Comparison of the Transcriptomic Signatures of Skin and Lung Fibroblasts from Patients with Systemic Sclerosis

Comparison of the Transcriptomic Signatures of Skin and Lung Fibroblasts from Patients with Systemic Sclerosis
Received: 24 March 2026 Revised: 15 April 2026 Accepted: 28 April 2026 Published: 15 May 2026

© 2026 The authors. This is an open access article under the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/).
Graphical Abstract
1. Introduction
Systemic Sclerosis (SSc, also known as scleroderma) is a chronic autoimmune disease that primarily affects connective tissues and often results in fibrosis due to the overproduction of extracellular matrix (ECM) components [1]. SSc predominantly affects the skin and internal organs such as the heart, kidneys, and lungs. SSc is more common in females than males, yet disease progression tends to be more severe in males [2,3].
SSc is categorized into diffuse cutaneous SSc (dcSSc), limited cutaneous SSc (lcSSc), and SSc sine scleroderma [4]. DcSSc is characterized by proximal skin fibrosis that can involve the trunk, upper arms, thighs and face, and shows rapid disease progression and severe organ involvement early in the course of the disease [5,6]. In lcSSc, skin thickening is progressive and restricted to the distal limbs and the face [7]. Inclusion of samples from patients with lcSSc can provide valuable information on skin fibrosis progression despite the low modified Rodnan skin score (mRSS) characteristic of lcSSc [8]. In SSc sine scleroderma, patients have internal organ involvement but no skin thickening.
SSc-associated interstitial lung disease (SSc-ILD) is observed in both dcSSc and lcSSc patients and results in impaired function [9]. SSc-ILD is currently the leading cause of death in SSc patients [10]. Because disease progression is similar and progressive in the skin of lcSSc patients, irrespective of disease duration, and the lungs of SSc-ILD patients, this study will focus on comparing the transcriptomic signatures of lcSSc skin fibroblasts (SSc skin) and SSc-ILD lung fibroblasts (SSc lung).
Fibroblasts are responsible for the excess production of ECM that leads to fibrosis [11,12]. Fibroblasts are a valuable tool for understanding the mechanisms of fibrosis and for testing pro-fibrotic triggers and anti-fibrotic therapies. Fibroblasts can be cultured from both lung and skin tissues of healthy and diseased donors and retain the phenotype of the original tissue [13]. We conducted several studies to capture disease-associated transcriptional differences of SSc skin and SSc lung fibroblasts by comparing them to their respective healthy control counterparts [14,15], but we have not previously compared transcriptional differences of SSc skin and SSc lung fibroblasts. Thus, the goal of this study is to compare the transcriptomic signatures of SSc skin and lung fibroblasts to identify transcriptomic similarities and differences.
2. Materials and Methods
2.1. Selection of Datasets for SSc Skin and SSc Lung Fibroblasts
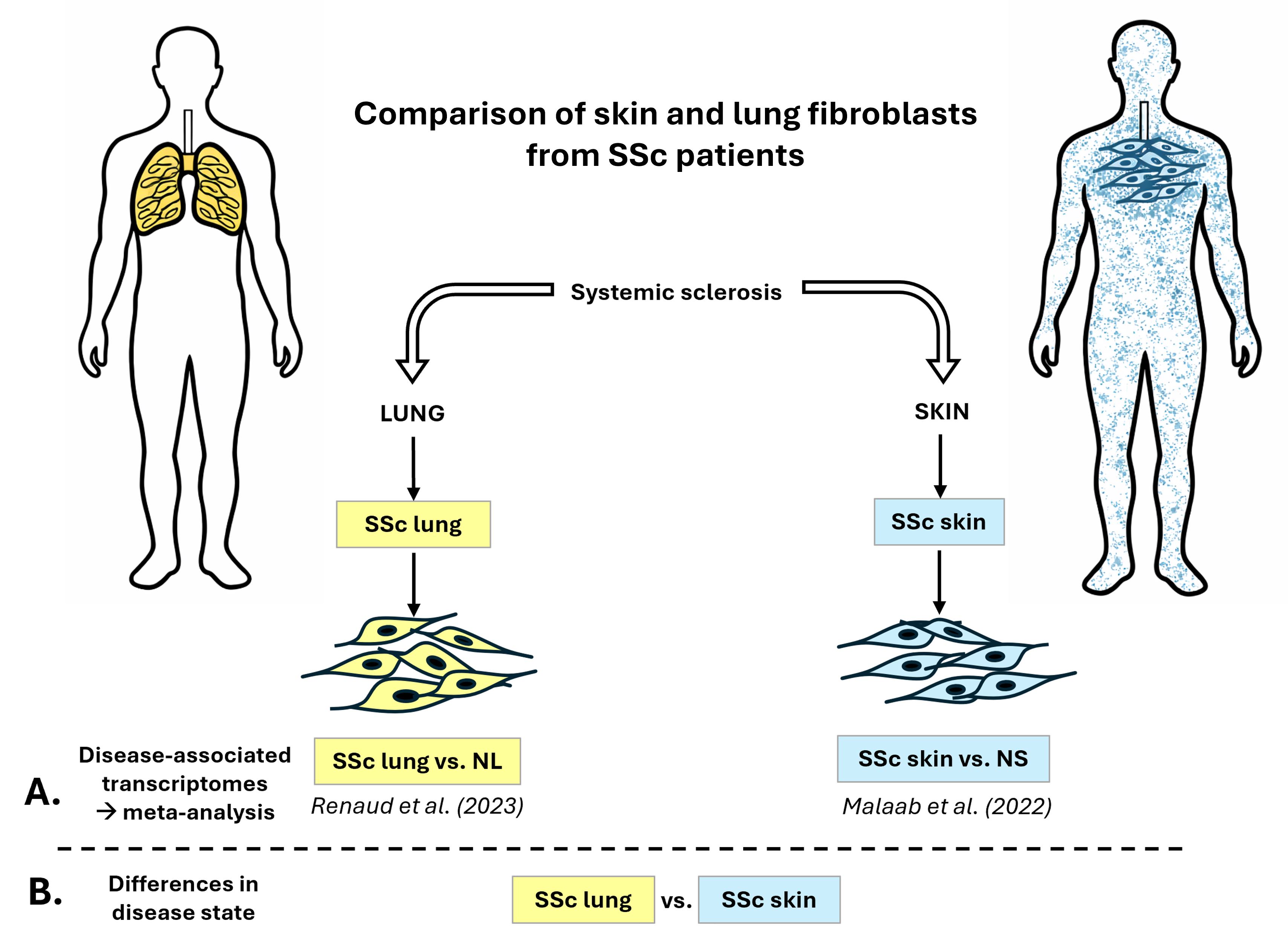
The differentially expressed (DE) genes used in this study were obtained from two of our published datasets (Figure 1A): (1) the SSc-ILD fibroblasts dataset (SSc lung) is from Renaud et al. [14], specifically from the European American cohort (NCBI GEO accession# GSE215841), and (2) the lcSSc skin fibroblasts dataset (SSc skin) is from Malaab et al. [15] (NCBI GEO accession# GSE153880).
