1. Introduction
Fibrosis is a pathological event that causes the increased deposition of collagen and other extracellular matrix (ECM) proteins into the tissues. More often than not, the cause of fibrosis is unknown. It becomes more prevalent with increasing age [
1,
2]. Estimates have suggested that about 45% of the deaths in the Western world can be attributed to fibrosis [
3]. Fibroblasts and myofibroblasts are key cell phenotypes required during wound healing, and they are pathologically involved during fibrosis. During routine wound healing, these cells migrate to the site of the tissue injury and secrete collagens and other ECM proteins that repair the damaged tissues. They use focal adhesions and the contractile properties of the cell’s cytoskeleton to pull the wound margins together [
4] and secrete ECM to fill in the damaged matrix. However, fibroblasts and myofibroblasts will also migrate to an area in the tissue with chronic inflammation and secrete abundant amounts of collagen. This event can cause fibrosis.
The origins of these migrating fibroblasts have been extensively studied, and they are thought to originate from various sources, including mesenchymal stem cells [
5], adipocytes [
6], epithelial and endothelial cells [
7], and the bone marrow [
5]. However, more recent and elegant studies investigating wound healing in deep excisional skin wounds identified the role of resident fibroblasts found in the fascia that migrate into the site of injury [
6,
7,
8]. Correa-Gallegos et al. showed that fibroblasts swarmed “en masse” from the fascia into the injury, dragging with them the surrounding ECM [
7]. They found that this pre-made wound sealant also contains vasculature, immune cells, and nerves. They believe this helps to accelerate wound closure but can contribute to scar formation [
7]. In other studies, Fisher et al. also found the transfer of ECM from outside the lung into the parenchyma. When the lung experiences injury or inflammation, the ECM from the pleuro-alveolar junctions migrates into the lung tissue, activating resident fibroblasts and causing fibrosis [
9]. This mechanism of fibroblast activation is substantially different from that found during deep excisional wound healing, where the fibroblasts drag the matrix into the wound bed, as no apparent cells were involved in this process. We speculate that a specialized, yet unidentified fibroblast may be involved in this process. We also assume that in other types of organ fibrosis, pleural ECM may migrate to the sites of inflammation in that organ, activating resident fibroblasts and causing fibrosis.
The ECM is not just a passive scaffold along which the fibroblasts crawl but plays a profoundly active and multifaceted role in regulating fibroblast migration [
10]. The ECM is comprised of more than collagen and contains other large molecular weight proteins, such as fibronectin, proteoglycans, and hyaluronic acid. There is an active interaction between the fibroblast and the ECM. The ECM provides a dynamic environment that sends physical and biochemical signals to guide, stimulate, or restrict the movement of fibroblasts [
10]. This intricate interplay is essential for processes such as wound healing, tissue development, and fibrosis. Fibroblasts respond to the stiffness of the ECM and migrate (durotaxis) towards stiffer matrices [
11,
12]. This process is crucial and aids in the process of wound healing as the nascent ECM is deposited and cross-linked, leading to an increase in ECM stiffness. However, intriguingly, there is an optimal stiffness range of the ECM for efficient fibroblast migration [
13]. If the ECM is too soft, the fibroblast cannot sufficiently grip the matrix to pull itself along. If the ECM is too stiff, this impedes fibroblast migration [
14]. This is because cells need a certain level of stiffness to generate the traction forces required for movement, while excessive stiffness makes it difficult to remodel the matrix or detach the trailing edge of the cell [
15]. In fibrosis, the ECM becomes excessively stiff due to the increased collagen deposition and cross-linking [
16]. This stiffness further activates fibroblasts, driving their differentiation into myofibroblasts and perpetuating the fibrotic cycle, including continued migration into and remodeling of the rigid matrix [
17]. The architecture and topography of the matrix are also essential and influence the migration of fibroblasts. The fibroblasts will align and migrate along collagen fibers or grooves and ridges in the ECM [
18,
19]. The porosity of the matrix can influence how fibroblasts navigate through the ECM, and the cells may need to degrade the matrix to move [
20]. Beyond static stiffness, the viscoelastic (time-dependent deformation) properties of the ECM can also influence fibroblast behavior [
21,
22]. More recent studies have demonstrated that viscoelasticity allows tissues to dissipate energy over time [
21], and studies have found that the fine-tuning of ECM viscoelasticity regulates fibroblast adhesion and migration dynamics [
23,
24]. This has implications for the development of fibrosis. Overall, there is a complex interaction between fibroblasts that modify the ECM while the ECM guides the fibroblast phenotype. In disease states like aging or fibrosis, changes in ECM composition, such as increased cross-linking and fragmentation of collagen, and the mechanics (increased stiffness) that the fibroblast encounters disrupt these crucial feedback loops, leading to impaired fibroblast function. This impaired fibroblast function is often reflected in reduced migration efficiency, resulting in pathological activation.
The role of fibroblast migration is an essential pathological factor in all fibrotic diseases, including systemic sclerosis (SSc). SSc is a rare autoimmune disease with fibrotic pathology manifesting in the skin and internal organs such as the lungs and heart [
25,
26,
27]. Approximately 30–50 years of age is most commonly seen for the onset of SSc; however, in rare instances, children can also get the disease [
28]. As Huang et al. mentioned, fibrosis is a common condition, yet pinpointing effective and long-lasting treatments has been challenging to date [
29]. This is likely due to a lack of understanding of the numerous overlapping pathways involved in the development of fibrosis.
This review has compiled essential aspects of fibroblast migration, especially discussing the cytoskeleton and actin stabilization. In addition, we discuss the relevance of focal adhesions and how they regulate the migration of fibroblasts. We will also discuss the little-known role of tropomyosin in fibroblast migration, as it is a critical protein that helps to stabilize actin filaments. We summarize the specific proteins and pathways discussed in cell migration and correlate their functional roles in the context of fibrosis. At the end of the review, we discuss novel areas that could be targeted therapeutically to mitigate myofibroblast activation and migration as much as possible means to abrogate fibrosis.
2. The Mechanics of Fibroblast Migration
Fibroblast migration is a critical factor in wound healing and fibrosis. Its mechanics are elegant and highly orchestrated, and they are centrally oriented around the fibroblast cytoskeleton. How fibroblasts migrate depends on ECM stiffness, the topography they encounter, and various growth factors and intracellular signaling pathways that regulate cytoskeletal reorganization and cell adhesion.
2.1. Fibroblast Polarity
The fibroblast polarity plays a vital role in the direction in which the fibroblast migrates. Myosins are motor proteins responding to cellular force and regulating many of the downstream intracellular processes as a result of this force. They are known to play a role in fibroblast migration and differentiation [
30,
31]. A recent study by Bun et al. [
32] investigated the polarity of fibroblasts by evaluating normal versus immortalized fibroblasts (high polarity versus low polarity, respectively). They previously found that normal fibroblasts could migrate in a straight direction without external stimuli and termed it intrinsic and directed migration [
32,
33]. Thus, the authors describe polarity-based fibroblast migration as a key characteristic, which is neither disrupted nor stimulated by environmental factors like chemoattractant concentration levels [
32]. They further examined this phenomenon and found that this intrinsic and direct migration was dependent on non-muscle myosin II (isoforms A and B) in embryonic fetal lung fibroblasts, which were the focus of this study [
33]. Their data shows that the two non-muscle myosin II isoforms affect the lengthening of the fibroblast, with A and B found at polar opposites. Non-muscle myosin IIB was seen to be critical in the forward momentum of the fibroblast because when it was knocked down, the fibroblasts traveled backward [
33]. Further defining the function of these isoforms, using a non-muscle myosin II ATPase inhibitor, they found that the fibroblasts altered their speed and direction during migration. Intriguingly, they found that the speed of the fibroblast increased while the direction of the fibroblast became erratic [
33]. They hypothesized that non-muscle myosin IIA constructs the forward propellers at the front of the fibroblast, while non-muscle myosin IIB blocks propeller construction at the rear of the cell [
33].
Utilizing decellularized lung tissues, it was found that non-muscle myosin II had different functions in the cell, depending on the matrix that the fibroblast encountered [
34]. Southern and colleagues found that non-muscle myosin II limited fibroblast protrusion and promoted polarized migration in human lung fibroblasts cultured on normal lungs. However, non-muscle myosin II activity enhanced cell protrusions but limited polarized migration of human lung fibroblasts cultured on decellularized fibrotic lung tissue, leading to enhanced myofibroblast differentiation [
34]. Overactivity of non-muscle myosin IIA has been found in keloid scars [
35]. Non-muscle myosin IIA regulates TGF-β signaling and a-smooth muscle actin expression, and inhibition of non-muscle myosin IIA downregulates TGF-β signaling and cell proliferation [
35]. The matrices on which the fibroblasts are studied influence non-muscle myosin II activity. Moreno-Arotzena et al. investigated fibroblast migration in 3D collagen versus fibrin matrices [
36]. The collagen gels had larger pore sizes compared to the fibrin gels, and the elastic shear modulus was approximately 20-fold higher in the fibrin gels. They observed that fibroblasts could migrate with the collagen gels more efficiently than in the fibrin gels. They were able to demonstrate that fibroblasts showed “contractile shaking”. They believed that this was due to cycles of plasma membrane protrusions as the cell explores its surroundings. In the presence of a chemoattractant, the cell uses traction forces that establish front-rear polarity. As a result of the polarity, the plasma membrane protrusions are restrained to the cell’s leading edge. As a consequence of polarity establishment, protrusion is mainly restricted to the leading edge of the cell [
36].
2.2. Focal Adhesions
For efficient fibroblast migration, there has to be a dynamic assembly and disassembly of focal adhesions [
37,
38]. Focal adhesion complexes are formed at the cell’s leading edge in the lamellipodia [
39,
40,
41]. The formation of a focal adhesion starts when integrins on the cell membrane bind to the ECM. This leads to their clustering and activation, which also induces a conformational change [
42,
43]. During fibroblast migration, many focal adhesions do not mature, and they are disassembled [
44]. However, when focal adhesions mature, they become more prominent, stable, and stationary on the plasma membrane. The cell uses them as anchors to move across the ECM, and the plasma membrane flows over the focal adhesion (), moving it closer to the trailing edge of the cell, where the focal adhesion is disassembled, and the proteins are recycled [
44]. The size of the focal adhesion correlates directly with the speed at which the cell travels across the ECM [
45]. The focal adhesions contain heterodimeric - and β-integrins combinations with other proteins, such as focal adhesion kinase, talin, paxillin, zyxin, and tensin [
46,
47,
48,
49,
50]. These additional proteins help to stabilize the adhesion complex [
51]. In addition to various integrins that the cell uses to engage the ECM, CD44 also interacts with ligands in the ECM, such as hyaluronan, collagen, fibronectin, and osteopontin. At the same time, its cytoplasmic domain connects to the actin cytoskeleton and functions to organize it, affecting the formation of the stress fibers and focal adhesion complexes, which are essential for cell migration and the development of fibrosis [
52,
53].
2.3. Cytoskeleton
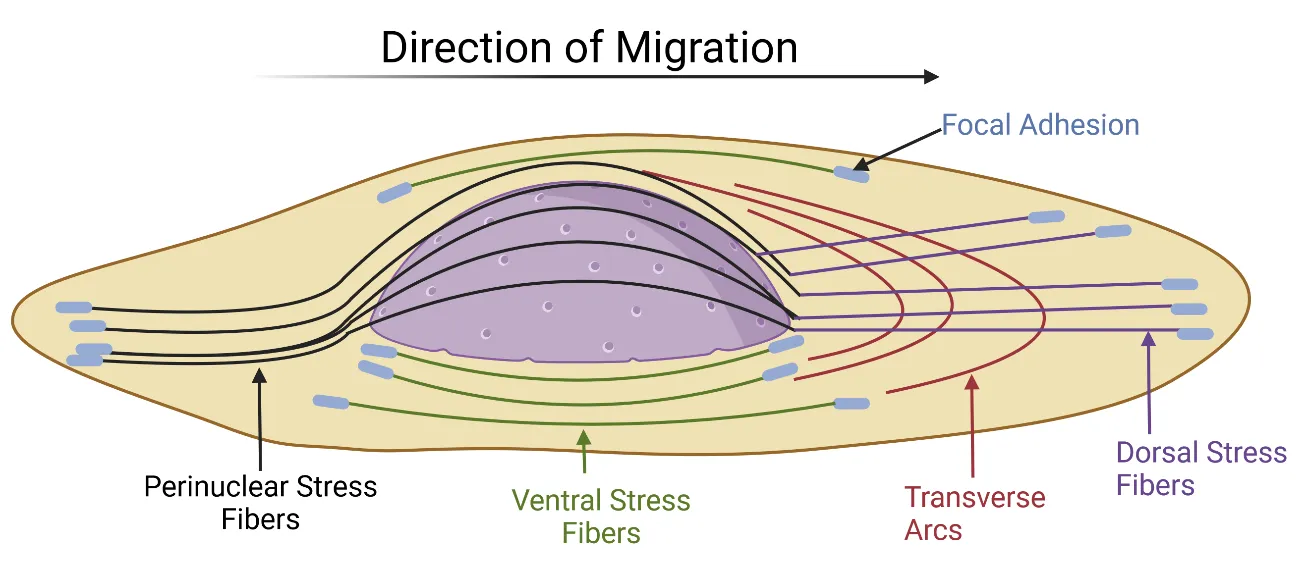
The fibroblast cytoskeleton is a crucial network of intracellular filaments called stress fibers, involved in cell migration, cell division, maintenance of cell shape, and intracellular trafficking [
54,
55,
56] (). Various actin-binding proteins help stabilize these filaments, influencing their function within the cell [
57,
58,
59]. Cytoskeleton consists of various types: dorsal, transverse, ventral, and perinuclear stress fibers [
60] (discussed in more detail below). All of these fibers function together to surround and orient the nuclear axis in line with the direction of fibroblast migration [
60,
61].
