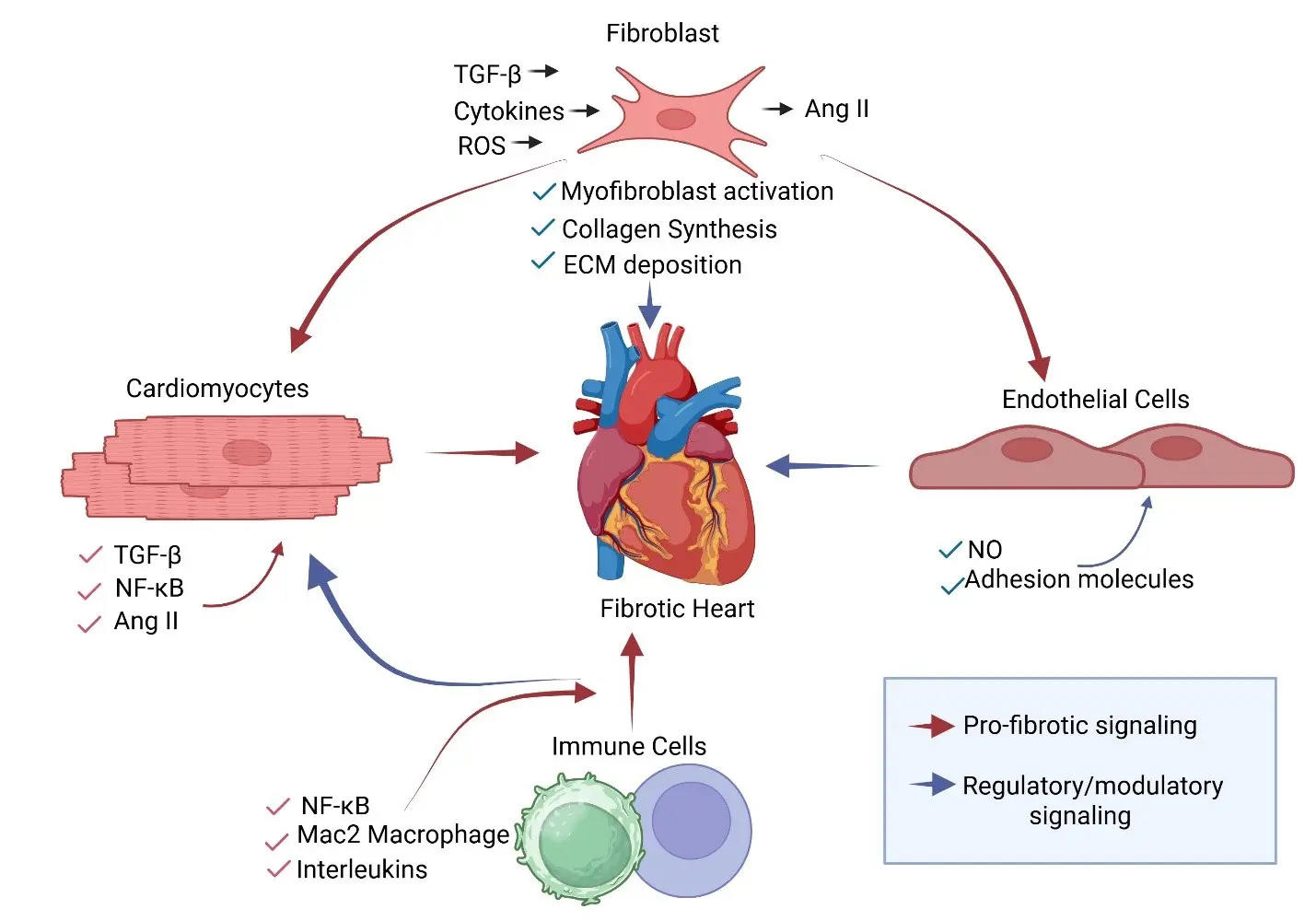
As a central metabolic and immune organ, the liver maintains a unique immune microenvironment which is crucial for sustaining health. When the immune balance in the liver is disrupted, it can drive the occurrence and progression of various chronic liver diseases, including liver fibrosis. Hepatic stellate cells (HSCs) are the key effector cells responsible for producing extracellular matrix (ECM) during liver fibrosis, and the hepatic immune microenvironment precisely regulates their activation. This review focuses on the complex bidirectional interaction network between HSCs and major immune cells in the liver, including macrophages, natural killer (NK) cells, and T cells. It systematically elucidates the central role of these interactions in maintaining hepatic homeostasis, mediating inflammatory responses, and driving the progression of fibrosis. A deeper understanding of the interaction between HSCs and immune cells is essential for elucidating the pathological mechanisms of liver fibrosis and will provide a theoretical basis for developing innovative therapeutic strategies targeting the immune microenvironment.
