-induced lung fibrosis and of humans with different fibrotic lung diseases identified a subset of fibroblasts expressing high levels of collagen triple helix repeat containing 1 (CTHRC1) [
138]. This subset increases during fibrotic tissue remodeling in both mice and humans and express the highest levels of collagens [
138]. Xie et al. classified the fibroblast populations in normal and fibrotic lungs in the experimental model of bleomycin-induced fibrosis [
139]. By single cell transcriptomic analysis they identified six different populations in normal murine lungs, and seven in fibrotic lungs. The additional population is defined as positive for PDGF receptor beta (PDGFRβ
+), but the authors could not identify the origin of this cell population emerging upon fibrotic stimuli [
139]. They also defined two matrix fibroblast populations identified by COL14A1 or COL13A1 as well as so-called lipofibroblasts (lipid-containing interstitial fibroblasts), which are positive for both ADRP (official name: perilipin2, PLIN2) and PPARγ [
139].
All of the different studies analyzing fibroblast populations demonstrate the heterogeneity of this cell type not only within single tissues, but also across different tissues and organs. One study trying to decipher the shared and distinct features of stromal cells across organs characterized two universal fibroblast subsets defined by the expression of COL15A1 and peptidase inhibitor 16 (PI16). The authors hypothesize that these two lineages might be progenitors of the tissue-specific fibroblast populations [
140].
. Summary of different fibroblast subsets identified in skin and lungs.
7. Conclusions
Regarding the findings in basic research, it is encouraging that the molecular mechanisms taking place during fibrogenesis are more and more discovered suggesting potential targets for therapeutic intervention. However, therapies targeting the ECM-releasing myofibroblasts and thus reversing fibrogenesis are still not available for clinical use.
One reason for this lack is that broad targeting of the core morphogen pathways is accompanied by severe adverse events as these pathways are also important for e.g. homeostatic stem cell renewal. Nevertheless, single signaling components might be targeted as for example for the Notch pathway, for which different drugs are actually developed and investigated in clinical trials [
141]. Also the components of mechanosignaling and ECM modification such as integrins, YAP, or lysyl oxydases are considered as potential targets for anti-fibrotic treatment [
142].
Taken together, the identification and characterization of different fibroblast subsets opened a new door to targeted therapies and personalized medicine. In addition, the use of spatial transcriptomics might further help in unravelling the mechanisms during fibrogenesis in more detail. Furthermore, by integrating different omics data, e.g. by combining gene expression (transcriptomics) with changes in metabolism (metabolomics), it might be possible to better predict the response to therapy.
Author Contributions
Writing – Original Draft Preparation, C.D, A.R.R; Writing – Review & Editing, C.D, A.R.R.
Ethics Statement
Not applicable.
Informed Consent Statement
Not applicable.
Funding
This work received no external funding.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
-
1.
Denton CP, Khanna D. Systemic sclerosis.
Lancet 2017,
390, 1685–1699.
[Google Scholar]
-
2.
Wollheim FA. Classification of systemic sclerosis. Visions and reality.
Rheumatology 2005,
44, 1212–1216.
[Google Scholar]
-
3.
Allanore Y, Simms R, Distler O, Trojanowska M, Pope J, Denton CP, et al. Systemic sclerosis.
Nat. Rev. Dis. Primers 2015,
1, 15002.
[Google Scholar]
-
4.
Matucci-Cerinic M, Kahaleh B, Wigley FM. Review: evidence that systemic sclerosis is a vascular disease.
Arthritis Rheum. 2013,
65, 1953–1962.
[Google Scholar]
-
5.
Wynn TA. Cellular and molecular mechanisms of fibrosis.
J. Pathol. 2008,
214, 199–210.
[Google Scholar]
-
6.
Pakshir P, Noskovicova N, Lodyga M, Son DO, Schuster R, Goodwin A, et al. The myofibroblast at a glance. J. Cell Sci. 2020, 133, doi:10.1242/jcs.227900.
-
7.
Distler JHW, Gyorfi AH, Ramanujam M, Whitfield ML, Konigshoff M, Lafyatis R. Shared and distinct mechanisms of fibrosis.
Nat. Rev. Rheumatol. 2019,
15, 705–730.
[Google Scholar]
-
8.
Wynn TA. Fibrotic disease and the T(H)1/T(H)2 paradigm.
Nat. Rev. Immunol. 2004,
4, 583–594.
[Google Scholar]
-
9.
Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA. Myofibroblasts and mechano-regulation of connective tissue remodelling.
Nat. Rev. Mol. Cell. Biol. 2002,
3, 349–363.
[Google Scholar]
-
10.
Bochaton-Piallat ML, Gabbiani G, Hinz B. The myofibroblast in wound healing and fibrosis: answered and unanswered questions. F1000Res 2016, 5, doi:10.12688/f1000research.8190.1.
-
11.
Hinz B, Phan SH, Thannickal VJ, Prunotto M, Desmouliere A, Varga J, et al. Recent developments in myofibroblast biology: paradigms for connective tissue remodeling.
Am. J. Pathol. 2012,
180, 1340–1355.
[Google Scholar]
-
12.
McAnulty RJ. Fibroblasts and myofibroblasts: their source, function and role in disease.
Int. J. Biochem. Cell Biol. 2007,
39, 666–671.
[Google Scholar]
-
13.
Mehal WZ, Iredale J, Friedman SL. Scraping fibrosis: expressway to the core of fibrosis.