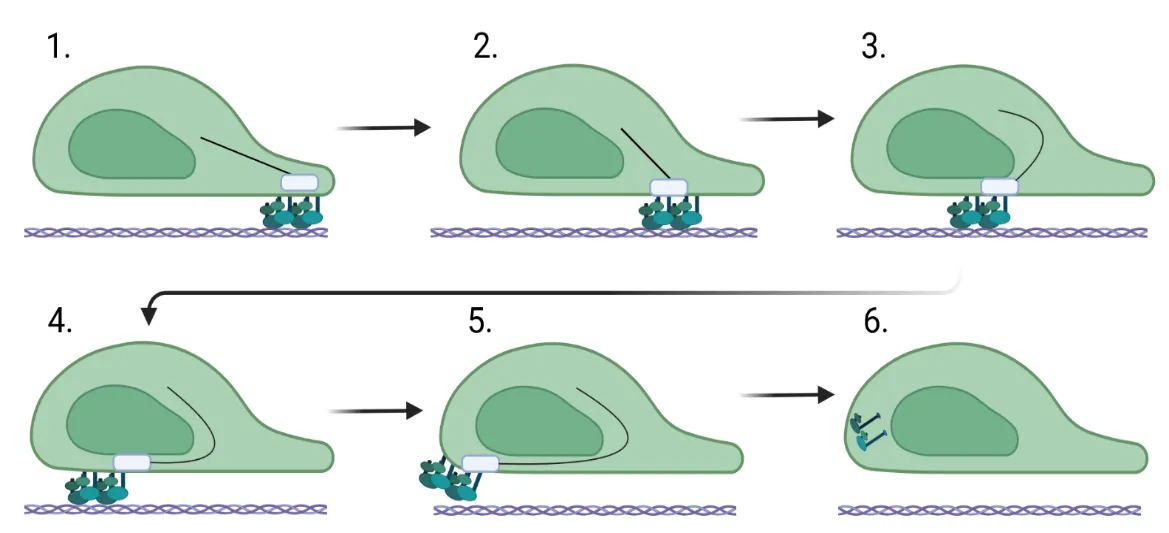
. Focal adhesion movement during fibroblast migration. (<b>1</b>) For the cell to migrate forward, focal adhesions bind to integrins and engage the ECM at the leading edge of the fibroblast. (<b>2</b>–<b>5</b>) The integrin/focal adhesion complex travels toward the back of the cell as it flows forward over them. (<b>6</b>) The integrins and focal adhesions disengage from the extracellular matrix and are recycled. Created in BioRender. Artlett C. (2025) https://BioRender.com/a86l650.
. Orientation of the four different cytoskeletal stress fibers in the fibroblast. The contractile forces that the nucleus undergoes during migration, force the nucleus to orient in the direction the fibroblast is migrating. Adapted from Maninova and Vomastek, 2016 [
60]. Created in BioRender. Artlett C. (2025) https://BioRender.com/m44p889.
2.3.1. Dorsal Stress Fibers
Approximately 10–30 F-actin filaments are associated together to form a dorsal stress fiber. These fibers do not contract due to their non-binding of myosin II [
62]. They are typically stabilized by a-actinin [
63], or tropomyosins [
64], among other proteins [
65]. The dorsal stress fibers connect focal adhesions in the lamellipodia and stretch centrally toward the nucleus. At their opposite end, closest to the nucleus, the fibers often interact with transverse arcs [
66,
67], which are often positioned at right angles to the proximal end of the dorsal surface [
66]. These stress fibers can also act as a platform for the assembly of other stress fibers within the cell [
62]. Dorsal stress fibers are short actin filament bundles that are prominent in lamellae [
66].
2.3.2. Transverse Arcs
The fibers comprising the transverse arcs are curved and surround the nucleus. They do not interact directly with focal adhesions [
66,
67,
68]. They are contractile and interact with α-actinin- and myosin II-associated actin bundles. They form an interconnected network with dorsal stress fibers rising toward the top of the cell [
66,
67,
68]. Non-muscle myosin IIA and IIB have different localization patterns. Non-muscle myosin IIA was found to localize in all transverse arcs as a punctate pattern, while non-muscle myosin IIB mainly localized to the proximal transverse arcs [
33]. Kuranno and colleagues were also able to show a temporal relationship between non-muscle myosin IIa and IIB. They showed in fibroblasts that non-muscle myosin IIA was present in newly formed transverse arcs, but as the transverse arcs matured, non-muscle myosin IIB became incorporated.
2.3.3. Ventral Stress Fibers
Ventral stress fibers are situated underneath the nucleus and extend toward the trailing edge of the cell [
66,
67,
69,
70]. The ventral fibers are discrete contractile fibers within the ventral plane, consisting of actin filaments stabilized by a-actinin and non-muscle myosin IIA [
33]. They are attached to the substrate through focal adhesions at each end. These fibers can vary in length and be oriented at various angles to the cell axis; however, the longer ventral fibers are aligned in the direction that the cell is traveling [
33]. These fibers produce most of the cellular traction and are involved in the retraction of the rear portion of the cell during migration [
58,
71,
72].
2.3.4. Perinuclear Stress Fibers
Perinuclear fibers are initiated at the leading edge of the cell and extend over the nucleus, where they terminate at the rear of the cell [
60]. Perinuclear fibers are stabilized by the linker of the nucleoskeleton and cytoskeleton complex (LINC), which is composed of Sun and Nesprin proteins [
73]. The perinuclear fibers and the LINC complex are crucial in regulating nuclear shape [
74,
75]. The nucleus rotates in the direction of migration when the perinuclear stress fibers are formed [
60]. This orientation was also found to require non-muscle II because the inhibition of its activity blocked the rotation of the nucleus [
60].
2.4. Tropomyosins
One of the lesser-studied proteins involved in fibroblast migration is the tropomyosin family. Tropomyosins are a group of rod-shaped proteins with various functions in muscle and non-muscle cells. Tropomyosins are a prominent family of proteins interacting with actin filaments [
64]. They are highly conserved and decorate most actin structures in a head-to-tail fashion [
64] in diverse organisms, ranging from yeast to humans [
64,
76]. They lie in the groove that forms when the actin molecule polymerizes [
77]. Tropomyosin isoforms function in muscle and non-muscle cells and are involved in various cellular pathways that control and regulate the cell’s cytoskeleton and other essential functions. Mammalian cells have four tropomyosin genes that give rise to more than forty alternatively spliced isoforms [
76,
78]. Only six of the forty tropomyosin splice variants encoded are essential to fibroblast actin stability. These include tropomyosins 1.6, 1.7, 2.1, 3.1, 3.2, and 4.2.
The primary function of tropomyosin in the fibroblast is to prevent actin cleavage by cofilin [
79,
80], as cofilin and tropomyosin are mutually exclusive [
80]. Tropomyosin 1.6 stabilizes a-smooth muscle actin incorporation into stress fibers [
81] and regulates myosin IB and myosin IC affinity to target actin and control fibroblast motor activity [
81,
82]. Tropomyosins are known regulators of cell migration, especially during wound healing [
83] and cancer metastases [
84]. Initial studies showed that focal adhesions contain three well-defined layers [
32,
44,
45]; however, recently, it has been shown that there are two additional layers within the focal adhesion, and these layers are enriched with tropomyosin 1.6 and tropomyosin 3.2 [
85]. In the absence of tropomyosin 1 and 3, Kumari et al. found that the actin filaments are thinner and less organized. They also identified the early localization of tropomyosin within the focal adhesions [
85]. Intriguingly, it has been shown that tropomyosin 3 is needed for the retraction of the tail at the rear of the cell and was found to be involved in focal adhesion disassembly [
85].
We speculate that specific tropomyosin isoforms, particularly Tpm1.6 and Tpm3.2, are key regulators of the persistent and directional migration of fibroblasts in fibrotic tissues. This observation would go beyond their general role in migration and could suggest a pathological influence in this spectrum of diseases. Their recently discovered enrichment in two additional layers within focal adhesions and their role in regulating myosin IB and IC affinity to actin suggest they modulate the precise dynamics of adhesion formation and turnover that is needed for persistent migration. Since tropomyosins organize actin filaments and stabilize stress fibers, which are the primary structures sensing mechanical cues, we propose that tropomyosins may be involved in the fibroblast’s ability to sense and respond to increased tissue stiffness. Their presence within focal adhesions, the primary interface for mechanosensing, positions them as critical players. Altered tropomyosin expression or function could therefore amplify pro-fibrotic mechanotransduction pathways, creating a vicious cycle of increasing stiffness and fibroblast activation.
3. Intracellular Signaling During Cell Migration
Intracellular signaling is another critical factor that helps to stabilize the cytoskeleton during fibroblast migration, and specific pathways have been strongly associated with this process.
3.1. ROCK/Rho Proteins
The Rho associated coil kinase (ROCK) proteins are essential regulators of the cytoskeleton. They act mainly by modulating actomyosin contraction [
86,
87,
88], a vital cell migration process. Their functional effect is phosphorylating motor protein myosin II or inhibiting phosphatase, dephosphorylating myosin. Collectively, this leads to increased myosin activity, resulting in actin shortening. The homologous Rho kinases, ROCK1 and ROCK2, are downstream targets of RhoA and RhoC, and collectively, they regulate the activity of myosin II [
89]. Myosin II contains a myosin light chain, and ROCK phosphorylates this chain [
90]. At the same time, ROCK inhibits the activity of the myosin light chain phosphatase [
90]. This ensures that the myosin light chain remains phosphorylated, promoting contraction of the actin filament, which helps drive cell migration [
91,
92]. The increased actomyosin contractility induced by ROCK also helps to promote cell contraction. This leads to increased cell adhesion to the ECM, allowing the cell to migrate. While ROCK1 and ROCK2 are involved in cell migration, they do so via distinct roles. ROCK1 destabilizes actin by regulating the myosin light chain, causing actin-myosin contraction [
90], while ROCK2 acts on cofilin to help stabilize the actin cytoskeleton. It does so by phosphorylating cofilin, which leads to its inactivation [
87], and this helps to stabilize the actin cytoskeleton. ROCK1 also plays a role in cell polarity, while ROCK2 regulates the contractile force [
93].
3.2. Sonic Hedgehog
The Sonic Hedgehog (SHH) protein is active during fibroblast migration. It enhances epithelial-to-mesenchymal transition, and while it can restrict migration, it has also been shown to enhance cell migration [
94,
95,
96]. SHH can activate cytoskeletal actin stability by activating T-Lymphoma Invasion and Metastasis-Inducing Protein 1/Rac Family Small GTPase 1, allowing the cells to migrate [
97]. It achieves this by binding to its protein receptors Patched and Smoothened, leading to the activation of genes involved in actin stability [
98]. More recent evidence shows that the transmembrane Smoothened is involved in fibroblast migration. This observation was supported by reduced fibroblast migration in cells treated with Smoothened siRNA [
99]. Basic fibroblast growth factor interacts with SHH, regulating the effects of downstream SHH gene activation and expression levels [
99]. A study by Hou et al., found that type II alveolar epithelial cells release SHH, which influences macrophage M2 activation through osteopontin production, causing pulmonary fibrosis. Further elucidating that by inhibiting SHH in bleomycin-mice fibrotic lungs, there was decreased macrophage M2 activation. It is important to note that other studies displayed either similar or conflicting results, pertaining to SHH’s direct inhibitory role versus its downstream effects [
100].
3.3. WNT/β-Catenin Signaling
The WNT (Wingless integration site)/β-catenin pathway is also involved in cell migration [
101], and various WNT proteins help to stabilize the cytoskeleton and define cell polarity [
102,
103]. WNT genes also enhance cell migration by increasing various matrix metalloproteinase genes that degrade the ECM, allowing more efficient cell migration through the matrix [
104]. WNT also interacts with SHH to regulate the differentiation of myofibroblasts [
105]. WNT expression helps to establish alignment of the cells within the tissues (called planar cell polarity) and orient the movement of the cells by affecting the cytoskeleton and microtubule formation [
102,
103]. In the study by Zhu and colleagues, they show that β-catenin and Glioma-Associated Oncogene Homolog 1 (Gli1) have a prominent role in this signaling pathway during fibroblast migration [
99]. They postulate that fibroblast migration is based on a modulating circuit with interactions between SHH signaling genes, Smoothened, Gli1, and β-catenin [
99].
3.4. PI3K/AKT Activation
PI3K (phosphatidylinositol 3-kinase) influences several key aspects involved in cell migration. It regulates the dynamics of the actin cytoskeleton, affects cell polarity, and increases cell adhesion to the ECM. PI3K signaling influences the formation and turnover of the actin cytoskeleton, and by affecting actin polymerization and depolymerization, it can facilitate the formation of lamellipodia and filopodia at the leading edge and affect the retraction of the rear of the cell during migration [
106]. PI3K/AKT signaling establishes and maintains cell polarity by regulating the localization and activation of proteins involved in cell polarity, such as those related to the formation of leading and trailing edges of the migrating cell [
107]. PI3K/AKT pathways can affect cell adhesion by regulating focal adhesion expression [
108,
109]. The activation of PI3K will trigger AKT activity, and this transcription factor helps to regulate the expression of proteins involved in cell survival and cell migration [
110]. The selective inhibition of P13K/AKT signaling has been further studied as a potential treatment in pulmonary fibrosis in an effort to counteract its role in increasing fibroblast’s life span [
111].
4. Migration and Fibrosis
Fibroblast migration is an essential feature of fibrosis [
112]. It has been associated with the excessive deposition of ECM proteins within the tissues. Fibroblast migration allows these cells to reach sites of inflammation and respond to this inflammation by depositing collagen and other ECM proteins; however, this can be detrimental in the case of fibrosis. While fibroblasts are normally quiescent when responding to injury or inflammation, they upregulate their cytoskeletal components and differentiate into highly contractile myofibroblasts [
113] and increase their deposition of collagen and ECM into the tissues.