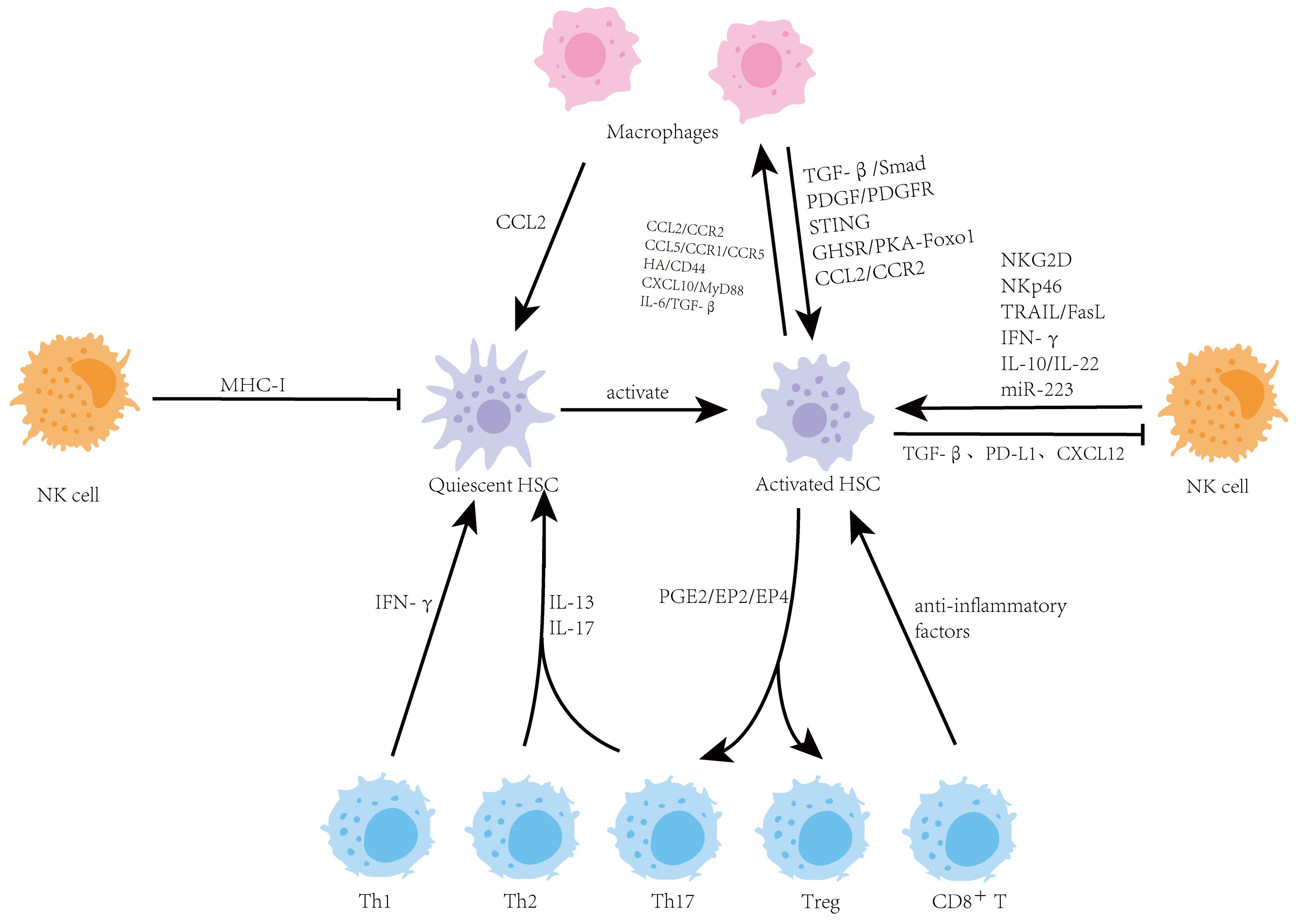
Cardiac fibrosis represents a global health crisis, observed in nearly all forms of heart disease, and contributes significantly to the progression of heart failure. Driven by diverse etiologies such as chronic hypertension, myocardial infarction, and metabolic disorders, cardiac fibrosis is characterized by the excessive deposition of extracellular matrix proteins. At the cellular level, the activation of cardiac fibroblasts into myofibroblasts serves as the primary mechanism for this structural remodelling. Excessive collagen deposition, crosslinking, and pathological scarring lead to increased ventricular stiffness, electrical arrhythmias, and a profound decline in cardiac function, affecting the quality of life for millions of patients worldwide. The review discusses the existing well-known profibrotic signals and molecular signalling pathways leading to cardiac fibroblast activation, collagen synthesis, and crosslinking. Mechanosensitive pathways, signalling mechanisms involved in collagen crosslinking, and epigenetic factors of cardiac fibrosis are also discussed along with their potential antifibrotic targets and therapeutic drugs. Further, small-molecule inhibitors, peptide-based therapies, natural compounds, and repurposed drugs for fibrosis are also discussed. This review concludes with recent approaches of chimeric antigen receptor (CAR)-T cell therapy for cardiac fibrosis.
Systemic Sclerosis (SSc) is a chronic autoimmune disease characterized by fibrosis in connective tissues. Fibroblasts are the effector cells of fibrosis since they contribute to the production of collagen and other extracellular matrix components. The goal of this study is to compare the transcriptomic profiles of primary human SSc skin and SSc lung fibroblasts. First, we conducted a meta-analysis of differentially expressed (DE) genes from two previously published differential analyses (SSc vs. normal) using skin and lung fibroblasts, observing 8.7% overlap in DE genes and 30% overlap in impacted pathways. Next, we characterized the signature of several genes of interest from the pro- and anti-fibrotic programs within the unique and overlap groups and explored overlapping drugs that are predicted to revert DE genes to “normal expression”. Finally, we identified 3760 DE genes between SSc lung and SSc skin fibroblasts, highlighting that fibroblasts in the disease state carry a tissue-specific signature that should be taken into consideration for therapeutic development. We also identified core genes that can serve as common targets for both skin and lung in SSc. To our knowledge, this is the first study to describe overlapping genes and pathways in primary human skin and lung fibroblasts from SSc patients.