Nat. Med. 2011,
17, 552–553.
[Google Scholar]
-
14.
Henderson NC, Arnold TD, Katamura Y, Giacomini MM, Rodriguez JD, McCarty JH, et al. Targeting of alphav integrin identifies a core molecular pathway that regulates fibrosis in several organs.
Nat. Med. 2013,
19, 1617–1624.
[Google Scholar]
-
15.
Kim KK, Sheppard D, Chapman HA. TGF-beta1 Signaling and Tissue Fibrosis.
Cold Spring Harb. Perspect. Biol. 2018,
10, a022293.
[Google Scholar]
-
16.
Mendoza FA, Jimenez SA. Serine/threonine kinase inhibition as antifibrotic therapy: transforming growth factor-beta and Rho kinase inhibitors.
Rheumatology 2022,
61, 1354–1365.
[Google Scholar]
-
17.
Luo K. Signaling Cross Talk between TGF-beta/Smad and Other Signaling Pathways.
Cold Spring Harb. Perspect. Biol. 2017,
9, a022137.
[Google Scholar]
-
18.
Akhmetshina A, Palumbo K, Dees C, Bergmann C, Venalis P, Zerr P, et al. Activation of canonical Wnt signalling is required for TGF-beta-mediated fibrosis.
Nat. Commun. 2012,
3, 735.
[Google Scholar]
-
19.
Dees C, Schlottmann I, Funke R, Distler A, Palumbo-Zerr K, Zerr P, et al. The Wnt antagonists DKK1 and SFRP1 are downregulated by promoter hypermethylation in systemic sclerosis.
Ann. Rheum. Dis. 2014,
73, 1232–1239.
[Google Scholar]
-
20.
Henderson J, Pryzborski S, Stratton R, O'Reilly S. Wnt antagonist DKK-1 levels in systemic sclerosis are lower in skin but not in blood and are regulated by microRNA33a-3p.
Exp. Dermatol. 2021,
30, 162–168.
[Google Scholar]
-
21.
Wei J, Fang F, Lam AP, Sargent JL, Hamburg E, Hinchcliff ME, et al. Wnt/beta-catenin signaling is hyperactivated in systemic sclerosis and induces Smad-dependent fibrotic responses in mesenchymal cells.
Arthritis Rheum. 2012,
64, 2734–2745.
[Google Scholar]
-
22.
Bergmann C, Hallenberger L, Chenguiti Fakhouri S, Merlevede B, Brandt A, Dees C, et al. X-linked inhibitor of apoptosis protein (XIAP) inhibition in systemic sclerosis (SSc).
Ann. Rheum. Dis. 2021,
80, 1048–1056.
[Google Scholar]
-
23.
Wei J, Melichian D, Komura K, Hinchcliff M, Lam AP, Lafyatis R, et al. Canonical Wnt signaling induces skin fibrosis and subcutaneous lipoatrophy: a novel mouse model for scleroderma?
Arthritis Rheum. 2011,
63, 1707–1717.
[Google Scholar]
-
24.
Shi C, Chen X, Yin W, Sun Z, Hou J, Han X. Wnt8b regulates myofibroblast differentiation of lung-resident mesenchymal stem cells via the activation of Wnt/beta-catenin signaling in pulmonary fibrogenesis.
Differentiation 2022,
125, 35–44.
[Google Scholar]
-
25.
Newman DR, Sills WS, Hanrahan K, Ziegler A, Tidd KM, Cook E, et al. Expression of WNT5A in Idiopathic Pulmonary Fibrosis and Its Control by TGF-beta and WNT7B in Human Lung Fibroblasts.
J. Histochem. Cytochem. 2016,
64, 99–111.
[Google Scholar]
-
26.
=Vuga LJ, Ben-Yehudah A, Kovkarova-Naumovski E, Oriss T, Gibson KF, Feghali-Bostwick C, et al. WNT5A is a regulator of fibroblast proliferation and resistance to apoptosis.
Am. J. Respir. Cell Mol. Biol. 2009,
41, 583–589.
[Google Scholar]
-
27.
Ingham PW, Nakano Y, Seger C. Mechanisms and functions of Hedgehog signalling across the metazoa.
Nat. Rev. Genet. 2011,
12, 393–406.
[Google Scholar]
-
28.
Chanda D, Otoupalova E, Smith SR, Volckaert T, De Langhe SP, Thannickal VJ. Developmental pathways in the pathogenesis of lung fibrosis.
Mol. Aspects Med. 2019,
65, 56–69.
[Google Scholar]
-
29.
Horn A, Palumbo K, Cordazzo C, Dees C, Akhmetshina A, Tomcik M, et al. Hedgehog signaling controls fibroblast activation and tissue fibrosis in systemic sclerosis.
Arthritis Rheum. 2012,
64, 2724–2733.
[Google Scholar]
-
30.
Liang R, Sumova B, Cordazzo C, Mallano T, Zhang Y, Wohlfahrt T, et al. Michalska-Jakubus, et al. The transcription factor GLI2 as a downstream mediator of transforming growth factor-beta-induced fibroblast activation in SSc.
Ann. Rheum. Dis. 2017,
76, 756–764.
[Google Scholar]
-
31.
Bray SJ. Notch signalling in context.
Nat. Rev. Mol. Cell. Biol. 2016,
17, 722–735.
[Google Scholar]
-
32.
