Molecular Targets and Emerging Therapeutics in Cardiac Fibrosis

Molecular Targets and Emerging Therapeutics in Cardiac Fibrosis

Received: 01 March 2026 Revised: 11 March 2026 Accepted: 07 April 2026 Published: 20 April 2026

© 2026 The authors. This is an open access article under the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/).
Graphical Abstract
1. Introduction
Cardiac fibrosis is a maladaptive structural consequence of various cardiovascular pathologies, including myocardial infarction, hypertension, cardiomyopathies, and valvular heart disease. It is characterised by pathological remodelling of extracellular matrix (ECM), particularly excessive deposition of collagen, within the interstitium, which progressively compromises myocardial elasticity. The resulting architectural distortion leads to arrhythmias and heart failure. Cardiac fibrosis can generally be classified as replacement fibrosis, interstitial fibrosis, and perivascular fibrosis. Replacement fibrosis is a reparative response to cardiomyocyte death and post-infarct scarring to maintain structural integrity. Replacement fibrosis leads to the accumulation of profibrotic proteins, collagen types I and III, and fibronectin. Further, increased crosslinking of the cardiac wall results in enhanced myocardial stiffness. This leads to impaired diastolic filling, disrupted electrical conduction by attenuating action potential propagation, life-threatening arrhythmias, and eventual systolic dysfunction. Interstitial fibrosis or reactive fibrosis is a diffuse expansion of the endomysium and perimysium, typically seen in pressure overload, metabolic syndrome, and dilated cardiomyopathy. In genetic cardiomyopathies, both replacement and interstitial fibrosis are observed. Perivascular fibrosis is characterised by an increased amount of connective tissue, particularly collagen, around the vessels. Perivascular and fine interstitial fibrosis are observed in fibrosis of heart failure with preserved ejection fraction and are often associated with systemic inflammation. Different forms of fibrosis (replacement, interstitial, and perivascular) observed in heart failure are schematically represented in Figure 1.
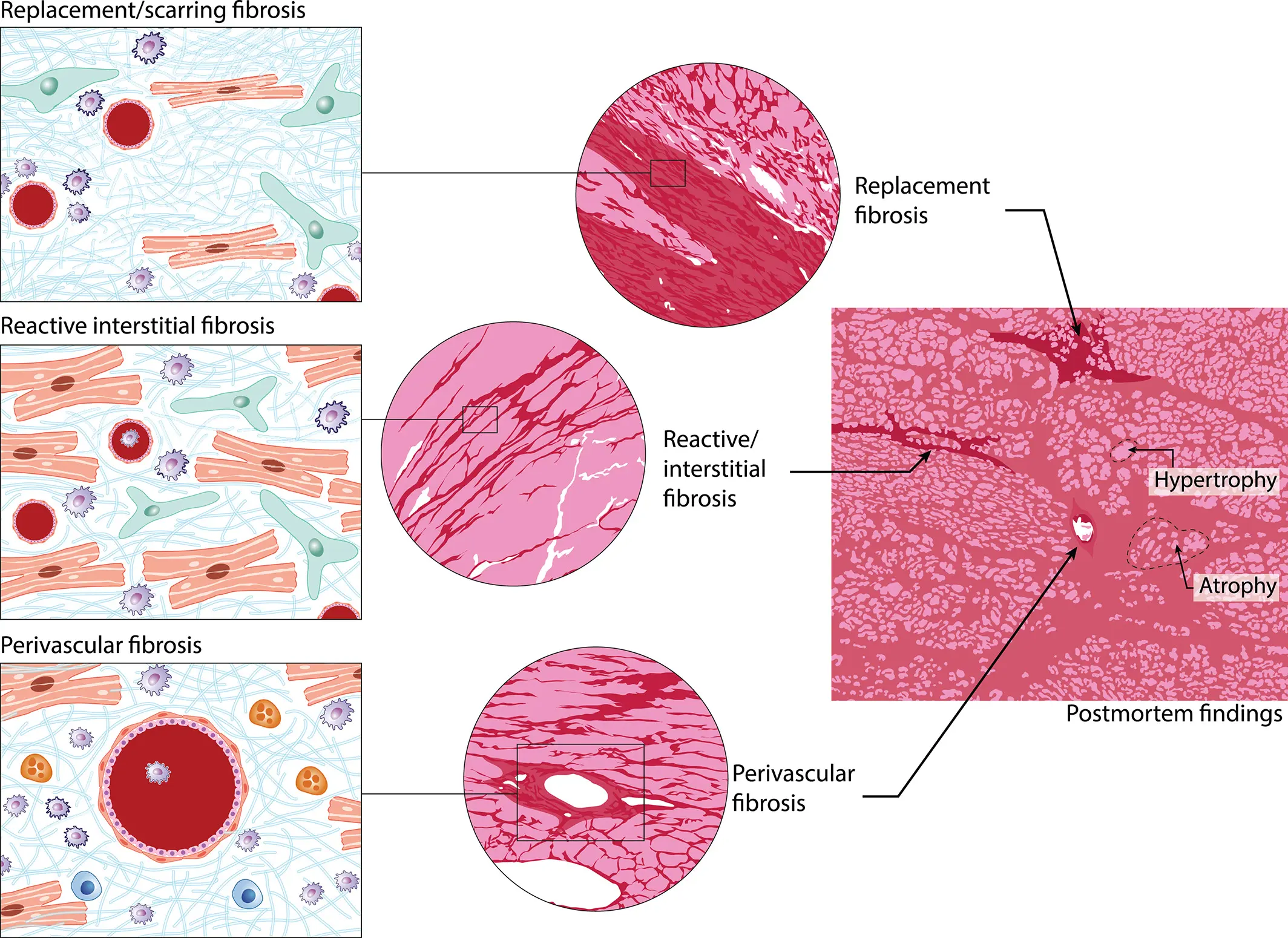
Figure 1. Schematic representation of different forms of fibrosis observed in heart failure. The major cell types: fibroblasts (green), inflammatory cells (blue and grey), and myocytes (orange-red), and their distribution within collagen fibrils are shown on the left. The right panel shows that all forms of fibrosis are observed in a failing heart. Adapted from Boer et al., 2019 [1], an open-access article distributed under the terms of the Creative Commons CC BY-NC 4 license.
A road map for defining, quantifying, and treating fibrosis in heart failure was defined by the Committee of Translational Research of the Heart Failure Association of the European Society of Cardiology [1]. Some of the therapeutic approaches for heart failure have direct or indirect anti-fibrotic activities. Current clinical management relies on repurposing agents that inhibit the renin-angiotensin-aldosterone system (RAAS), such as angiotensin converting enzyme (ACE) inhibitors, angiotensin receptor blockers (ARBs), and mineralocorticoid receptor antagonists (MRAs), which exhibit protective effects to hypertension and heart failure. Newer therapies, like sodium-glucose cotransporter 2 (SGLT2) inhibitors and angiotensin receptor-neprilysin inhibitor (ARNI) (sacubitril/valsartan), have shown secondary anti-fibrotic effects. These therapies manage symptoms or slow down the progression of heart failure, but they often fail to stop the underlying scarring process. Direct inhibition of transforming growth factor-β1 (TGF-β1) signalling has largely failed in clinical trials due to its pleiotropic, context-dependent roles in tissue homeostasis and repair, combined with pathway redundancy and a lack of cell-specific targeting, resulting in limited efficacy and unacceptable on-target toxicity [2]. Clinical trials using inhibitors of the main driver of fibrosis, the pro-fibrotic cytokines, TGF-β1, have also not shown completely satisfactory results, emphasising a critical need to identify drug targets and specific drugs to prevent the progression of fibrosis. In this review, we revisit classical signalling pathways and profibrotic signals that promote fibroblast activation and collagen production. Additionally, we discuss current strategies for identifying more precise molecular signalling mechanisms and the newer targets, and discuss mechanosensitive and epigenetic mechanisms in cardiac fibrosis. Small-molecule inhibitors of these drug targets, peptide-based therapies for cardiac fibrosis, natural compounds used as antifibrotic agents, and important drugs repurposed for cardiac fibrosis are also reviewed. Further, chimeric antigen receptor (CAR) T-cell therapy, designed to selectively deplete activated myofibroblasts, is also briefly reviewed.
Myocardial Remodelling Post-Cardiac Injury and Progression to Fibrosis
Prolonged ischemia generated during myocardial infarction results in cardiomyocyte death. The loss of functional tissue triggers an inherent wound-healing response aimed at maintaining the structural integrity of the heart. The wound-healing process post-myocardial injury can be categorized into three distinct, but overlapping phases: the inflammatory phase, the proliferative or reparative phase, and the maturation/remodelling phase. The inflammatory phase is a tightly regulated response that is essential for clearing necrotic debris before initiating the ECM remodelling. This process is triggered by the release of damage-associated molecular patterns (DAMPs) from dying cardiomyocytes and fragments of degraded ECM components. These signals activate resident macrophages, which secrete cytokines such as tumor necrosis factor α (TNF-α), interleukin-1β (IL-1β), and interleukin-1α (IL-1α), which in turn attract circulating monocytes and neutrophils via Toll-like receptors (TLRs), and other resident cells, including endothelial cells and fibroblasts. Monocytes and neutrophils clear cell debris by phagocytosis. During the resolution phase, the anti-inflammatory cytokines are secreted, promoting M1-M2 macrophage polarisation and programmed neutrophil apoptosis. Cardiac fibroblasts play an important role in the ECM remodelling. During the pro-inflammatory phase, the quiescent cardiac fibroblasts that reside within the interstitial space and perivascular space are activated by DAMPs, ROS, and cytokines. Activated fibroblasts proliferate, migrate, transdifferentiate into myofibroblasts, and secrete ECM to remodel the matrix. Myofibroblasts are migratory, secretory, and contractile cells, expressing α-smooth muscle actin (α-SMA) stress fibres, matricellular protein periostin, and predominantly secrete collagen type 1. While the resident fibroblasts are the major source of myofibroblasts, studies indicate that heterogeneous cell types, including endothelial, epicardial-derived cells, smooth muscle cells, and circulating bone marrow-derived fibrocytes, transdifferentiate to myofibroblasts. The remodelled ECM, particularly collagen type I, protects the cardiac wall from rupture. However, the sustained activation of various signaling pathways, such as the RAAS, the overproduction of TGF-β, dysregulated immune response, elevated proinflammatory cytokines, and imbalance in regulators of ECM remodeling, all lead to persistent activation of myofibroblasts and excessive deposition of collagen type I and its crosslinking, resulting in pathological remodeling. The signalling pathways that drive cellular responses and the progression to fibrosis are discussed in detail in Section 2.
2. Signalling Pathways Regulating Wound Healing and Cardiac Fibroblast Response
2.1. Inflammatory Signalling Pathways
The early inflammatory phase is characterized by the recruitment of innate immune cells to the injured myocardium. Neutrophils are among the first responders, clearing necrotic debris and releasing proteolytic enzymes and reactive oxygen species [3]. These cells, along with subsequently recruited immune cells, secrete pro-inflammatory cytokines such as IL-1 and TNF-α, which amplify inflammation and promote leukocyte recruitment [4]. IL-1 signalling plays a key role in linking the inflammation to fibroblast activation, as IL-1α activates multiple intracellular pathways in cardiac fibroblasts, including extracellular signal-regulated kinase (ERK1/2), p38 mitogen-activated protein kinase (MAPK), c-Jun N-terminal kinase (JNK), phosphoinositide 3-kinase (PI3K)/AKT serine/threonine kinase 1 (Akt), and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), thereby regulating fibroblast function and matrix remodelling [5]. IL-1β further contributes to fibrotic remodelling by upregulating angiotensin II type 1 (AT1) receptor expression and enhancing cardiac fibroblast migration, while also inducing matrix metalloproteinases (MMP) expression in cardiac fibroblasts [6]. Following neutrophil infiltration, circulating monocytes are recruited to the injured myocardium and differentiate into macrophages, which orchestrate both inflammatory and reparative processes [7]. In addition to their role as inflammatory mediators, monocytic fibroblast precursors have been shown to directly contribute to fibrotic remodelling, as demonstrated in angiotensin II–induced cardiac hypertrophy, where these cells promote ECM deposition and fibrosis [8].
IL-6 family cytokines, acting primarily through the Janus kinase/signal transducer and activator of transcription (JAK/STAT3) signalling pathway, are key mediators linking inflammation to cardiac fibroblast activation and fibrosis [9]. In cultured cardiac fibroblasts, IL-6 enhances STAT3 phosphorylation, whereas STAT3 inhibition attenuates IL-6–induced collagen synthesis and reverses cardiac hypertrophy [10]. Moreover, genetic deletion of IL-6 has reduced myocardial STAT3 phosphorylation and has improved adverse remodelling in a mouse model of coxsackievirus B3–induced dilated cardiomyopathy [11]. Clinically, elevated circulating levels of IL-6, together with TNF-α, have been shown to predict the future development of heart failure in asymptomatic elderly individuals, highlighting the translational relevance of IL-6–STAT3 signalling in cardiac disease [12].
TNF-α is a key pro-inflammatory cytokine that signals through the TNF receptors (TNFR1 and TNFR2), with soluble TNF-α preferentially activating TNFR1 and membrane-bound TNF-α signalling through TNFR2 [13]. TNF-α exerts context-dependent and sometimes opposing effects during cardiac remodelling. TNF-α has promoted pro-fibrotic responses by driving endothelial-to-mesenchymal transition [14] and increasing lysyl oxidase expression through TGF-β and PI3K signalling [15]. However, transmembrane TNF-α signalling via TNFR2 has exerted protective effects by attenuating pressure overload–induced cardiac hypertrophy [16] and limiting myocarditis and cardiac death through suppression of activated heart-reactive CD4+ T cell expansion [17].
Innate immune receptors, particularly TLRs, play a critical role in sensing tissue damage and initiating inflammatory responses in cardiac fibroblasts [3]. Cardiac fibroblasts have been identified as significant TLR9-responsive cells with important functional roles in myocardial inflammatory signalling [18]. Cell-autonomous TLR4 signalling has been shown to modulate TGF-β-induced activation of human cardiac fibroblasts [19]. Recent studies have further highlighted the contribution of inflammasome signalling to fibroblast activation, demonstrating that inflammasome activation in cardiac fibroblasts is essential for myocardial ischemia–reperfusion injury [20], and establishing immune–fibroblast cross-talk as an important amplification loop in chronic cardiac inflammation.
2.2. Transforming Growth Factor-β Signalling
TGF-β is the most extensively studied and central mediator of cardiac fibroblast activation and pathological cardiac fibrosis [21]. In mammals, the TGF-β subfamily comprises three isoforms: TGF-β1, TGF-β2, and TGF-β3. Among these, TGF-β1 is the predominant and most intensively investigated isoform [22]. In the healthy myocardium, TGF-β is stored in a latent complex bound to latency-associated peptide and latent TGF-β-binding proteins within the ECM. Following cardiac injury, latent TGF-β is rapidly activated through proteolytic cleavage by MMPs, MMP-2, MMP-9, plasmin, integrins, and matricellular proteins such as thrombospondin-1 [23].
Multiple cardiac cell types contribute to the TGF-β pool in the injured heart, including macrophages, monocytes, endothelial cells, cardiomyocytes, platelets, pericytes, and resident fibroblasts [24]. Macrophage-derived TGF-β plays an important paracrine role in shaping the fibrotic niche [7], while endothelial cell-specific activation of TGF-β promotes microvascular fibrosis [25]. Cardiomyocyte-specific TGF-β signalling has also emerged as a contributor to fibrotic remodelling, as cardiomyocyte-specific deletion of TGF-βRII has reduced fibrosis in pressure-overload models [26]. Nevertheless, accumulating genetic and lineage-tracing evidence indicates that fibroblast-intrinsic TGF-β signalling represents the dominant driver of pathological ECM deposition and scar formation [27].
TGF-β exerts its profibrotic effects through both canonical intracellular signalling proteins that act as key transducers for TGF-β, suppressor of mother against decapentaplegic (SMAD)-dependent, and non-canonical SMAD-independent signalling pathways. Canonical signalling is initiated when TGF-β binds to the type II TGF-β receptor (TGF-βRII), leading to recruitment and activation of the type I receptor, anaplastic lymphoma kinase-5 (ALK5) (TGF-βRI), a serine/threonine kinase present on the cell membrane. Activated ALK5 phosphorylates receptor-regulated SMADs, SMAD2 and SMAD3, which subsequently form complexes with the common mediator SMAD4 and translocate into the nucleus to regulate transcription of profibrotic target genes [22]. Although both SMAD2 and SMAD3 are involved in canonical TGF-β signalling, in vivo evidence demonstrates that SMAD3 plays a critical and indispensable role in cardiac fibrosis, whereas SMAD2 appears largely dispensable [28]. SMAD3 null mice have shown attenuated α-SMA expression, reduced cardiac fibroblast migration, and decreased synthesis of ECM proteins synthesis including collagens I, III, and fibronectin [29].