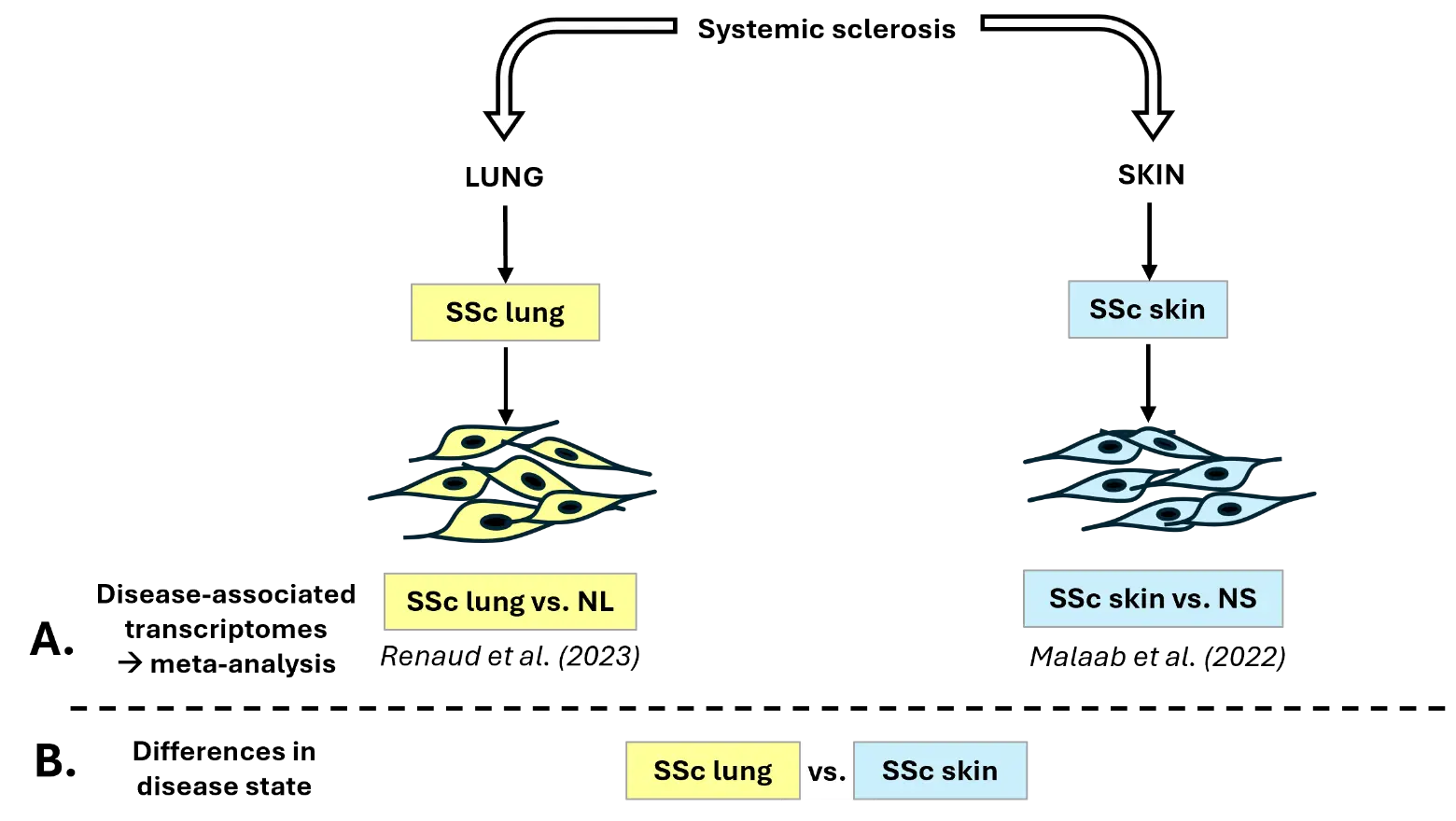
Figure 1. Experimental design. (A) Experimental design for the meta-analysis comparing disease-associated transcriptional differences in SSc lung and SSc skin fibroblasts [14,15]. (B) Experimental design for the differential expression analysis “SSc lung vs. SSc skin” fibroblasts. NL: normal lung. NS: normal skin.
2.2. Differential Expression Analysis & Meta-Analysis
The differential expression analysis “SSc vs. normal” was conducted on both skin and lung fibroblasts to determine disease-associated transcriptional differences using DESeq2 package [16] (Figure 1A). DE genes were determined based on 2 criteria: FDR adjusted p-value (q-value) < 0.1 and log2 fold change (log2FC) > |0.6|. The meta-analysis was performed on iPathwayGuide (Advaita), generating Venn diagrams for genes, pathways, and Gene Ontology terms, including Biological Process (BP), Molecular Function (MF), and Cellular Component (CC). The significance cutoff for pathways and Gene Ontology is using the p-value (p-value < 0.05).
A new differential expression analysis was conducted, “SSc lung vs. SSc skin” (q-value < 0.1, log2FC > |0.6|), to determine differences in disease state by comparing SSc lung and SSc skin fibroblasts with the same package and criteria of significance (Figure 1B). The same library preparations were performed by Novogene (NEBNext® UltraTM RNA library prep kit for Illumina, NEB, Ipswich, MA, USA) on the two RNAseq datasets. To account for possible batch effect, selected DE genes were validated by qPCR using the same RNA used for RNA sequencing.
2.3. cDNA and qPCR Validation
A NanoDrop Lite spectrophotometer (ThermoFisher Scientific, Waltham, MA, USA) was used to quantify RNA concentration and quality. To synthesize cDNA, 1 μg of RNA per 20 μL was used with random hexamers and the SuperScript IV reverse transcriptase (Invitrogen, Carlsbad, CA, USA) on a C1000 Touch Thermal Cycler (Bio-Rad, Hercules, CA, USA). Quantitative PCR (qPCR) was performed to measure mRNA expression levels using the TaqMan real-time PCR system (Life Technologies, Carlsbad, CA, USA) according to the manufacturer’s protocol on a TaqMan Gene Expression Assays Step One Plus real-time PCR system (Life Technologies) using the following TaqMan human primers: ACTA2 (Hs00426835_g1), COL3A1 (Hs00943809_m1), CTHRC1 (Hs00298917_m1), IL6 (Hs00985639_m1), and LOX (Hs00184700_m1). The housekeeping genes GAPDH (FAM Hs02758991_g1, abbreviated GAP) and B2M (VIC Hs00187842_m1) were duplexed. The target gene $$2^{-\Delta \mathrm{CT}}$$ values normalized to GAP or B2M were statistically analyzed in GraphPad Prism version 10.4.1 (GraphPad Software Inc., La Jolla, CA, USA).
2.4. Heatmaps
A list of genes of interest (GOI) was generated with genes that are known to be markers of alveolar or inflammatory fibroblasts, pro-fibrotic genes, ECM genes, or anti-fibrotic genes (Supplemental Table S1). This list of GOI was then merged with the DE genes that are (1) unique to SSc-ILD fibroblasts, (2) unique to lcSSc fibroblasts, or (3) overlapping. A second “merge” was performed to obtain the counts from the original differential expression analysis. Morpheus was then used to generate heatmaps (https://software.broadinstitute.org/morpheus/, accessed on 1 February 2026) using the “Kmeans clustering” method (metric: one minus Pearson correlation; clustering first on columns, then on rows; maximum iterations: 1000). For the overlapping DE genes, since 2 sets of counts were available for SSc-ILD pulmonary and lcSSc dermal fibroblasts, 2 heatmaps were generated.