D’Souza B, Meloty-Kapella L, Weinmaster G. Canonical and non-canonical Notch ligands.
Curr. Top. Dev. Biol. 2010,
92, 73–129.
[Google Scholar]
-
33.
Dees C, Tomcik M, Zerr P, Akhmetshina A, Horn A, Palumbo K, et al. Notch signalling regulates fibroblast activation and collagen release in systemic sclerosis.
Ann. Rheum. Dis. 2011,
70, 1304–1310.
[Google Scholar]
-
34.
Dees C, Zerr P, Tomcik M, Beyer C, Horn A, Akhmetshina A, et al. Inhibition of Notch signaling prevents experimental fibrosis and induces regression of established fibrosis.
Arthritis Rheum. 2011,
63, 1396–1404.
[Google Scholar]
-
35.
Kavian N, Servettaz A, Mongaret C, Wang A, Nicco C, Chereau C, et al. Targeting ADAM-17/notch signaling abrogates the development of systemic sclerosis in a murine model.
Arthritis Rheum. 2010,
62, 3477–3487.
[Google Scholar]
-
36.
Kiyokawa H, Morimoto M. Notch signaling in the mammalian respiratory system, specifically the trachea and lungs, in development, homeostasis, regeneration, and disease.
Dev. Growth Differ. 2020,
62, 67–79.
[Google Scholar]
-
37.
Hu B, Wu Z, Bai D, Liu T, Ullenbruch MR, Phan SH. Mesenchymal deficiency of Notch1 attenuates bleomycin-induced pulmonary fibrosis.
Am. J. Pathol. 2015,
185, 3066–3075.
[Google Scholar]
-
38.
Wang YC, Chen Q, Luo JM, Nie J, Meng QH, Shuai W, et al. Notch1 promotes the pericyte-myofibroblast transition in idiopathic pulmonary fibrosis through the PDGFR/ROCK1 signal pathway.
Exp. Mol. Med. 2019,
51, 1–11.
[Google Scholar]
-
39.
Weikum ER, Liu X, Ortlund EA. The nuclear receptor superfamily: A structural perspective.
Protein Sci. 2018,
27, 1876–1892.
[Google Scholar]
-
40.
Derrett-Smith E, Clark KEN, Shiwen X, Abraham DJ, Hoyles RK, Lacombe O, et al. The pan-PPAR agonist lanifibranor reduces development of lung fibrosis and attenuates cardiorespiratory manifestations in a transgenic mouse model of systemic sclerosis.
Arthritis Res. Ther. 2021,
23, 234.
[Google Scholar]
-
41.
Ghosh AK, Bhattacharyya S, Lakos G, Chen SJ, Mori Y, Varga J. Disruption of transforming growth factor beta signaling and profibrotic responses in normal skin fibroblasts by peroxisome proliferator-activated receptor gamma.
Arthritis Rheum. 2004,
50, 1305–1318.
[Google Scholar]
-
42.
Wei, A, Gao Q, Chen F, Zhu X, Chen X, Zhang L, et al. Inhibition of DNA methylation de-represses peroxisome proliferator-activated receptor-gamma and attenuates pulmonary fibrosis.
Br. J. Pharmacol. 2022,
179, 1304–1318.
[Google Scholar]
-
43.
Wei J, Ghosh AK, Sargent JL, Komura K, Wu M, Huang QQ, et al. PPARgamma downregulation by TGFss in fibroblast and impaired expression and function in systemic sclerosis: a novel mechanism for progressive fibrogenesis.
PLoS ONE 2010,
5, e13778.
[Google Scholar]
-
44.
Ghosh AK, Bhattacharyya S, Wei J, Kim S, Barak Y, Mori Y, et al. Peroxisome proliferator-activated receptor-gamma abrogates Smad-dependent collagen stimulation by targeting the p300 transcriptional coactivator.
FASEB J. 2009,
23, 2968–2977.
[Google Scholar]
-
45.
Zerr P, Vollath S, Palumbo-Zerr K, Tomcik M, Huang J, Distler A, et al. Vitamin D receptor regulates TGF-beta signalling in systemic sclerosis.
Ann. Rheum. Dis. 2015,
74, e20.
[Google Scholar]
-
46.
Tzilas V, Bouros E, Barbayianni I, Karampitsakos T, Kourtidou S, Ntassiou M, et al. Vitamin D prevents experimental lung fibrosis and predicts survival in patients with idiopathic pulmonary fibrosis.
Pulm. Pharmacol. Ther. 2019,
55, 17–24.
[Google Scholar]
-
47.
Avouac J, Pezet S, Gonzalez V, Baudoin L, Cauvet A, Ruiz B, et al. Estrogens Counteract the Profibrotic Effects of TGF-beta and their Inhibition Exacerbates Experimental Dermal Fibrosis.
J. Invest. Dermatol. 2020,
140, 593–601.
[Google Scholar]
-
48.
Elhai M, Avouac J, Walker UA, Matucci-Cerinic M, Riemekasten G, Airo P, Hachulla E, et al. A gender gap in primary and secondary heart dysfunctions in systemic sclerosis: a EUSTAR prospective study.
Ann. Rheum. Dis. 2016,
75, 163–169.
[Google Scholar]
-
49.
Aida-Yasuoka K, Peoples C, Yasuoka H, Hershberger P, Thiel K, Cauley JA, et al. Estradiol promotes the development of a fibrotic phenotype and is increased in the serum of patients with systemic sclerosis.