In parallel, TGF-β activates multiple non-canonical signalling cascades, including p38 MAPK, ERK1/2, JNK, transforming growth factor-β-activated kinase 1 (TAK1), and Rho family guanosine triphosphatases (GTPases) [30,31]. These SMAD-independent routes regulate fibroblast activation and proliferation, cytoskeletal dynamics, and epithelial to mesenchymal transition (EMT), thereby reinforcing the activated myofibroblast phenotype. Although these kinases integrate signals from numerous cytokines and mechanical stimuli, experimental inhibition of TAK1 or p38α MAPK has attenuated TGF-β-induced ECM production and myofibroblast differentiation, underscoring their functional relevance downstream of TGF-β [32,33]. Together, genetic, pharmacological, and single-cell transcriptomic evidence indicate that TGF-β signalling, particularly fibroblast-intrinsic SMAD3-dependent pathways, acts as a central molecular determinant of myofibroblast activation and cardiac fibrotic remodelling [34,35,36].
Studies have shown that cardiac fibroblast-specific deletion of SMAD2/3 or TGF-β receptors (TGF-βR1/2) from cardiac fibroblasts inhibited the profibrotic genes and ECM remodelling [35]. Several studies have reported the potential of Pirfenidone (5-methyl-1-phenyl-2-[1H]-pyridone), a synthetic small-molecule drug, to inhibit cardiac fibrosis by targeting the TGF-β1/SMAD pathway or by reducing both collagen expression and vascular permeability [37,38]. Further, bioinformatics analysis and preclinical analysis have shown that Pirfenidone targeted p38γ-MAPK12, the TGF-β1-SMAD2/3 pathway, and other effector proteins such as MMP2, MMP14, PDGF-α/β, and IGF-1 in the post-MI LV remodelling [39].
2.3. The Renin–Angiotensin–Aldosterone Signalling
The RAAS signalling is a complex hormonal network that plays a crucial role in cardiovascular homeostasis. Although it is classically known for controlling blood pressure, blood volume, and sodium balance, RAAS activity also influences inflammation, ECM turnover, and cardiac remodelling [40,41,42]. Renin, an aspartyl protease, cleaves angiotensinogen, synthesized primarily in the liver and expressed in cardiac tissue, into angiotensin I. Angiotensin I is subsequently converted by angiotensin-converting enzyme (ACE) into angiotensin II (Ang II).
In the setting of myocardial injury, cardiomyocytes, immune cells, and fibroblasts locally generate renin and ACE, leading to increased interstitial Ang II levels [43]. Ang II acts via autocrine, paracrine, and endocrine mechanisms, triggering intracellular signalling pathways involving MAPK, ERK, PKC, and Smad proteins. These pathways promote cardiac fibroblast proliferation, migration, resistance to apoptosis, differentiation into myofibroblasts, and excessive synthesis of collagen and fibronectin, ultimately contributing to cardiac hypertrophy, fibrosis, and adverse ventricular remodelling [44,45,46]. In addition, Ang II further stimulates myocardial aldosterone production, and aldosterone further contributes to cardiac fibrosis by directly promoting fibroblast activation and matrix synthesis, as well as by modulating cardiomyocyte, immune, and vascular cell function [43].
The profibrotic actions of the classical RAAS are counterbalanced by protective mechanisms, including signalling through the AT2 receptor and the alternative ACE2/angiotensin-(1–7)/Mas pathway. ACE2 converts Ang II into angiotensin-(1–7), which exerts vasodilatory, anti-inflammatory, and antifibrotic effects through Mas receptor activation and downstream pathways involving nitric oxide, cyclic AMP–dependent protein kinase, and reduced MAPK activity [47]. Experimental studies have shown that ACE2 deficiency has worsened cardiac remodelling [48], whereas ACE2 upregulation has attenuated fibrosis [49]. The complexity of RAAS signalling is further increased by additional angiotensin peptides, including Ang III, Ang IV, Ang A, and alamandine, which act through multiple receptors in a tissue and dose-dependent manner [50]. Collectively, these mechanisms explain the central role of RAAS in cardiac remodelling and fibrosis and provide the biological basis for therapeutic strategies aimed at restoring balance between the detrimental classical RAAS axis and its protective counter-regulatory pathways.
2.4. The Wnt/β-Catenin Signalling
Canonical Wnt signalling is initiated when Wnt ligands bind Frizzled family transmembrane receptors and the co-receptors LRP5/6, leading to dissociation of the β-catenin destruction complex. This process results in stabilization and cytoplasmic accumulation of β-catenin, followed by nuclear translocation and interaction with T-cell factor/lymphoid enhancer-binding factor (TCF/LEF) transcription factors and co-activators to regulate gene expression [51]. Wnt ligands may also activate non-canonical β-catenin-independent pathways, highlighting the complexity of Wnt signalling responses in the injured heart.
In the uninjured myocardium, Wnt signalling is tightly regulated at multiple levels by endogenous inhibitors and intracellular mediators. Members of the Dickkopf (Dkk) family inhibit Wnt signalling by preventing the formation of the Frizzled–LRP receptor complex, while soluble frizzled-related proteins (sFRPs) act extracellularly by binding Wnt ligands and limiting their availability for receptor activation, thereby reducing β-catenin–dependent transcription [52,53]. In addition to these extracellular regulators, Disheveled functions as a key intracellular signalling intermediate that integrates both canonical and non-canonical Wnt pathways [54].
Wnt signalling is reactivated following myocardial injury. In mouse models of myocardial infarction, myocardial injury has induced transcriptional upregulation of multiple Wnt ligands and antagonists, including Wnt2, Wnt4, Wnt10b, Wnt11, Dkk1, and Dkk2, within the injured myocardium [55]. Concomitantly, acute ischaemic cardiac injury has triggered organ-wide Wnt-dependent epicardial activation that has promoted epithelial to mesenchymal transition and expansion of cardiac fibroblast populations, driving profibrotic remodelling [56]. Consistent with these experimental findings, nuclear accumulation of β-catenin and its transcriptional partner, T cell factor (TCF-4), has been detected in cardiac fibroblasts of failed paediatric heart allografts [57] and conditional deletion of β-catenin in cardiac fibroblasts has attenuated interstitial fibrosis and preserved cardiac function [58].
Extensive crosstalk between Wnt/β-catenin and TGF-β signalling pathways coordinates cardiac fibroblast activation and fibrotic remodelling [59]. At the molecular level, β-catenin cooperates with Smad 2 downstream of TGF-β receptors to co-regulate the expression of α-SMA, thereby reinforcing the myofibroblast phenotype [60]. Disruption of either pathway attenuates fibroblast activation, underscoring the functional interdependence of Wnt and TGF-β signalling in driving persistent fibroblast activation and pathological cardiac fibrosis.
2.5. MAPK Signalling (ERK, p38, JNK)
Among the intracellular signalling cascades that govern fibroblast activation, MAPK pathways, including ERK, p38 MAPK, and JNK, play critical and functionally distinct roles [61]. ERK1/2 signalling is predominantly activated downstream of extracellular cues sensed by three major classes of membrane receptors: integrins, G-protein-coupled receptors (GPCRs), and receptor tyrosine kinases. Following cardiac injury, growth factors and neurohormones such as platelet-derived growth factor (PDGF), endothelin 1, and Ang II are released and activate receptor tyrosine kinases and GPCRs, leading to ERK activation. In parallel, integrin-mediated mechanotransduction provides an additional route for ERK1/2 activation [62]. Beyond the canonical Smad-dependent pathway, TGF-β further amplifies fibroblast activation through non-canonical signalling cascades, including ERK, JNK, and p38 MAPK pathways, thereby reinforcing profibrotic responses and positioning ERK as a central signalling hub [63]. Sustained or aberrant activation of ERK and JNK MAPK signalling promotes excessive cardiac fibroblast proliferation and matrix-remodelling, thereby driving pathological myocardial fibrosis and contributing to the progression of heart failure [64,65]. In cardiac fibroblasts, p38 MAPK, particularly the p38α isoform, plays a central role in the reactive oxygen species and cytokine-dependent induction of IL-6 and MMP-3, highlighting the involvement of MAPK pathways in inflammatory signalling [66,67].
JNK signalling represents a stress-activated MAPK pathway that contributes to cardiac fibroblast activation and broader pathological remodelling in the heart. Beyond canonical activation, post-translational modulation of JNK, including S-nitrosylation of c-Jun N-terminal kinase, has been implicated in pressure overload–induced cardiac dysfunction and fibrosis [68], highlighting the importance of context-specific regulation of this pathway. In human atrial fibroblasts, Ang II–induced differentiation has been mediated through p21-activated kinase 1–dependent activation of the JNK/c-Jun axis [69], further linking JNK signalling to fibroblast phenotypic conversion. Similarly, resistin has promoted cardiac fibroblast differentiation through coordinated activation of JAK/STAT3 and JNK/c-Jun pathways [70], and Wnt4-dependent cardiac repair has been shown to involve phospho-JNK/JNK signalling [71]. Collectively, these findings establish MAPK pathways as key signalling hubs that integrate mechanical stress, inflammatory mediators, and neurohormonal stimuli to sustain persistent fibroblast activation and pathological cardiac fibrosis.
TGF-Wntβ/-catenin and MAPK pathways intersect to drive cardiac fibroblast activation, differentiation into myofibroblasts, and collagen production, promoting fibrosis. TGF-triggers Smad-dependent profibrotic responses, while canonical Wntβ/-catenin signalling synergises with TGF-β to enhance IL-11 production, increasing ECM deposition. Additionally, non-coding microRNAs that regulate proteins in the above pathways are also targets for fibrosis. The important miRNAs miR-101a, miR-145, miR-675, miR-10a, miR-15, MiR-323a-3p, miR-202-3p, miR-433, miR-29b, miR-503 targeting TGF-β and Wnt pathways are reviewed in detail [72].
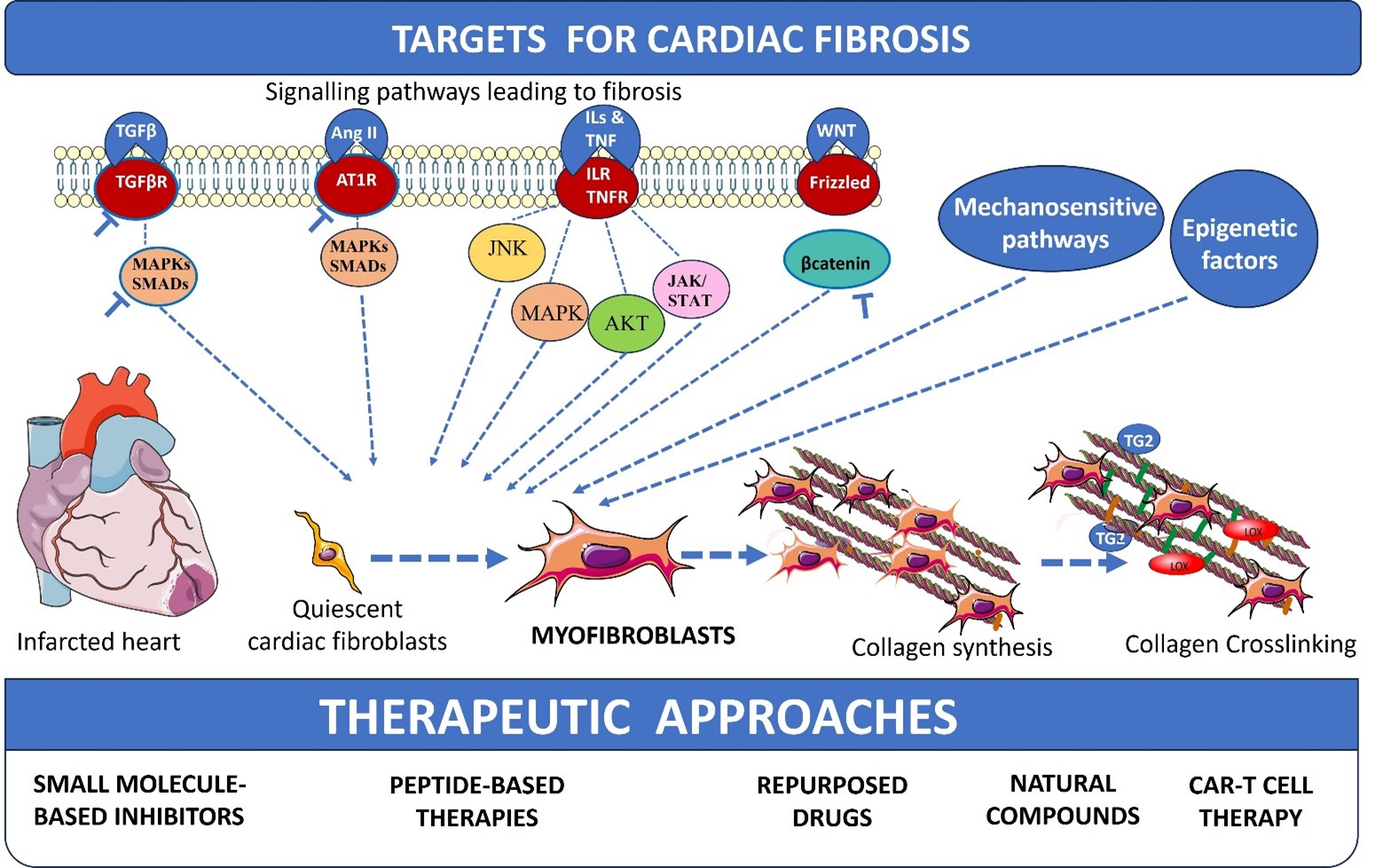
A schematic of pathways, targeting cardiac fibroblast activation, transdifferentiation to myofibroblasts, collagen synthesis, ECM remodelling, and crosslinking, is represented in Figure 2. Targeting general TGF-β, inflammatory, and Wnt pathways for cardiac fibrosis is challenging due to their pleiotropic nature, as they play essential roles in normal tissue homeostasis, immune regulation, and repair, leading to significant off-target effects and toxicity when blocked systematically. Targeting specific proteins or effectors downstream of these signalling pathways would be a more appropriate approach for cardiac fibrosis.
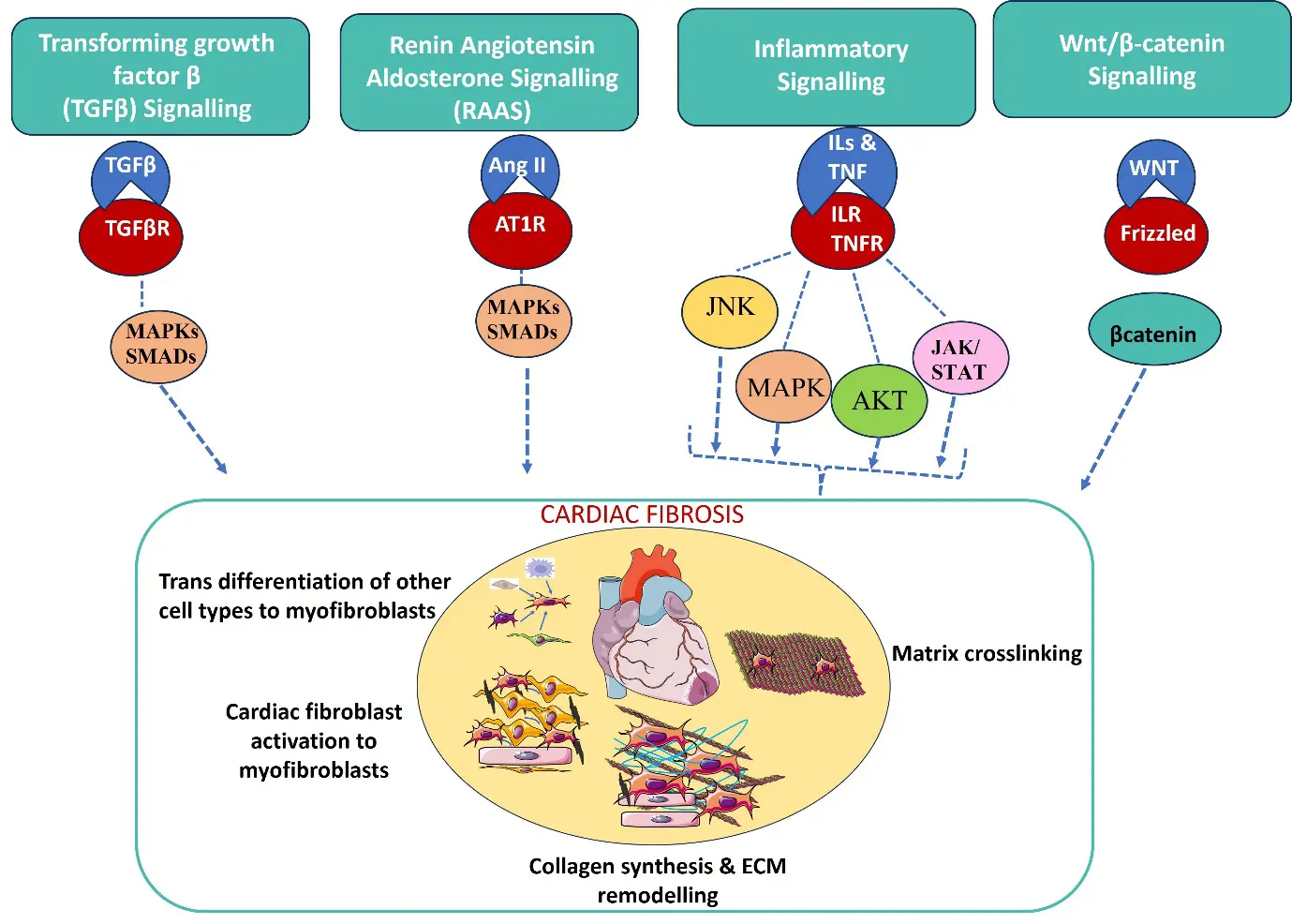
Figure 2. Schematic of signalling pathways involved in cardiac fibrosis. Profibrotic stimuli, receptors, and a few proteins in signalling pathways, and their regulators are
all
targets for cardiac fibrosis. The pathways regulate the transdifferentiation of cardiac fibroblasts and other cell types to myofibroblasts, matrix remodelling, collagen synthesis and crosslinking.