2.5. Statistical Analysis
GraphPad Prism 10 version 10.4.1 (GraphPad Software, Inc.) was used for statistical analysis. To identify outliers, the ROUT method was used. A two-tailed unpaired t-test with 95% confidence level was performed on the qPCR validation dataset. p < 0.05 was considered significant: * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001. Error bars = SEM.
3. Results
3.1. Meta-Analysis: Disease-Associated Transcriptomes of SSc Skin and SSc Lung Fibroblasts
We previously performed the differential expression analysis “SSc vs. normal” in (1) lung fibroblasts of SSc-ILD patients (SSc lung) versus normal lung (NL) fibroblasts from healthy control donors, and (2) skin fibroblasts of lcSSc patients (SSc skin) versus site-matched normal skin (NS) of healthy control donors (Figure 1A). The number of DE genes for each differential expression analysis is shown in Table 1. The meta-analysis shows that 467 DE genes overlap between SSc skin and SSc lung while 1328 are unique to SSc skin fibroblasts and 1558 are unique to SSc lung fibroblasts (Figure 2A). Out of the 467 overlapping DE genes between SSc skin and SSc lung fibroblasts, 235 are commonly upregulated while 57 are commonly downregulated, representing 8.7% of the total DE genes (Figure 2B, Supplemental Table S2). Note also that amongst the 467 overlapping DE genes, 59 are upregulated in SSc lung while downregulated in SSc skin fibroblasts, and 93 DE genes are downregulated in SSc lung while upregulated in SSc skin fibroblasts, emphasizing that “overlapping DE genes” can also have opposite regulation depending on the tissue of origin.
Table 1. Summary of the DE genes returned by the differential expression analysis previously performed.
|
Comparison |
# of DE Genes |
|---|---|
|
SSc lung vs. NL |
2025 |
|
SSc skin vs. NS |
1795 |
3.2. Meta-Analysis: Impacted Pathways in SSc Skin and SSc Lung Fibroblasts
Focusing on impacted pathways, the meta-analysis (Supplemental Table S3) revealed that: (1) 48 pathways are unique to SSc skin fibroblasts (Figure 2C), including “Alcoholic liver disease”, “Renin secretion”, “TGFβ signaling pathway”, “MAPK signaling pathway”, “IL17 signaling pathway”, “ErbB signaling pathway”, “GABAergic synapse”, “Vascular smooth muscle contraction”, “Gastric acid secretion”, and “Estrogen signaling pathway”, pointing to a biological state characterized by intense immune and inflammatory activation, rewired cell signaling, and metabolic and mitochondrial reprogramming, all of which suggest a highly stressed or disease-altered cellular environment. (2) 29 pathways are unique to SSc lung fibroblasts (Figure 2D), including “Olfactory transduction”, “cAMP signaling pathway”, “Steroid biosynthesis”, “Retinol metabolism”, “Renin-angiotensin system”, “Nicotine addiction”, “Arachidonic acid metabolism”, “Metabolism of xenobiotics by cytochrome P450”, “Choline metabolism in cancer”, and “Complement and coagulation cascades”, highlighting metabolic reprogramming (especially lipid and steroid metabolism), activation of detoxification/xenobiotic pathways, cancer-associated pathways and hormone-driven signaling, suggesting a system under chemical stress with strong endocrine and metabolic adaptation. (3) 33 pathways overlapped between SSc skin and SSc lung fibroblasts, representing 30% of all pathways (Figure 2E). Among these, the top 10 most impacted were “AGE-RAGE signaling pathway in diabetic complications”, “Arrhythmogenic right ventricular cardiomyopathy”, “Axon Guidance”, “Calcium signaling pathway”, “Cell adhesion molecules”, “Cytokine-cytokine receptor interaction”, “Cytoskeleton in muscle cells”, “Dilated cardiomyopathy”, “ECM-receptor interaction”, and “Focal adhesion”, reflecting a broad remodeling of core cell-signaling networks, altered cell–cell/matrix communication, and disease-associated and immune-linked pathways, pointing to a system undergoing structural and regulatory remodeling.
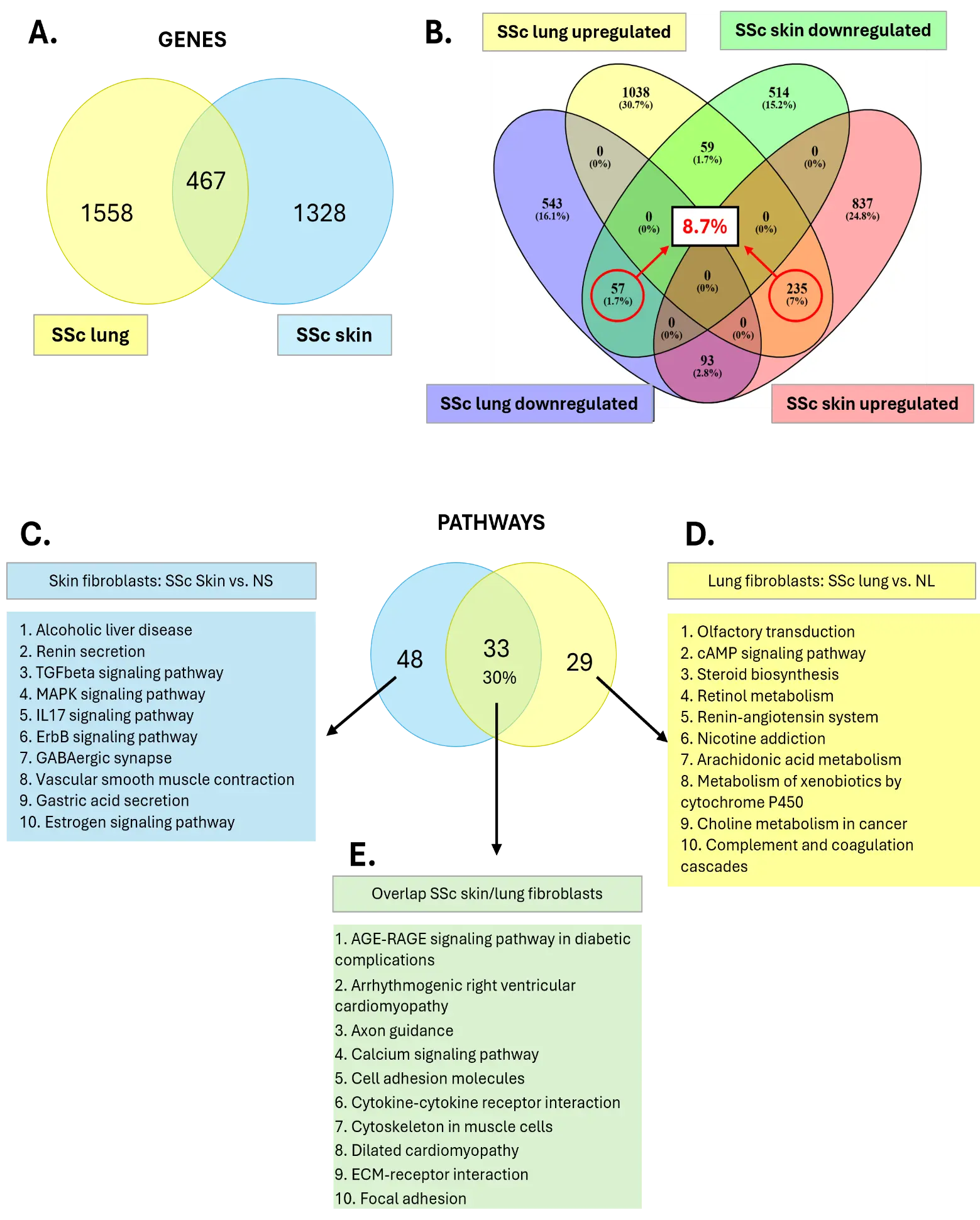
Figure 2. Meta-analysis: genes and pathways. (A) Venn diagram (2 entries) of genes unique or overlapping in SSc lung and SSc skin fibroblasts. (B) Venn diagram (4 entries) of upregulated or downregulated genes in SSc lung and SSc skin fibroblasts. (C) Top 10 impacted pathways unique to SSc skin fibroblasts. (D) Top 10 impacted pathways unique to SSc lung fibroblasts. (E) Top 10 impacted pathways overlapping between SSc skin and SSc lung fibroblasts.