Arthritis Res. Ther. 2013,
15, R10.
[Google Scholar]
-
50.
Soldano S, Montagna P, Brizzolara R, Sulli A, Parodi A, Seriolo B, et al. Effects of estrogens on extracellular matrix synthesis in cultures of human normal and scleroderma skin fibroblasts.
Ann. N. Y. Acad. Sci. 2010,
1193, 25–29.
[Google Scholar]
-
51.
Brown M, O’Reilly S. The immunopathogenesis of fibrosis in systemic sclerosis.
Clin. Exp. Immunol. 2019,
195, 310–321.
[Google Scholar]
-
52.
Mutsaers SE, Miles T, Prele CM, Hoyne GF. Emerging role of immune cells as drivers of pulmonary fibrosis.
Pharmacol. Ther. 2023,
252, 108562.
[Google Scholar]
-
53.
Fertin C, Nicolas JF, Gillery P, Kalis B, Banchereau J, Maquart FX. Interleukin-4 stimulates collagen synthesis by normal and scleroderma fibroblasts in dermal equivalents.
Cell. Mol. Biol. 1991,
37, 823–829.
[Google Scholar]
-
54.
Postlethwaite AE, Holness MA, Katai H, Raghow R. Human fibroblasts synthesize elevated levels of extracellular matrix proteins in response to interleukin 4.
J. Clin. Invest. 1992,
90, 1479–1485.
[Google Scholar]
-
55.
Sempowski GD, Derdak S, Phipps RP. Interleukin-4 and interferon-gamma discordantly regulate collagen biosynthesis by functionally distinct lung fibroblast subsets.
J. Cell. Physiol. 1996,
167, 290–296.
[Google Scholar]
-
56.
Wegrowski Y, Paltot V, Gillery P, Kalis B, Randoux A, Maquart FX. Stimulation of sulphated glycosaminoglycan and decorin production in adult dermal fibroblasts by recombinant human interleukin-4.
Biochem. J. 1995,
307, 673–678.
[Google Scholar]
-
57.
Chakraborty D, Sumova B, Mallano T, Chen CW, Distler A, Bergmann C, et al. Activation of STAT3 integrates common profibrotic pathways to promote fibroblast activation and tissue fibrosis.
Nat. Commun. 2017,
8, 1130.
[Google Scholar]
-
58.
Dees C, Tomcik M, Palumbo-Zerr K, Distler A, Beyer C, Lang V, et al. JAK-2 as a novel mediator of the profibrotic effects of transforming growth factor beta in systemic sclerosis.
Arthritis Rheum. 2012,
64, 3006–3015.
[Google Scholar]
-
59.
O’Reilly S, Ciechomska M, Cant R, van Laar JM. Interleukin-6 (IL-6) trans signaling drives a STAT3-dependent pathway that leads to hyperactive transforming growth factor-beta (TGF-beta) signaling promoting SMAD3 activation and fibrosis via Gremlin protein.
J. Biol. Chem. 2014,
289, 9952–9960.
[Google Scholar]
-
60.
Van Linthout S, Miteva K, Tschope C. Crosstalk between fibroblasts and inflammatory cells.
Cardiovasc. Res. 2014,
102, 258–269.
[Google Scholar]
-
61.
van Putten S, Shafieyan Y, Hinz B. Mechanical control of cardiac myofibroblasts.
J. Mol. Cell. Cardiol. 2016,
93, 133–142.
[Google Scholar]
-
62.
Ezzo M, Hinz B. Novel approaches to target fibroblast mechanotransduction in fibroproliferative diseases.
Pharmacol. Ther. 2023,
250, 108528.
[Google Scholar]
-
63.
Deng Z, Fear MW, Suk Choi Y, Wood FM, Allahham A, Mutsaers SE, et al. The extracellular matrix and mechanotransduction in pulmonary fibrosis.
Int. J. Biochem. Cell. Biol. 2020,
126, 105802.
[Google Scholar]
-
64.
Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, et al. Role of YAP/TAZ in mechanotransduction.
Nature 2011,
474, 179–183.
[Google Scholar]
-
65.
Liang M, Yu M, Xia R, Song K, Wang J, Luo J, et al. Yap/Taz Deletion in Gli(+) Cell-Derived Myofibroblasts Attenuates Fibrosis.
J. Am. Soc. Nephrol. 2017,
28, 3278–3290.
[Google Scholar]
-
66.
Aravamudhan A, Haak AJ, Choi KM, Meridew JA, Caporarello N, Jones DL, et al. Tschumperlin. TBK1 regulates YAP/TAZ and fibrogenic fibroblast activation.
Am. J. Physiol. Lung Cell. Mol. Physiol. 2020,
318, L852–L863.
[Google Scholar]
-
67.
Shi-Wen X, Racanelli M, Ali A, Simon A, Quesnel K, Stratton RJ, et al. Verteporfin inhibits the persistent fibrotic phenotype of lesional scleroderma dermal fibroblasts.
J. Cell Commun. Signal 2021,
15, 71–80.
[Google Scholar]
-
68.
Chitturi P, Xu S, Ahmed Abdi B, Nguyen J, Carter DE, Sinha S, et al. Tripterygium wilfordii derivative celastrol, a YAP inhibitor, has antifibrotic effects in systemic sclerosis.
Ann. Rheum. Dis. 2023,
82, 1191–1204.