3. Proteins and Signalling Pathways That Regulate Collagen Crosslinking and Myocardial Stiffness, and Their Drug Targets
The relative abundance of collagen types I and III, and their extent of cross-linking, dictate the passive mechanical properties of the heart [73]. Collagen cross-linking in the cardiac ECM is a fundamental post-translational modification that dictates myocardial stiffness and structural integrity. The structural stability of the cardiac wall is maintained by a delicate equilibrium involving MMPs and tissue inhibitors of metalloproteinases (TIMPs). The process is primarily driven by the enzyme lysyl oxidase (LOX), secreted by cardiac fibroblasts. LOX catalyses the oxidative deamination of lysine and hydroxylysine residues in collagen. This process leads to the formation of aldehyde groups, which then crosslink adjacent collagen fibrils, thereby increasing tensile strength. We have investigated the role of periostin, a matricellular protein, in the regulation of LOX in activated cardiac fibroblasts in response to Ang II and TGFβ1. Our results indicate that periostin regulates LOX via ERK1/2 MAPK signalling and serum response factor (SRF) in myofibroblasts in vitro. The expression of periostin is also correlated with upregulated expression of collagen and LOX in the infarcted myocardium in a rat model of MI [64]. Transglutaminase 2 (TG2) functions as a calcium-dependent acyltransferase that catalyses the formation of stable crosslink ε(γ-glutamyl)lysine isopeptide bonds, providing an alternative enzymatic pathway for collagen cross-linking independent of the LOX-mediated oxidative deamination. TG2 is involved in early ECM remodelling, while LOX contributes to crosslinking at later stages. The degree of cross-linking determines the solubility, stiffness, and resistance to degradation of the resulting fibrils. Enhanced matrix stiffness in turn leads to myofibroblast activation, creating a loop for fibrosis. The imbalance in the equilibrium of collagen synthesis, MMPs, TIMP production, and upregulation of LOX, often observed in the progression of hypertensive heart disease and heart failure, results in a stiff, non-compliant ventricle. The different phases of cardiac ECM remodelling post myocardial infarction, the role of Lysyl oxidase and TG2 in collagen crosslinking, and the role of periostin in the regulation of Lysyl oxidase are schematically represented in Figure 3.
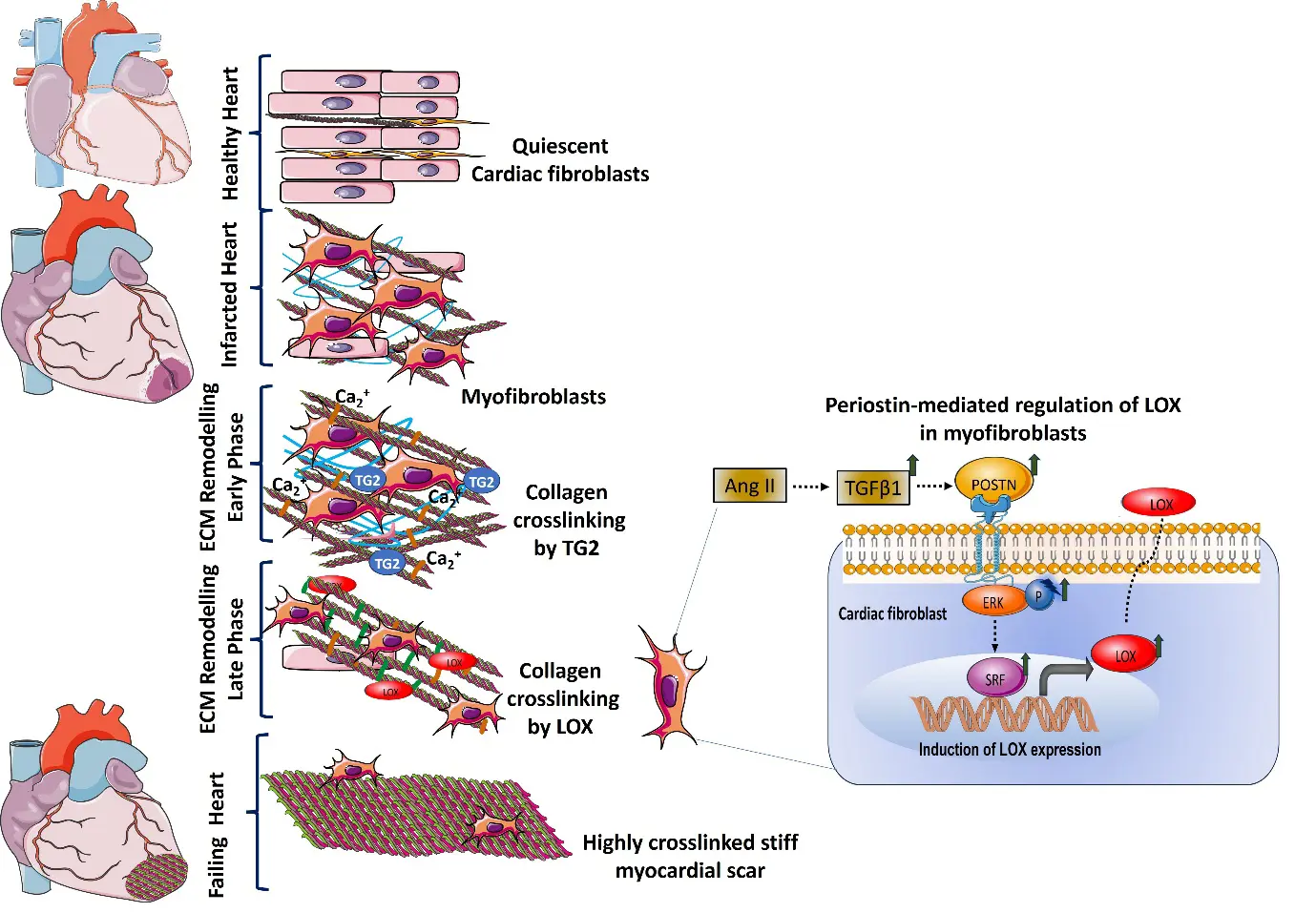
Figure 3. Schematic representation of different phases of ECM remodelling in an infarcted heart, the role of LOX and TG2 in collagen crosslinking, and the role of periostin in the regulation of LOX. Periostin regulates LOX via ERK signalling through the involvement of serum-responsive factor (SRF) in myofibroblasts in response to profibrotic stimuli such as Angiotensin II and TGFβ1.
While collagen synthesis can be targeted by regulating RAAS and other signalling pathways, collagen crosslinking requires targeting of specific proteins like LOX and transglutaminase. LOX was inhibited using β-aminopropionitrile (BAPN) in a volume overload model in rats, which attenuated collagens, MMPs, tissue inhibitor expression, and ventricular wall stress, partially attenuated ventricular hypertrophy, and decreased the decline in cardiac function. BAPN had a direct effect on collagen crosslinking; however, its effect on MMPs and TIMP could be secondary [74]. In an MI model, SNT-5382, a potent, selective lysyl oxidase-like 2 (LOXL2), exhibited reduced collagen volume fraction at the border zone of the infarct region. SNT-5382 reduced mature collagen crosslinks, while the total collagen content was unaffected. Further, a randomised, double-blind, placebo-controlled Phase 1 clinical trial (ACTRN12617001564347) conducted in healthy subjects showed that the LOXL2 inhibitor was very well tolerated, with no serious adverse events [75]. Since periostin regulates LOX, periostin is also a potent target for collagen crosslinking. A highly selective TG2 small-molecule inhibitor, 1–155, reduced both TG2 activity and its export into the ECM by blocking its cell-surface interaction with its binding partner, syndecan, thereby reducing secretion. The antifibrotic ability of this inhibitor was tested in two in vivo models, such as an interstitial cardiac fibrosis model of pressure overload using angiotensin II infusion and an acute myocardial infarction model induced by ligation of the left anterior descending coronary artery. In both models, there was a 50% significant reduction in infarct size, Sirius red staining for collagen deposition, and in levels of the TG2-mediated protein crosslink ε(γ-glutamyl)lysine [76]. In another study, the tissue transglutaminase (tTG) inhibitor ERW1041E, an irreversible pharmacological inhibitor of enzymatically active tTG, attenuated cardiomyocyte hypertrophy and interstitial fibrosis by reducing collagen protein levels and collagen crosslinking in a mouse model of pressure-overload; however, it did not affect ejection fraction. In vitro, the inhibitor did not affect collagen transcription but induced MMP3 and TIMP1 synthesis in cardiac fibroblasts through non-enzymatic action [77]. Complete inhibition of collagen crosslinking is lethal, as it may lead to cardiac wall rupture and other serious complications, such as aortic aneurysm. Hence, the timing of targeting collagen crosslinking is crucial for reducing myocardial stiffness while maintaining cardiac wall integrity.
Matricellular Proteins as Therapeutic Targets for Cardiac Fibrosis
Matricellular proteins are a class of non-structural ECM proteins that function as dynamic signalling adapters and regulate cell-matrix interactions by binding to cell-surface receptors, growth factors, and other structural proteins. They act as molecular “switches” that are rapidly upregulated following heart injury to orchestrate the transformation of quiescent fibroblasts into active, collagen-secreting myofibroblasts. They also play an important role in collagen chain alignment and crosslinking. Cellular communication network (CCN) family proteins, CCN1, CCN2, CCN5, and periostin are key matricellular proteins involved in cardiac fibrosis. Endogenous CCN1 was essential for scar formation and had a role in the arrangement of collagen fibrils in the maturing scar. CCN1 knockout animals exhibited reduced alignment and heightened tortuosity of collagen fibres, and reduced organizational coherency, packing, and size of collagen fibrils [78]. Cardiac remodelling and fibrosis are also observed in chronic kidney disorders (CKD); CCN1 expression was markedly enhanced in the serum and heart, accompanied by an increase in COL1, TGF-β, heavy-chain cardiac myosin, ANP, disordered myocardial arrangement, cardiac fibrosis, hypertrophy, and decreased cardiac systolic function in a mouse model of CKD. Knockdown of CCN1 reversed these effects, reduced MAPK signalling and fibrosis [79]. Connective tissue growth factor (CTGF, CCN2) is an autocrine regulator of cardiac fibrosis. TGF-β, Ang II, and ET-1 induce CCN2, which is a downstream mediator of these proteins. CCN2 was upregulated in cardiac fibroblasts in response to Ang II, and CCN2 deletion from activated fibroblasts inhibited the fibrotic remodelling [80]. CCN2 silencing inhibits the activation of the MAPK signalling pathway and alleviates myocardial fibrosis and left ventricular hypertrophy [81]. Both CCN1 and CCN2 deletions led to decreased fibrosis. CCN5 is a secreted matricellular protein that mediates anti-fibrotic activity by inhibiting cardiac fibroblast-to-myofibroblast transition and TGF-β1 signalling pathway. Serum CCN5 level decreased in hypertensive patients, but significantly increased in hypertensive patients on an ACE inhibitor, indicating a negative association between CCN5 and Ang II-induced hypertension, in heart failure patients [82]. A therapeutic target stimulating endogenous CCN5 production could reduce cardiac fibrosis.
Periostin is another matricellular protein that is not present in the healthy adult heart; it is expressed after myocardial infarction and plays a role in the progression of fibrosis. Studies from our lab indicate that, in vitro, periostin is upregulated in cardiac fibroblasts in response to Ang II and TGF-β1 treatment, and that periostin is expressed in the infarct scar post-myocardial injury. Further, we have shown that knockdown of periostin decreases collagen crosslinking enzyme LOX, and that periostin regulates LOX via ERK-MAPK, involving SRF [64]. Single-cell RNA sequencing revealed that Postn-positive fibroblasts played a significant role in the progression of myocardial fibrosis, and knocking down Postn decreased cardiac fibroblast activity, inhibited their migration, and induced apoptosis [83]. Targeted ablation of periostin-expressing cardiac fibroblasts in chronic AngII-exposed mice and in the MI model resulted in reduced fibrosis without compromising scar stability and cardiac function [84]. A recent study showed that periostin is a pivotal target of microRNA-150-5p in primary adult human cardiac fibroblast activation and chronic MI mouse model [85].
4. Mechanosensitive Pathways in Cardiac Fibroblasts and the Drug Targets
The stiffness of the heart is contributed to by the adult cardiomyocyte and the cardiac ECM. Cardiac fibroblasts function as sophisticated mechanosensors that respond to physical cues such as stretch, compression, and stiffness. At the cellular level, force is transmitted from the ECM or neighbouring cells to cardiac fibroblasts through specific transmembrane proteins, focal adhesion molecules, GPCRs, stretch-activated ion channels, and cytoskeletal proteins, which sense the mechanical signals (stretch or ECM stiffness) into specific signal transduction pathways. In cardiac fibroblasts (α1β1, α2β1, and α11β1), integrins that bind to collagen, and β3 integrins that bind to fibronectin, are the sensors for changes in extracellular matrix stiffness [86]. Integrins and focal adhesion complexes (paxillin and vinculin) transmit mechanical signals across the membrane to the cytoskeleton. Cytoskeletal actin polymerization phosphorylates Rho kinases and Lin-11, Isl-1, and Mec-3 kinases (LIM kinases), further promoting nuclear translocation of transcription factors, myocardin-related transcription factors (MRTF), a cofactor for transcription factor SRF, that binds the promoter region of α-SMA and SM22. Actin polymerisation during cytoskeletal dynamics modulates the Hippo signalling pathway, regulating nuclear translocation of the Yes-Associated Protein (YAP). Cyclic strain and ECM stiffness directly promote cardiac fibroblast activation and proliferation through YAP activation [86]. Studies have shown that YAP promotes myofibroblast differentiation and associated ECM gene expression through engagement of TEA domain transcription factor 1 and subsequent de novo expression of MRTF-A. Cardiac fibroblast YAP is a promising therapeutic target to prevent fibrotic remodelling and heart failure [87]. YAP inhibitor, Verteporfin, reduced profibrotic activation of cardiac stromal cells induced by mechanical cues, even in the presence of TGFβ. Further, verteporfin significantly reduced fibrosis and remodelling in an ischemic in vivo model; however, it did not improve cardiac functions [88]. Z11, a small-molecule inhibitor of nucleosome assembly protein 1-like 1 (NAP1L1), is found to prevent the interaction between NAP1L1 and YAP1, promote ubiquitination degradation of YAP1, and alleviate cardiac fibrosis by inhibiting the AKT/mTOR signalling, cardiac fibroblasts activation, and collagen deposition. Z11 decreased the expression of α-SMA, NAP1L1, and YAP1, collagen I, and collagen deposition in a mouse MI model [89].
Increased ECM Stretch can mechanically open the latent complex, activate TGF-β, and release active TGF-β from the ECM. DAMPs are released into the extracellular space during cellular stress, and ECM fragments of hyaluronic acid, and ECM-localized small leucine-rich proteoglycans (SLRPs) are released during tissue damage. This further activates TLRs and transcription factor nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB), which in turn leads to enhanced fibrosis. Mechanical stress can also activate TLRs. Syndecan-4 also has a role in mechanotransduction in the myocardium. Syndecan binds to GAG chains to extracellular molecules and to the cytoplasmic domain to the cytoskeleton. Syndecan-4 signalling via nuclear factor of activated T-cells (NFAT) regulates ECM production and cardiac myofibroblast differentiation in response to mechanical stress. The mechanotransduction pathways in cardiac fibroblasts are reviewed in detail by Herum et al. [90].