3.3. Balance Between Pro and Anti-Fibrotic Programs in SSc Skin and SSc Lung Fibroblasts
To focus on the deregulation of genes of interest (GOI, Supplemental Table S1) implicated in fibrosis and inflammation collated from the scientific literature, we generated heatmaps specific to the DE genes that are unique to SSc skin fibroblasts and SSc lung fibroblasts (Figure 3). In SSc skin fibroblasts (Figure 3A), many ECM genes, markers of inflammatory fibroblasts and pro-fibrotic genes are upregulated as expected, including COL10A1, COL13A1, COL18A1, COL23A1, COLL27A1, COL6A1, COL6A2, COL7A1, COL9A3, TFAP2A, HLA-DRB1, PTGDS, DNMT3B and IL6, but several ECM and pro-fibrotic genes are also downregulated, such as COL14A1, COL21A1, COL6A6, COL9A2, HOXD11, PCSK9 and TGFB3. Additionally, markers of alveolar fibroblasts and anti-fibrotic genes were upregulated (GPM6B, NPNT, PLAUR) while others were downregulated (SAA1, INMT, HOXD10, KLF4, MMP10, ID1, GLI3) in SSc skin fibroblasts, suggesting that the balance between the pro- and anti-fibrotic programs is likely balanced in these fibroblasts, but inflammation and immune responses are active.
In SSc lung fibroblasts (Figure 3B), most of the markers of inflammatory fibroblasts, ECM, and pro-fibrotic genes from our list of GOI are upregulated, including CCL2, IGF1, IGFBP5, LOX, TGFB2, IGFBP3, POSTN, COL1A1, COL8A1, COL11A1, COL11A2, COL15A1, COL24A1, COL4A1, COL4A2, COL4A3, COL4A4, COL5A1 and COL5A2. Only MMP28, APOE, CXCL14, ELOVL6, IL33 and TNC are downregulated genes from the pro-fibrotic and inflammatory program in these fibroblasts. Additionally, the anti-fibrotic genes CTSL, HOPX and PLAU are downregulated in SSc lung fibroblasts, except for IGFBP4, which is upregulated, along with the markers of alveolar fibroblasts FMO2 and TCF21. Together, these results emphasize that the balance is tipped in favor of the pro-fibrotic program in the disease-associated transcriptional signature of SSc lung fibroblasts than it is in the SSc skin fibroblast signature, with both the IGF and TGFβ signaling pathways represented.
As shown above, among the 467 overlapping DE genes between SSc skin and SSc lung fibroblasts, only 292 had the same direction of regulation, representing 8.7% of the total DE genes (Figure 2B). In Figure 3C we show that, included in these 292 DE genes, are (i) the upregulated pro-fibrotic genes IGF2, PDGFA, TGFBR1, THBS1 and WNT5A, and (ii) the downregulated anti-fibrotic gene MMP9 along with the marker of alveolar fibroblasts GPX3 and the long non-coding RNA H19. Also present in the overlap are pro-fibrotic genes with opposite regulation in SSc skin and SSc lung fibroblasts, including (iii) ACTA2, COL3A1, FGF9 and TGFBI that are downregulated in SSc skin fibroblasts while upregulated in SSc lung fibroblasts, and (iv) CD74 and COL22A1 that are upregulated in SSc skin fibroblasts while downregulated in SSc lung fibroblasts. Note also that the anti-fibrotic genes MMP1 and MMP15 are upregulated in SSc skin fibroblasts while downregulated in SSc lung fibroblasts.
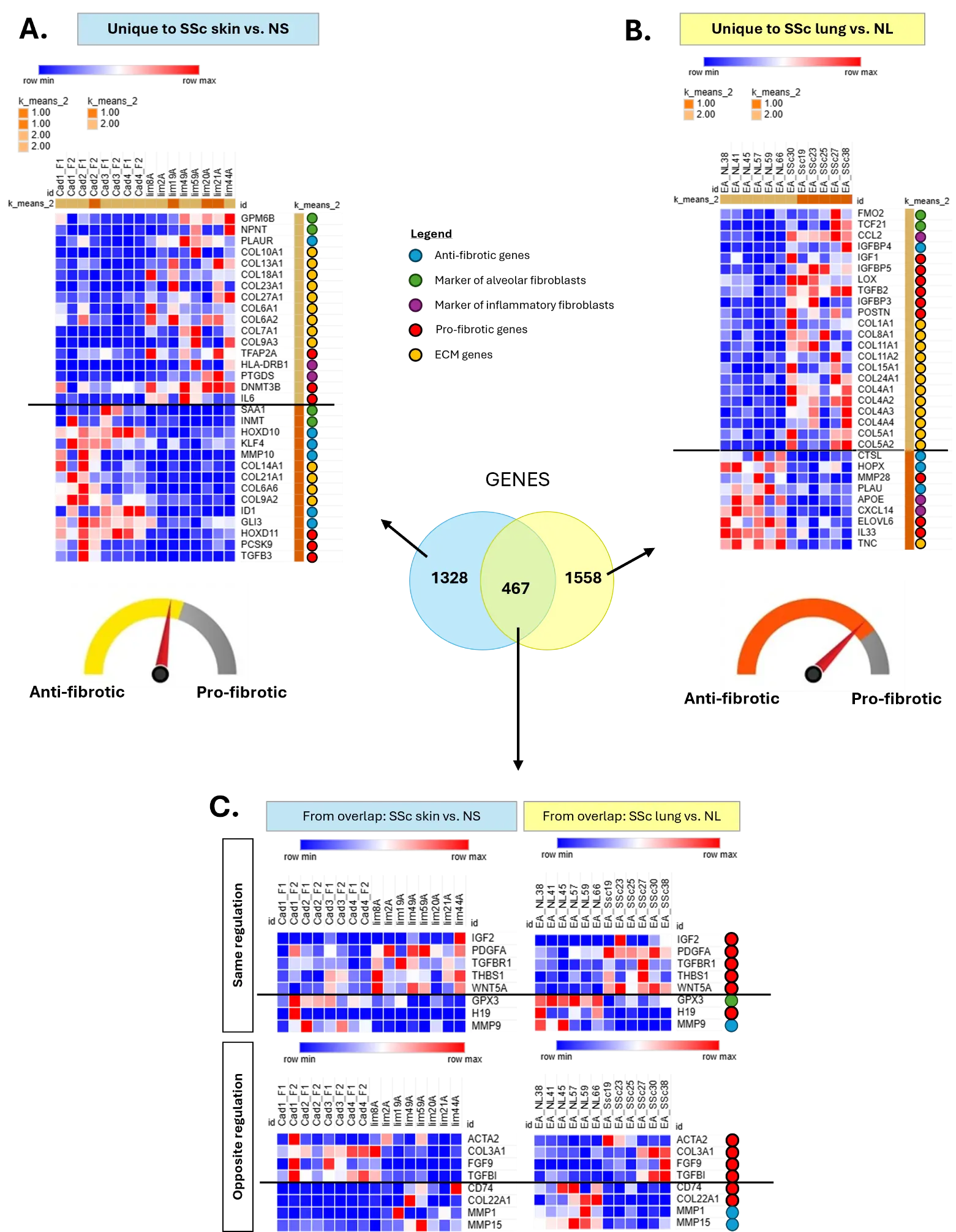
Figure 3. Signature of GOI in SSc skin and SSc lung fibroblasts. (A) Heatmap of GOI in SSc skin fibroblasts. (B) Heatmap of GOI in SSc lung fibroblasts. (C) Heatmaps of GOI in the overlap between SSc skin and SSc lung fibroblasts.