[Google Scholar]
-
69.
Wu D, Wang W, Li X, Yin B, Ma Y. Single-cell sequencing reveals the antifibrotic effects of YAP/TAZ in systemic sclerosis.
Int. J. Biochem. Cell. Biol. 2022,
149, 106257.
[Google Scholar]
-
70.
Southern BD, Li H, Mao H, Crish JF, Grove LM, Scheraga RG, et al. A novel mechanoeffector role of fibroblast S100A4 in myofibroblast transdifferentiation and fibrosis.
J. Biol. Chem. 2023,
300, 105530.
[Google Scholar]
-
71.
Xu Y, Huang Y, Cheng X, Hu B, Jiang D, Wu L, et al. Mechanotransductive receptor Piezo1 as a promising target in the treatment of fibrosis diseases.
Front. Mol. Biosci. 2023,
10, 1270979.
[Google Scholar]
-
72.
Chen H, Qu J, Huang X, Kurundkar A, Zhu L, Yang N, et al. Mechanosensing by the alpha6-integrin confers an invasive fibroblast phenotype and mediates lung fibrosis.
Nat. Commun. 2016,
7, 12564.
[Google Scholar]
-
73.
Sawant M, Wang F, Koester J, Niehoff A, Nava MM, Lundgren-Akerlund E, et al. Ablation of integrin-mediated cell-collagen communication alleviates fibrosis.
Ann. Rheum. Dis. 2023,
82, 1474–1486.
[Google Scholar]
-
74.
Yue B. Biology of the extracellular matrix: an overview.
J. Glaucoma 2014,
23, S20–S23.
[Google Scholar]
-
75.
Huang M, Cai G, Baugh LM, Liu Z, Smith A, Watson M, et al. Systemic Sclerosis Dermal Fibroblasts Induce Cutaneous Fibrosis Through Lysyl Oxidase-like 4: New Evidence From Three-Dimensional Skin-like Tissues.
Arthritis Rheumatol. 2020,
72, 791–801.
[Google Scholar]
-
76.
Espindola MS, Habiel DM, Coelho AL, Parimon T, Chen P, Mikels-Vigdal A, Hogaboam CM. Translational Studies Reveal the Divergent Effects of Simtuzumab Targeting LOXL2 in Idiopathic Pulmonary Fibrosis.
Fibrosis 2023,
1, 10007.
[Google Scholar]
-
77.
Muir AJ, Levy C, Janssen HLA, Montano-Loza AJ, Shiffman ML, Caldwell S, et al. Simtuzumab for Primary Sclerosing Cholangitis: Phase 2 Study Results With Insights on the Natural History of the Disease.
Hepatology 2019,
69, 684–698.
[Google Scholar]
-
78.
Johnson TS, El-Koraie AF, Skill NJ, Baddour NM, El Nahas AM, Njloma M, et al. Tissue transglutaminase and the progression of human renal scarring.
J. Am. Soc. Nephrol. 2003,
14, 2052–2062.
[Google Scholar]
-
79.
Johnson TS, Skill NJ, El Nahas AM, Oldroyd SD, Thomas GL, Douthwaite JA, et al. Transglutaminase transcription and antigen translocation in experimental renal scarring.
J. Am. Soc. Nephrol. 1999,
10, 2146–2157.
[Google Scholar]
-
80.
Olsen KC, Sapinoro RE, Kottmann RM, Kulkarni AA, Iismaa SE, Johnson GV, et al. Transglutaminase 2 and its role in pulmonary fibrosis.
Am. J. Respir. Crit. Care. Med. 2011,
184, 699–707.
[Google Scholar]
-
81.
Zhou X, Trinh-Minh T, Matei AE, Gyorfi AH, Hong X, Bergmann C, et al. Amelioration of Fibrotic Remodeling of Human 3-Dimensional Full-Thickness Skin by Transglutamase 2 Inhibition.
Arthritis Rheumatol. 2023,
75, 1619–1627.
[Google Scholar]
-
82.
Hasaneen NA, Cao J, Pulkoski-Gross A, Zucker S, Foda HD. Extracellular Matrix Metalloproteinase Inducer (EMMPRIN) promotes lung fibroblast proliferation, survival and differentiation to myofibroblasts.
Respir. Res. 2016,
17, 17.
[Google Scholar]
-
83.
Liu F, Yu F, Lu YZ, Cheng PP, Liang LM, Wang M, et al. Crosstalk between pleural mesothelial cell and lung fibroblast contributes to pulmonary fibrosis.
Biochim. Biophys. Acta Mol. Cell. Res. 2020,
1867, 118806.
[Google Scholar]
-
84.
Yanaba K, Asano Y, Tada Y, Sugaya M, Kadono T, Hamaguchi Y, et al. Increased serum soluble CD147 levels in patients with systemic sclerosis: association with scleroderma renal crisis.
Clin. Rheumatol. 2012,
31, 835–839.
[Google Scholar]
-
85.
Niwa H, Kanno Y, Shu E, Seishima M. Decrease in matrix metalloproteinase‑3 activity in systemic sclerosis fibroblasts causes alpha2‑antiplasmin and extracellular matrix deposition, and contributes to fibrosis development.
Mol. Med. Rep. 2020,
22, 3001–3007.
[Google Scholar]
-
86.
Cinetto F, Ceccato J, Caputo I, Cangiano D, Montini B, Lunardi F, et al. GSK-3 Inhibition Modulates Metalloproteases in a Model of Lung Inflammation and Fibrosis.