In cardiac fibroblasts, the intracellular transmission of mechanical cues can be elicited through mechanically responsive ion channels. During mechanosensing, deformation of the cell membrane induces changes in ion influx, particularly Ca2+ ions. Though not electrically excitable, cardiac fibroblasts are sensitive to membrane potential oscillations that happen following myocardial infarction and ECM remodelling. Transient receptor potential canonical type 6 (TRPC6), a nonselective receptor-operated cation channel, and Piezo 1 are the most studied mechanosensitive ion channels in cardiac fibroblasts. TRPC6 is sensitive to stretch, stress, and osmotic pressure and is highly permeable to Ca2+ during cardiac pathology, and is a mediator of fibrosis [91]. In vitro, cardiac fibroblasts showed upregulation of TRCP6 in response to TGF-β1 and Angiotensin II, via p38 MAPK activation and by serum response factor transcriptional regulation of the TRPC6 promoter. This, in turn, increased Ca2+ permeability in cultured cardiac fibroblasts, which induced myofibroblast transformation via calcineurin–NFAT signalling. Overexpression of TRCP6 induced spontaneous differentiation of fibroblasts to myofibroblasts, while knockdown attenuated myofibroblast differentiation induced by TGF-β1, confirming its role in myofibroblast activation [92]. Further, orally bioavailable TRPC6 antagonist BI 749327, selectively inhibited TRPC6 expression of profibrotic genes and interstitial fibrosis in mice subjected to sustained pressure overload and improved cardiac function [93]. Piezo 1 is a mechanosensitive cation channel that plays an important role in cardiac fibroblast activation and progression of fibrosis. Cardiac fibroblasts express mechanically activated Piezo1 channels, which stimulate paracrine IL-6 secretion via the p38 mitogen-activated protein kinase downstream of Ca2+ entry [94]. Emig et al., have shown that Piezo1-overexpression in human atrial fibroblasts induced cytoskeletal reorganisation by affecting the architecture of actin stress fibres. The study showed that in human atrial fibroblasts seeded on soft and stiff hydrogels, Piezo1 was required to adapt the cell stiffness to the substrate stiffness. Further, Piezo-induced cell stiffening is mediated by an autocrine mechanism via IL-6 secretion, and can transmit cell stiffening to neighbouring cells in a paracrine manner mediated by IL-6 [95], as depicted in Figure 4. Braidotti et al. reviewed Piezo1-induced autocrine, paracrine, and paratensile effectors in activated fibroblasts, highlighting the role of Piezo1 in mechanosensing in cardiac fibroblasts and suggesting Piezo1 as a potential target for cardiac fibrotic remodelling [96].
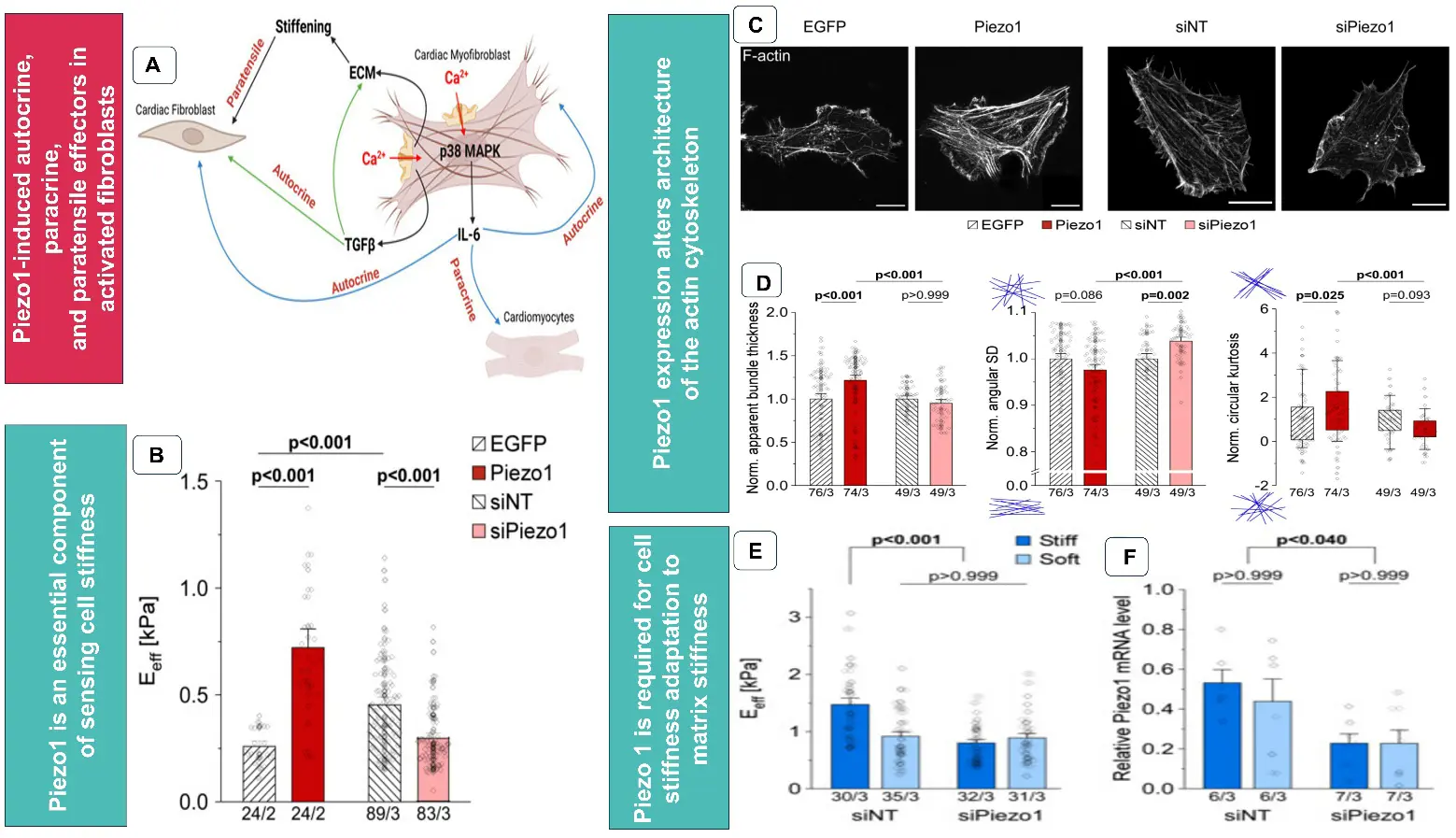
Figure 4. (A) Schematic representation of Piezo1-induced autocrine, paracrine, and paratensile effectors in activated fibroblasts. (Adapted from Braidotti et al., [96] an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license). (B) Stiffness of human atrial fibroblast line, 3 days after overexpression of EGFP, Piezo1, non-targeting siRNA (siNT), and siRNA mediated knock-down of Piezo1 (siPiezo1), (C) Phalloidin staining of actin cytoskeleton in human atrial fibroblasts 3 days post-transfection (D) apparent thickness, angular standard deviation (SD), and circular kurtosis of actin bundles, (E,F) average stiffness and mRNA levels of Piezo 1 of cells after 4 days of culture on stiff (~6 kPa) or soft (~3 kPa) hydrogels respectively. (B–F) Adapted from Emig et al., [95] an open-access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.
5. Targeting Epigenetic Factors
Epigenetic modifications are changes in DNA, 3D chromatin architecture, mRNA, and post-translational modifications of histone (acetylation, methylation, ubiquitination, and phosphorylation). The epigenetic modifiers (DNA modifiers, histone protein modifiers, and mRNA modifiers) are broadly classified as writers, readers, and erasers. Writers are enzymes that catalyse the addition of chemical groups to DNA or histone tails, leading to gene silencing or activation. DNA methyl transferases (DNMTs), histone acetyltransferases (HATs), RNA methylases, acetylases, and protein arginine methyltransferase (PRMTs) are examples of “Writer” enzymes. Readers are proteins that recognise, bind, and interpret the covalent modifications laid down by writers, leading to transcription or chromatin remodelling (e.g., Bromodomains). Erasers are enzymes that remove the chemical tags deposited by writers and modulate gene expression (e.g., histone deacetylases (HDACs) and demethylases). Histone acetylation increases gene expression while histone deacetylation suppresses gene transcription. Histone acetylation mainly occurs at the ε-amino groups of lysine residues in the N-terminal tails of histones H3 and H4 and is associated with transcriptional activation by promoting chromatin relaxation. HMTs or histone methyl transferases induce methylation, which, depending on the type of methylation, is associated with both gene repression and active transcription of genes. Perez et al. recently reviewed the epigenetic modifications in cardiac fibrosis in detail and how chromatin-modifying enzymes orchestrate the transcriptional changes in cardiac fibroblasts [97].
A study in a hypertensive model showed that H3K9 acetylation in myocardial tissue correlated with increased α-SMA expression. Inhibition of HAT p300 using the small-molecule inhibitor L002 reduced myofibroblast differentiation and fibrosis in a mouse model of hypertension [98]. Curcumin, a natural compound, inhibited p300 and reduced fibrosis, BNP expression, perivascular fibrosis, and downregulated acetylation levels of GATA4 and pro-hypertrophic gene expression in a chronic heart failure Dahl salt-sensitive rat model [99]. However, a clinical trial (UMIN000014232) showed curcumin intake in patients with initial signs of hypertensive heart disease with left ventricular ejection fraction ≥60%, decreased plasma BNP levels without altering ventricular diastolic function [100]. DNMTS also has a role in regulating α-SMA activation. DNMT3a promotes CFs activation and fibrosis through the ERK1/2 pathway [101]. DNMT3a inhibition using low-intensity pulsed ultrasound (LIPUS) prevents hypoxia-induced cardiac fibroblast activation and cardiac fibrosis [102].
Recent studies indicate that epigenetic modifications affecting RNA are also involved in cardiac fibroblast activation and profibrotic gene expression. N6-methyladenosine (m6A) is the most common modification observed in mRNA. Methyltransferase-like protein 3 (METTL3) is the primary catalytic “writer” enzyme that makes m6A modifications on mRNA. METTL3 overexpression upregulated cardiac fibroblast activation to myofibroblast and collagen deposition, and its downregulation attenuated these changes. Transcriptome and m6A profiling analyses revealed that the expression and m6A levels of collagen-related genes were altered [103]. A recent study showed that inhibition of METTL3-METTL14 using controlled delivery of its inhibitor S-adenosyl-l-homocysteine from microneedles reduced mitochondrial fragmentation, differentiation to myofibroblasts, and expression of collagen I and collagen III [104]. FTO (Fat mass and obesity-associated) protein is an m6A-demethylase of mRNA. Ang II is found to suppress FTO expression, and leonurine, a natural compound with a cardioprotective role, reverses these effects [105]. Overexpression of protein arginine methyltransferase 5 (PRMT5) induces cardiac fibroblast differentiation to myofibroblasts, and its inhibition reverses the effects [106]. Further, PRMT5 inhibition downregulates Col1A1 and Acta2, and its interaction with Smad3. EPZ015666, a selective inhibitor of PRMT5, improved fibrosis and left ventricular dysfunction in an in vivo mouse model [107].
Bromodomain-containing protein 4 (BRD4), an epigenetic reader, has been identified as a central regulator of the pro-fibrotic cardiac fibroblast phenotype induced in response to TGF-β stimulation. Chromatin targeting of BRD4 is controlled via a p38-dependent signalling circuit for epigenetic reprogramming [108]. JQ1, a specific BET (bromodomain and extraterminal) bromodomain inhibitor, decreased cardiac fibrosis in a pressure-overload or MI model. JQ1, blocked the expression of periostin, a marker of myofibroblasts [109]. HDACs induce chromatin condensation and suppress gene transcription. HDAC8 has a role in cardiac fibrosis. HDAC8 interacts with signal transducer and activator of transcription 3 (STAT3), facilitating its binding to the MMP12 promoter. An HDAC inhibitor, YAK577, inhibited HDAC8, thereby reducing Col1A1 and fibronectin gene expression and MMP12 mRNA levels in an isoproterenol-induced heart failure model [110]. A small-molecule activator of SIRT3 (a class3 HDAC), (2-APQC), alleviates isoproterenol-induced cardiac hypertrophy and myocardial fibrosis in vitro and in vivo rat models. It inhibited the activation of AKT, mTOR, (mTOR)-p70 ribosomal protein S6 kinase (p70S6K), c-jun N-terminal kinase (JNK), TGF-β, and Smad3 pathways to improve isoproterenol-induced cardiac hypertrophy and myocardial fibrosis [111]. Although several in vitro and preclinical studies in animals have identified epigenetic enzymes as potential therapeutic targets for cardiac fibrosis, clinical studies are limited.
6. Small Molecule Inhibitors and Peptide-Based Therapies for Cardiac Fibrosis
Small-molecule inhibitors are low-molecular-weight organic compounds for pharmacological approaches targeting various signalling pathways. Unlike larger biologics, these low-molecular-weight compounds easily penetrate cell membranes to interact with specific intracellular targets. Their increased bioavailability, reduced immune response, and ability to precisely target proteins and pathways make them a good candidate to develop as a drug. Several small-molecule inhibitors have been explored in targeting collagen crosslinking, mechanosensing pathways, and epigenetic factors regulating cardiac fibrosis. Some of the key small-molecule inhibitors targeting signalling pathways discussed in Sections 3–5 are summarised in Table 1. While these molecules show promising results in preclinical trials, the toxicity of a few inhibitors has to be carefully addressed. Another disadvantage of small molecules in treating cardiac fibrosis is that while they specifically target one pathway, they do not target other pathways that promote fibrosis.
Peptide-based therapies represent an emerging class of precision medicine for cardiac fibrosis, utilising short chains of amino acids to modulate cellular signalling with higher specificity than small molecules. They disrupt specific protein-protein interactions and exhibit more specificity than small-molecule inhibitors. Glucagon-like peptide-1 receptor (GLP-1R) agonists, a class of glucose-lowering drugs, exhibit cardioprotective effects. Liraglutide, a glucagon-like peptide-1 receptor agonist, decreased interstitial cardiac fibrosis and expression of inflammatory and oxidative stress markers in Ang II-induced hypertension and age-induced cardiac fibrosis mice model [112]. Results from STEP-HFpEF (NCT04788511) and STEP-HFpEF-DM (NCT04916470) trials indicate that Semaglutide, a GLP-1R agonist, targets the underlying inflammatory processes and contributes to better overall cardiovascular outcomes in patients with HFpEF [113]. Obesity-driven systemic inflammation can cause coronary microvascular dysfunction, leading to myocardial fibrosis, stiffness, and diastolic dysfunction. A meta-analysis also indicates that GLPRAs exhibit protective effects in obese patients with chronic heart failure [114]. Natriuretic peptides, atrial natriuretic peptide (ANP), brain natriuretic peptide (BNP), and C-type natriuretic peptide (CNP), cardiac-derived hormones, regulate the renin–angiotensin–aldosterone–vasopressin and sympathetic nervous systems and exhibit pleiotropic effects on myocardial and vascular remodelling, and immune responses. ANP and BNP are synthesised by myocytes in response to myocardial stretch, volume overload, and during heart failure, while CNP is predominantly produced by endothelial cells in response to shear stress and inflammation. They exhibit cardioprotective effects by inhibiting RAAS and have anti-fibrotic agents in the heart by increasing cyclic guanosine monophosphate (cGMP) in cardiac fibroblasts, which inhibit collagen synthesis. The decrease in the level of CNP, secreted by endothelial cells, is positively correlated to increased cardiac fibrosis in mice [115]. Another study showed that Angiotensin-receptor-neprilysin inhibitors increase the bioavailability of ANP. This decreased myocardial fibrosis by increasing protein kinase G; by inhibiting SMAD signalling in cultured cardiac fibroblasts and cardiac fibrosis in Type 2 diabetic rats [116]. Cenderitide is a designer CNP, fused to the C-terminus of Dendroaspis natriuretic peptide (DNP). Cenderitide was engineered to co-activate the two NP receptors, particulate guanylyl cyclase (pGC)-A and pGC-B, to enhance the anti-fibrotic properties of dual-receptor activation. Cenderitide significantly attenuated the development of cardiac fibrosis and preserved diastolic function by enhancing pGC-A-mediated suppression of the potent pro-fibrotic factor aldosterone and by inhibiting fibroblast proliferation and collagen synthesis linked to pGC-B activation [117,118]. Relaxin, a heterodimeric peptide and insulin-like hormone, was found to decrease cardiac fibroblast to myofibroblast transition by increasing Wnt ligands and Wnt signalling, and decreasing fibrosis, in a rat model of ageing [119]. Based on these findings, recombinant human relaxin or longer-acting analogues of relaxin [120] are currently being evaluated in various clinical trials as treatments for HFpEF [121]. Another study reported that the recombinant human relaxin reduced chronic NLRP3 inflammasome activation which promotes fibrosis through IL-1β. Relaxin reduced myofibroblast NLRP3 inflammasome through the involvement of relaxin family peptide receptor-1(RXFP1), angiotensin II type 2 receptor (AT2R), ATP receptor-P2X7R, and by the inhibition of TLR-4, ROS, and caspase-1 [122]. A recent update on a clinical trial (NCT05592275) using volenrelaxin showed improvement in left atrial function at a low dose; however, the treatment with this long-acting form of human relaxin was associated with worsening congestion in patients with recently decompensated HFpEF [123].
Humanin, an endogenous mitochondria-derived peptide, has been used to reverse cardiac fibrosis and apoptosis in the ageing heart in a mouse model. Humanin reduced oxidative stress, cardiac fibroblast proliferation, decreased TGF-β1, collagen deposition, and apoptosis of cells [124]. Cathelicidin-related antimicrobial peptide (CRAMP) is another peptide that reduced hyperglycemia-induced endothelial-to-mesenchymal transition (EndMT) in mouse heart endothelial cells and reduced fibrosis in diabetic mice by attenuating TGF-β/Smad signalling and AMP-activated protein kinase (AMPKa1) and mechanistic Target of Rapamycin (mTOR) signalling [125]. Peptides that exhibited therapeutic benefits for cardiac fibrosis are summarized in Table 1.