3.4. Predicted Drugs in the Overlap Between SSc Skin and SSc Lung Fibroblasts
The iPathwayGuide “upstream regulators” tool returned 10 chemicals out of which 3 are classified as “drugs” predicted as absent/insufficient (Figure 4). These drugs, if introduced in the cells, are predicted to bring levels of expression closer to the phenotypic signatures of non-fibrotic fibroblasts: the bioactive natural product “acteoside”, the calcium channel blocker “manidipine”, and “medroxyprogesterone acetate”. The small overlap in drugs between SSc skin and SSc lung fibroblasts reinforces the differentiation between the two subtypes and highlights the need for tissue-specific therapeutic development.
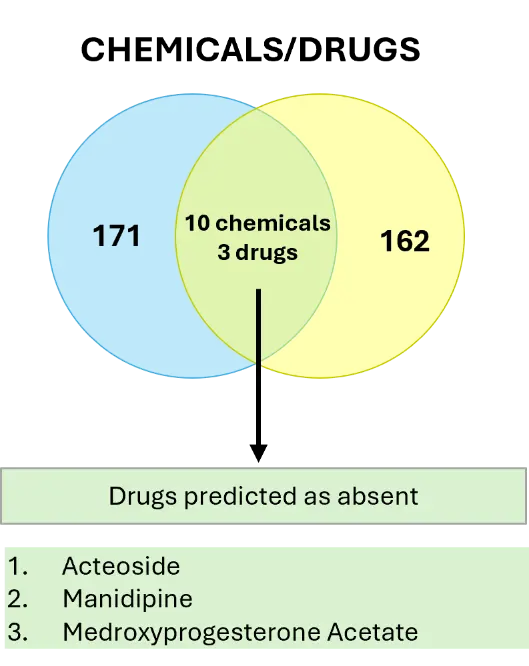
Figure 4. Meta-analysis—Upstream Regulators, Chemicals & Drugs. Drugs overlapping between SSc skin and SSc lung fibroblasts.
3.5. Differences in Disease State: “SSc Lung vs. SSc Skin” Fibroblasts
To compare the SSc phenotype of lung and skin fibroblasts, we performed a new differential expression analysis using the raw counts of SSc lung and SSc skin fibroblasts from the 2 differential expression analyses previously published and used for the meta-analysis described above. The differential expression analysis “SSc lung vs. SSc skin” fibroblasts returned 3760 DE genes, out of which 1724 are upregulated and 2036 are downregulated in SSc lung compared to SSc skin fibroblasts (Figure 5A,B, Supplemental Table S4). This gene signature impacted 95 pathways (Figure 5C, Supplemental Table S5), including “Cell adhesion molecules”, “ECM-receptor interaction”, “Viral protein interaction with cytokine and cytokine receptor”, “Focal adhesion”, “Neuroactive ligand-receptor interaction”, and “Regulation of actin cytoskeleton”. Selected DE genes were validated by qPCR (Figure 5D) and confirmed the upregulation of ACTA2 and COL3A1 mRNA levels and the downregulation of CTHRC1, IL6 and LOX in SSc lung fibroblasts relative to SSc skin fibroblasts.
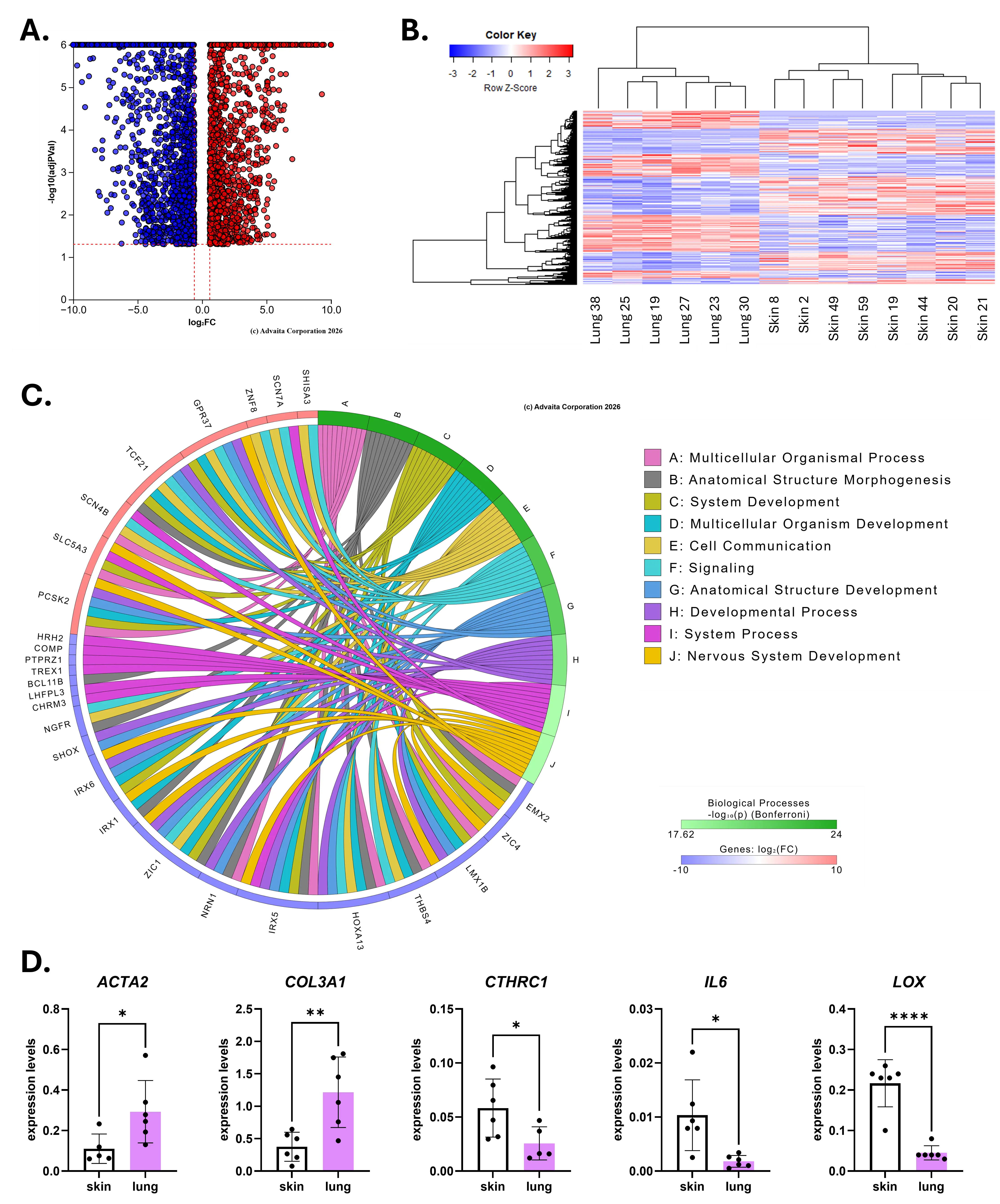
Figure 5. Differences in disease state: “SSc lung vs. SSc skin” fibroblasts. (A) Volcano plot for the differential expression analysis “SSc lung vs. SSc skin” fibroblasts (q-value < 0.1, log2FC > |0.6|). (B) Heatmap generated by DEseq2 for all genes with a q-value < 0.1. (C) Ribbon plot generated by iPathwayGuide for the top 10 enriched pathways showing the top 10 DE genes contributing to each pathway. For panels (B–D): downregulated DE genes are shown in blue, upregulated DE genes are shown in red. (D) Validation of selected DE genes in SSc skin and SSc lung fibroblasts by qPCR. * p < 0.05, ** p < 0.01, **** p < 0.0001 relative to skin.