Front. Mol. Biosci. 2021,
8, 633054.
[Google Scholar]
-
87.
Peng Z, Konai MM, Avila-Cobian LF, Wang M, Mobashery S, Chang M. MMP-1 and ADAM10 as Targets for Therapeutic Intervention in Idiopathic Pulmonary Fibrosis.
ACS Pharmacol. Transl. Sci. 2022,
5, 548–554.
[Google Scholar]
-
88.
Hermann A, Goyal R, Jeltsch A. The Dnmt1 DNA-(cytosine-C5)-methyltransferase methylates DNA processively with high preference for hemimethylated target sites.
J. Biol. Chem. 2004,
279, 48350–48359.
[Google Scholar]
-
89.
Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond.
Nat. Rev. Genet. 2012,
13, 484–492.
[Google Scholar]
-
90.
Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development.
Cell 1999,
99, 247–257.
[Google Scholar]
-
91.
Bechtel W, McGoohan S, Zeisberg EM, Muller GA, Kalbacher H, Salant DJ, et al. Methylation determines fibroblast activation and fibrogenesis in the kidney.
Nat. Med. 2010,
16, 544–550.
[Google Scholar]
-
92.
Dees C, Potter S, Zhang Y, Bergmann C, Zhou X, Luber M, et al. TGF-beta-induced epigenetic deregulation of SOCS3 facilitates STAT3 signaling to promote fibrosis.
J. Clin. Invest. 2020,
130, 2347–2363.
[Google Scholar]
-
93.
Henderson J, Brown M, Horsburgh S, Duffy L, Wilkinson S, Worrell J, et al. Methyl cap binding protein 2: a key epigenetic protein in systemic sclerosis.
Rheumatology 2019,
58, 527–535.
[Google Scholar]
-
94.
Zhang Y, Potter S, Chen CW, Liang R, Gelse K, Ludolph I, et al. Poly(ADP-ribose) polymerase-1 regulates fibroblast activation in systemic sclerosis.
Ann. Rheum. Dis. 2018,
77, 744–751.
[Google Scholar]
-
95.
Malaab M, Renaud L, Takamura N, Zimmerman KD, da Silveira WA, Ramos PS, et al. Antifibrotic factor KLF4 is repressed by the miR-10/TFAP2A/TBX5 axis in dermal fibroblasts: insights from twins discordant for systemic sclerosis.
Ann. Rheum. Dis. 2022,
81, 268–277.
[Google Scholar]
-
96.
Baker Frost D, da Silveira W, Hazard ES, Atanelishvili I, Wilson RC, Flume J, et al. Differential DNA Methylation Landscape in Skin Fibroblasts from African Americans with Systemic Sclerosis.
Genes 2021,
12, 129.
[Google Scholar]
-
97.
Barnes PJ, Adcock IM, Ito K. Histone acetylation and deacetylation: importance in inflammatory lung diseases.
Eur. Respir. J. 2005,
25, 552–563.
[Google Scholar]
-
98.
Bhattacharyya S, Ghosh AK, Pannu J, Mori Y, Takagawa S, Chen G, et al. Fibroblast expression of the coactivator p300 governs the intensity of profibrotic response to transforming growth factor beta.
Arthritis Rheum. 2005,
52, 1248–1258.
[Google Scholar]
-
99.
Ghosh AK, Bhattacharyya S, Lafyatis R, Farina G, Yu J, Thimmapaya B, et al. p300 is elevated in systemic sclerosis and its expression is positively regulated by TGF-beta: epigenetic feed-forward amplification of fibrosis.
J. Invest. Dermatol. 2013,
133, 1302–1310.
[Google Scholar]
-
100.
Zehender A, Li YN, Lin NY, Stefanica A, Nuchel J, Chen CW, et al. TGFbeta promotes fibrosis by MYST1-dependent epigenetic regulation of autophagy.
Nat. Commun. 2021,
12, 4404.
[Google Scholar]
-
101.
Kramer M, Dees C, Huang J, Schlottmann I, Palumbo-Zerr K, Zerr P, et al. Inhibition of H3K27 histone trimethylation activates fibroblasts and induces fibrosis.
Ann. Rheum. Dis. 2013,
72, 614–620.
[Google Scholar]
-
102.
Maurer B, Distler JH, Distler O. The Fra-2 transgenic mouse model of systemic sclerosis.
Vascul. Pharmacol. 2013,
58, 194–201.
[Google Scholar]
-
103.
Bergmann C, Brandt A, Merlevede B, Hallenberger L, Dees C, Wohlfahrt T, et al. The histone demethylase Jumonji domain-containing protein 3 (JMJD3) regulates fibroblast activation in systemic sclerosis.
Ann. Rheum. Dis. 2018,
77, 150–158.
[Google Scholar]
-
104.
Wohlfahrt T, Rauber S, Uebe S, Luber M, Soare A, Ekici A, et al. PU.1 controls fibroblast polarization and tissue fibrosis.
Nature 2019,
566, 344–349.
[Google Scholar]
-
105.
Liu G, Friggeri A, Yang Y, Milosevic J, Ding Q, Thannickal VJ, et al. miR-21 mediates fibrogenic activation of pulmonary fibroblasts and lung fibrosis.
J. Exp. Med. 2010,
207, 1589–1597.
[Google Scholar]
-
106.