The direction of fibroblast migration in fibrotic tissues plays a role in the overall recruitment of the fibroblasts into the lesions. Tisler and colleagues found that fibroblasts migrated on collagen isolated from fibrotic lungs with enhanced directionality, while fibroblasts did not have this feature when cultured on collagen from normal lungs [
114]. Intriguingly, in the Tisler study, they also found that the cytoskeleton in the fibroblast was aligned with the direction of the collagen in the ECM, and they discovered that fibroblasts exhibited a faster migration speed on collagen derived from fibrotic lung [
114]. To aid in this increased speed of the fibroblasts, focal adhesion kinase is significantly elevated in fibrotic tissue, and targeting the activity of focal adhesion kinase has been shown to reduce fibrosis [
115,
116]. In fibrotic fibroblasts, focal adhesion kinase is persistently activated, and this leads to the elevated deposition of collagens in the tissues [
115]. While little is known about the tropomyosin family in fibrosis, this family of proteins is known to regulate cell migration [
117] and is found to be elevated in fibrosis [
118,
119,
120]. Inhibitors of tropomyosin are being actively developed in the context of cancer. Investigators aim to inhibit tumor progression by impeding or limiting cell migration and invasion while sensitizing cancer cells to other treatments [
121]. Synergistic effects of the two tropomyosin inhibitors TR100 or AMT-3507 have been observed in combination with the anti-microtubule drug vincristine, which also targets the cytoskeleton [
122]. This led to enhanced apoptosis and mitotic arrest in cancer cells. These inhibitors could be utilized to limit cell migration in fibrotic settings. Preclinical studies with inhibitors such as TR100 and ATM-3507, which aim to inhibit specific tropomyosin isoforms, have shown some promising results in cancer models. In animal studies, the combination showed minimal weight loss when used in conjunction with other chemotherapies. However, these were preliminary findings in animal models, and a comprehensive human toxicity profile has yet to be determined.
Structural changes to the fibroblast cytoskeleton play a critical role in fibrotic diseases, and this cytoskeleton reorganization is mediated by RhoA/ROCK signaling [
123]. Due to the increased matrix stiffness found in these tissues, ROCK1 and ROCK2 are elevated [
124], and these proteins also influence the differentiation of myofibroblasts [
125], their adhesion to the ECM [
126], and their polarity [
127]. The ROCK/Rho pathway drives actomyosin contraction [
128], allowing the cells to migrate. The importance of these studies is underscored by the success of ROCK inhibitors in reducing fibrosis. Using a non-specific ROCK inhibitor in the bleomycin mouse model, there was decreased differentiation of macrophages into the M2 class, which significantly reduced the progression of fibrosis in the lungs [
129]. In contrast, selective ROCK inhibitors were more efficacious with fewer adverse events than the non-selective ROCK inhibitors [
130]. Bei et al., showed that the specific ROCK inhibitor Fasudil suppressed hypochlorous acid dermal fibrosis in mice by inhibiting TGF- downstream signaling proteins Smad2/3 and ERK1/2 [
131]. However, it has been reported that non-selective ROCK inhibitors can cause adverse events such as hypotension [
132]. Selective versus non-selective ROCK inhibitors need further examination for the treatment of pulmonary fibrosis.
PI3K/AKT also has many overlapping functions in fibrosis and is found to regulate cell adhesion [
133], cell polarity and directional migration [
134,
135], and actin cytoskeleton dynamics [
136]. WNT gene expression is strongly associated with fibrotic tissues [
137] and has been extensively studied in lung fibrosis [
105,
138,
139,
140]. Of the WNT genes, WNT7A and WNT10A are more likely to be elevated [
137,
141,
142], although in the Tsk1 mouse model of skin fibrosis, Wnt2, Wnt9a, Wnt10b, and Wnt11 are increased [
143]. Using bleomycin to induce lung fibrosis in a mouse model, Chen and colleagues found that Wnt7a and Wnt10b were significantly elevated over several other elevated Wnt genes in myofibroblasts [
105]. Inhibition of the transcription factor Gli1 limited the conversion of lung mesenchymal stem cells to myofibroblasts, and as a result, there was less fibrosis with reduced Wnt gene expression [
105]. The canonical WNT/β-catenin axis is activated in fibrosis and influences changes in the cytoskeleton [
144] as the stabilization of β-catenin, upregulates the transcription of genes that are involved in the remodeling of the cytoskeleton [
144]. WNT/β-catenin can also signal non-canonically via the WNT/Ca
2+ and WNT/planar cell polarity pathways to affect the dynamics of the cytoskeleton [
145,
146]. This pathway influences matrix degradation by allowing fibroblasts to move through the matrix [
147]. SHH is especially important in epithelial-to-mesenchymal transition [
148], actin stability [
149], and profoundly influences the WNT/β-catenin pathway [
150].
5. Beyond the Traditional Views of the Dynamic Nature of Fibroblast Migration
As discussed above, there is complexity in fibroblast migration that is influenced by the dynamic interactions between the fibroblasts and the ECM. Fibroblast migration is a pivotal process in wound healing because it plays a role in orchestrating tissue repair, ECM deposition, and wound contraction. However, in a pathological setting, fibroblast migration also plays a pathogenic role in the development of fibrosis. Traditionally, fibroblast movement has been understood primarily through direct cellular interactions, as fibroblasts sense the ECM and modify their responses to soluble growth factors. However, recent groundbreaking discoveries have expanded this view. These discoveries have revealed sophisticated mechanisms of intercellular communication and environmental sensing that profoundly influence fibroblast motility and overall wound resolution and fibrosis. One of the most exciting developments is the recognition of extracellular vesicles as critical mediators of cell-to-cell communication. These small vesicles are secreted from fibroblasts (and other cells) and are central orchestrators of wound healing [
151] and fibrosis [
152]. At the same time, direct evidence shows that fibroblast migration is modulated through extracellular vesicles derived from macrophages [
153], as the vesicles carry a rich cargo of growth factors, cytokines, and microRNAs that can alter the inflammatory milieu, promote angiogenesis, and directly or indirectly stimulate fibroblast proliferation and activation [
151,
153,
154].
Additional unusual migratory processes include blebbisomes [
155] and migrasomes [
156,
157], which are novel forms of communication and cell-derived material exchange. The recently discovered blebbisome is a large (approximately 20 µm) organelle-rich vesicle that forms during active cell blebbing. They contain functional mitochondria and other intracellular organelles. Their exact role in fibroblast migration and wound healing is still under intense investigation [
155]. However, it is thought that blebbisomes can transfer metabolic support (via functional mitochondria) to recipient fibroblasts, thereby reinvigorating less active or damaged fibroblasts. Jeppson et al. speculated that this could enhance the fibroblast energy status required for migration or deliver specific pro-migratory enzymes or growth factors directly to the wound site, where they actively promote tissue remodeling [
155]. Migrasomes are smaller extracellular vesicle structures (0.5–3 µm) that are also left behind by migrating cells [
156,
158]. They are characterized by the presence of the transmembrane protein tetraspanin [
159], and they have been found to encapsulate a diverse range of cargo, including cytosolic proteins, RNA, and mitochondrial DNA [
160,
161]. Cutting-edge research suggests that migrasomes can also transmit signals to recipient cells. For example, migrasomes derived from young fibroblasts can lessen the senescence phenotype found in aged keratinocytes and enhance wound healing in aged skin [
156]. This finding suggests that migrasomes transfer factors that have a direct role in rejuvenating the microenvironment of the wound bed. Migrasomes have been found to promote fibrosis [
160], suggesting that they also have an active role in this process. Beyond these intrinsic cellular and vesicular mechanisms, fibroblast migration is still exquisitely sensitive to a diverse array of stimuli. This can also be influenced by biochemical growth factors, such as TGF-β, PDGF, and FGF, etc. These cytokines function as potent chemoattractants and activators of fibroblast proliferation. They also enhance matrix synthesis. Furthermore, the dynamic interplay with other resident and infiltrating cell types influences the migration of fibroblasts. These include keratinocytes, which secrete many essential factors that profoundly affect fibroblast behavior [
162]. In addition, endothelial cells are involved in angiogenesis and mediate crosstalk between fibroblasts and various immune cells, and this crosstalk modulates fibroblast responses and the inflammatory and reparative microenvironment [
163]. All these cells are paramount and influence fibroblast migration in wound healing, and they have also been shown to be active in the development of fibrosis. The mechanical properties of the extracellular matrix itself, including its stiffness and topographical cues, as discussed above, also exert profound guidance over fibroblast movement, directing them towards areas of injury and remodeling.
The core interactive pathways in fibrosis that govern fibroblast migration are shown in .
. Core interactive pathways governing fibroblast migration in fibrosis. Many of the key pathways in the fibroblast are overlapping as they alter the structural dynamics of the fibroblast during migration. The activation of these pathways is often enhanced in fibrosis. References used to create the figure and discussed in more detail below. RhoA/ROCK Pathway: [
141,
142,
143,
144,
145,
146] PI3K/AKT Pathway: [
70,
147,
148,
149] WNT/β-catenin Pathway: [
124,
150,
151,
152,
153,
154,
155,
157,
158,
159,
160] SHH Pathway: [
161,
162,
163]. Created in BioRender. Artlett C. (2025) https://BioRender.com/mpenb3i.
6. Addressing the Clinical Need for Selective Inhibitors of Fibrosis
A critical unmet medical need in the field of fibrotic disease that remains is the development of successful therapeutic interventions that can effectively resolve pathological fibrosis. For this to be successful, it should not compromise the essential roles of quiescent fibroblasts, as these are required for normal tissue maintenance, architecture, and repair. This has been very challenging to date, as it is underscored by the understanding that the complete abrogation of fibroblast activity could impair beneficial wound healing and tissue homeostasis. Therefore, unique perspectives that point towards strategic shifts in therapeutic development are warranted. Strategies that will selectively target or alter myofibroblast function or differentiation while preserving the vital regenerative capacity of fibroblasts are needed. The core challenge in the clinic with anti-fibrotic therapy lies in disarming the ‘pro-fibrotic’ switch without halting the ‘pro-healing’ function of the fibroblast. Because myofibroblasts are distinguished from quiescent fibroblasts by several key features, these features could make them vulnerable to therapeutic modulation. For example, they exhibit enhanced contractility [
164,
165], sustained production of excessive ECM deposition, and heightened resistance to apoptosis [
166,
167]. These characteristics are driven by specific molecular pathways, including persistent TGF-β signaling and activation of the RhoA/ROCK pathway [
124,
168]. Myofibroblasts also exhibit altered mechanosensing, a process that is often dysregulated in chronic fibrotic conditions but is less active or more tightly controlled in quiescent fibroblasts [
169]. Therefore, successful therapies must selectively interfere with the myofibroblast-specific functional traits rather than broadly inhibiting all fibroblast activities. As mentioned, myofibroblasts have persistently activated TGF-β; however, broadly inhibiting this cytokine failed to be therapeutically beneficial in the treatment of fibrosis [
170]. However, targeting downstream effectors or co-factors of TGF-β signaling, such as those that are only active during myofibroblast differentiation, may be a more successful approach. For example, targeting the activation of ROCK has shown some promise by attenuating myofibroblast differentiation and contractility while sparing fibroblast proliferation [
171]. However, in contrast, the targeting of focal adhesion kinase on myofibroblasts was detrimental as it delayed wound healing by suppressing the expression of p38 and Akt [
172].
Targeting tropomyosins could be another route for successful therapeutic intervention, as this family is involved in stabilizing the actin cytoskeleton, which is essential for mediating fibroblast migration. Specific isoforms are upregulated in fibrosis, and their targeting lowers collagen deposition. For example, the knocking down of tropomyosin 2.1 abrogated collagen gel contraction [
118] and slowed fibroblast migration [
173], highlighting this tropomyosin isoform as a possible therapeutic target for lung fibrosis. In cardiac fibrosis, the ectopic administration of a small circular RNA (circYap) was found to attenuate fibrosis by binding to tropomyosin 4 and -actin. This resulted in decreased actin polymerization and reduced ECM deposition. This suggests that modulating tropomyosin activity could be beneficial in this context.
An additional critical shift in thinking involves exploring pathways that actively reverse the differentiation of myofibroblasts or promote myofibroblast quiescence (or apoptosis) rather than merely preventing myofibroblast differentiation. Oral mucosa and embryonic wound healing models [
174,
175] offer valuable insights into this selective approach, and these insights can be drawn from natural regenerative healing models. Unlike tissues in other organs, these systems exhibit scarless or minimal-scar healing [
176]. This is characterized by a unique fibroblast behavior profile where myofibroblast differentiation is either attenuated or transient because the inflammation within the wound site is more temporary than in other tissues [
177,
178]. In these models, fibroblasts efficiently migrate, proliferate, and deposit matrix components to facilitate initial wound closure, yet they do not persistently differentiate into highly contractile, matrix-overproducing myofibroblasts. This physiological balance suggests that these systems possess intrinsic mechanisms that fine-tune myofibroblast activation, drive efficient resolution, and provide invaluable blueprints for therapeutic intervention [
179,
180,
181].
The exploitation of myofibroblast cell surface markers [
175,
182] or targeting their metabolic vulnerabilities [
183] would be a unique approach around which more precise drug delivery methods could be developed. Myofibroblasts exhibit increased glycolysis [
184,
185] and glutaminolysis [
186,
187]. Inhibiting mitochondrial pyruvate import impedes the development of myofibroblasts [
188,
189]. Myofibroblasts also have unique cell surface markers such as CD49e (alpha 5 integrin) [
190], CD29 (beta 1 integrin) [
191], and CD9 (tetraspanin glycoprotein) [
192] are found on various subsets of myofibroblasts, and they could be leveraged in targeted antibody-drug conjugates while sparing quiescent fibroblasts.