4. Discussion
The goal of this study was to compare the transcriptomic signatures of SSc skin and SSc lung fibroblasts. First, our meta-analysis identified 292 overlapping DE genes between SSc skin and SSc lung fibroblasts, representing 8.7% overlap, and 33 (30%) commonly impacted pathways. From the signature of selected GOI, we defined that SSc lung fibroblasts seem to be in a more active state of fibrosis, while inflammatory activation is more pronounced in SSc skin fibroblasts. We also explored drugs predicted to restore the overlapping DE genes to “normal expression” when introduced to the fibroblasts. Finally, we identified 3760 DE genes between SSc lung fibroblasts and SSc skin fibroblasts, highlighting that fibroblasts in SSc disease state carry a tissue-specific signature that should be taken into consideration for therapeutic development.
4.1. Meta-Analysis: Overlapping DE Genes and Pathways
Several studies have explored targets that impact fibrosis in either SSc skin or SSc lung fibroblasts, but to our knowledge, this study is the first meta-analysis to report disease-associated genes and pathways that are either unique or overlapping between SSc skin and SSc lung fibroblasts. At the gene level, our data show minimal overlap, only 8.7% (292 DE genes), between SSc skin and SS lung fibroblasts. However, the impacted pathway analysis revealed a 30% overlap. This emphasizes how difficult it would be to predict lung involvement based on SSc skin transcriptome or to identify therapies that would be equally effective in skin and lung fibroblasts. However, within the 30% overlapping pathways, several fundamental pathways are present and are discussed below.
The most enriched pathway in the overlap is the “AGE-RAGE signaling pathway in diabetic complications”, known to elicit activation of multiple intracellular signal pathways involving NADPH oxidase, protein kinase C, and MAPKs, leading to NFkB activity that promotes the expression of pro-inflammatory cytokines such as IL1, IL6, and TNFα. Moreover, AGE-RAGE interaction induces JAK-STAT and PI3K-Akt dependent pathways that regulate cell proliferation and apoptosis, respectively, as well as hypoxia-mediated induction of EGR1.
The “Calcium signaling pathway” is also in the overlap between SSc skin and SSc lung fibroblasts. This pathway is a major contributor to fibrosis as calcium oscillations activate fibroblast differentiation into myofibroblasts, increase ECM production and collagen secretion, and activate fibrotic signaling [17]. Blocking calcium signals as a therapeutic strategy to prevent fibrosis has been considered in dermal, pulmonary, and cardiac fibroblasts [18,19,20,21].
Cell adhesion is a central driver of SSc-related fibrosis as it controls how fibroblasts interact with their environment and neighboring cells, and how they respond to stiffening of the ECM [22]. The “Cell adhesion molecules” pathway is enriched in both SSc skin and SSc lung fibroblasts in our meta-analysis. These molecules are (glyco)proteins expressed on the cell surface, including integrins, immunoglobulins, selectins, and cadherins, and play a critical role in a wide array of biologic processes, including hemostasis, the immune response, inflammation, embryogenesis, and the development of neuronal tissue [23]. Another axis of communication between cells and their environment is the “Cytokine-cytokine receptor interaction” pathway, also enriched in the overlap. Cytokines can activate innate and adaptive inflammatory host defenses, cell growth and death, differentiation, angiogenesis, and developmental and repair processes aimed at restoring homeostasis. TGFβ and several cytokines are implicated in SSc [24,25] as they activate fibroblasts, amplify ECM production, and sustain a chronic inflammatory–fibrotic loop involving Th2 cells, growth-factor pathways, and altered fibroblast signaling [26]. Additionally, the “ECM-receptor interaction” pathway contributes to control of cellular activities such as adhesion, migration, differentiation, proliferation, and apoptosis by allowing interactions between cells and the ECM via transmembrane molecules.
Our meta-analysis revealed that once skin and lung fibroblasts have reached SSc disease state, they show little gene and pathway overlap, highlighting that fibroblasts take on a particular molecular phenotype depending on the tissue of origin. It has been shown that adult fibroblasts maintain the embryonic gene signature of their organ of origin, explaining fibroblast heterogeneity across different homeostatic tissues [27]. We show here that this specific phenotype remains distinct even in disease states, reinforcing the need for organ-specific strategies for targeted control of fibrosis.
4.2. A Gene Signature of Active Fibrosis in SSc Lung Fibroblasts
The heatmaps generated for well-known factors involved in fibrosis and all DE collagens showed that SSc lung fibroblasts are likely in a more active state of inflammation and fibrosis compared to SSc skin fibroblasts, as shown by the upregulation of several pro-inflammatory and pro-fibrotic factors. Noticeably, several factors belonging to the IGF signaling pathway are expressed at higher levels in SSc lung fibroblasts; IGF1, which is known for stimulating fibroblasts differentiation into myofibroblasts [28], and IGFBP3/IGFPB5/LOX, which have been described as central mediators of fibrosis by contributing to ECM deposition and inducing fibroblast migration [29,30,31,32]. Other pro-fibrotic pathways are also represented, including the TGFβ pathway via upregulation of TGFB2 [33,34]. Unlike TGFB1, which is significantly upregulated in both IPF and SSc lung fibroblasts, TGFB2 is only upregulated in SSc lung fibroblasts in our analysis, and is induced by another pro-fibrotic growth factor—IGF2 [34]. Note that IGF2 and TGFBR1 are both overlapping upregulated genes in skin and lung fibroblasts, but the upregulation of TGFB2 is only observed in SSc lung fibroblasts, and surprisingly, TGFB3 is uniquely downregulated in SSc skin compared to lung fibroblasts in our dataset. This is not consistent with a previous report [35], however, our analysis uaws dermal fibroblasts from lcSSc patients of different disease duration while most studies use dermal fibroblasts from early disease dcSSc patients [36]. This could also explain why we identified POSTN as exclusively upregulated in SSc lung fibroblasts, whereas most of the literature reports it as upregulated in SSc skin fibroblasts [37,38]. Taken together, our findings show that the IGF and TGFβ signaling pathways are active in SSc lung fibroblasts.