Henderson J, Wilkinson S, Przyborski S, Stratton R, O’Reilly S. microRNA27a-3p mediates reduction of the Wnt antagonist sFRP-1 in systemic sclerosis.
Epigenetics 2021,
16, 808–817.
[Google Scholar]
-
107.
Maurer B, Stanczyk J, Jungel A, Akhmetshina A, Trenkmann M, Brock M, et al. MicroRNA-29, a key regulator of collagen expression in systemic sclerosis.
Arthritis Rheum. 2010,
62, 1733–1743.
[Google Scholar]
-
108.
Yao Q, Xing Y, Wang Z, Liang J, Lin Q, Huang M, et al. MiR-16-5p suppresses myofibroblast activation in systemic sclerosis by inhibiting NOTCH signaling.
Aging 2020,
13, 2640–2654.
[Google Scholar]
-
109.
Wang Z, Jinnin M, Nakamura K, Harada M, Kudo H, Nakayama W, et al. Long non-coding RNA TSIX is upregulated in scleroderma dermal fibroblasts and controls collagen mRNA stabilization.
Exp. Dermatol. 2016,
25, 131–136.
[Google Scholar]
-
110.
Takata M, Pachera E, Frank-Bertoncelj M, Kozlova A, Jungel A, Whitfield ML, et al. OTUD6B-AS1 Might Be a Novel Regulator of Apoptosis in Systemic Sclerosis.
Front. Immunol. 2019,
10, 1100.
[Google Scholar]
-
111.
Wasson CW, Abignano G, Hermes H, Malaab M, Ross RL, Jimenez SA, et al. Long non-coding RNA HOTAIR drives EZH2-dependent myofibroblast activation in systemic sclerosis through miRNA 34a-dependent activation of NOTCH.
Ann. Rheum. Dis. 2020,
79, 507–517.
[Google Scholar]
-
112.
Wasson CW, Ross RL, Wells R, Corinaldesi C, Georgiou IC, Riobo-Del Galdo NA, et al. Long non-coding RNA HOTAIR induces GLI2 expression through Notch signalling in systemic sclerosis dermal fibroblasts.
Arthritis Res. Ther. 2020,
22, 286.
[Google Scholar]
-
113.
Pachera E, Assassi S, Salazar GA, Stellato M, Renoux F, Wunderlin A, et al. Long noncoding RNA H19X is a key mediator of TGF-beta-driven fibrosis.
J. Clin. Invest. 2020,
130, 4888–4905.
[Google Scholar]
-
114.
Quiros PM, Mottis A, Auwerx J. Mitonuclear communication in homeostasis and stress.
Nat. Rev. Mol. Cell. Biol. 2016,
17, 213–226.
[Google Scholar]
-
115.
Braga PC, Alves MG, Rodrigues AS, Oliveira PF. Mitochondrial Pathophysiology on Chronic Kidney Disease.
Int. J. Mol. Sci. 2022,
23, 1776.
[Google Scholar]
-
116.
Bueno M, Lai YC, Romero Y, Brands J, St Croix CM, Kamga C, et al. PINK1 deficiency impairs mitochondrial homeostasis and promotes lung fibrosis.
J. Clin. Invest. 2015,
125, 521–538.
[Google Scholar]
-
117.
Jaeger VK, Lebrecht D, Nicholson AG, Wells A, Bhayani H, Gazdhar A, et al. Mitochondrial DNA mutations and respiratory chain dysfunction in idiopathic and connective tissue disease-related lung fibrosis.
Sci. Rep. 2019,
9, 5500.
[Google Scholar]
-
118.
Cantanhede IG, Liu H, Liu H, Balbuena Rodriguez V, Shiwen X, Ong VH, et al. Exploring metabolism in scleroderma reveals opportunities for pharmacological intervention for therapy in fibrosis.
Front. Immunol. 2022,
13, 1004949.
[Google Scholar]
-
119.
Zhou X, Trinh-Minh T, Tran-Manh C, Giessl A, Bergmann C, Gyorfi AH, et al. Impaired Mitochondrial Transcription Factor A Expression Promotes Mitochondrial Damage to Drive Fibroblast Activation and Fibrosis in Systemic Sclerosis.
Arthritis Rheumatol. 2022,
74, 871–881.
[Google Scholar]
-
120.
Zhang M, Tong Z, Wang Y, Fu W, Meng Y, Huang J, et al. Relationship between ferroptosis and mitophagy in renal fibrosis: a systematic review.
J. Drug Target. 2023,
31, 858–866.
[Google Scholar]
-
121.
Patel AS, Song JW, Chu SG, Mizumura K, Osorio JC, Shi Y, et al. Epithelial cell mitochondrial dysfunction and PINK1 are induced by transforming growth factor-beta1 in pulmonary fibrosis.
PLoS ONE 2015,
10, e0121246.
[Google Scholar]
-
122.
Gazdhar A, Lebrecht D, Roth M, Tamm M, Venhoff N, Foocharoen C, et al. Time-dependent and somatically acquired mitochondrial DNA mutagenesis and respiratory chain dysfunction in a scleroderma model of lung fibrosis.
Sci. Rep. 2014,
4, 5336.
[Google Scholar]
-
123.
Xiao H, Xie Y, Xi K, Xie J, Liu M, Zhang Y, et al. Targeting Mitochondrial Sirtuins in Age-Related Neurodegenerative Diseases and Fibrosis.