In addition, as myofibroblasts are sensitive to matrix stiffness, developing strategies that reduce the pathological stiffness or alter the mechanical cues of the ECM in a way that deactivates myofibroblasts without disrupting ECM scaffolding needed for fibroblast migration and functional integrity. This approach could be transformative and is currently under investigation [
193,
194]. The intriguing observation by Locy et al. [
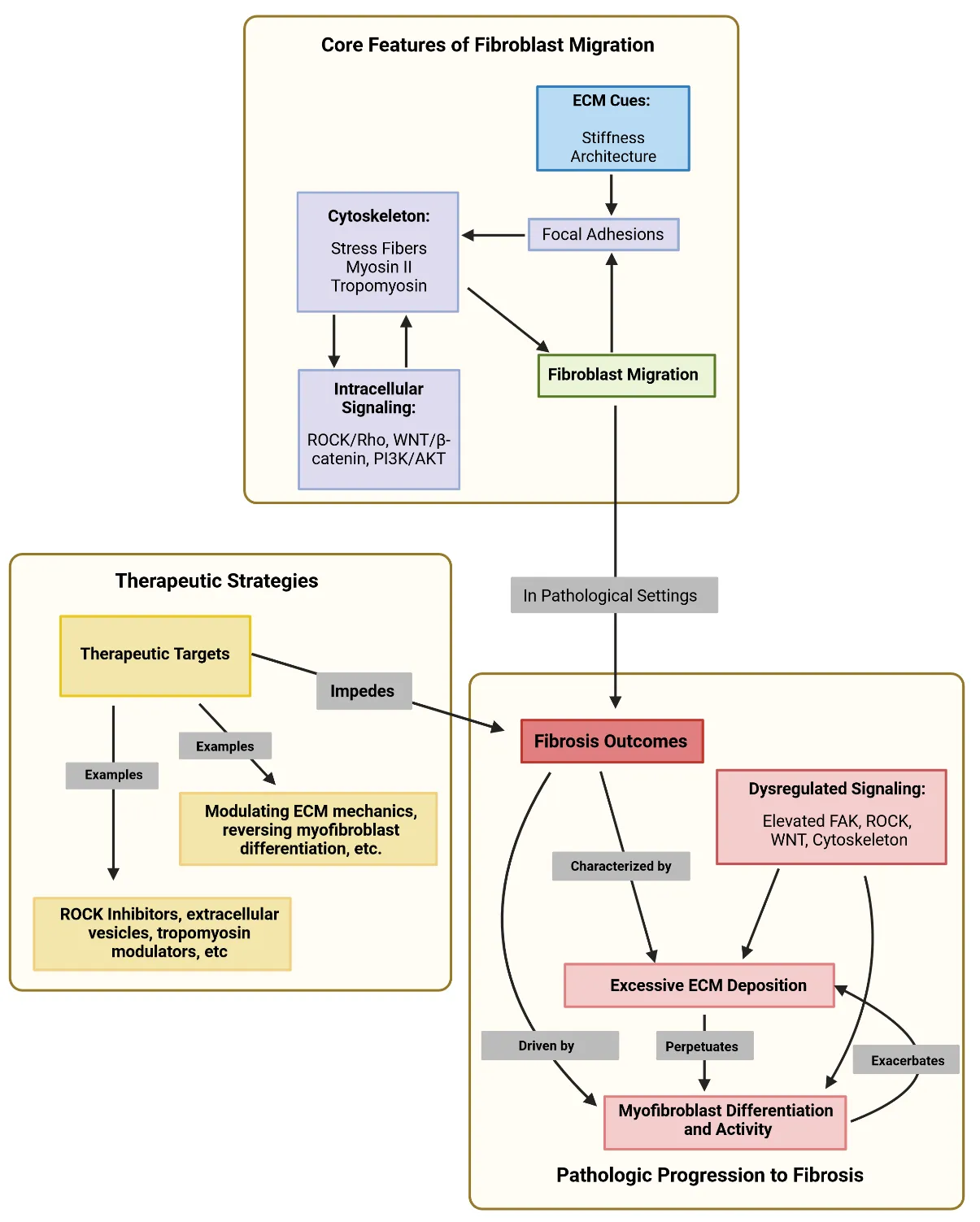
194], demonstrated that oxidative crosslinking of fibronectin attenuated fibroblast migration, supporting this hypothesis. outlines some of the core features that we have discussed about fibroblast migration and their dysregulation in fibrosis.
. Core pathways of fibroblast migration that are dysregulated during fibrosis and their potential therapeutic targets. Created in BioRender. Artlett C. (2025) https://BioRender.com/v5arbrp.
7. Conclusions
The recent and novel discoveries made by migrating fibroblasts could pave the way for innovative therapies that enhance fibroblast migration in wound healing or mitigate fibrosis. For example, harnessing the power of extracellular vesicles or designing migrasomes or blebbisomes to deliver reparative signals directly into the wound could represent a cutting-edge cell-free therapeutic approach to enhance fibroblast migration, leading to faster wound healing rates. These approaches would circumvent some of the difficulties and challenges associated with cell transplantation. As migrasomes have been found to enhance healing in aged skin, this approach would be pertinent to the elderly with chronic venous stasis ulcers, as these ulcers are notoriously difficult to heal. Furthermore, by precisely modulating or controlling the content and release of wound healing factors by the vesicles or by mimicking their functional capabilities, future interventions could achieve more precise and effective stimulation of fibroblast migration into the wound. This could improve wound closure and could significantly mitigate the pathological consequences of dysregulated healing. In addition, by manipulating extracellular vesicles to carry molecules that inhibit fibrosis, it would be an effective means to target this pathology. Indeed, the use of myofibroblast targeting extracellular vesicles to control fibrosis is currently under development [
195].
However, as fibrosis is a complex pathological event, ultimately resolving this complex pathology will require a multi-pronged approach that moves far beyond single-target drugs. Future therapies will likely aim to specifically “disarm” myofibroblasts at various stages. This could be achieved by preventing their differentiation and promoting their de-differentiation or apoptosis while simultaneously neutralizing their pro-fibrotic output. This should all be in an environment that fosters and supports the beneficial, regenerative functions of fibroblasts. This necessitates a continued and more profound understanding of myofibroblast heterogeneity, their unique molecular signatures, and the intricate interplay of cellular and extracellular signals that drive fibrotic progression and cell migration, particularly through the lens of regenerative healing models.
Author Contributions
B.T.J.: writing the manuscript, reviewing and editing the manuscript, and approving the final version. C.M.A.: writing the manuscript, reviewing and editing the manuscript, and approving the final version.
Ethics Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Funding
This research received no external funding.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
-
1.
Murtha LA, Morten M, Schuliga MJ, Mabotuwana NS, Hardy SA, Waters DW, et al. The Role of Pathological Aging in Cardiac and Pulmonary Fibrosis.
Aging Dis. 2019,
10, 419–428. doi:10.14336/ad.2018.0601.
[Google Scholar]
-
2.
Puglisi S, Torrisi SE, Giuliano R, Vindigni V, Vancheri C. What We Know About the Pathogenesis of Idiopathic Pulmonary Fibrosis.
Semin. Respir. Crit. Care Med. 2016,
37, 358–367. doi:10.1055/s-0036-1580693.
[Google Scholar]
-
3.
Wynn TA. Fibrotic disease and the T(H)1/T(H)2 paradigm.
Nat. Reviews. Immunol. 2004,
4, 583–594. doi:10.1038/nri1412.
[Google Scholar]
-
4.
Shin D, Minn KW. The effect of myofibroblast on contracture of hypertrophic scar.
Plast. Reconstr. Surg. 2004,
113, 633–640. doi:10.1097/01.Prs.0000101530.33096.5b.
[Google Scholar]
-
5.
Forbes SJ, Russo FP, Rey V, Burra P, Rugge M, Wright NA, et al. A significant proportion of myofibroblasts are of bone marrow origin in human liver fibrosis.
Gastroenterology 2004,
126, 955–963. doi:10.1053/j.gastro.2004.02.025.
[Google Scholar]
-
6.
Jiang D, Christ S, Correa-Gallegos D, Ramesh P, Kalgudde Gopal S, Wannemacher J, et al. Injury triggers fascia fibroblast collective cell migration to drive scar formation through N-cadherin.
Nat. Commun. 2020,
11, 5653. doi:10.1038/s41467-020-19425-1.
[Google Scholar]
-
7.
Correa-Gallegos D, Jiang D, Christ S, Ramesh P, Ye H, Wannemacher J, et al. Patch repair of deep wounds by mobilized fascia.
Nature 2019,
576, 287–292. doi:10.1038/s41586-019-1794-y.
[Google Scholar]
-
8.
Wan L, Jiang D, Correa-Gallegos D, Ramesh P, Zhao J, Ye H, et al. Connexin43 gap junction drives fascia mobilization and repair of deep skin wounds.
MatrixBiol. J. Int. Soc. Matrix Biol. 2021,
97, 58–71. doi:10.1016/j.matbio.2021.01.005.
[Google Scholar]
-
9.
Fischer A, Han W, Hu S, Häusl MM, Wannemacher J, Kadri S, et al. Targeting pleuro-alveolar junctions reverses lung fibrosis in mice.
Nat. Commun. 2025,
16, 173. doi:10.1038/s41467-024-55596-x.
[Google Scholar]
-
10.
Crossley RM, Johnson S, Tsingos E, Bell Z, Berardi M, Botticelli M, et al. Modeling the extracellular matrix in cell migration and morphogenesis: a guide for the curious biologist.
Front. Cell Dev. Biol. 2024,
12, 1354132. doi:10.3389/fcell.2024.1354132.
[Google Scholar]
-
11.
Yip AK, Zhang S, Chong LH, Cheruba E, Woon JYX, Chua TX, et al. Zyxin Is Involved in Fibroblast Rigidity Sensing and Durotaxis.
Front. Cell Dev. Biol. 2021,
9, 735298. doi:10.3389/fcell.2021.735298.
[Google Scholar]
-
12.
Raab M, Swift J, Dingal PC, Shah P, Shin JW, Discher DE. Crawling from soft to stiff matrix polarizes the cytoskeleton and phosphoregulates myosin-II heavy chain.
J. Cell Biol. 2012,
199, 669–683. doi:10.1083/jcb.201205056.
[Google Scholar]
-
13.
Mierke CT. Extracellular Matrix Cues Regulate Mechanosensing and Mechanotransduction of Cancer Cells.
Cells 2024,
13, 96. doi:10.3390/cells13010096.
[Google Scholar]
-
14.
Rens EG, Merks RMH. Cell Shape and Durotaxis Explained from Cell-Extracellular Matrix Forces and Focal Adhesion Dynamics.
iScience 2020,
23, 101488. doi:10.1016/j.isci.2020.101488.
[Google Scholar]
-
15.
Asano S, Ito S, Takahashi K, Furuya K, Kondo M, Sokabe M, et al. Matrix stiffness regulates migration of human lung fibroblasts.
Physiol. Rep. 2017,
5, e13281. doi:10.14814/phy2.13281.
[Google Scholar]
-
16.
Levental KR, Yu H, Kass L, Lakins JN, Egeblad M, Erler JT, et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling.
Cell 2009,
139, 891–906. doi:10.1016/j.cell.2009.10.027.
[Google Scholar]
-
17.
Wu Y, Millender J, Padgett B, Marx M, Madnick S, Puterbaugh R, et al. An in vitro model to measure the strength and stiffness of the extracellular matrix synthesized de novo by human fibroblasts.
In Vitro Model. 2025,
4, 59–69. doi:10.1007/s44164-025-00081-y.
[Google Scholar]
-
18.
Wang WY, Pearson AT, Kutys ML, Choi CK, Wozniak MA, Baker BM, et al. Extracellular matrix alignment dictates the organization of focal adhesions and directs uniaxial cell migration.
APL Bioeng. 2018,
2, 046107. doi:10.1063/1.5052239.
[Google Scholar]
-
19.
Doyle AD, Kutys ML, Conti MA, Matsumoto K, Adelstein RS, Yamada KM. Micro-environmental control of cell migration--myosin IIA is required for efficient migration in fibrillar environments through control of cell adhesion dynamics.
J. Cell Sci. 2012,
125, 2244–2256. doi:10.1242/jcs.098806.
[Google Scholar]
-
20.
Inoue A, Obayashi K, Sonoda Y, Nakamura A, Ueno T, Kuhara S, et al. Regulation of matrix metalloproteinase-1 and alpha-smooth muscle actin expression by interleukin-1 alpha and tumour necrosis factor alpha in hepatic stellate cells.
Cytotechnology 2017,
69, 461–468. doi:10.1007/s10616-016-9948-3.
[Google Scholar]
-
21.
Liu Z, Ling SD, Liang K, Chen Y, Niu Y, Sun L, et al. Viscoelasticity of ECM and cells-origin, measurement and correlation.
Mechanobiol. Med. 2024,
2, 100082. doi:10.1016/j.mbm.2024.100082.
[Google Scholar]
-
22.
Bauer A, Gu L, Kwee B, Li WA, Dellacherie M, Celiz AD, et al. Hydrogel substrate stress-relaxation regulates the spreading and proliferation of mouse myoblasts.
Acta Biomater. 2017,
62, 82–90. doi:10.1016/j.actbio.2017.08.041.
[Google Scholar]
-
23.
Chester D, Kathard R, Nortey J, Nellenbach K, Brown AC. Viscoelastic properties of microgel thin films control fibroblast modes of migration and pro-fibrotic responses.
Biomaterials 2018,
185, 371–382. doi:10.1016/j.biomaterials.2018.09.012.
[Google Scholar]
-
24.
Vining KH, Stafford A, Mooney DJ. Sequential modes of crosslinking tune viscoelasticity of cell-instructive hydrogels.