MMP28, the most recently identified MMP, has pro-fibrotic properties. MMP28-deficient mice are protected from bleomycin-induced lung fibrosis, and overexpression of MMP28 accelerated epithelial cell proliferation, protected them against apoptosis, enhanced cell migration, and decreased E-cadherin expression while increasing fibronectin [39]. Another unique characteristic of MMP28 is that it can be found in both the cytoplasm and the nucleus of lung epithelial cells. We show here that this pro-fibrotic MMP is uniquely downregulated in SSc lung fibroblasts as compared to healthy controls.
We also captured the downregulation of many anti-fibrotic genes in SSc lung fibroblasts, including CTSL, PLAU, and HOPX. CTSL is considered an anti-fibrotic proteinase because it is necessary for the cleavage of collagen type XVIII to release endostatin, a potent anti-fibrotic protein that regulates ECM proteins, LOX, and EGR1 [40,41,42]. PLAU is another ECM-remodeling proteinase that promotes fibrosis resolution and activates MMPs, and we have previously shown that PLAU (uPA) and its receptor PLAUR (uPAR) mediate the anti-fibrotic effects of an anti-fibrotic peptide derived from endostatin [43]. Downregulation of HOPX has also been reported in IPF alveolar epithelial cells and may contribute to the loss of regenerative processes in end-stage IPF lungs [44]. The overall fingerprint we observed in SSc lung fibroblasts suggests that anti-fibrotic molecules are downregulated while several pro-fibrotic factors are upregulated, indicating that the fibrosis is progressive despite the severity of disease in explanted SSc lungs.
4.3. A Signature of Persistent Inflammation in SSc Skin Fibroblasts
In SSc skin fibroblasts, the upregulation of IL6, HLA-DRB1, and PTGDS [25,45,46,47], together with the downregulation of SAA1 [48], indicates a shift toward an IL6-predominant, antigen-presenting inflammatory state, with reduced acute-phase signaling and enhanced prostaglandin D2-linked modulation. Increased expression of PLAUR [43] and the markers of alveolar fibroblasts, NPNT and GPM6B [49], combined with reduced MMP10, suggest reorganization of the uPA/uPAR pericellular niche. This combination would favor stable cell–matrix anchoring by limiting uPA driven plasmin generation and downstream MMP activation, thereby constraining matrix degradation and suppressing the acquisition of an invasive fibroblast phenotype [43,50,51]. Developmental and pro-fibrotic transcriptional programs are also altered as shown by the downregulation of pro-fibrotic factors (TGFB3, HOXD11) [52], and anti-fibrotic factors (ID1, GLI3, HOXD10, KLF4) [15,53,54,55], alongside the upregulation of pro-fibrotic genes (TFAP2A, DNMT3B) [15,56] suggesting attenuation of canonical TGFβ, EMT, Hedgehog/GLI, and HOX pathways, despite ongoing epigenetic remodeling. Finally, decreased PCSK9 and INMT, together with increased PTGDS, point to shifts in lipid and metabolic signaling that may modulate inflammatory and remodeling responses. Collectively, the signature of SSc skin fibroblasts is characterized by persistent inflammation and matrix engagement, but reduced TGFβ/EMT developmental signaling compared to lung fibroblasts, indicating a distinct remodeling phenotype within the lcSSc skin microenvironment.
4.4. The Overlapping Signature of SSc Skin and SSc Lung Fibroblasts
Among our selected GOI (Supplemental Table S1), only 8 showed deregulation in the same direction in SSc skin and lung fibroblasts. Of these, several growth factors were increased in SSc in both skin and lung fibroblasts, including PDGFA, WNT5A, FGF9, and IGF2. Interestingly, PDGFA has been shown to increase TGFβ1 levels in human lung fibroblasts [57], and IGF2 is increased in SSc lung fibroblasts [58]. IGF2 increases collagen type I, collagen type III, and fibronectin production in fibroblasts [59], and the fact that its expression is upregulated in both SSc skin and lung fibroblasts suggests that IGF2, similarly to PDGFA and WNT5A, is a therapeutic target for both skin and lung in SSc patients. Most genes that show general involvement in inflammation and extracellular matrix regulation share similar directionality trends in both SSc skin and SSc lung fibroblasts, i.e., downregulation of MMP9, and upregulation of THBS1 and WNT5A, suggesting that, similar to PDGFA, WNT5A, FGF9, and IGF2, these are core genes that should be considered when developing therapies to target both skin and lung fibrosis.
4.5. Different Collagens Are Deregulated in SSc Skin and SSc Lung Fibroblasts
We examined the profile of collagens, given that 44 collagen genes which encode 28 collagen proteins exist in humans [60]. We consulted ColPTMScape, a collagen post-translational modification (PTM) database managed by the Proteomics Lab that offers “human” and “lung” queries, and the enrichment of the well-known COL1A1 along with COL4A1, COL4A2, COL5A1 and COL5A2 was reported in human lungs. In our results, we identified 12 collagen genes uniquely upregulated in SSc lung fibroblasts, including COL1A1, 4 isoforms of collagen type IV (COL4A1, COL4A2, COL4A3, COL4A4), 2 isoforms of collagen type V (COL5A1, COL5A2), and 2 isoforms of collagen type XI (COL11A1, COL11A2). Collagen type IV is a major component of the basement membrane that is poorly studied in the context of pulmonary fibrosis [45]. Collagen type V is also found in basement membranes, where it plays a role in cell adhesion and matrix repair, and it can polymerize with collagen type I in lungs to create heterotypic fibrils [46,47]. Others have reported increased expression of collagen type V in early skin disease in SSc patients [48], but in our study focused on lcSSc skin fibroblasts, we did not capture that signature. Collagen type V and XI are structurally and functionally closely related and can form rare hybrid collagen molecules [49]. Interestingly, COL11A1 and COL11A2 are also exclusively upregulated in SSc lung fibroblasts. Together, they seem to play a fundamental role in fibrillogenesis by creating a core buried within major collagen fibrils, yet they can still regulate cell adhesion and wound healing [49].
The SSc skin fibroblast heatmap shows 14 DE collagen genes, including multiple members of the collagen type VI and type IX families. The signature of DE genes from the pro- and anti-fibrotic programs is more equally distributed between the upregulated and downregulated groups. For example, from the collagen type VI family of non-fibrillar collagens that are implicated in ECM organization and regulation of dermal matrix assembly, composition, and fibroblast behavior [47], we report upregulation of COL6A1 and COL6A2, accompanied by the downregulation of COL6A6, in SSc skin fibroblasts. Interestingly, COL6A6 silencing resulted in an increase in MMP1 [48], which could infer anti-fibrotic properties, an unexpected feature for a collagen type VI.