Table 1. (A) Small molecule inhibitors targeting collagen crosslinking, mechanosensing pathway proteins, and epigenetic modifiers of cardiac fibrosis. Their mechanism of action and effects on cardiac fibroblasts and cardiac fibrosis are detailed based on in vitro and preclinical in vivo models of fibrosis. (B) Peptides targeting signalling pathways that activate cardiac fibroblasts and fibrosis.
|
Sl No. |
Inhibitors That Target Cardiac Fibrosis |
Target |
Mechanism |
In Vitro/In Vivo Effects on Cardiac Fibrosis |
Refs. |
|---|---|---|---|---|---|
|
(A). Small molecule inhibitors |
|||||
|
1 |
1–155 |
transglutaminase 2 (TG2) |
Inhibits TG2 activity and the export of TG2 to the ECM by blocking cell surface interaction with its binding partner, syndecan, and reducing its secretion, thereby decreasing collagen crosslinking by TG2 |
Significant reduction in infarct size, collagen deposition and in the levels of the TG2-mediated protein crosslinks in the interstitial fibrosis model and in the MI model |
[76] |
|
2 |
ERW1041E |
tissue transglutaminase (tTG) |
Inhibits enzymatically active tTG and reduces collagen crosslinking |
In vitro, the inhibitor did not affect collagen transcription but induced MMP3 and TIMP1 synthesis in cardiac fibroblasts through non-enzymatic action. It attenuated interstitial fibrosis by reducing collagen protein levels and collagen crosslinking in a pressure-overloaded model of mice; however, it did not affect ejection fraction. |
[77] |
|
3 |
β-aminopropionitrile (BAPN) |
lysyl oxidase (LOX) |
BAPN inhibits collagen crosslinking and has secondary effects by inhibiting MMPs and TIMP |
In a volume overload model in rats, BAPN attenuated collagen, MMPs, TIMP, and ventricular wall stress. It partially attenuated ventricular hypertrophy and decreased the decline in cardiac function. |
[74] |
|
4 |
SNT-5382 |
lysyl oxidase-like 2 |
Reduce mature collagen crosslinks |
Reduced collagen volume fraction at the border zone of the infarct region in an MI model. |
[75] |
|
5 |
Verteporfin |
Yes-associated protein 1 (YAP) |
Decrease profibrotic activation of cardiac stromal cells induced by mechanical cues |
Significantly reduced fibrosis and remodelling in an ischemic in vivo model; however, it did not improve cardiac functions. |
[88] |
|
6 |
Z1149421873 |
nucleosome assembly protein 1-like 1 (NAP1L1) |
Prevents the interaction between NAP1L1 and YAP1, promotes ubiquitination and degradation of YAP1 and inhibits the AKT/mTOR signalling |
Inhibited cardiac fibroblast activation and collagen deposition. Decreased the expression of α-SMA, NAP1L1, and YAP1, collagen I and collagen deposition in a mouse MI model. |
[89] |
|
7 |
BI 749327 |
transient receptor potential canonical 6 (TRPC6) antagonist |
Prevents Ca2+ overload, has 85- and 42-fold selectivity over related channels, TRPC3 and TRPC7 and prevent activation of nuclear factor of activated T cells (NFAT) |
Inhibited TRPC6 expression of profibrotic genes and interstitial fibrosis in mice subjected to sustained pressure overload, and improved cardiac function. |
[93] |
|
8 |
L002 and C646 |
histone acetyltransferase p300 (HAT p300) |
Binds to the acetyl-CoA binding pocket of the p300 catalytic domain (specifically targeting the Factor Acetyltransferase or FAT domain) |
Reduced myofibroblast differentiation and fibrosis in a mouse model of hypertension. |
[98] |
|
9 |
S-Adenosyl-l-homocysteine (SAH) |
methyltransferase-like 3 (METTL3)-methyltransferase-like 14 (METTL14) |
Inhibits m6A methylation levels |
Reduced cardiac fibroblast proliferation, α-SMA expression, differentiation to myofibroblasts, and expression of FGF2, Fn1, Col3a1, and Col6a3, as well as myofibroblast migration. There was a lower degree of myocardial fibrosis in SAH-treated rats compared to MI rats |
[104] |
|
10 |
EPZ015666 |
protein arginine methyltransferase 5 (PRMT5) |
PRMT5 forms a protein complex with transcription factors, and its binding to promoter sites |
Suppressed α-SMA expression induced by TGF-β in cultured cardiac fibroblasts. Improved fibrosis and left ventricular dysfunction in an in vivo mouse model of pressure overload. |
[107] |
|
11 |
JQ1 |
bromodomain-containing protein 4 |
Inhibits transactivation of a common pathologic gene regulatory program for NFκB and TGF-β signalling networks regulating inflammatory and profibrotic proteins |
Blocked the expression of periostin, a marker of myofibroblasts, decreased cardiac fibrosis in a pressure-overload or MI model. |
[109] |
|
12 |
YAK577 |
histone deacetylase 8 (HDAC8) |
Reduce HDAC8 mRNA levels |
Reduced Col1A1 and fibronectin gene expression and MMP12 mRNA levels in an isoproterenol-induced heart failure model. |
[110] |
|
(B). Peptides therapies for cardiac fibrosis |
|||||
|
13 |
Liraglutide (Glucagon-like peptide-1 receptor agonist) |
glucagon-like peptide-1 receptor |
Attenuates NFκB activity and translocation |
Decreased interstitial cardiac fibrosis and expression of inflammatory and oxidative stress markers in Ang II-induced hypertension and age-induced cardiac fibrosis mouse model. |
[112] |
|
14 |
Cenderitide |
natriuretic peptide receptors |
Enhance particulate guanylyl cyclase(pGC-A)-mediated suppression of aldosterone and pGC-B-mediated collagen synthesis |
Attenuated the development of cardiac fibrosis and preserved diastolic function. |
|
|
15 |
Relaxin |
relaxin family peptide receptor-1(RXFP1), angiotensin II type 2 receptor ATP receptor (P2X7R), Wnt signalling |
Increase Wnt ligands and Wnt signalling, reduced myofibroblast NLRP3 inflammasome through RXFP1, P2X7R |
Decrease cardiac fibroblast to myofibroblast transition, decreased fibrosis in a rat model of ageing, inhibit ROS and caspase-1. |
[122] |
|
16 |
Humanin, an endogenous mitochondria-derived peptide |
Akt and glycogen synthase kinase 3β |
Attenuates fibrosis by activating the Akt/glycogen synthase kinase 3β pathway |
Reduced oxidative stress, cardiac fibroblast proliferation, TGF-β1, collagen deposition and apoptosis of cells, reversed cardiac fibrosis and apoptosis in the ageing heart in a mouse model. |
[124] |
|
17 |
CRAMP |
TGFβ/Smad, AMPKa1 and mTOR |
Attenuates TGF-β/Smad signalling, AMPKa1 and mTOR signalling |
Reduced hyperglycemia-induced endothelial-to-mesenchymal transition and reduced fibrosis in diabetic mice. |
[125] |
7. Repurposing of Drugs for Cardiac Fibrosis
Many promising anti-fibrotic candidates that were successful in preclinical trials fail in human trials due to their toxicity, off-target effects, or other reasons. By leveraging existing pharmacokinetic and safety profiles, drug repurposing significantly truncates the bench-to-bedside timeline, offering an accelerated pathway to address the profound unmet need of treating cardiac fibrosis in heart failure. Hence, several FDA-approved drugs developed for liver, lung, and pulmonary fibrosis are also being researched for their effectiveness in cardiac fibrosis. Metformin, an FDA-approved anti-diabetic drug derived from Galega officinalis, has been researched for cardioprotective effects. In vitro studies have shown that metformin decreased the proliferation and migration of cardiac fibroblasts treated under high-glucose conditions. Metformin decreased p-Stat3 expression and upregulated mitochondrial complex I protein Grim-19 and Sirt1 expression to inhibit proliferation and migration [126]. In vitro and in vivo studies in animals have shown that metformin, via activation of the AMPK pathway, inhibits mitochondrial complex I, increases AMP concentration, and modulates energy metabolism and cardiac contraction. Metformin also inhibited proinflammatory cytokines, TGF-β, reduced cardiac fibrosis, and improved ventricular remodelling. Additionally, metformin modulated eNOS-mediated vascular relaxation [127]. Another study in diabetic db/db mice showed that a combination of exercise and metformin inhibits the TGF-β1/Smad pathway, attenuating myocardial fibrosis. Metformin reduced NF-κB–mediated inflammatory response to enhance the inhibition of the positive feedback loop in the TGF-β1/Smad pathway, which in turn attenuated myofibroblast activation and ECM deposition in the diabetic mice model [128]. Based on the promising cardiovascular outcomes in preclinical studies, a Met-HeFT trial (NCT03514108) (Metformin in Patients with Chronic Heart Failure), a Danish, randomised, double-blind, placebo-controlled study (DANHEART), is examining the efficacy and safety of metformin in patients with heart failure (HFrEF) and diabetes, insulin resistance, or obesity [129]. A meta-analysis showed that metformin improved cardiovascular outcomes, including reduced all-cause mortality, lower rates of myocardial infarctions, and improved heart failure outcomes [130].
Mesalazine (5-aminosalicylic acid) is structurally similar to aspirin and is also repurposed for cardiac fibrosis. It is used to treat inflammatory bowel disease and ulcerative colitis. Mesalazine reduced proliferation, myofibroblast differentiation, and collagen deposition in the TGF-β-treated human cardiac fibroblast line. Further, it decreased cell stiffness, stress relaxation, and cell adhesion. The molecular mechanisms of antifibrotic effects of mesalazine have been identified as dual inhibition of SMAD2/3 and ERK1/2phosphorylation [131]. Pirfenidone, an FDA-approved antifibrotic medicine for idiopathic pulmonary fibrosis, is repurposed for cardiac fibrosis. In the mouse model of pressure overload, pirfenidone reduced fibrosis via repressing TGF-β1/Smad3 signalling. In the PIROUETTE trial, a randomised phase 2 clinical trial (NCT02932566), treatment with pirfenidone over 52 weeks in patients with heart failure with preserved ejection fraction led to a reduction in myocardial extracellular volume, a marker for cardiac fibrosis [132]. However, larger trials are required to prove the clinical benefits. Nintedanib is an FDA-approved tyrosine kinase inhibitor used for idiopathic pulmonary fibrosis and chronic fibrotic interstitial lung diseases. In vitro, it reduced the expression of profibrotic genes in cardiac fibroblasts and modulated hypertrophy in cardiomyocytes [133]. Nintedanib-treated mice subjected to transverse aortic constriction did not show cardiac functional decline at 4 weeks. Further, it decreased the expression of collagen Type 1A and collagen Type 3A genes at 8 weeks. Additionally, it reduced myocardial and systemic inflammation by inhibiting pro-inflammatory subsets and promoting regulatory T cells.
8. Natural Compounds
Resveratrol, a non-flavonoid polyphenol isolated from grapes, giant knotweed, peanuts, and other plants, is the most widely explored plant compound that has anti-fibrotic effects. Studies indicate it acts by targeting multiple proteins and signalling pathways. Several in vitro studies in cardiac fibroblasts and in vivo studies in small animal models have shown that resveratrol has improved myocardial fibrosis and pulmonary fibrosis induced by isoproterenol, streptozotocin-induced diabetes, doxorubicin, and viral myocarditis [134]. Following these reports, a clinical trial (ChiCTR1800016122) investigated whether supplementation of resveratrol improved remodelling in patients with hypertension. The group treated with resveratrol had lower biomarkers indicating cardiac fibrosis, evidenced by decreased procollagen type I C-peptide and galectin-3 at 6 months. The study suggested that resveratrol could be used as an adjunct to conventional therapies for hypertensive heart disease [135].
Several products derived from natural compounds used in traditional Chinese medicine have been investigated for their antifibrotic effects. Evodiamine, a quinolone alkaloid isolated from Evodia rutaecarpa, a traditional Chinese medicine, attenuated TGF-β1-induced activation of primary rat cardiac fibroblasts into myofibroblasts and activation of Smad2, Smad3, ERK1/2, and Akt, and the nuclear translocation of Smad4. The drug also prevented endothelial to mesenchymal transition in vitro, highlighting its ability to target other cell types that contribute to fibrosis [136]. A cardiac glycoside bufalin derived from Chinese toad venom (ChanSu or Bufo gargarizans) with known anti-cancer and anti-inflammatory properties, is reported to inhibit the proliferation of primary human cardiac fibroblasts, decrease collagen type I, α1 protein levels in vitro, with significant reductions in interstitial fibrosis, perivascular fibrosis, and diastolic dysfunction in an Ang II infusion model in mice. The mechanism of action of this glycoside was shown by regulating miRNA miR-671-5p, which targets selenoprotein P1 and modulates myocardial fibrosis and stiffness [137]. Qifu yixin prescription (QYP), a traditional Chinese medicine formula used in the treatment of heart failure, was tested for its antifibrotic effects in vitro and in vivo. QYP effectively suppresses cardiac fibroblast activation by stimulating soluble guanylate cyclase activation and inhibition of ERK phosphorylation. In a mouse model of heart failure by thoracic aortic constriction, QYP reduced interstitial and perivascular fibrosis [138]. Salvianolic acid B, an active component of the Chinese drug Salvia miltiorrhiza, is used to treat angina pectoris, hyperlipidemia, and coronary heart disease is reported to decrease myocardial fibrosis in diabetic cardiomyopathy by decreasing collagen and regulating TGF-β1/Smad7 expression. In vitro the drug inhibited cardiac fibroblast proliferation, migration, phenotypic transformation, and collagen secretion, in a high glucose-induced diabetic fibrosis model [139]. Tongxinluo, a traditional Chinese drug made from 12 distinct herbs, is found to inhibit myocardial fibrosis by inhibiting endothelial-mesenchymal transition through activating the NRG-1/ErbB- PI3K/AKT signalling cascade, in a rat reperfusion injury model [140]. The oral administration of ethanolic extract of Boerhavia diffusa, a leafy vegetable, which has quercetin, boeravinone B, kaempferol, and caffeic acid as bioactive components, decreased myocardial oxidative stress, increased endogenous antioxidant enzymes, by regulating the metabolism of glutathione, and decreased myocardial collagen content in an Ang II-induced MI rats [141]. Forskolin, derived from the roots of Coleus forskohlii, decreased the expression of fibronectin, collagen I, TGF-β, and α-SMA, reduced myocardial fibrosis, and improved cardiac diastolic function through its antioxidative stress ability [142]. Plumbagin, derived from Plumbagin zeylanica, partially inhibited TGF-β1-induced myocardial fibroblast fibrosis by promoting autophagy activation and by suppressing the AKT/mTOR pathway both in vitro and in vivo in an animal MI model [143].
These in vitro and in vivo studies in small animals clearly indicate that many of these phyto compounds exhibit cardioprotective effects, including antifibrotic effects by regulating multiple pathways, molecular mechanisms, and multiple cell types of the heart, such as fibroblasts, immune cells, endothelial cells, and by autophagy. However, more in-depth investigations, toxicity analysis, and efficacy studies in large animals are required before exploration in clinical trials.
9. Chimeric Antigen Receptor (CAR)-T Cell Therapy
Chimeric antigen receptor (CAR)-T cell therapy is used to target activated cardiac fibroblasts and treat cardiac fibrosis. CAR-T cells were designed to recognize and kill cells expressing fibroblast activation protein (FAP) alpha. This approach led to a significant reduction in cardiac fibrosis and restored cardiac function after injury in mice [144]. The clinical use of CAR T cells, based on viral-based gene therapy, is limited by safety concerns, cytokine release syndrome, cardiotoxicity, and sustained transgene expression. Since FAP is required for wound healing to prevent cardiac wall rupture, sustained transgene expression of FAP in CAR-T cells might interfere with normal wound healing [145]. Later studies demonstrated that injection of lipid nanoparticles (LNPs) encoding mRNA for CAR against FAP produced functional, transient antifibrotic CAR-T cells in vivo, which reduced fibrosis and restored cardiac function after injury [146]. A recent study used a CD3-targeted LNP system enabling efficient mRNA delivery into T cells for in vivo CAR-T generation. This study has shown that FAP-CAR-T cells selectively target FAP-positive cells with high FAP expression without harming normal cardiomyocytes. In vivo-generated FAP-CAR-T cells significantly alleviated myocardial fibrosis and effectively improved cardiac function [147].
Another study explored the use of FAP-targeted CAR-T cells and tested their efficacy in autoimmune (EAM) and viral (CVB3) myocarditis models. Human cardiac organoids treated with interleukin-17A (IL-17A) were used to model inflammatory fibrosis. Cardiac organoids treated with FAP-CAR-T cells showed 55% less fibrosis. In EAM and CVB3 models, FAP-CAR-T cells reduced fibrosis by 65% and 55%, respectively, and restored ejection fraction [148]. In yet another study, a reactive oxygen species (ROS)-responsive hydrogel was designed for intrapericardial delivery of FAP-specific CAR-T cells to treat post-myocardial infarction fibrosis. The hydrogel selectively degrades in the ROS-enriched microenvironment, enabling the controlled release of encapsulated FAP-CAR-T cells. In murine models of MI-induced fibrosis, the hydrogel-based delivery enhanced FAP-CAR-T cell infiltration, persistence, and effector function, resulting in significant depletion of activated fibroblasts, attenuating fibrotic remodelling, and preserving cardiac structure and left ventricular function [149].