Aging Dis. 2023,
14, 1583–1605.
[Google Scholar]
-
124.
Sosulski ML, Gongora R, Feghali-Bostwick C, Lasky JA, Sanchez CG. Sirtuin 3 Deregulation Promotes Pulmonary Fibrosis.
J. Gerontol. A Biol. Sci. Med. Sci. 2017,
72, 595–602.
[Google Scholar]
-
125.
Zhou T, Lin W, Lin S, Zhong Z, Luo Y, Lin Z, et al. Association of Nuclear Receptor Coactivators with Hypoxia-Inducible Factor-1alpha in the Serum of Patients with Chronic Kidney Disease.
Biomed. Res. Int. 2020,
2020, 1587915.
[Google Scholar]
-
126.
Tarin D, Croft CB. Ultrastructural features of wound healing in mouse skin.
J. Anat. 1969,
105, 189–190.
[Google Scholar]
-
127.
LeBleu VS, Neilson EG. Origin and functional heterogeneity of fibroblasts.
FASEB J. 2020,
34, 3519–3536.
[Google Scholar]
-
128.
Franks JM, Martyanov V, Cai G, Wang Y, Li Z, Wood TA, et al. A Machine Learning Classifier for Assigning Individual Patients With Systemic Sclerosis to Intrinsic Molecular Subsets.
Arthritis Rheumatol. 2019,
71, 1701–1710.
[Google Scholar]
-
129.
Driskell RR, Lichtenberger BM, Hoste E, Kretzschmar K, Simons BD, Charalambous M, et al. Distinct fibroblast lineages determine dermal architecture in skin development and repair.
Nature 2013,
504, 277–281.
[Google Scholar]
-
130.
Korosec A, Frech S, Gesslbauer B, Vierhapper M, Radtke C, Petzelbauer P, et al. Lineage Identity and Location within the Dermis Determine the Function of Papillary and Reticular Fibroblasts in Human Skin.
J. Invest. Dermatol. 2019,
139, 342–351.
[Google Scholar]
-
131.
Mastrogiannaki M, Lichtenberger BM, Reimer A, Collins CA, Driskell RR, Watt FM. beta-Catenin Stabilization in Skin Fibroblasts Causes Fibrotic Lesions by Preventing Adipocyte Differentiation of the Reticular Dermis.
J. Invest. Dermatol. 2016,
136, 1130–1142.
[Google Scholar]
-
132.
Marangoni RG, Datta P, Paine A, Duemmel S, Nuzzo M, Sherwood L, et al. Thy-1 plays a pathogenic role and is a potential biomarker for skin fibrosis in scleroderma.
JCI Insight. 2022,
7, 149426.
[Google Scholar]
-
133.
Tan C, Jiang M, Wong SS, Espinoza CR, Kim C, Li X, et al. Soluble Thy-1 reverses lung fibrosis via its integrin-binding motif.
JCI Insight. 2019,
4, 131152.
[Google Scholar]
-
134.
Rinkevich Y, Walmsley GG, Hu MS, Maan ZN, Newman AM, Drukker M, et al. Skin fibrosis. Identification and isolation of a dermal lineage with intrinsic fibrogenic potential.
Science 2015,
348, aaa2151.
[Google Scholar]
-
135.
Jiang D, Correa-Gallegos D, Christ S, Stefanska A, Liu J, Ramesh P, et al. Two succeeding fibroblastic lineages drive dermal development and the transition from regeneration to scarring.
Nat. Cell. Biol. 2018,
20, 422–431.
[Google Scholar]
-
136.
Gyorfi AH, Matei AE, Fuchs M, Liang C, Rigau AR, Hong X, et al. Engrailed 1 coordinates cytoskeletal reorganization to induce myofibroblast differentiation.
J. Exp. Med. 2021,
218, e20201916.
[Google Scholar]
-
137.
Tabib T, Morse C, Wang T, Chen W, Lafyatis R. SFRP2/DPP4 and FMO1/LSP1 Define Major Fibroblast Populations in Human Skin.
J. Invest. Dermatol. 2018,
138, 802–810.
[Google Scholar]
-
138.
Tsukui T, Sun KH, Wetter JB, Wilson-Kanamori JR, Hazelwood LA, Henderson NC, et al. Collagen-producing lung cell atlas identifies multiple subsets with distinct localization and relevance to fibrosis.
Nat. Commun. 2020,
11, 1920.
[Google Scholar]
-
139.
Xie T, Wang Y, Deng N, Huang G, Taghavifar F, Geng Y, et al. Single-Cell Deconvolution of Fibroblast Heterogeneity in Mouse Pulmonary Fibrosis.
Cell Rep. 2018,
22, 3625–3640.
[Google Scholar]
-
140.
Buechler MB, Pradhan RN, Krishnamurty AT, Cox C, Calviello AK, Wang AW, et al. Cross-tissue organization of the fibroblast lineage.
Nature 2021,
593, 575–579.
[Google Scholar]
-
141.
Seguro Paula F, Delgado Alves J. The role of the Notch pathway in the pathogenesis of systemic sclerosis: clinical implications. Expert.
Rev. Clin. Immunol. 2021,
17, 1257–1267.
[Google Scholar]
-
142.
Leask A, Naik A, Stratton RJ. Back to the future: targeting the extracellular matrix to treat systemic sclerosis.
Nat. Rev. Rheumatol. 2023,
19, 713–723.
[Google Scholar]