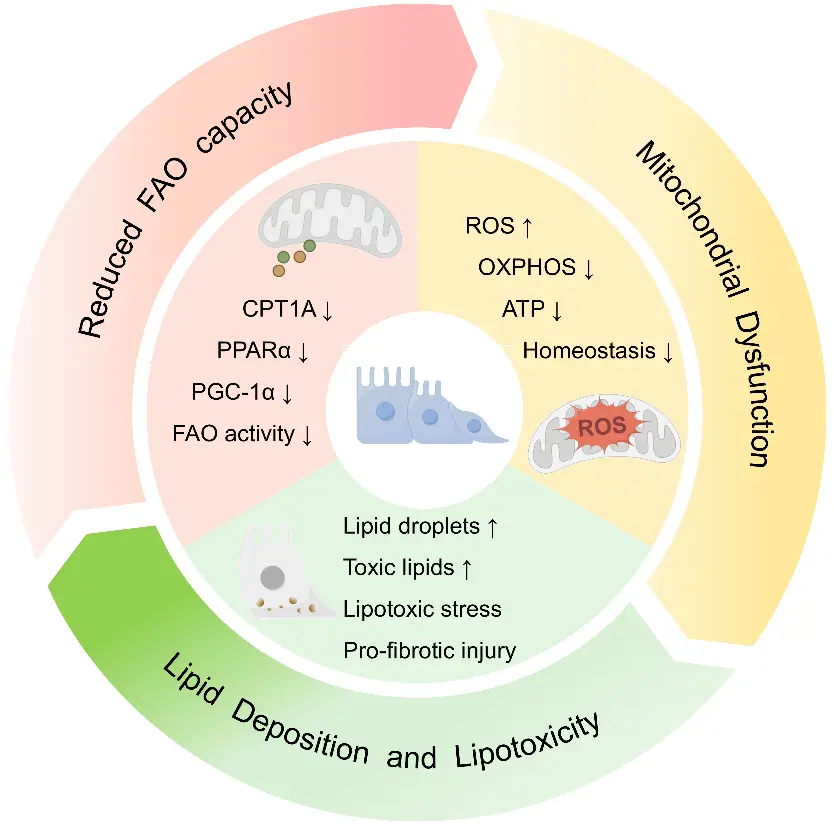
Tubulointerstitial fibrosis is a central pathological basis for the persistent progression of chronic kidney disease. Its initiation and progression involve multiple mechanisms, including disordered energy metabolism, lipid accumulation, inflammatory responses, and abnormal extracellular matrix deposition. As a major energy source for renal tubular epithelial cells, mitochondrial fatty acid oxidation (FAO) is essential for maintaining tubular metabolic homeostasis. Impaired FAO leads to insufficient ATP production, aggravated lipotoxicity, and mitochondrial homeostasis disruption, thereby further activating oxidative stress, inflammatory pathways, and profibrotic signaling, which, in turn, promote tubular injury and the progression of interstitial fibrosis. This review summarizes the basic physiological processes of mitochondrial FAO and its pathological role in tubulointerstitial fibrosis, with particular emphasis on the mechanisms by which FAO impairment drives metabolic reprogramming, lipotoxicity, and abnormalities in mitochondrial quality control. It also outlines recent advances in therapeutic strategies aimed at restoring FAO, improving mitochondrial function, and alleviating lipotoxicity and secondary profibrotic responses. Current evidence suggests that targeting FAO impairment may offer a promising therapeutic approach for delaying the progression of renal fibrosis; however, further efforts are needed to strengthen clinical translation.
Cardiac fibrosis is a central pathological feature of heart failure and contributes to myocardial stiffening, impaired electrical conduction, and progressive ventricular dysfunction. Traditionally, fibrotic remodeling has been viewed as a fibroblast-driven process in which activated fibroblasts deposit excessive extracellular matrix following cardiac injury. However, emerging evidence indicates that fibrosis arises from coordinated interactions among multiple cardiac cell populations, including cardiomyocytes, endothelial cells, immune cells, pericytes, and fibroblasts. In this review, we discuss the role of cardiomyocytes and their interactions with other cell types in the heart in facilitating cardiac fibrosis. We discuss how interactions among cardiomyocytes, immune cells, endothelial cells, pericytes, and fibroblasts contribute to fibrotic remodeling in both ischemic and non-ischemic heart disease. Our signaling emphasis is on transforming growth factor-β (TGF-β)-mediated cardiac fibrosis in the context of cellular interplay. We posit that a better understanding of these integrated signaling networks may reveal new opportunities to prevent or reverse pathological cardiac fibrosis.