10. Conclusions
Targeting cardiac fibrosis is a critical clinical necessity, an unmet medical need to prevent progression into heart failure. However, cardiac fibrosis is a complex molecular mechanism with multi-layered crosstalk between various cell types and signalling pathways. This makes it nearly impossible for a single-target drug to prevent the progression of tissue scarring. Current research is focused on identifying specific molecular “brakes” that can be applied at three distinct stages of the scarring process: myofibroblast transformation, collagen synthesis, and collagen crosslinking. The window for targeting cardiac fibrosis is very crucial and depends on the progression of fibrosis. Too early intervention following myocardial infarction may result in cardiac wall rupture, and too late intervention often results in the formation of a mature crosslinked ECM scar that is difficult to degrade. The small-molecule inhibitors of specific protein targets and signalling pathways have shown better selectivity, minimise off-target toxicity, and improve efficiency in reversing fibrosis. Small-molecule inhibitors of epigenetic regulators can restore normal, quiescent fibroblast function rather than permanently deleting a gene or protein, providing a safer, tunable therapeutic approach. Epigenetic inhibitors act as master switches that simultaneously suppress a wide network of profibrotic and proinflammatory genes. However, the challenge with epigenetic modifiers as drug targets is the lack of selectivity for specific genes, leading to unintended activation or silencing of genes throughout the genome. Safety concerns about the long-term consequences of using these drugs, which fundamentally alter gene regulation, also need to be addressed. Drug repurposing for cardiac fibrosis is a more favourable strategy, which accelerates clinical translation by bypassing early-stage safety trials, reducing clinical trial costs, and utilising medications with already-proven pharmacological profiles. The use of advanced platforms enables high-throughput screening for toxicity, with high-content imaging and functional analysis, allowing for the simultaneous measurement of multiple, highly relevant phenotypes. Beyond pharmacological and repurposed approaches, the FAP-targeted CAR-T cell therapy offers a strategy for selectively identifying and eliminating activated myofibroblasts and reversing cardiac fibrosis.
Statement of the Use of Generative AI and AI-Assisted Technologies in the Writing Process
During the preparation of this manuscript, the author(s) used Grammarly to improve grammar correction and clarity of writing. After using this tool/service, the authors reviewed and edited the content as needed and take full responsibility for the content of the published article.
Acknowledgments
N.M. acknowledges the general research support provided to the laboratory by the Indian Council of Medical Research, Government of India.
Author Contributions
N.M. and S.R. performed the design concept, bibliographic search and manuscript drafting. N.M. prepared the figures and revised the draft critically for its intellectual content. Both authors have read and agreed to the final version of the manuscript.
Ethics Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
Funding
No specific grant supported the preparation of this manuscript.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
-
De Boer RA, De Keulenaer G, Bauersachs J, Brutsaert D, Cleland JG, Diez J, et al. Towards Better Definition, Quantification and Treatment of Fibrosis in Heart Failure. A Scientific Roadmap by the Committee of Translational Research of the Heart Failure Association (HFA) of the European Society of Cardiology. Eur. J. Heart Fail. 2019, 21, 272–285. DOI:10.1002/ejhf.1406 [Google Scholar]
-
Akhurst RJ, Hata A. Targeting the TGFβ signalling pathway in disease. Nat. Rev. Drug Discov. 2012, 11, 790–811. DOI:10.1038/nrd3810 [Google Scholar]
-
Mann DL. Innate immunity and the failing heart: The cytokine hypothesis revisited. Circ. Res. 2015, 116, 1254–1268. DOI:10.1161/CIRCRESAHA.116.302317 [Google Scholar]
-
Frangogiannis NG. The inflammatory response in myocardial injury, repair and remodeling. Nat. Rev. Cardiol. 2014, 11, 255–265. DOI:10.1038/nrcardio.2014.28 [Google Scholar]
-
Turner NA, Das A, Warburton P, O’Regan DJ, Ball SG, Porter KE. Interleukin-1α stimulates proinflammatory cytokine expression in human cardiac myofibroblasts. Am. J. Physiol.-Heart Circ. Physiol. 2009, 297, H1117–H1127. DOI:10.1152/ajpheart.00372.2009 [Google Scholar]
-
Bujak M, Frangogiannis NG. The role of Interleukin-1 in the pathogenesis of heart disease. Arch. Immunol. Et Ther. Exp. 2009, 57, 165–176. DOI:10.1007/s00005-009-0024-y [Google Scholar]
-
Wynn TA, Vannella KM. Macrophages in tissue repair, regeneration, and fibrosis. Immunity 2016, 44, 450–462. DOI:10.1016/j.immuni.2016.02.015 [Google Scholar]
-
Haudek SB, Cheng J, Du J, Wang Y, Hermosillo-Rodriguez J, Trial J, et al. Monocytic fibroblast precursors mediate fibrosis in angiotensin-II-induced cardiac hypertrophy. J. Mol. Cell Cardiol. 2010, 49, 499–507. DOI:10.1016/j.yjmcc.2010.05.005 [Google Scholar]
-
Jiang H, Yang J, Li T, Wang X, Fan Z, Ye Q, et al. JAK/STAT3 signaling in cardiac fibrosis: A promising therapeutic target. Front. Pharmacol. 2024, 15, 1336102. DOI:10.3389/fphar.2024.1336102 [Google Scholar]
-
Mir SA, Chatterjee A, Mitra A, Pathak K, Mahata SK, Sarkar S. Inhibition of Signal Transducer and Activator of Transcription 3 (STAT3) Attenuates Interleukin-6 (IL-6)-induced Collagen Synthesis and Resultant Hypertrophy in Rat Heart. J. Biol. Chem. 2012, 287, 2666–2677. DOI:10.1074/jbc.M111.246173 [Google Scholar]
-
Li Q, Ye WX, Huang ZJ, Zhang Q, He YF. Effect of IL-6-mediated STAT3 signaling pathway on myocardial apoptosis in mice with dilated cardiomyopathy. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 3042–3050. DOI:10.26355/eurrev_201904_17586 [Google Scholar]
-
Braunwald E. Biomarkers in Heart Failure. N. Engl. J. Med. 2008, 358, 2148–2159. DOI:10.1056/NEJMra0800239 [Google Scholar]
-
Rolski F, Błyszczuk P. Complexity of TNF-α Signaling in Heart Disease. J. Clin. Med. 2020, 9, 3267. DOI:10.3390/jcm9103267 [Google Scholar]
-
Hu H, Huang J, Zhang S, Zhang B, Li W, Sun K. Tumor necrosis factor-α stimulation endothelial-to-mesenchymal transition during cardiac fibrosis via endothelin-1 signaling. J. Biochem. Mol. Toxicol. 2023, 37, e23411. DOI:10.1002/jbt.23411 [Google Scholar]
-
Voloshenyuk TG, Hart AD, Khoutorova E, Gardner JD. TNF-α increases cardiac fibroblast lysyl oxidase expression through TGF-β and PI3Kinase signaling pathways. Biochem. Biophys. Res. Commun. 2011, 413, 370–375. DOI:10.1016/j.bbrc.2011.08.109 [Google Scholar]
-
Miao K, Zhou L, Ba H, Li C, Gu H, Yin B, et al. Transmembrane tumor necrosis factor alpha attenuates pressure-overload cardiac hypertrophy via tumor necrosis factor receptor 2. PLoS Biol. 2020, 18, e3000967. DOI:10.1371/journal.pbio.3000967 [Google Scholar]
-
Rolski F, Tkacz K, Węglarczyk K, Kwiatkowski G, Pelczar P, Jaźwa-Kusior A, et al. TNF-α protects from exacerbated myocarditis and cardiac death by suppressing expansion of activated heart-reactive CD4+ T cells. Cardiovasc. Res. 2023, 120, 82–94. DOI:10.1093/cvr/cvad158 [Google Scholar]
-
Ohm IK, Alfsnes K, Belland Olsen M, Ranheim T, Sandanger Ø, Dahl TB, et al. Toll-like receptor 9 mediated responses in cardiac fibroblasts. PLoS ONE 2014, 9, e104398. DOI:10.1371/journal.pone.0104398 [Google Scholar]
-
Vijayakumar G, Latha A, Anil AP, Surve Y, Nair BG, Pillai IC. Cell autonomous TLR4 signaling modulates TGF-β induced activation of human cardiac fibroblasts. Heliyon 2025, 11, e42452. DOI:10.1016/j.heliyon.2025.e42452 [Google Scholar]
-
Kawaguchi M, Takahashi M, Hata T, Kashima Y, Usui F, Morimoto H, et al. Inflammasome activation of cardiac fibroblasts is essential for myocardial ischemia/reperfusion injury. Circulation 2011, 123, 594–604. DOI:10.1161/CIRCULATIONAHA.110.982777 [Google Scholar]
-
Travers JG, Kamal FA, Robbins J, Yutzey KE, Blaxall BC. Cardiac Fibrosis: The Fibroblast Awakens. Circ. Res. 2016, 118, 1021–1040. DOI:10.1161/CIRCRESAHA.115.306565 [Google Scholar]
-
Massagué J. TGFβ signalling in context. Nat. Rev. Mol. Cell Biol. 2012, 13, 616–630. DOI:10.1038/nrm3434 [Google Scholar]
-
Bujak M, Frangogiannis N. The role of TGF-β signaling in myocardial infarction and cardiac remodeling. Cardiovasc. Res. 2007, 74, 184–195. DOI:10.1016/j.cardiores.2006.10.002 [Google Scholar]
-
Frangogiannis NG. Transforming growth factor–β in tissue fibrosis. J. Exp. Med. 2020, 217, e20190103. DOI:10.1084/jem.20190103 [Google Scholar]
-
Wermuth PJ, Carney KR, Mendoza FA, Piera-Velazquez S, Jimenez SA. Endothelial cell-specific activation of transforming growth factor-β signaling in mice induces cutaneous, visceral, and microvascular fibrosis. Lab. Investig. 2017, 97, 806–818. DOI:10.1038/labinvest.2017.23 [Google Scholar]
-
Koitabashi N, Danner T, Zaiman AL, Pinto YM, Rowell J, Mankowski J, et al. Pivotal role of cardiomyocyte TGF-β signaling in the murine pathological response to sustained pressure overload. J. Clin. Investig. 2011, 121, 2301–2312. DOI:10.1172/JCI44824 [Google Scholar]
-
Kanisicak O, Khalil H, Ivey MJ, Karch J, Maliken BD, Correll RN, et al. Genetic lineage tracing defines myofibroblast origin and function in the injured heart. Nat. Commun. 2016, 7, 12260. DOI:10.1038/ncomms12260 [Google Scholar]
-
Huang S, Chen B, Humeres C, Alex L, Hanna A, Frangogiannis NG. The role of Smad2 and Smad3 in regulating homeostatic functions of fibroblasts in vitro and in adult mice. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118703. DOI:10.1016/j.bbamcr.2020.118703 [Google Scholar]
-
Dobaczewski M, Bujak M, Li N, Gonzalez-Quesada C, Mendoza LH, Wang XF, et al. Smad3 signaling critically regulates fibroblast phenotype and function in healing myocardial infarction. Circ. Res. 2010, 107, 418–428. DOI:10.1161/CIRCRESAHA.109.216101 [Google Scholar]
-
Chen PY, Qin L, Simons M. TGFβ signaling pathways in human health and disease. Front. Mol. Biosci. 2023, 10, 1113061. DOI:10.3389/fmolb.2023.1113061 [Google Scholar]
-
Zhang YE. Non-Smad pathways in TGF-β signaling. Cell Res. 2009, 19, 128–139. DOI:10.1038/cr.2008.328 [Google Scholar]
-
Bale S, Verma P, Yalavarthi B, Scarneo SA, Hughes P, Amin MA, et al. Pharmacological inhibition of TAK1 prevents and induces regression of experimental organ fibrosis. JCI Insight 2023, 8, e165358. DOI:10.1172/jci.insight.165358 [Google Scholar]
-
Meyer-ter-Vehn T, Gebhardt S, Sebald W, Buttmann M, Grehn F, Schlunck G, et al. p38 Inhibitors Prevent TGF-β–Induced Myofibroblast Transdifferentiation in Human Tenon Fibroblasts. Investig. Ophthalmol. Vis. Sci. 2006, 47, 1500–1509. DOI:10.1167/iovs.05-0361 [Google Scholar]
-
Bujak M, Ren G, Kweon HJ, Dobaczewski M, Reddy A, Taffet G, et al. Essential Role of Smad3 in Infarct Healing and in the Pathogenesis of Cardiac Remodeling. Circulation 2007, 116, 2127–2138. DOI:10.1161/CIRCULATIONAHA.107.704197 [Google Scholar]
-
Khalil H, Kanisicak O, Prasad V, Correll RN, Fu X, Schips T, et al. Fibroblast-specific TGF-β–Smad2/3 signaling underlies cardiac fibrosis. J. Clin. Investig. 2017, 127, 3770–3783. DOI:10.1172/JCI94753 [Google Scholar]
-
Russell-Hallinan A, Cappa O, Kerrigan L, Tonry C, Edgar K, Glezeva N, et al. Single-Cell RNA Sequencing Reveals Cardiac Fibroblast-Specific Transcriptomic Changes in Dilated Cardiomyopathy. Cells 2024, 13, 752. DOI:10.3390/cells13090752 [Google Scholar]
-
Li N, Hang W, Shu H, Zhou N. Pirfenidone alleviates cardiac fibrosis induced by pressure overload via inhibiting TGF-β1/Smad3 signalling pathway. J. Cell Mol. Med. 2022, 26, 4548–4555. DOI:10.1111/jcmm.17478 [Google Scholar]
-
Yamagami K, Oka T, Wang Q, Ishizu T, Lee JK, Miwa K, et al. Pirfenidone exhibits cardioprotective effects by regulating myocardial fibrosis and vascular permeability in pressure-overloaded hearts. Am. J. Physiol.-Heart Circ. Physiol. 2015, 309, H512–H522. DOI:10.1152/ajpheart.00137.2015 [Google Scholar]
-
Aimo A, Iborra-Egea O, Martini N, Galvez-Monton C, Burchielli S, Panichella G, et al. Cardiac protection by pirfenidone after myocardial infarction: A bioinformatic analysis. Sci. Rep. 2022, 12, 4691. DOI:10.1038/s41598-022-08523-3 [Google Scholar]
-
Siragy HM. Special Issue “The Angiotensin in Human. Health and Diseases”. Int. J. Mol. Sci. 2025, 26, 11940. DOI:10.3390/ijms262411940 [Google Scholar]
-
Suzuki Y, Ruiz-Ortega M, Egido J. Angiotensin II: A double-edged sword in inflammation. J. Nephrol. 2000, 13 (Suppl. S3), S101-10. Available online: https://europepmc.org/article/med/11132026 (accessed on 1 January 2026).