Biomaterials 2019,
188, 187–197. doi:10.1016/j.biomaterials.2018.10.013.
[Google Scholar]
-
25.
Moazedi-Fuerst FC, Lackner A, Kreuzer SM, Eller K, Odler B, Kovacs G, et al. Successful long-term systemic sclerosis treatment by high-frequent low-dose B cell-depleting therapy.
J. Autoimmun. 2024,
147, 103246. doi:10.1016/j.jaut.2024.103246.
[Google Scholar]
-
26.
Tyndall AJ, Bannert B, Vonk M, Airò P, Cozzi F, Carreira PE, et al. Causes and risk factors for death in systemic sclerosis: a study from the EULAR Scleroderma Trials and Research (EUSTAR) database.
Ann. Rheum. Dis. 2010,
69, 1809–1815. doi:10.1136/ard.2009.114264.
[Google Scholar]
-
27.
Denton CP, Khanna D. Systemic sclerosis.
Lancet 2017,
390, 1685–1699. doi:10.1016/S0140-6736(17)30933-9.
[Google Scholar]
-
28.
Adrovic A, Karatemiz G, Esatoglu SN, Yildiz M, Sahin S, Barut K, et al. Juvenile and adult-onset scleroderma: Different clinical phenotypes.
Semin. Arthritis Rheum. 2023,
60, 152197. doi:10.1016/j.semarthrit.2023.152197.
[Google Scholar]
-
29.
Huang Y, Zhao H, Zhang Y, Tang Y, Shi X, Jiang S, et al. Enhancement of Zyxin Promotes Skin Fibrosis by Regulating FAK/PI3K/AKT and TGF-beta Signaling Pathways via Integrins.
Int. J. Biol. Sci. 2023,
19, 2394–2408. doi:10.7150/ijbs.77649.
[Google Scholar]
-
30.
Bond JE, Ho TQ, Selim MA, Hunter CL, Bowers EV, Levinson H. Temporal spatial expression and function of non-muscle myosin II isoforms IIA and IIB in scar remodeling.
Lab. Invest. 2011,
91, 499–508. doi:10.1038/labinvest.2010.181.
[Google Scholar]
-
31.
Lo CM, Buxton DB, Chua GC, Dembo M, Adelstein RS, Wang YL. Nonmuscle myosin IIb is involved in the guidance of fibroblast migration.
Mol. Biol. Cell 2004,
15, 982–989. doi:10.1091/mbc.e03-06-0359.
[Google Scholar]
-
32.
Bun T, Sato Y, Futami H, Tagawa Y, Murakami Y, Takahashi M. Cytoskeletal fractionation identifies LMO7 as a positive regulator of fibroblast polarization and directed migration.
Biochem. Biophys. Res. Commun. 2023,
638, 58–65. doi:10.1016/j.bbrc.2022.11.048.
[Google Scholar]
-
33.
Kuragano M, Murakami Y, Takahashi M. Nonmuscle myosin IIA and IIB differentially contribute to intrinsic and directed migration of human embryonic lung fibroblasts.
Biochem. Biophys. Res. Commun. 2018,
498, 25–31. doi:10.1016/j.bbrc.2018.02.171.
[Google Scholar]
-
34.
Southern BD, Grove LM, Rahaman SO, Abraham S, Scheraga RG, Niese KA, Sun H, Herzog EL, Liu F, Tschumperlin DJ, et al. Matrix-driven Myosin II Mediates the Pro-fibrotic Fibroblast Phenotype.
J. Biol. Chem. 2016,
291, 6083–6095. doi:10.1074/jbc.M115.712380.
[Google Scholar]
-
35.
Lu YY, Fang CC, Hong CH, Wu CH, Lin YH, Chang KL, Lee CH. Nonmuscle Myosin II Activation Regulates Cell Proliferation, Cell Contraction, and Myofibroblast Differentiation in Keloid-Derived Fibroblasts.
Adv. Wound Care (New Rochelle) 2020,
9, 491–501. doi:10.1089/wound.2019.0944.
[Google Scholar]
-
36.
Moreno-Arotzena O, Borau C, Movilla N, Vicente-Manzanares M, Garcia-Aznar JM. Fibroblast Migration in 3D is Controlled by Haptotaxis in a Non-muscle Myosin II-Dependent Manner.
Ann. Biomed. Eng. 2015,
43, 3025–3039. doi:10.1007/s10439-015-1343-2.
[Google Scholar]
-
37.
Smilenov LB, Mikhailov A, Pelham RJ, Marcantonio EE, Gundersen GG. Focal adhesion motility revealed in stationary fibroblasts.
Science 1999,
286, 1172–1174. doi:10.1126/science.286.5442.1172.
[Google Scholar]
-
38.
Zamir E, Katz M, Posen Y, Erez N, Yamada KM, Katz BZ, et al. Dynamics and segregation of cell-matrix adhesions in cultured fibroblasts.
Nat. Cell Biol. 2000,
2, 191–196. doi:10.1038/35008607.
[Google Scholar]
-
39.
Petroll WM, Ma L, Jester JV. Direct correlation of collagen matrix deformation with focal adhesion dynamics in living corneal fibroblasts.
J. Cell Sci. 2003,
116, 1481–1491. doi:10.1242/jcs.00357.
[Google Scholar]
-
40.
Bertolucci CM, Guibao CD, Zheng J. Structural features of the focal adhesion kinase-paxillin complex give insight into the dynamics of focal adhesion assembly.
Protein Sci. 2005,
14, 644–652.
[Google Scholar]
-
41.
Plotnikov SV, Pasapera AM, Sabass B, Waterman CM. Force fluctuations within focal adhesions mediate ECM-rigidity sensing to guide directed cell migration.
Cell 2012,
151, 1513–1527. doi:10.1016/j.cell.2012.11.034.
[Google Scholar]
-
42.
Kechagia JZ, Ivaska J, Roca-Cusachs P. Integrins as biomechanical sensors of the microenvironment.
Nat. Reviews. Mol. Cell Biol. 2019,
20, 457–473. doi:10.1038/s41580-019-0134-2.
[Google Scholar]
-
43.
Shattil SJ, Kim C, Ginsberg MH. The final steps of integrin activation: the end game.
Nat. Reviews. Mol. Cell Biol. 2010,
11, 288–300. doi:10.1038/nrm2871.
[Google Scholar]
-
44.
Mavrakis M, Juanes MA. The compass to follow: Focal adhesion turnover.
Curr. Opin. Cell Biol. 2023,
80, 102152. doi:10.1016/j.ceb.2023.102152.
[Google Scholar]
-
45.
Kim DH, Wirtz D. Focal adhesion size uniquely predicts cell migration.
FASEBJ. : Off. Publ. Fed. Am. Soc. Exp. Biol. 2013,
27, 1351–1361. doi:10.1096/fj.12-220160.
[Google Scholar]
-
46.
Pernier J, Cardoso Dos Santos M, Souissi M, Joly A, Narassimprakash H, Rossier O, et al. Talin and kindlin cooperate to control the density of integrin clusters.
J. Cell Sci. 2023,
136, jcs260746. doi:10.1242/jcs.260746.
[Google Scholar]
-
47.
Geiger B, Boujemaa-Paterski R, Winograd-Katz SE, Balan Venghateri J, Chung WL, Medalia O. The Actin Network Interfacing Diverse Integrin-Mediated Adhesions.
Biomolecules 2023,
13, 294. doi:10.3390/biom13020294.
[Google Scholar]
-
48.
Holland EN, Fernández-Yagüe MA, Zhou DW, O’Neill EB, Woodfolk AU, Mora-Boza A, et al. FAK, vinculin, and talin control mechanosensitive YAP nuclear localization.
Biomaterials 2024,
308, 122542. doi:10.1016/j.biomaterials.2024.122542.
[Google Scholar]
-
49.
Malik-Sheriff RS, Imtiaz S, Grecco HE, Zamir E. Diverse patterns of molecular changes in the mechano-responsiveness of focal adhesions.
Sci. Rep. 2018,
8, 2187. doi:10.1038/s41598-018-20252-0.
[Google Scholar]
-
50.
Bernau K, Torr EE, Evans MD, Aoki JK, Ngam CR, Sandbo N. Tensin 1 Is Essential for Myofibroblast Differentiation and Extracellular Matrix Formation.
Am. J. Respir. Cell Mol. Biol. 2017,
56, 465–476. doi:10.1165/rcmb.2016-0104OC.
[Google Scholar]
-
51.
Ripamonti M, Wehrle-Haller B, de Curtis I. Paxillin: A Hub for Mechano-Transduction from the β3 Integrin-Talin-Kindlin Axis.
Front. Cell Dev. Biol. 2022,
10, 852016. doi:10.3389/fcell.2022.852016.
[Google Scholar]
-
52.
Acharya N, Sharma SK, Mishra D, Dhooria S, Dhir V, Jain S. Efficacy and safety of pirfenidone in systemic sclerosis-related interstitial lung disease-a randomised controlled trial.
Rheumatol. Int. 2020,
40, 703–710. doi:10.1007/s00296-020-04565-w.
[Google Scholar]
-
53.
Ghatak S, Hascall VC, Markwald RR, Feghali-Bostwick C, Artlett CM, Gooz M, Bogatkevich GS, et al. TGF beta-1 induced CD44v6-NOX4 signaling in pathogenesis of idiopathic pulmonary fibrosis.
J. Biol. Chem. 2017. doi:10.1074/jbc.M116.752469.
[Google Scholar]
-
54.
Bear JE, Haugh JM. Directed migration of mesenchymal cells: where signaling and the cytoskeleton meet.
Curr. Opin. Cell Biol. 2014,
30, 74–82. doi:10.1016/j.ceb.2014.06.005.
[Google Scholar]
-
55.
Sandbo N, Smolyaninova LV, Orlov SN, Dulin NO. Control of Myofibroblast Differentiation and Function by Cytoskeletal Signaling.
Biochemistry (Moscow) 2016,
81, 1698–1708. doi:10.1134/s0006297916130071.
[Google Scholar]
-
56.
Rogers SL, Gelfand VI. Membrane trafficking, organelle transport, and the cytoskeleton.
Curr. Opin. Cell Biol. 2000,
12, 57–62. doi:10.1016/s0955-0674(99)00057-5.
[Google Scholar]
-
57.
Hartwig JH, Kwiatkowski DJ. Actin-binding proteins.
Curr. Opin. Cell Biol. 1991,
3, 87–97. doi:10.1016/0955-0674(91)90170-4.
[Google Scholar]
-
58.
Burridge K, Guilluy C. Focal adhesions, stress fibers and mechanical tension.
Exp. Cell Res. 2016,
343, 14–20. doi:10.1016/j.yexcr.2015.10.029.
[Google Scholar]
-
59.
Mangeat P, Burridge K. Actin-membrane interaction in fibroblasts: what proteins are involved in this association?
J. Cell Biol. 1984,
99, 95s–103s. doi:10.1083/jcb.99.1.95s.
[Google Scholar]
-
60.
Maninová M, Vomastek T. Dorsal stress fibers, transverse actin arcs, and perinuclear actin fibers form an interconnected network that induces nuclear movement in polarizing fibroblasts.
FEBS J. 2016,
283, 3676–3693. doi:10.1111/febs.13836.
[Google Scholar]
-
61.
Zhu R, Liu C, Gundersen GG. Nuclear positioning in migrating fibroblasts.
Semin. Cell Dev. Biol. 2018,
82, 41–50. doi:10.1016/j.semcdb.2017.11.006.
[Google Scholar]
-
62.
Kassianidou E, Kumar S. A biomechanical perspective on stress fiber structure and function.
Biochim. Et Biophys. Acta 2015,
1853, 3065–3074. doi:10.1016/j.bbamcr.2015.04.006.
[Google Scholar]
-
63.
Lazarides E, Burridge K. Alpha-actinin: immunofluorescent localization of a muscle structural protein in nonmuscle cells.
Cell 1975,
6, 289–298. doi:10.1016/0092-8674(75)90180-4.
[Google Scholar]
-
64.
Gunning PW, Hardeman EC, Lappalainen P, Mulvihill DP. Tropomyosin—master regulator of actin filament function in the cytoskeleton.
J. Cell Sci. 2015,
128, 2965–2974. doi:10.1242/jcs.172502.
[Google Scholar]
-
65.
Tojkander S, Gateva G, Lappalainen P. Actin stress fibers--assembly, dynamics and biological roles.
J. Cell Sci. 2012,
125, 1855–1864. doi:10.1242/jcs.098087.
[Google Scholar]
-
66.
Hotulainen P, Lappalainen P. Stress fibers are generated by two distinct actin assembly mechanisms in motile cells.
J. Cell Biol. 2006,
173, 383–394. doi:10.1083/jcb.200511093.
[Google Scholar]
-
67.
Kovac B, Teo JL, Mäkelä TP, Vallenius T. Assembly of non-contractile dorsal stress fibers requires α-actinin-1 and Rac1 in migrating and spreading cells.
J. Cell Sci. 2013,
126, 263–273. doi:10.1242/jcs.115063.
[Google Scholar]
-
68.
Heath JP. Behaviour and structure of the leading lamella in moving fibroblasts. I. Occurrence and centripetal movement of arc-shaped microfilament bundles beneath the dorsal cell surface.
J. Cell Sci. 1983,
60, 331–354. doi:10.1242/jcs.60.1.331.
[Google Scholar]
-
69.
Ang SF, Zhao ZS, Lim L, Manser E. DAAM1 is a formin required for centrosome re-orientation during cell migration.
PLoS ONE 2010,
5, e13064. doi:10.1371/journal.pone.0013064.
[Google Scholar]
-
70.
Small JV, Rottner K, Kaverina I, Anderson KI. Assembling an actin cytoskeleton for cell attachment and movement.
Biochim. Et Biophys. Acta 1998,
1404, 271–281. doi:10.1016/s0167-4889(98)00080-9.
[Google Scholar]
-
71.
Gardel ML, Schneider IC, Aratyn-Schaus Y, Waterman CM. Mechanical integration of actin and adhesion dynamics in cell migration.
Annu. Rev. Cell Dev. Biol. 2010,
26, 315–333. doi:10.1146/annurev.cellbio.011209.122036.
[Google Scholar]
-
72.
Galkin VE, Orlova A, Egelman EH. Actin filaments as tension sensors.
Curr. Biol. 2012,
22, R96–R101. doi:10.1016/j.cub.2011.12.010.
[Google Scholar]
-
73.
Starr DA, Fridolfsson HN. Interactions between nuclei and the cytoskeleton are mediated by SUN-KASH nuclear-envelope bridges.
Annu. Rev. Cell Dev. Biol. 2010,
26, 421–444. doi:10.1146/annurev-cellbio-100109-104037.
[Google Scholar]
-
74.
Khatau SB, Hale CM, Stewart-Hutchinson PJ, Patel MS, Stewart CL, Searson PC, et al. A perinuclear actin cap regulates nuclear shape.
Proc. Natl. Acad. Sci. United States Am. 2009,
106, 19017–19022. doi:10.1073/pnas.0908686106.
[Google Scholar]
-
75.
Vishavkarma R, Raghavan S, Kuyyamudi C, Majumder A, Dhawan J, Pullarkat PA. Role of actin filaments in correlating nuclear shape and cell spreading.
PLoS ONE 2014,
9, e107895. doi:10.1371/journal.pone.0107895.
[Google Scholar]
-
76.
Hitchcock-DeGregori SE, Barua B. Tropomyosin Structure, Function, and Interactions: A Dynamic Regulator.
Subcell. Biochem. 2017,
82, 253–284. doi:10.1007/978-3-319-49674-0_9.
[Google Scholar]
-
77.
Gunning P, O’Neill G, Hardeman E. Tropomyosin-based regulation of the actin cytoskeleton in time and space.
Physiol. Rev. 2008,
88, 1–35. doi:10.1152/physrev.00001.2007.
[Google Scholar]
-
78.
Meiring JCM, Bryce NS, Niño JLG, Gabriel A, Tay SS, Hardeman EC, et al. Tropomyosin concentration but not formin nucleators mDia1 and mDia3 determines the level of tropomyosin incorporation into actin filaments.
Sci. Rep. 2019,
9, 6504. doi:10.1038/s41598-019-42977-2.
[Google Scholar]
-
79.
Ostrowska-Podhorodecka Z, Śliwinska M, Reisler E, Moraczewska J. Tropomyosin isoforms regulate cofilin 1 activity by modulating actin filament conformation.
Arch. Biochem. Biophys. 2020,
682, 108280. doi:10.1016/j.abb.2020.108280.
[Google Scholar]
-
80.
Gateva G, Kremneva E, Reindl T, Kotila T, Kogan K, Gressin L, et al. Tropomyosin Isoforms Specify Functionally Distinct Actin Filament Populations In Vitro.
Curr. Biol. 2017,
27, 705–713. doi:10.1016/j.cub.2017.01.018.
[Google Scholar]
-
81.
Kee AJ, Yang L, Lucas CA, Greenberg MJ, Martel N, Leong GM, et al. An actin filament population defined by the tropomyosin Tpm3.1 regulates glucose uptake.
Traffic 2015,
16, 691–711. doi:10.1111/tra.12282.
[Google Scholar]
-
82.
Tang N, Ostap EM. Motor domain-dependent localization of myo1b (myr-1).
Curr. Biol. 2001,
11, 1131–1135. doi:10.1016/s0960-9822(01)00320-7.
[Google Scholar]
-
83.
Lees JG, Ching YW, Adams DH, Bach CT, Samuel MS, Kee AJ, et al. Tropomyosin regulates cell migration during skin wound healing.
J. Investig. Dermatol. 2013,
133, 1330–1339. doi:10.1038/jid.2012.489.
[Google Scholar]
-
84.
Xu T, Verhagen MP, Teeuwssen M, Sun W, Joosten R, Sacchetti A, et al. Tropomyosin1 isoforms underlie epithelial to mesenchymal plasticity, metastatic dissemination, and resistance to chemotherapy in high-grade serous ovarian cancer.
Cell Death Differ. 2024,
31, 360–377. doi:10.1038/s41418-024-01267-9.
[Google Scholar]
-
85.
Kumari R, Ven K, Chastney M, Kokate SB, Peränen J, Aaron J, et al. Focal adhesions contain three specialized actin nanoscale layers.
Nat. Commun. 2024,
15, 2547. doi:10.1038/s41467-024-46868-7.
[Google Scholar]
-
86.
Srinivasan S, Das S, Surve V, Srivastava A, Kumar S, Jain N, et al. Blockade of ROCK inhibits migration of human primary keratinocytes and malignant epithelial skin cells by regulating actomyosin contractility.
Sci. Rep. 2019,
9, 19930. doi:10.1038/s41598-019-56447-2.
[Google Scholar]
-
87.
Shi J, Wu X, Surma M, Vemula S, Zhang L, Yang Y, et al. Distinct roles for ROCK1 and ROCK2 in the regulation of cell detachment.
Cell Death Dis. 2013,
4, e483. doi:10.1038/cddis.2013.10.
[Google Scholar]
-
88.
Marshall-Burghardt S, Migueles-Ramírez RA, Lin Q, El Baba N, Saada R, Umar M, et al. Excitable Rho dynamics control cell shape and motility by sequentially activating ERM proteins and actomyosin contractility.
Sci. Adv. 2024,
10,
eadn6858. doi:10.1126/sciadv.adn6858.
[Google Scholar]
-
89.
Autenrieth TJ, Frank SC, Greiner AM, Klumpp D, Richter B, Hauser M, et al. Actomyosin contractility and RhoGTPases affect cell-polarity and directional migration during haptotaxis.
Integr. Biol. 2016,
8, 1067–1078. doi:10.1039/c6ib00152a.
[Google Scholar]
-
90.
Guan G, Cannon RD, Coates DE, Mei L. Effect of the Rho-Kinase/ROCK Signaling Pathway on Cytoskeleton Components.
Genes 2023,
14, 272. doi:10.3390/genes14020272.
[Google Scholar]
-
91.
Lawson CD, Ridley AJ. Rho GTPase signaling complexes in cell migration and invasion.
J. Cell Biol. 2018,
217, 447–457. doi:10.1083/jcb.201612069.
[Google Scholar]
-
92.
Vega FM, Fruhwirth G, Ng T, Ridley AJ. RhoA and RhoC have distinct roles in migration and invasion by acting through different targets.
J. Cell Biol. 2011,
193, 655–665. doi:10.1083/jcb.201011038.
[Google Scholar]
-
93.
Newell-Litwa KA, Badoual M, Asmussen H, Patel H, Whitmore L, Horwitz AR. ROCK1 and 2 differentially regulate actomyosin organization to drive cell and synaptic polarity.
J. Cell Biol. 2015,
210, 225–242. doi:10.1083/jcb.201504046.
[Google Scholar]
-
94.
Zhu S, Ye Y, Shi Y, Dang J, Feng X, Chen Y, et al. Sonic Hedgehog Regulates Proliferation, Migration and Invasion of Synoviocytes in Rheumatoid Arthritis via JNK Signaling.
Front. Immunol. 2020,
11, 1300. doi:10.3389/fimmu.2020.01300.
[Google Scholar]
-
95.
Hu Z, Chen Y, Zhu S, Feng X, Zhang B, Huang J. Sonic Hedgehog Promotes Proliferation and Migration of Fibroblast-Like Synoviocytes in Rheumatoid Arthritis via Rho/ROCK Signaling.
J. Immunol. Res. 2022,
2022, 3423692. doi:10.1155/2022/3423692.
[Google Scholar]
-
96.
Liu F, Feng XX, Zhu SL, Huang HY, Chen YD, Pan YF, et al. Sonic Hedgehog Signaling Pathway Mediates Proliferation and Migration of Fibroblast-Like Synoviocytes in Rheumatoid Arthritis via MAPK/ERK Signaling Pathway.
Front. Immunol. 2018,
9, 2847. doi:10.3389/fimmu.2018.02847.
[Google Scholar]
-
97.
Sasaki N, Kurisu J, Kengaku M. Sonic hedgehog signaling regulates actin cytoskeleton via Tiam1-Rac1 cascade during spine formation.
Mol. Cell Neurosci. 2010,
45, 335–344. doi:10.1016/j.mcn.2010.07.006.
[Google Scholar]
-
98.
Villavicencio EH, Walterhouse DO, Iannaccone PM. The sonic hedgehog-patched-gli pathway in human development and disease.
Am. J. Hum. Genet. 2000,
67, 1047–1054. doi:10.1016/s0002-9297(07)62934-6.
[Google Scholar]
-
99.
Zhu ZX, Sun CC, Ting Zhu Y, Wang Y, Wang T, Chi LS, et al. Hedgehog signaling contributes to basic fibroblast growth factor-regulated fibroblast migration.
Exp. Cell Res. 2017,
355, 83–94. doi:10.1016/j.yexcr.2017.03.054.
[Google Scholar]
-
100.
Hou J, Ji J, Chen X, Cao H, Tan Y, Cui Y, et al. Alveolar epithelial cell-derived Sonic hedgehog promotes pulmonary fibrosis through OPN-dependent alternative macrophage activation.
FEBS J. 2021,
288, 3530–3546. doi:10.1111/febs.15669.
[Google Scholar]
-
101.
Mi Y, Zhong L, Lu S, Hu P, Pan Y, Ma X, et al. Quercetin promotes cutaneous wound healing in mice through Wnt/β-catenin signaling pathway.
J. Ethnopharmacol. 2022,
290, 115066. doi:10.1016/j.jep.2022.115066.
[Google Scholar]
-
102.
Čada Š, Bryja V. Local Wnt signalling in the asymmetric migrating vertebrate cells.
Semin. Cell Dev. Biol. 2022,
125, 26–36. doi:10.1016/j.semcdb.2021.11.020.
[Google Scholar]
-
103.
Ji Y, Hao H, Reynolds K, McMahon M, Zhou CJ. Wnt Signaling in Neural Crest Ontogenesis and Oncogenesis.
Cells 2019,
8, 1173. doi:10.3390/cells8101173.
[Google Scholar]
-
104.
Wu B, Crampton SP, Hughes CC. Wnt signaling induces matrix metalloproteinase expression and regulates T cell transmigration.
Immunity 2007,
26, 227–239. doi:10.1016/j.immuni.2006.12.007.
[Google Scholar]
-
105.
Chen X, Shi C, Cao H, Chen L, Hou J, Xiang Z, et al. The hedgehog and Wnt/beta-catenin system machinery mediate myofibroblast differentiation of LR-MSCs in pulmonary fibrogenesis.
Cell Death Dis. 2018,
9, 639. doi:10.1038/s41419-018-0692-9.
[Google Scholar]
-
106.
Wu CY, Lin MW, Wu DC, Huang YB, Huang HT, Chen CL. The role of phosphoinositide-regulated actin reorganization in chemotaxis and cell migration.
Br. J. Pharmacol. 2014,
171, 5541–5554. doi:10.1111/bph.12777.
[Google Scholar]
-
107.
Schymeinsky J, Then C, Sindrilaru A, Gerstl R, Jakus Z, Tybulewicz VL, Scharffetter-Kochanek K, Walzog B. Syk-mediated translocation of PI3Kdelta to the leading edge controls lamellipodium formation and migration of leukocytes.
PLoS ONE 2007,
2, e1132. doi:10.1371/journal.pone.0001132.
[Google Scholar]
-
108.
Trepat X, Chen Z, Jacobson K. Cell migration.
Compr. Physiol. 2012,
2, 2369–2392. doi:10.1002/cphy.c110012.
[Google Scholar]
-
109.
Welf ES, Haugh JM. Signaling pathways that control cell migration: models and analysis.
Wiley Interdiscip. Rev. Syst. Biol. Med. 2011,
3, 231–240. doi:10.1002/wsbm.110.
[Google Scholar]
-
110.
Henderson V, Smith B, Burton LJ, Randle D, Morris M, Odero-Marah VA. Snail promotes cell migration through PI3K/AKT-dependent Rac1 activation as well as PI3K/AKT-independent pathways during prostate cancer progression.
Cell Adh Migr. 2015,
9, 255–264. doi:10.1080/19336918.2015.1013383.
[Google Scholar]
-
111.
Pan L, Cheng Y, Yang W, Wu X, Zhu H, Hu M, et al. Nintedanib Ameliorates Bleomycin-Induced Pulmonary Fibrosis, Inflammation, Apoptosis, and Oxidative Stress by Modulating PI3K/Akt/mTOR Pathway in Mice.
Inflammation 2023,
46, 1531–1542. doi:10.1007/s10753-023-01825-2.
[Google Scholar]
-
112.
Cen R, Wang L, He Y, Yue C, Tan Y, Li L, et al. Dermal Fibroblast Migration and Proliferation Upon Wounding or Lipopolysaccharide Exposure is Mediated by Stathmin.
Front. Pharmacol. 2021,
12, 781282. doi:10.3389/fphar.2021.781282.
[Google Scholar]
-
113.
D’Urso M, Kurniawan NA. Mechanical and Physical Regulation of Fibroblast-Myofibroblast Transition: From Cellular Mechanoresponse to Tissue Pathology.
Front. Bioeng. Biotechnol. 2020,
8, 609653. doi:10.3389/fbioe.2020.609653.
[Google Scholar]
-
114.
Tisler M, Alkmin S, Chang HY, Leet J, Bernau K, Sandbo N, et al. Analysis of fibroblast migration dynamics in idiopathic pulmonary fibrosis using image-based scaffolds of the lung extracellular matrix.