-
Simko F, Hrenak J, Adamcova M, Paulis L. Renin–Angiotensin–Aldosterone System: Friend or Foe—The Matter of Balance. Insight on History, Therapeutic Implications and COVID-19 Interactions. Int. J. Mol. Sci. 2021, 22, 3217. DOI:10.3390/ijms22063217 [Google Scholar]
-
Frangogiannis NG. Cardiac fibrosis. Cardiovasc. Res. 2021, 117, 1450–1488. DOI:10.1093/cvr/cvaa324 [Google Scholar]
-
de Boer RA, Pokharel S, Flesch M, van Kampen DA, Suurmeijer AJH, Boomsma F, et al. Extracellular signal regulated kinase and SMAD signaling both mediate the angiotensin II driven progression towards overt heart failure in homozygous TGR(mRen2)27. J. Mol. Med. 2004, 82, 678–687. DOI:10.1007/s00109-004-0579-3 [Google Scholar]
-
Bhullar SK, Dhalla NS. Angiotensin II-Induced Signal Transduction Mechanisms for Cardiac Hypertrophy. Cells 2022, 11, 3336. DOI:10.3390/cells11213336 [Google Scholar]
-
Verma K, Pant M, Paliwal S, Dwivedi J, Sharma S. An Insight on Multicentric Signaling of Angiotensin II in Cardiovascular system: A Recent Update. Front. Pharmacol. 2021, 12, 734917. DOI:10.3389/fphar.2021.734917 [Google Scholar]
-
Santos RAS, Sampaio WO, Alzamora AC, Motta-Santos D, Alenina N, Bader M, et al. The ACE2/Angiotensin-(1–7)/MAS Axis of the Renin-Angiotensin System: Focus on Angiotensin-(1–7). Physiol. Rev. 2018, 98, 505–553. DOI:10.1152/physrev.00023.2016 [Google Scholar]
-
Kassiri Z, Zhong J, Guo D, Basu R, Wang X, Liu PP, et al. Loss of Angiotensin-Converting Enzyme 2 Accelerates Maladaptive Left Ventricular Remodeling in Response to Myocardial Infarction. Circ. Heart Fail. 2009, 2, 446–455. DOI:10.1161/CIRCHEARTFAILURE.108.840124 [Google Scholar]
-
Ferreira AJ, Castro CH, Guatimosim S, Almeida PWM, Gomes ERM, Dias-Peixoto MF, et al. Attenuation of isoproterenol-induced cardiac fibrosis in transgenic rats harboring an angiotensin-(1-7)-producing fusion protein in the heart. Ther. Adv. Cardiovasc. Dis. 2010, 4, 83–96. DOI:10.1177/1753944709353426 [Google Scholar]
-
Valentini A, Heilmann RM, Kühne A, Biagini L, De Bellis D, Rossi G. The Renin–Angiotensin–Aldosterone System (RAAS): Beyond Cardiovascular Regulation. Vet. Sci. 2025, 12, 777. DOI:10.3390/vetsci12080777 [Google Scholar]
-
MacDonald BT, He X. Frizzled and LRP5/6 Receptors for Wnt/β-Catenin Signaling. Cold Spring Harb. Perspect. Biol. 2012, 4, a007880. DOI:10.1101/cshperspect.a007880 [Google Scholar]
-
Clevers H, Nusse R. Wnt/β-Catenin Signaling and Disease. Cell 2012, 149, 1192–1205. DOI:10.1016/j.cell.2012.05.012 [Google Scholar]
-
Liu LB, Chen XD, Zhou XY, Zhu Q. The Wnt antagonist and secreted frizzled-related protein 5: Implications on lipid metabolism, inflammation, and type 2 diabetes mellitus. Biosci. Rep. 2018, 38, BSR20180011. DOI:10.1042/BSR20180011 [Google Scholar]
-
Sharma M, Castro-Piedras I, Simmons GE, Pruitt K. Dishevelled: A masterful conductor of complex Wnt signals. Cell Signal. 2018, 47, 52–64. DOI:10.1016/j.cellsig.2018.03.004 [Google Scholar]
-
Aisagbonhi O, Rai M, Ryzhov S, Atria N, Feoktistov I, Hatzopoulos AK. Experimental myocardial infarction triggers canonical Wnt signaling and endothelial-to-mesenchymal transition. Dis. Models Mech. 2011, 4, 469–483. DOI:10.1242/dmm.006510 [Google Scholar]
-
Duan J, Gherghe C, Liu D, Hamlett E, Srikantha L, Rodgers L, et al. Wnt1/βcatenin injury response activates the epicardium and cardiac fibroblasts to promote cardiac repair. EMBO J. 2012, 31, 429–442. DOI:10.1038/emboj.2011.418 [Google Scholar]
-
Ye B, Ge Y, Perens G, Hong L, Xu H, Fishbein MC, et al. Canonical Wnt/β-catenin signaling in epicardial fibrosis of failed pediatric heart allografts with diastolic dysfunction. Cardiovasc. Pathol. 2013, 22, 54–57. DOI:10.1016/j.carpath.2012.03.004 [Google Scholar]
-
Xiang FL, Fang M, Yutzey KE. Loss of β-catenin in resident cardiac fibroblasts attenuates fibrosis induced by pressure overload in mice. Nat. Commun. 2017, 8, 712. DOI:10.1038/s41467-017-00840-w [Google Scholar]
-
Piersma B, Bank RA, Boersema M. Signaling in Fibrosis: TGF-β, WNT, and YAP/TAZ Converge. Front. Med. 2015, 2, 59. DOI:10.3389/fmed.2015.00059 [Google Scholar]
-
Carthy JM, Garmaroudi FS, Luo Z, McManus BM. Wnt3a Induces Myofibroblast Differentiation by Upregulating TGF-β Signaling Through SMAD2 in a β-Catenin-Dependent Manner. PLoS ONE 2011, 6, e19809. DOI:10.1371/journal.pone.0019809 [Google Scholar]
-
Rose BA, Force T, Wang Y. Mitogen-activated protein kinase signaling in the heart: Angels versus demons in a heart-breaking tale. Physiol. Rev. 2010, 90, 1507–1546. DOI:10.1152/physrev.00054.2009 [Google Scholar]
-
Gilbert CJ, Longenecker JZ, Accornero F. ERK1/2: An Integrator of Signals That Alters Cardiac Homeostasis and Growth. Biology 2021, 10, 346. DOI:10.3390/biology10040346 [Google Scholar]
-
Dobaczewski M, Chen W, Frangogiannis NG. Transforming growth factor (TGF)-β signaling in cardiac remodeling. J. Mol. Cell Cardiol. 2011, 51, 600–606. DOI:10.1016/j.yjmcc.2010.10.033 [Google Scholar]
-
Radhakrishnan S, Shenoy SJ, Devidasan I, Shaji BV, Gopal S, Sreekumaran S, et al. Periostin regulates lysyl oxidase through ERK1/2 MAPK-dependent serum response factor in activated cardiac fibroblasts. Cell Biochem. Funct. 2024, 42, e4066. DOI:10.1002/cbf.4066 [Google Scholar]
-
Zhang Z, Yang Z, Wang S, Wang X, Mao J. Targeting MAPK-ERK/JNK pathway: A potential intervention mechanism of myocardial fibrosis in heart failure. Biomed. Pharmacother. 2024, 173, 116413. DOI:10.1016/j.biopha.2024.116413 [Google Scholar]
-
Sinfield JK, Das A, O’Regan DJ, Ball SG, Porter KE, Turner NA. p38 MAPK alpha mediates cytokine-induced IL-6 and MMP-3 expression in human cardiac fibroblasts. Biochem. Biophys. Res. Commun. 2013, 430, 419–424. DOI:10.1016/j.bbrc.2012.11.071 [Google Scholar]
-
Sano M, Fukuda K, Sato T, Kawaguchi H, Suematsu M, Matsuda S, et al. ERK and p38 MAPK, but not NF-kappaB, are critically involved in reactive oxygen species-mediated induction of IL-6 by angiotensin II in cardiac fibroblasts. Circ. Res. 2001, 89, 661–669. DOI:10.1161/hh2001.098873 [Google Scholar]
-
Zhou M, Chen JY, Chao ML, Zhang C, Shi ZG, Zhou XC, et al. S-nitrosylation of c-Jun N-terminal kinase mediates pressure overload-induced cardiac dysfunction and fibrosis. Acta Pharmacol. Sin. 2022, 43, 602–612. DOI:10.1038/s41401-021-00674-9 [Google Scholar]
-
Zhou Y, Xie Y, Li T, Zhang P, Chen T, Fan Z, et al. P21-activated kinase 1 mediates angiotensin II-induced differentiation of human atrial fibroblasts via the JNK/c-Jun pathway. Mol. Med. Rep. 2021, 23, 207. DOI:10.3892/mmr.2021.11846 [Google Scholar]
-
Singh R, Kaundal RK, Zhao B, Bouchareb R, Lebeche D. Resistin induces cardiac fibroblast-myofibroblast differentiation through JAK/STAT3 and JNK/c-Jun signaling. Pharmacol. Res. 2021, 167, 105414. DOI:10.1016/j.phrs.2020.105414 [Google Scholar]
-
Dong W, Zhao Y, Wen D, Lin Y, Zeng C, Gu J, et al. Wnt4 is crucial for cardiac repair by regulating mesenchymal-endothelial transition via the phospho-JNK/JNK. Theranostics 2022, 12, 4110–4126. DOI:10.7150/thno.71392 [Google Scholar]
-
Yousefi F, Shabaninejad Z, Vakili S, Derakhshan M, Movahedpour A, Dabiri H, et al. TGF-β and WNT signaling pathways in cardiac fibrosis: Non-coding RNAs come into focus. Cell Commun. Signal. 2020, 18, 87. DOI:10.1186/s12964-020-00555-4 [Google Scholar]
-
Chaturvedi RR, Herron T, Simmons R, Shore D, Kumar P, Sethia B, et al. Passive Stiffness of Myocardium from Congenital Heart Disease and Implications for Diastole. Circulation 2010, 121, 979–988. DOI:10.1161/CIRCULATIONAHA.109.850677 [Google Scholar]
-
El Hajj EC, El Hajj MC, Ninh VK, Gardner JD. Inhibitor of lysyl oxidase improves cardiac function and the collagen/MMP profile in response to volume overload. Am. J. Physiol.-Heart Circ. Physiol. 2018, 315, H463–H473. DOI:10.1152/ajpheart.00086.2018 [Google Scholar]
-
Perryman L, Findlay A, Baskar J, Charlton B, Foot J, Hamilton R, et al. The small molecule LOXL2 inhibitor SNT-5382 reduces cardiac fibrosis and achieves strong clinical target engagement. Sci. Rep. 2025, 15, 22653. DOI:10.1038/s41598-025-06312-2 [Google Scholar]
-
Wang Z, Stuckey DJ, Murdoch CE, Camelliti P, Lip GYH, Griffin M. Cardiac fibrosis can be attenuated by blocking the activity of transglutaminase 2 using a selective small-molecule inhibitor. Cell Death Dis. 2018, 9, 613. DOI:10.1038/s41419-018-0573-2 [Google Scholar]
-
Shinde AV, Su Y, Palanski BA, Fujikura K, Garcia MJ, Frangogiannis NG. Pharmacologic inhibition of the enzymatic effects of tissue transglutaminase reduces cardiac fibrosis and attenuates cardiomyocyte hypertrophy following pressure overload. J. Mol. Cell Cardiol. 2018, 117, 36–48. DOI:10.1016/j.yjmcc.2018.02.016 [Google Scholar]
-
Fischer AG, Elliott EM, Brittian KR, Garrett L, Sadri G, Aebersold J, et al. Matricellular protein CCN1 promotes collagen alignment and scar integrity after myocardial infarction. Matrix Biol. J. Int. Soc. Matrix Biol. 2024, 133, 14–32. DOI:10.1016/j.matbio.2024.08.001 [Google Scholar]
-
Zhao Y, Gu L, Chen Y, Lin Y, Xing J, Xu D, et al. Cysteine-Rich Protein 61 (CCN1) Deficiency Alleviated Cardiac Remodeling in 5/6 Nephrectomized Mice by Suppressing the MAPK Signaling Pathway. Cardiovasc. Ther. 2025, 2025, 6813183. DOI:10.1155/cdr/6813183 [Google Scholar]
-
Dorn LE, Petrosino JM, Wright P, Accornero F. CTGF/CCN2 is an autocrine regulator of cardiac fibrosis. J. Mol. Cell Cardiol. 2018, 121, 205–211. DOI:10.1016/j.yjmcc.2018.07.130 [Google Scholar]
-
Hong L, Lai H, Fang Y, Tao Y, Qiu Y. Silencing CTGF/CCN2 inactivates the MAPK signaling pathway to alleviate myocardial fibrosis and left ventricular hypertrophy in rats with dilated cardiomyopathy. J. Cell Biochem. 2018, 119, 9519–9531. DOI:10.1002/jcb.27268 [Google Scholar]
-
Huang A, Li H, Zeng C, Chen W, Wei L, Liu Y, et al. Endogenous CCN5 Participates in Angiotensin II/TGF-β1 Networking of Cardiac Fibrosis in High Angiotensin II-Induced Hypertensive Heart Failure. Front. Pharmacol. 2020, 11, 1235. DOI:10.3389/fphar.2020.01235 [Google Scholar]
-
Nie W, Zhao Z, Xiahou Z, Zhang J, Liu Y, Wang Y, et al. Single-cell RNA sequencing reveals the potential role of Postn(+) fibroblasts in promoting the progression of myocardial fibrosis after myocardial infarction. Sci. Rep. 2025, 15, 22390. DOI:10.1038/s41598-025-04990-6 [Google Scholar]
-
Kaur H, Takefuji M, Ngai CY, Carvalho J, Bayer J, Wietelmann A, et al. Targeted Ablation of Periostin-Expressing Activated Fibroblasts Prevents Adverse Cardiac Remodeling in Mice. Circ. Res. 2016, 118, 1906–1917. DOI:10.1161/CIRCRESAHA.116.308643 [Google Scholar]
-
Hayasaka T, Moukette B, Sepúlveda MN, Kawaguchi S, Aonuma T, Moustapha H, et al. Periostin is a Pivotal Target of microRNA-150-5p in Cardiac Fibroblast Activation and Chronic Myocardial Infarction. JACC Basic. Transl. Sci. 2025, 10, 101330. DOI:10.1016/j.jacbts.2025.101330 [Google Scholar]
-
Pesce M, Duda GN, Forte G, Girao H, Raya A, Roca-Cusachs P, et al. Cardiac fibroblasts and mechanosensation in heart development, health and disease. Nat. Rev. Cardiol. 2023, 20, 309–324. DOI:10.1038/s41569-022-00799-2 [Google Scholar]
-
Francisco J, Zhang Y, Jeong JI, Mizushima W, Ikeda S, Ivessa A, et al. Blockade of Fibroblast YAP Attenuates Cardiac Fibrosis and Dysfunction Through MRTF-A Inhibition. JACC Basic. Transl. Sci. 2020, 5, 931–945. DOI:10.1016/j.jacbts.2020.07.009 [Google Scholar]
-
Garoffolo G, Casaburo M, Amadeo F, Salvi M, Bernava G, Piacentini L, et al. Reduction of Cardiac Fibrosis by Interference With YAP-Dependent Transactivation. Circ. Res. 2022, 131, 239–257. DOI:10.1161/CIRCRESAHA.121.319373 [Google Scholar]
-
Yu T, Wang H, Gong J, Ma J, Kong C, He Y, et al. Z11, a small-molecule inhibitor of NAP1L1, alleviates cardiac fibrosis by regulating AKT/mTOR pathway. Eur. J. Pharmacol. 2026, 1011, 178440. DOI:10.1016/j.ejphar.2025.178440 [Google Scholar]
-
Herum K, Lunde I, McCulloch A, Christensen G. The Soft- and Hard-Heartedness of Cardiac Fibroblasts: Mechanotransduction Signaling Pathways in Fibrosis of the Heart. J. Clin. Med. 2017, 6, 53. DOI:10.3390/jcm6050053 [Google Scholar]
-
Yamaguchi Y, Iribe G, Nishida M, Naruse K. Role of TRPC3 and TRPC6 channels in the myocardial response to stretch: Linking physiology and pathophysiology. Prog. Biophys. Mol. Biol. 2017, 130, 264–272. DOI:10.1016/j.pbiomolbio.2017.06.010 [Google Scholar]
-
Davis J, Burr AR, Davis GF, Birnbaumer L, Molkentin JD. A TRPC6-Dependent Pathway for Myofibroblast Transdifferentiation and Wound Healing In Vivo. Dev. Cell. 2012, 23, 705–715. DOI:10.1016/j.devcel.2012.08.017 [Google Scholar]
-
Lin BL, Matera D, Doerner JF, Zheng N, Del Camino D, Mishra S, et al. In vivo selective inhibition of TRPC6 by antagonist BI 749327 ameliorates fibrosis and dysfunction in cardiac and renal disease. Proc. Natl. Acad. Sci. USA 2019, 116, 10156–10161. DOI:10.1073/pnas.1815354116 [Google Scholar]
-
Blythe NM, Muraki K, Ludlow MJ, Stylianidis V, Gilbert HTJ, Evans EL, et al. Mechanically activated Piezo1 channels of cardiac fibroblasts stimulate p38 mitogen-activated protein kinase activity and interleukin-6 secretion. J. Biol. Chem. 2019, 294, 17395–17408. DOI:10.1074/jbc.RA119.009167 [Google Scholar]
-
Emig R, Knodt W, Krussig MJ, Zgierski-Johnston CM, Gorka O, Groß O, et al. Piezo1 Channels Contribute to the Regulation of Human Atrial Fibroblast Mechanical Properties and Matrix Stiffness Sensing. Cells 2021, 10, 663. DOI:10.3390/cells10030663 [Google Scholar]
-
Braidotti N, Chen SN, Long CS, Cojoc D, Sbaizero O. Piezo1 Channel as a Potential Target for Hindering Cardiac Fibrotic Remodeling. Int. J. Mol. Sci. 2022, 23, 8065. DOI:10.3390/ijms23158065 [Google Scholar]
-