Am. J. Physiology. Lung Cell. Mol. Physiol. 2020,
318, L276–l286. doi:10.1152/ajplung.00087.2019.
[Google Scholar]
-
115.
Lagares D, Busnadiego O, García-Fernández RA, Kapoor M, Liu S, Carter DE, et al. Inhibition of focal adhesion kinase prevents experimental lung fibrosis and myofibroblast formation.
Arthritis Rheum. 2012,
64, 1653–1664. doi:10.1002/art.33482.
[Google Scholar]
-
116.
Zhao XK, Cheng Y, Liang Cheng M, Yu L, Mu M, Li H, et al. Focal Adhesion Kinase Regulates Fibroblast Migration via Integrin beta-1 and Plays a Central Role in Fibrosis.
Sci. Rep. 2016,
6, 19276. doi:10.1038/srep19276.
[Google Scholar]
-
117.
Mitchell CB, Black B, Sun F, Chrzanowski W, Cooper-White J, Maisonneuve B, et al. Tropomyosin Tpm 2.1 loss induces glioblastoma spreading in soft brain-like environments.
J. Neurooncol 2019,
141, 303–313. doi:10.1007/s11060-018-03049-z.
[Google Scholar]
-
118.
Bradbury P, Nader CP, Cidem A, Rutting S, Sylvester D, He P, et al. Tropomyosin 2.1 collaborates with fibronectin to promote TGF-β(1)-induced contraction of human lung fibroblasts.
Respir. Res. 2021,
22, 129. doi:10.1186/s12931-021-01730-y.
[Google Scholar]
-
119.
Li L, Gu L, Yao Z, Wang Y, Tang Z, Wu X. Arecoline suppresses epithelial cell viability by upregulating tropomyosin-1 through the transforming growth factor-β/Smad pathway.
Pharm. Biol. 2020,
58, 1244–1251. doi:10.1080/13880209.2020.1851729.
[Google Scholar]
-
120.
Shibata T, Shibata S, Ishigaki Y, Kiyokawa E, Ikawa M, Singh DP, et al. Tropomyosin 2 heterozygous knockout in mice using CRISPR-Cas9 system displays the inhibition of injury-induced epithelial-mesenchymal transition, and lens opacity.
Mech. Ageing Dev. 2018,
171, 24–30. doi:10.1016/j.mad.2018.03.001.
[Google Scholar]
-
121.
Stehn JR, Haass NK, Bonello T, Desouza M, Kottyan G, Treutlein H, et al. A novel class of anticancer compounds targets the actin cytoskeleton in tumor cells.
Cancer Res. 2013,
73, 5169–5182. doi:10.1158/0008-5472.Can-12-4501.
[Google Scholar]
-
122.
Currier MA, Stehn JR, Swain A, Chen D, Hook J, Eiffe E, et al. Identification of Cancer-Targeted Tropomyosin Inhibitors and Their Synergy with Microtubule Drugs.
Mol. Cancer Ther. 2017,
16, 1555–1565. doi:10.1158/1535-7163.Mct-16-0873.
[Google Scholar]
-
123.
Ni J, Dong Z, Han W, Kondrikov D, Su Y. The role of RhoA and cytoskeleton in myofibroblast transformation in hyperoxic lung fibrosis.
Free. Radic. Biol. Med. 2013,
61, 26–39. doi:10.1016/j.freeradbiomed.2013.03.012.
[Google Scholar]
-
124.
Hadzipasic M, Sten MS, Massaad E, Kiapour A, Connolly ID, Esposito E, et al. ROCK-dependent mechanotransduction of macroscale forces drives fibrosis in degenerative spinal disease.
Nat. Biomed. Eng. 2025. doi:10.1038/s41551-025-01396-7.
[Google Scholar]
-
125.
Htwe SS, Cha BH, Yue K, Khademhosseini A, Knox AJ, Ghaemmaghami AM. Role of Rho-Associated Coiled-Coil Forming Kinase Isoforms in Regulation of Stiffness-Induced Myofibroblast Differentiation in Lung Fibrosis.
Am. J. Respir. Cell Mol. Biol. 2017,
56, 772–783. doi:10.1165/rcmb.2016-0306OC.
[Google Scholar]
-
126.
Li Z, Bratlie KM. Fibroblasts treated with macrophage conditioned medium results in phenotypic shifts and changes in collagen organization.
Mater. Sci. Eng. C Mater. Biol. Appl. 2021,
122, 111915. doi:10.1016/j.msec.2021.111915.
[Google Scholar]
-
127.
Shimizu Y, Dobashi K, Iizuka K, Horie T, Suzuki K, Tukagoshi H, et al. Contribution of small GTPase Rho and its target protein rock in a murine model of lung fibrosis.
Am. J. Respir. Crit. Care Med. 2001,
163, 210–217. doi:10.1164/ajrccm.163.1.2001089.
[Google Scholar]
-
128.
Rezaei M, Mehta JL, Zadeh GM, Khedri A, Rezaei HB. Myosin light chain phosphatase is a downstream target of Rho-kinase in endothelin-1-induced transactivation of the TGF-β receptor.
Cell Biochem. Biophys. 2024,
82, 1109–1120. doi:10.1007/s12013-024-01262-4.
[Google Scholar]
-
129.
Li Q, Cheng Y, Zhang Z, Bi Z, Ma X, Wei Y, et al. Inhibition of ROCK ameliorates pulmonary fibrosis by suppressing M2 macrophage polarisation through phosphorylation of STAT3.
Clin. Transl. Med. 2022,
12, e1036. doi:10.1002/ctm2.1036.
[Google Scholar]
-
130.
Knipe RS, Probst CK, Lagares D, Franklin A, Spinney JJ, Brazee PL, et al. The Rho Kinase Isoforms ROCK1 and ROCK2 Each Contribute to the Development of Experimental Pulmonary Fibrosis.
Am. J. Respir. Cell Mol. Biol. 2018,
58, 471–481. doi:10.1165/rcmb.2017-0075OC.
[Google Scholar]
-
131.
Bei Y, Hua-Huy T, Nicco C, Duong-Quy S, Le-Dong NN, Tiev KP, et al. RhoA/Rho-kinase activation promotes lung fibrosis in an animal model of systemic sclerosis.
Exp. Lung Res. 2016,
42, 44–55. doi:10.3109/01902148.2016.1141263.
[Google Scholar]
-
132.
Grisk O. Potential benefits of rho-kinase inhibition in arterial hypertension.
Curr. Hypertens. Rep. 2013,
15, 506–513. doi:10.1007/s11906-013-0373-0.
[Google Scholar]
-
133.
Zhang X, Yang F, Han L, Su Q, Gao Y, Wu R, et al. Identification of pivotal genes and crucial pathways in liver fibrosis through WGCNA analysis.
Technol. Health Care 2025,
33, 431–448. doi:10.3233/thc-241142.
[Google Scholar]
-
134.
Li H, Xin G, Zhou Q, Yu X, Wan C, Wang Y, et al. Qingkailing granule alleviates pulmonary fibrosis by inhibiting PI3K/AKT and SRC/STAT3 signaling pathways.
Bioorg Chem. 2024,
146, 107286. doi:10.1016/j.bioorg.2024.107286.
[Google Scholar]
-
135.
Zhang X, Zhang J, Liu Y, Wang Q, Chen Z. Pirfenidone Inhibits Fibroblast Proliferation, Migration or Adhesion and Reduces Epidural Fibrosis in Rats via the PI3k/AKT Signaling Pathway.
Biochem. Biophys. Res. Commun. 2021,
547, 183–191. doi:10.1016/j.bbrc.2021.01.055.
[Google Scholar]
-
136.
Xue R, Zhou J, Wu J, Meng Q, Gong J, Shen L. P-element-Induced Wimpy-Testis-Like Protein 1 Regulates the Activation of Pancreatic Stellate Cells Through the PI3K/AKT/mTOR Signaling Pathway.
Dig. Dis. Sci. 2023,
68, 1339–1350. doi:10.1007/s10620-022-07605-6.
[Google Scholar]
-
137.
Frost J, Estivill X, Ramsay M, Tikly M. Dysregulation of the Wnt signaling pathway in South African patients with diffuse systemic sclerosis.
Clin. Rheumatol. 2019,
38, 933–938. doi:10.1007/s10067-018-4298-5.
[Google Scholar]
-
138.
Staal FJ, Clevers HC. WNT signalling and haematopoiesis: a WNT-WNT situation.
Nat. Rev. Immunol. 2005,
5, 21–30. doi:10.1038/nri1529.
[Google Scholar]
-
139.
Morrisey EE. Wnt signaling and pulmonary fibrosis.
Am. J. Pathol. 2003,
162, 1393–1397. doi:10.1016/S0002-9440(10)64271-X.
[Google Scholar]
-
140.
Moon RT, Kohn AD, De Ferrari GV, Kaykas A. WNT and beta-catenin signalling: diseases and therapies.
Nat. Rev. Genet. 2004,
5, 691–701. doi:10.1038/nrg1427.
[Google Scholar]
-
141.
Meuten T, Hickey A, Franklin K, Grossi B, Tobias J, Newman DR, et al. WNT7B in fibroblastic foci of idiopathic pulmonary fibrosis.
Respir. Res. 2012,
13, 62. doi:10.1186/1465-9921-13-62.
[Google Scholar]
-
142.
Oda K, Yatera K, Izumi H, Ishimoto H, Yamada S, Nakao H, et al. Profibrotic role of WNT10A via TGF-β signaling in idiopathic pulmonary fibrosis.
Respir. Res. 2016,
17, 39. doi:10.1186/s12931-016-0357-0.
[Google Scholar]
-
143.
Bayle J, Fitch J, Jacobsen K, Kumar R, Lafyatis R, Lemaire R. Increased expression of Wnt2 and SFRP4 in Tsk mouse skin: role of Wnt signaling in altered dermal fibrillin deposition and systemic sclerosis.
J.Invest. Dermatol. 2008,
128, 871–881.
[Google Scholar]
-
144.
Akhmetshina A, Palumbo K, Dees C, Bergmann C, Venalis P, Zerr P, et al. Activation of canonical Wnt signalling is required for TGF-β-mediated fibrosis.
Nat. Commun. 2012,
3, 735. doi:10.1038/ncomms1734.
[Google Scholar]
-
145.
Landin Malt A, Hogan AK, Smith CD, Madani MS, Lu X. Wnts regulate planar cell polarity via heterotrimeric G protein and PI3K signaling.
J. Cell Biol. 2020,
219, e201912071. doi:10.1083/jcb.201912071.
[Google Scholar]
-
146.
Vargas JY, Loria F, Wu YJ, Córdova G, Nonaka T, Bellow S, et al. The Wnt/Ca(
2+) pathway is involved in interneuronal communication mediated by tunneling nanotubes.
Embo J. 2019,
38, e101230. doi:10.15252/embj.2018101230.
[Google Scholar]
-
147.
Chen X, Ma T, Chen Y, Sun Q, Wang H, Wang Y. USP14 promotes osteoarthritis progression by deubiquitinating FZD8 to activate the Wnt/β-catenin signaling pathway.
Immunobiology 2025,
230, 152905. doi:10.1016/j.imbio.2025.152905.
[Google Scholar]
-
148.
Bai Y, Lu H, Lin C, Xu Y, Hu D, Liang Y, et al. Sonic hedgehog-mediated epithelial-mesenchymal transition in renal tubulointerstitial fibrosis.
Int. J. Mol. Med. 2016,
37, 1317–1327. doi:10.3892/ijmm.2016.2546.
[Google Scholar]
-
149.
Xiao C, Ogle SA, Schumacher MA, Schilling N, Tokhunts RA, Orr-Asman MA, et al. Hedgehog signaling regulates E-cadherin expression for the maintenance of the actin cytoskeleton and tight junctions.
Am. J. Physiology. Gastrointest. Liver Physiol. 2010,
299, G1252–G1265. doi:10.1152/ajpgi.00512.2009.
[Google Scholar]
-
150.
Maimets M, Pedersen MT, Guiu J, Dreier J, Thodberg M, Antoku Y, et al. Mesenchymal-epithelial crosstalk shapes intestinal regionalisation via Wnt and Shh signalling.
Nat. Commun. 2022,
13, 715. doi:10.1038/s41467-022-28369-7.
[Google Scholar]
-
151.
Zhang Y, Wang Z, Wu L, Zhu L, Lu M, Zhang Z, et al. Rapamycin-induced small extracellular vesicles under GelMA scaffolds facilitate diabetic wound repair through accelerating angiogenesis and alleviating macrophage-mediated inflammation via PI3K/Akt signaling pathway.
Mater. Today Bio. 2025,
32, 101846. doi:10.1016/j.mtbio.2025.101846.
[Google Scholar]
-
152.
Bhandari R, Yang H, Kosarek NN, Smith AE, Garlick JA, Hinchcliff M, et al. Human dermal fibroblast-derived exosomes induce macrophage activation in systemic sclerosis.
Rheumatology 2023,
62, Si114–si124. doi:10.1093/rheumatology/keac453.
[Google Scholar]
-
153.
Lei R, da Silva TB, Cui Z, Ye H. Small extracellular vesicles from human bone marrow mesenchymal stromal cells enhance migration and regulate reparative gene expression in dermal fibroblasts.
Sci. Rep. 2025,
15, 19383. doi:10.1038/s41598-025-04057-6.
[Google Scholar]
-
154.
Argentino G, Olivieri B, Morandi M, Bonisoli G, Beri R, Tinazzi E, et al. Lymphomonocytic Extracellular Vesicles Influence Fibroblast Proliferation and Collagen Production in Systemic Sclerosis.
Int. J. Mol. Sci. 2025,
26, 2699 doi:10.3390/ijms26062699.
[Google Scholar]
-
155.
Jeppesen DK, Sanchez ZC, Kelley NM, Hayes JB, Ambroise J, Koory EN, et al. Blebbisomes are large, organelle-rich extracellular vesicles with cell-like properties.