Pérez M, Gómez M, Castellar-López J, Araos P, Mendoza-Torres E, Bolívar S. Epigenetic modifications in cardiac fibrosis: Recent evidence of new pharmacological targets. Front. Mol. Biosci. 2025, 12, 1583446. DOI:10.3389/fmolb.2025.1583446 [Google Scholar]
-
Rai R, Sun T, Ramirez V, Lux E, Eren M, Vaughan DE, et al. Acetyltransferase p300 inhibitor reverses hypertension-induced cardiac fibrosis. J. Cell. Mol. Med. 2019, 23, 3026–3031. DOI:10.1111/jcmm.14162 [Google Scholar]
-
Sunagawa Y, Funamoto M, Shimizu K, Shimizu S, Sari N, Katanasaka Y, et al. Curcumin, an Inhibitor of p300-HAT Activity, Suppresses the Development of Hypertension-Induced Left Ventricular Hypertrophy with Preserved Ejection Fraction in Dahl Rats. Nutrients 2021, 13, 2608. DOI:10.3390/nu13082608 [Google Scholar]
-
Funamoto M, Sunagawa Y, Katanasaka Y, Kato T, Funada J, Ajiro Y, et al. Effects of high-absorption curcumin for the prevention of hypertensive heart disease: A double-blind, placebo-controlled, randomized clinical study. Eur. Heart J. Open 2022, 2, oeac057. DOI:10.1093/ehjopen/oeac057 [Google Scholar]
-
Tao H, Yang JJ, Chen ZW, Xu SS, Zhou X, Zhan HY, et al. DNMT3A silencing RASSF1A promotes cardiac fibrosis through upregulation of ERK1/2. Toxicology 2014, 323, 42–50. DOI:10.1016/j.tox.2014.06.006 [Google Scholar]
-
Zhao K, Weng L, Xu T, Yang C, Zhang J, Ni G, et al. Low-intensity pulsed ultrasound prevents prolonged hypoxia-induced cardiac fibrosis through HIF-1α/DNMT3a pathway via a TRAAK-dependent manner. Clin. Exp. Pharmacol. Physiol. 2021, 48, 1500–1514. DOI:10.1111/1440-1681.13562 [Google Scholar]
-
Li T, Zhuang Y, Yang W, Xie Y, Shang W, Su S, et al. Silencing of METTL3 attenuates cardiac fibrosis induced by myocardial infarction via inhibiting the activation of cardiac fibroblasts. FASEB J. 2021, 35, e21162. DOI:10.1096/fj.201903169R [Google Scholar]
-
Huang B, Xie L, Ke M, Fan Y, Tan J, Ran J, et al. Programmed Release METTL3-14 Inhibitor Microneedle Protects Myocardial Function by Reducing Drp1 m6A Modification-Mediated Mitochondrial Fission. ACS Appl. Mater. Interfaces 2023, 15, 46583–46597. DOI:10.1021/acsami.3c06318 [Google Scholar]
-
Meng Y, Xi T, Fan J, Yang Q, Ouyang J, Yang J. The inhibition of FTO attenuates the antifibrotic effect of leonurine in rat cardiac fibroblasts. Biochem. Biophys. Res. Commun. 2024, 693, 149375. DOI:10.1016/j.bbrc.2023.149375 [Google Scholar]
-
Dong XL, Yuan BH, Yu SZ, Liu H, Pan XH, Sun J, et al. Adriamycin induces cardiac fibrosis in mice via PRMT5-mediated cardiac fibroblast activation. Acta Pharmacol. Sin. 2023, 44, 573–583. DOI:10.1038/s41401-022-00963-x [Google Scholar]
-
Katanasaka Y, Yabe H, Murata N, Sobukawa M, Sugiyama Y, Sato H, et al. Fibroblast-specific PRMT5 deficiency suppresses cardiac fibrosis and left ventricular dysfunction in male mice. Nat. Commun. 2024, 15, 2472. DOI:10.1038/s41467-024-46711-z [Google Scholar]
-
Stratton MS, Bagchi RA, Felisbino MB, Hirsch RA, Smith HE, Riching AS, et al. Dynamic Chromatin Targeting of BRD4 Stimulates Cardiac Fibroblast Activation. Circ. Res. 2019, 125, 662–677. DOI:10.1161/CIRCRESAHA.119.315125 [Google Scholar]
-
Duan Q, McMahon S, Anand P, Shah H, Thomas S, Salunga HT, et al. BET bromodomain inhibition suppresses innate inflammatory and profibrotic transcriptional networks in heart failure. Sci. Transl. Med. 2017, 9, eaah5084. DOI:10.1126/scitranslmed.aah5084 [Google Scholar]
-
Zhou H, Kee HJ, Wan L, Asfaha Y, Fischer F, Kassack MU, et al. YAK577 Attenuates Cardiac Remodeling and Fibrosis in Isoproterenol-Infused Heart Failure Mice by Downregulating MMP12. Korean Circ. J. 2025, 55, 231. DOI:10.4070/kcj.2024.0093 [Google Scholar]
-
Peng F, Liao M, Jin W, Liu W, Li Z, Fan Z, et al. 2-APQC, a small-molecule activator of Sirtuin-3 (SIRT3), alleviates myocardial hypertrophy and fibrosis by regulating mitochondrial homeostasis. Signal Transduct. Target. Ther. 2024, 9, 133. DOI:10.1038/s41392-024-01816-1 [Google Scholar]
-
Gaspari T, Brdar M, Lee HW, Spizzo I, Hu Y, Widdop RE, et al. Molecular and cellular mechanisms of glucagon-like peptide-1 receptor agonist-mediated attenuation of cardiac fibrosis. Diabetes Vasc. Dis. Res. 2016, 13, 56–68. DOI:10.1177/1479164115605000 [Google Scholar]
-
Bonfioli GB, Rodella L, Metra M, Vizzardi E. GLP-1 receptor agonists as promising anti-inflammatory agents in heart failure with preserved ejection fraction. Heart Fail. Rev. 2024, 30, 131–136. DOI:10.1007/s10741-024-10450-6 [Google Scholar]
-
Jia A, Yang M, Wang T, Hua Y, Lu H. Efficacy and safety of GLP-1 receptor agonists in the treatment of obese patients with chronic heart failure: A meta-analysis. Front. Cardiovasc. Med. 2025, 12, 1633114. DOI:10.3389/fcvm.2025.1633114 [Google Scholar]
-
Richards AM. C-Type Natriuretic Peptide and Cardiac Fibrosis. Hypertension 2011, 57, 154–155. DOI:10.1161/HYPERTENSIONAHA.110.163865 [Google Scholar]
-
Hatem SN, Bakhos JJ, Saliba Y, Hajal J, Achkouty G, Semaan A, et al. The anti-fibrotic effect of atrial natriuretic peptide depends on myocardial accumulation of the peptide in rat heart. Eur. Heart J. 2023, 44 (Suppl. S2), ehad655.3157. DOI:10.1093/eurheartj/ehad655.3157 [Google Scholar]
-
Martin FL, Sangaralingham SJ, Huntley BK, McKie PM, Ichiki T, Chen HH, et al. CD-NP: A Novel Engineered Dual Guanylyl Cyclase Activator with Anti-Fibrotic Actions in the Heart. PLoS ONE 2012, 7, e52422. DOI:10.1371/journal.pone.0052422 [Google Scholar]
-
Ichiki T, Dzhoyashvili N, Burnett JC. Natriuretic peptide based therapeutics for heart failure: Cenderitide: A novel first-in-class designer natriuretic peptide. Int. J. Cardiol. 2019, 281, 166–171. DOI:10.1016/j.ijcard.2018.06.002 [Google Scholar]
-
Romero G, Martin B, Gabris B, Salama G. Relaxin suppresses atrial fibrillation, reverses fibrosis and reduces inflammation in aged hearts. Biochem. Pharmacol. 2024, 227, 116407. DOI:10.1016/j.bcp.2024.116407 [Google Scholar]
-
Wołowiec Ł, Jaśniak A, Osiak-Gwiazdowska J, Czaplińska D, Szymczak A, Pęcherz JA, et al. Long-acting relaxin analogues: A novel tool in cardiology. Front. Pharmacol. 2025, 16, 1626469. DOI:10.3389/fphar.2025.1626469 [Google Scholar]
-
Connolly K, George R, Omar S, Matsson E, Åstrand M, Althage M, et al. Novel Relaxin Receptor RXFP1 Agonist AZD3427 in the Treatment of Heart Failure: A Phase 1a/b, First-in-Human, Randomized, Single-Blind, Placebo-Controlled Study. J. Am. Heart Assoc. 2024, 13, e034067. DOI:10.1161/JAHA.123.034067 [Google Scholar]
-
Tapia Cáceres F, Gaspari TA, Hossain MA, Samuel CS. Relaxin Inhibits the Cardiac Myofibroblast NLRP3 Inflammasome as Part of Its Anti-Fibrotic Actions via the Angiotensin Type 2 and ATP (P2X7) Receptors. Int. J. Mol. Sci. 2022, 23, 7074. DOI:10.3390/ijms23137074 [Google Scholar]
-
Borlaug BA, Testani JM, Petrie MC, Wang Z, Cunningham J, Adams KF, et al. Effects of volenrelaxin in worsening heart failure with preserved ejection fraction: A phase 2 randomized trial. Nat. Med. 2025, 31, 3853–3861. DOI:10.1038/s41591-025-03939-6 [Google Scholar]
-
Qin Q, Mehta H, Yen K, Navarrete G, Brandhorst S, Wan J, et al. Chronic treatment with the mitochondrial peptide humanin prevents age-related myocardial fibrosis in mice. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H1127–H1136. DOI:10.1152/ajpheart.00685.2017 [Google Scholar]
-
Zheng X, Peng M, Li Y, Wang X, Lu W, Wang X, et al. Cathelicidin-related antimicrobial peptide protects against cardiac fibrosis in diabetic mice heart by regulating endothelial-mesenchymal transition. Int. J. Biol. Sci. 2019, 15, 2393–2407. DOI:10.7150/ijbs.35736 [Google Scholar]
-
Li Y, Liu X, Wan L, Han B, Ma S, Pan H, et al. Metformin suppresses cardiac fibroblast proliferation under high-glucose conditions via regulating the mitochondrial complex I protein Grim-19 involved in the Sirt1/Stat3 signaling pathway. Free Radic. Biol. Med. 2023, 206, 1–12. DOI:10.1016/j.freeradbiomed.2023.06.013 [Google Scholar]
-
Pillai AA, Melo L, Frishman WH, Aronow WS. The Effects of Metformin on Weight Loss, Cardiovascular Health, and Longevity. Cardiol. Rev. 2024, 10-1097. DOI:10.1097/CRD.0000000000000832 [Google Scholar]
-
Liu J, Lu J, Zhang L, Liu Y, Zhang Y, Gao Y, et al. The combination of exercise and metformin inhibits TGF-β1/Smad pathway to attenuate myocardial fibrosis in db/db mice by reducing NF-κB–mediated inflammatory response. Biomed. Pharmacother. 2023, 157, 114080. DOI:10.1016/j.biopha.2022.114080 [Google Scholar]
-
Wiggers H. A Randomized, Double-Blind, Placebo Controlled Study (DANHEART): Hydralazine-ISDN in Patients with Chronic Heart Failure—Hydralazine Heart Failure Trial (H-HeFT) and Metformin in Patients with Chronic Heart Failure and Diabetes or Insulin Resistance—Metformin Heart Failure Trial (Met-HeFT). 2025. Clinical Trial Registration No.: NCT03514108. Available online: https://clinicaltrials.gov/study/NCT03514108 (accessed on 11 February 2026).
-
Han Y, Xie H, Liu Y, Gao P, Yang X, Shen Z. Effect of metformin on all-cause and cardiovascular mortality in patients with coronary artery diseases: A systematic review and an updated meta-analysis. Cardiovasc. Diabetol. 2019, 18, 96. DOI:10.1186/s12933-019-0900-7 [Google Scholar]
-
Hoffmann M, Kant TA, Emig R, Rausch JSE, Newe M, Schubert M, et al. Repurposing mesalazine against cardiac fibrosis in vitro. Naunyn-Schmiedeberg's Arch. Pharmacol. 2021, 394, 533–543. DOI:10.1007/s00210-020-01998-9 [Google Scholar]
-
Lewis GA, Dodd S, Clayton D, Bedson E, Eccleson H, Schelbert EB, et al. Pirfenidone in heart failure with preserved ejection fraction: A randomized phase 2 trial. Nat. Med. 2021, 27, 1477–1482. DOI:10.1038/s41591-021-01452-0 [Google Scholar]
-
Umbarkar P, Singh AP, Tousif S, Zhang Q, Sethu P, Lal H. Repurposing Nintedanib for pathological cardiac remodeling and dysfunction. Pharmacol. Res. 2021, 169, 105605. DOI:10.1016/j.phrs.2021.105605 [Google Scholar]
-
Yu D, Tang Z, Li B, Yu J, Li W, Liu Z, et al. Resveratrol against Cardiac Fibrosis: Research Progress in Experimental Animal Models. Molecules 2021, 26, 6860. DOI:10.3390/molecules26226860 [Google Scholar]
-
Zheng X, Hai J, Yang Y, Zhang C, Ma X, Kong B, et al. Effects of resveratrol supplementation on cardiac remodeling in hypertensive patients: A randomized controlled clinical trial. Hypertens. Res. 2023, 46, 1493–1503. DOI:10.1038/s41440-023-01231-z [Google Scholar]
-
Wu QQ, Xiao Y, Jiang XH, Yuan Y, Yang Z, Chang W, et al. Evodiamine attenuates TGF-β1-induced fibroblast activation and endothelial to mesenchymal transition. Mol. Cell Biochem. 2017, 430, 81–90. DOI:10.1007/s11010-017-2956-6 [Google Scholar]
-
Schimmel K, Jung M, Foinquinos A, José GS, Beaumont J, Bock K, et al. Natural Compound Library Screening Identifies New Molecules for the Treatment of Cardiac Fibrosis and Diastolic Dysfunction. Circulation 2020, 141, 751–767. DOI:10.1161/CIRCULATIONAHA.119.042559 [Google Scholar]
-
Xu Z, Yang J, Hu Y, Wan Q, Wang X, Lu C, et al. Qifu yixin prescription ameliorates cardiac fibrosis by activating soluble guanylate cyclase (sGC) in heart failure. J. Ethnopharmacol. 2025, 340, 119229. DOI:10.1016/j.jep.2024.119229 [Google Scholar]
-
Luo H, Fu L, Wang X, Xu Y, Tao L, Shen X. Salvianolic acid B ameliorates myocardial fibrosis in diabetic cardiomyopathy by deubiquitinating Smad7. Chin. Med. 2023, 18, 161. DOI:10.1186/s13020-023-00868-9 [Google Scholar]
-
Yin Y, Zhang Q, Zhao Q, Ding G, Wei C, Chang L, et al. Tongxinluo Attenuates Myocardiac Fibrosis after Acute Myocardial Infarction in Rats via Inhibition of Endothelial-to-Mesenchymal Transition. BioMed Res. Int. 2019, 2019, 6595437. DOI:10.1155/2019/6595437 [Google Scholar]
-
Prathapan A, Varghese MV, Abhilash S, Mathew AK, Nair A, Nair RH, et al. Polyphenol rich ethanolic extract from Boerhavia diffusa L. mitigates angiotensin II induced cardiac hypertrophy and fibrosis in rats. Biomed. Pharmacother. 2017, 87, 427–436. DOI:10.1016/j.biopha.2016.12.114 [Google Scholar]
-
Zhang X, Ke PX, Yuan X, Zhang GP, Chen WL, Zhang GS. Forskolin Protected against Streptozotocin-Induced Diabetic Cardiomyopathy via Inhibition of Oxidative Stress and Cardiac Fibrosis in Mice. BioMed Res. Int. 2021, 2021, 8881843. DOI:10.1155/2021/8881843 [Google Scholar]
-
Guo S, Qi X, Zhang L, Lu K, Li X, Zhu J, et al. Plumbagin improves myocardial fibrosis after myocardial infarction by inhibiting the AKT/mTOR pathway to upregulate autophagy levels. Int. Immunopharmacol. 2025, 148, 114086. DOI:10.1016/j.intimp.2025.114086 [Google Scholar]
-
Aghajanian H, Kimura T, Rurik JG, Hancock AS, Leibowitz MS, Li L, et al. Targeting cardiac fibrosis with engineered T cells. Nature 2019, 573, 430–433. DOI:10.1038/s41586-019-1546-z [Google Scholar]
-
Abdalla AME, Miao Y, Ahmed AIM, Meng N, Ouyang C. CAR-T cell therapeutic avenue for fighting cardiac fibrosis: Roadblocks and perspectives. Cell Biochem. Funct. 2024, 42, e3955. DOI:10.1002/cbf.3955 [Google Scholar]
-
Rurik JG, Tombácz I, Yadegari A, Méndez Fernández PO, Shewale SV, Li L, et al. CAR T cells produced in vivo to treat cardiac injury. Science 2022, 375, 91–96. DOI:10.1126/science.abm0594 [Google Scholar]
-
Liu X, Du Z, Zhang Y, Lv X, Zhang C, Li D. CAR-T cells generated in vivo for the targeted treatment of myocardial fibrosis. Int. Immunopharmacol. 2026, 168, 115840. DOI:10.1016/j.intimp.2025.115840 [Google Scholar]
-
Hua X, Sun Z, Liang Z, Huang Y, Mo H, Dong F, et al. Engineered T cell therapy for the treatment of cardiac fibrosis during chronic phase of myocarditis. Theranostics 2026, 16, 2037–2051. DOI:10.7150/thno.116749 [Google Scholar]
-
Cao B, Liu M, Anwaier S, Long X, You T, He Z, et al. Intrapericardial delivery of FAP-CAR-T cells via a ROS-responsive hydrogel to treat cardiac fibrosis. J. Control. Release 2026, 392, 114697. DOI:10.1016/j.jconrel.2026.114697 [Google Scholar]