Nat. Cell Biol. 2025,
27, 438–448. doi:10.1038/s41556-025-01621-0.
[Google Scholar]
-
156.
Tu H, Shi Y, Guo Y, Zou Z, He Y, Zhou J, et al. Young fibroblast-derived migrasomes alleviate keratinocyte senescence and enhance wound healing in aged skin.
J. Nanobiotechnol. 2025,
23, 200. doi:10.1186/s12951-025-03293-2.
[Google Scholar]
-
157.
Zhang F, Liu W, Mao Y, Yang Y, Ling C, Liu Y, et al. Migrasome, a migration-dependent organelle.
Front. Cell Dev. Biol. 2024,
12, 1417242. doi:10.3389/fcell.2024.1417242.
[Google Scholar]
-
158.
Zucker B, Dharan R, Wang D, Yu L, Sorkin R, Kozlov MM. Migrasome formation is initiated preferentially in tubular junctions by membrane tension.
Biophys. J. 2025,
124, 604–619. doi:10.1016/j.bpj.2024.12.029.
[Google Scholar]
-
159.
Dharan R, Huang Y, Cheppali SK, Goren S, Shendrik P, Wang W, et al. Tetraspanin 4 stabilizes membrane swellings and facilitates their maturation into migrasomes.
Nat. Commun. 2023,
14, 1037. doi:10.1038/s41467-023-36596-9.
[Google Scholar]
-
160.
Peng Y, Mei S, Qi X, Tang R, Yang W, Feng J, et al. PGC-1α mediates migrasome secretion accelerating macrophage-myofibroblast transition and contributing to sepsis-associated pulmonary fibrosis.
Exp. Mol. Med. 2025,
57, 759–774. doi:10.1038/s12276-025-01426-z.
[Google Scholar]
-
161.
Ma L, Li Y, Peng J, Wu D, Zhao X, Cui Y, et al. Discovery of the migrasome, an organelle mediating release of cytoplasmic contents during cell migration.
Cell Res. 2015,
25, 24–38. doi:10.1038/cr.2014.135.
[Google Scholar]
-
162.
Terlecki-Zaniewicz L, Pils V, Bobbili MR, Lämmermann I, Perrotta I, Grillenberger T, et al. Extracellular Vesicles in Human Skin: Cross-Talk from Senescent Fibroblasts to Keratinocytes by miRNAs.
J. Investig. Dermatol. 2019,
139, 2425–2436.e2425. doi:10.1016/j.jid.2019.05.015.
[Google Scholar]
-
163.
Liu Q, Niu Y, Pei Z, Yang Y, Xie Y, Wang M, et al. Gas6-Axl signal promotes indoor VOCs exposure-induced pulmonary fibrosis via pulmonary microvascular endothelial cells-fibroblasts cross-talk.
J. Hazard. Mater. 2024,
474, 134786. doi:10.1016/j.jhazmat.2024.134786.
[Google Scholar]
-
164.
Torr EE, Ngam CR, Bernau K, Tomasini-Johansson B, Acton B, Sandbo N. Myofibroblasts exhibit enhanced fibronectin assembly that is intrinsic to their contractile phenotype.
J. Biol. Chem. 2015,
290, 6951–6961. doi:10.1074/jbc.M114.606186.
[Google Scholar]
-
165.
Huang X, Gai Y, Yang N, Lu B, Samuel CS, Thannickal VJ, et al. Relaxin regulates myofibroblast contractility and protects against lung fibrosis.
Am. J. Pathol. 2011,
179, 2751–2765. doi:10.1016/j.ajpath.2011.08.018.
[Google Scholar]
-
166.
Jin L, Bao B, Huang XT, Tao JH, Duan JX, Zhong WJ, et al. MEOX1 triggers myofibroblast apoptosis resistance, contributing to pulmonary fibrosis in mice.
J. Cell Physiol. 2024,
239, e31442. doi:10.1002/jcp.31442.
[Google Scholar]
-
167.
Rehan M, Deskin B, Kurundkar AR, Yadav S, Matsunaga Y, Manges J, et al. Nicotinamide N-methyltransferase mediates lipofibroblast-myofibroblast transition and apoptosis resistance.
J. Biol. Chem. 2023,
299, 105027. doi:10.1016/j.jbc.2023.105027.
[Google Scholar]
-
168.
Luo C, Huang C, Zhu Y, Zhou Y, Qiao Y, Shi C, et al. Inhibition of Rho GEFs attenuates pulmonary fibrosis through suppressing myofibroblast activation and reprogramming profibrotic macrophages.
Cell Death Dis. 2025,
16, 278. doi:10.1038/s41419-025-07573-5.
[Google Scholar]
-
169.
Berry CE, Downer M, Morgan AG, Griffin M, Liang NE, Kameni L, et al. The effects of mechanical force on fibroblast behavior in cutaneous injury.
Front. Surg. 2023,
10, 1167067. doi:10.3389/fsurg.2023.1167067.
[Google Scholar]
-
170.
Metropulos AE, Munshi HG, Principe DR. The difficulty in translating the preclinical success of combined TGFβ and immune checkpoint inhibition to clinical trial.
EBioMedicine 2022,
86, 104380. doi:10.1016/j.ebiom.2022.104380.
[Google Scholar]
-
171.
Ali F, Ilyas A. Belumosudil with ROCK-2 inhibition: chemical and therapeutic development to FDA approval for the treatment of chronic graft-versus-host disease.
Curr. Res. Transl. Med. 2022,
70, 103343. doi:10.1016/j.retram.2022.103343.
[Google Scholar]
-
172.
Bildyug N. Inhibition of Integrin-Associated Kinases FAK and ILK on the In Vitro Model of Skin Wound Healing.
Appl. Biochem. Biotechnol. 2024,
196, 5604–5615. doi:10.1007/s12010-023-04842-x.
[Google Scholar]
-
173.
Sao K, Jones TM, Doyle AD, Maity D, Schevzov G, Chen Y, et al. Myosin II governs intracellular pressure and traction by distinct tropomyosin-dependent mechanisms.
Mol. Biol. Cell 2019,
30, 1170–1181. doi:10.1091/mbc.E18-06-0355.
[Google Scholar]
-
174.
Walraven M, Akershoek JJ, Beelen RH, Ulrich MM. In vitro cultured fetal fibroblasts have myofibroblast-associated characteristics and produce a fibrotic-like environment upon stimulation with TGF-β1: Is there a thin line between fetal scarless healing and fibrosis?
Arch. Dermatol. Res. 2017,
309, 111–121. doi:10.1007/s00403-016-1710-3.
[Google Scholar]
-
175.
Yan Y, Liu H, Yi J, Chen Z, Chen J, Zhang J, et al. Identification of differentially expressed proteins involved in fetal scarless wound healing using a rat model of cleft lip.
Mol. Med. Rep. 2021,
24, 596. doi:10.3892/mmr.2021.12235.
[Google Scholar]
-
176.
Griffin MF, Fahy EJ, King M, Guardino N, Chen K, Abbas DB, et al. Understanding Scarring in the Oral Mucosa.
Adv. Wound Care 2022,
11, 537–547. doi:10.1089/wound.2021.0038.
[Google Scholar]
-
177.
Mak K, Manji A, Gallant-Behm C, Wiebe C, Hart DA, Larjava H, et al. Scarless healing of oral mucosa is characterized by faster resolution of inflammation and control of myofibroblast action compared to skin wounds in the red Duroc pig model.
J. Dermatol. Sci. 2009,
56, 168–180. doi:10.1016/j.jdermsci.2009.09.005.
[Google Scholar]
-
178.
Glim JE, Beelen RH, Niessen FB, Everts V, Ulrich MM. The number of immune cells is lower in healthy oral mucosa compared to skin and does not increase after scarring.
Arch. Oral Biol. 2015,
60, 272–281. doi:10.1016/j.archoralbio.2014.10.008.
[Google Scholar]
-
179.
Roh JL, Lee J, Kim EH, Shin D. Plasticity of oral mucosal cell sheets for accelerated and scarless skin wound healing.
Oral. Oncol. 2017,
75, 81–88. doi:10.1016/j.oraloncology.2017.10.024.
[Google Scholar]
-
180.
Knight R, Board-Davies E, Brown H, Clayton A, Davis T, Karatas B, et al. Oral Progenitor Cell Line-Derived Small Extracellular Vesicles as a Treatment for Preferential Wound Healing Outcome.
Stem Cells Transl. Med. 2022,
11, 861–875. doi:10.1093/stcltm/szac037.
[Google Scholar]
-
181.
Sezgin B, Tatar S, Karahuseyinoglu S, Sahin GN, Ergun Y, Meric G, et al. The effects of oral mucosa-derived heterotopic fibroblasts on cutaneous wound healing.
J. Plast. Reconstr. Aesthet. Surg. 2021,
74, 2751–2758. doi:10.1016/j.bjps.2021.02.011.
[Google Scholar]
-
182.
Tarzemany R, Jiang G, Jiang JX, Gallant-Behm C, Wiebe C, Hart DA, Larjava H, Häkkinen L. Connexin 43 regulates the expression of wound healing-related genes in human gingival and skin fibroblasts.
Exp. Cell Res. 2018,
367, 150–161. doi:10.1016/j.yexcr.2018.03.031.
[Google Scholar]
-
183.
Bernard K, Logsdon NJ, Ravi S, Xie N, Persons BP, Rangarajan S, et al. Metabolic Reprogramming Is Required for Myofibroblast Contractility and Differentiation.
J. Biol. Chem. 2015,
290, 25427–25438. doi:10.1074/jbc.M115.646984.
[Google Scholar]
-
184.
Sun W, Zhou S, Peng L, Wang W, Liu Y, Wang T, et al. Fatty Acid Oxidation-Glycolysis Metabolic Transition Affects ECM Homeostasis in Silica-Induced Pulmonary Fibrosis.
Adv. Sci. 2025,
12, e2407134. doi:10.1002/advs.202407134.
[Google Scholar]
-
185.
Lin MH, Lee YC, Liao JB, Chou CY, Yang YF. PTGES is involved in myofibroblast differentiation via HIF-1α-dependent glycolysis pathway.
J. Cell. Mol. Med. 2024,
28, e70157. doi:10.1111/jcmm.70157.
[Google Scholar]
-
186.
Shan B, Guo C, Zhou H, Chen J. Tanshinone IIA alleviates pulmonary fibrosis by modulating glutamine metabolic reprogramming based on [U-(13)C(5)]-glutamine metabolic flux analysis.
J. Adv. Res. 2025,
70, 531–544. doi:10.1016/j.jare.2024.04.029.
[Google Scholar]
-
187.
Gibb AA, Huynh AT, Gaspar RB, Ploesch TL, Lombardi AA, Lorkiewicz PK, et al. Glutamine uptake and catabolism is required for myofibroblast formation and persistence.
J. Mol. Cell. Cardiol. 2022,
172, 78–89. doi:10.1016/j.yjmcc.2022.08.002.
[Google Scholar]
-
188.
Jeon KI, Kumar A, Brookes PS, Nehrke K, Huxlin KR. Manipulating mitochondrial pyruvate carrier function causes metabolic remodeling in corneal myofibroblasts that ameliorates fibrosis.
Redox Biol. 2024,
75, 103235. doi:10.1016/j.redox.2024.103235.
[Google Scholar]
-
189.
Contento G, Wilson JA, Selvarajah B, Platé M, Guillotin D, Morales V, et al. Pyruvate metabolism dictates fibroblast sensitivity to GLS1 inhibition during fibrogenesis.
JCI Insight 2024,
9. doi:10.1172/jci.insight.178453.
[Google Scholar]
-
190.
Shochet GE, Brook E, Bardenstein-Wald B, Grobe H, Edelstein E, Israeli-Shani L, et al. Integrin alpha-5 silencing leads to myofibroblastic differentiation in IPF-derived human lung fibroblasts.
Ther. Adv. Chronic Dis. 2020,
11, 2040622320936023. doi:10.1177/2040622320936023.
[Google Scholar]
-
191.
McAndrews KM, Miyake T, Ehsanipour EA, Kelly PJ, Becker LM, McGrail DJ, et al. Dermal αSMA(+) myofibroblasts orchestrate skin wound repair via β1 integrin and independent of type I collagen production.
Embo J. 2022,
41, e109470. doi:10.15252/embj.2021109470.
[Google Scholar]
-
192.
Huang W, Zhang Z, Li X, Zheng Q, Wu C, Liu L, et al. CD9 promotes TβR2-TβR1 association driving the transition of human dermal fibroblasts to myofibroblast under hypoxia.
Mol. Med. (Camb. Mass.) 2024,
30, 162. doi:10.1186/s10020-024-00925-5.
[Google Scholar]
-
193.
Yue W, Zhang H, Gao Y, Ding J, Xue R, Dong C, et al. Procollagen-lysine 2-oxoglutarate 5-dioxygenase 2 promotes collagen cross-linking and ECM stiffening to induce liver fibrosis.
Biochim. Biophys. Acta Mol. Basis Dis. 2024,
1870, 167205. doi:10.1016/j.bbadis.2024.167205.
[Google Scholar]
-
194.
Locy ML, Rangarajan S, Yang S, Johnson MR, Bernard K, Kurundkar A, et al. Oxidative cross-linking of fibronectin confers protease resistance and inhibits cellular migration.
Sci. Signal. 2020,
13,
eaau2803. doi:10.1126/scisignal.aau2803.
[Google Scholar]
-
195.
Wang Y, Jiang H, Chen Q, Guo F, Zhang B, Hu L, et al. Myofibroblast-Targeting Extracellular Vesicles: A Promising Platform for Cardiac Fibrosis Drug Delivery.
Biomater. Res. 2025,
29, 0179. doi:10.34133/bmr.0179.
[Google Scholar]