A noticeable trend in SSc skin fibroblasts is the split of the “fibril-associated collagens with interrupted triple helices” (FACIT) that includes types IX, XII, and XIV. Collagens in this subclass share unique structural characteristics such as short triple-helical domains, multiple interruptions in the triple helix, large non-collagenous domains, and flexible hinge regions [49]. They do not form fibrils, but instead bind, stabilize, and regulate the fibrils made by other collagens, and are crucial for the stability of the ECM. Since the creation of this subclass, new collagens have been added, including COL21A1 [50]. Here we report that COL9A3 and COL22A1 are upregulated in SSc skin fibroblasts, while COL9A2, COL12A1, and COL14A1 are downregulated. We previously reported increased expression of COL22A1 in SSc dermal fibroblasts and induction of its expression by TGFβ in skin in organ culture [61]. Note that in SSc lung fibroblasts, only one FACIT, COL22A1, was reported as downregulated as compared to healthy controls. We also observed the upregulation of two collagens that have anti-fibrotic properties in SSc skin fibroblasts: COL7A1 and COL18A1. As mentioned earlier, cleavage of COL18A1 by CTSL produces the anti-fibrotic matrikine endostatin [43,44,45]. The COL7A1 gene encodes anchoring fibrils, which are essential for the stability of the skin and other epithelial organs, and genetic loss results in dystrophic epidermolysis bullosa with chronic skin fragility and fibrosis [51,52].
4.6. Drugs Predicted to Return SSc Skin and SSc Lung Fibroblasts to “Normal State”
Leveraging our meta-analysis, 292 overlapping DE genes and two candidate drugs were identified as potential upstream regulators in SSc skin and SSc lung fibroblasts. Acteoside is a natural phenylethanoid glycoside from plants that has anti-inflammatory, anti-oxidant, and anti-tumor properties and is reported to treat cardiovascular disease, diabetes, and cancer [62,63]. In both murine and human lung fibroblasts, acteoside (aka verbascoside) inhibited TGFβ1-induced collagen type I expression via downregulation of Smad/non-Smad pathway and oxidative stress [64]. It also delayed the progression of renal interstitial fibrosis in diabetic nephropathy by anti-oxidation and regulating the autophagy-lysosome pathway [65].
Manidipine, a 3rd generation dihydropyridine calcium channel blocker used to treat mild to moderate hypertension, is also a predicted drug in the overlap of our meta-analysis. In fibrosis-related studies, manidipine (a) inhibited the expression of fibrillar collagens type I, type III, type IV, and the metallopeptidase inhibitor 2 TIMP2, (b) boosted the expression of matrix metallopeptidases 2 and 7 (MMP2, MMP7), and (c) regulated autophagy and inflammation, contributing to the reduction in renal fibrosis [66,67]. In mesangial cells, manidipine inhibited the transcription of several cytokines, including interleukin 1β (IL1β), but it also increased IL6, a known pro-fibrotic factor overexpressed in SSc [25,68]. Taken together, the upstream regulators analysis led to the identification of two potential drugs of interest that could have a beneficial impact on both skin and lung fibrosis, neither one of these drugs has been tested in patients with SSc or related fibrosing diseases.
4.7. SSc Lung vs. SSc Skin Fibroblasts: Differences in Disease State
More than 3500 DE genes were identified in “SSc lung vs. SSc skin” analysis, highlighting the considerable phenotypic differences in disease state in these fibroblasts. We validated that mRNA levels of ACTA2 and COL3A1 are higher in SSc lung fibroblasts, while levels of CTHRC1, IL6, and LOX are lower compared to SSc skin fibroblasts. Amongst the top 10 impacted pathways in SSc lung fibroblasts compared to SSc skin fibroblasts are the fundamental pathways “Cell adhesion molecules”, “ECM-receptor interaction”, “Focal adhesion” and “Cytokine-cytokine receptor interaction”. These were also reported in the meta-analysis that examined DE genes between healthy and diseased fibroblasts, but here we show profound differences in diseased fibroblasts originating from SSc-ILD lungs and lcSSc skin. As mentioned earlier, the fibroblast phenotype in homeostatic or disease state reflects the embryonic gene signature of the organ of origin [27], but we should also consider disease status as another factor generating differences, as SSc-ILD lungs were from severe pulmonary fibrosis, while lcSSc skin fibroblasts were collected from patients with different disease duration.
4.8. Limitations
The study has several limitations. Fibroblasts were from patients of varying disease duration, treatment history, and ages, which may all impact gene expression. In addition, lung fibroblasts are from explanted lungs, which reflect severe end-stage disease and thus might not reflect earlier disease stages. Lastly, skin and lung fibroblasts were from different donors.
5. Conclusions
To our knowledge, this is the first study comparing gene expression and pathways in human primary skin and lung fibroblasts from SSc patients. Our meta-analysis revealed that the overlap in DE genes, including collagens, and pathways is small, highlighting how different the phenotypic signatures of fibroblasts are when derived from different organs, and the need to take these signatures into consideration when developing therapeutic strategies to reverse fibrosis. SSc skin fibroblasts are dominated by immune and cytokine-driven activation, with strong inflammatory and pro-fibrotic signaling, as well as mitochondrial and metabolic stress. In contrast, SSc lung fibroblasts exhibit activation of the IGF and TGFβ signaling pathways, deep metabolic rewiring, particularly in lipid and steroid pathways, together with strong xenobiotic detoxification and hormone-linked/cancer-like signaling. These differences in skin and lung fibroblast signatures suggest that currently available treatments might not be equally effective in both tissues. From the overlap, we captured that SSc skin and lung fibroblasts share a conserved pathogenic signature defined by rewired core signaling pathways, altered cell–matrix communication, and immune-driven transcriptional dysregulation. The overlapping pathways reveal that, despite tissue-specific pressures, SSc fibroblasts converge on a common activated, stress-adapted, and pro-fibrotic state that underlies the systemic nature of the disease. Further, genes common to skin and lung fibroblasts, including WNT5A, PDGFA, and IGF2, can serve as common targets for the treatment of both skin and lung fibrosis in SSc.
Supplementary Materials
The following supporting information can be found at: www.sciepublish.com/xxx/s1, Table S1: Genes of interest; Table S2: Meta-analysis DE genes; Table S3: Meta-analysis pathways; Table S4: DE genes in SSc lung vs. SSc skin fibroblasts; Table S5: Pathways in SSc lung vs. SSc skin fibroblasts.
Statement of the Use of Generative AI and AI-Assisted Technologies in the Writing Process
During the preparation of this manuscript, the authors used Copilot in order to summarize pathways and Gene Ontology terms from the lists generated by iPathwayGuide and to generate human body outline for the graphical abstract. After using this tool/service, the authors reviewed and edited the content as needed and take full responsibility for the content of the published article.
Acknowledgments
We thank the patients who graciously agreed to donate skin and lung tissues for this research.
Author Contributions
Conceptualization, C.A.F.-B. and L.R.; Validation, S.E.K. and L.R.; Formal Analysis, S.E.K. and L.R.; Investigation, S.E.K. and L.R.; Resources, C.A.F.-B. and L.R.; Data Curation, S.E.K. and L.R.; Writing—Original Draft Preparation, S.E.K. and L.R.; Writing—Review & Editing, C.A.F.-B., S.E.K. and L.R.; Visualization, S.E.K. and L.R.; Supervision, C.A.F.-B. and L.R.; Project Administration C.A.F.-B. and L.R.; Funding Acquisition, C.A.F.-B. and L.R.
Ethics Statement
The study was conducted according to the guidelines of the Declaration of Helsinki and approved by the Institutional Review Board of the University of Pittsburgh under IRB# 970946 (for lung tissues, renewed annually since 1997) and IRB# 0403072 (for skin tissues, renewed annually since 2004).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
Data is available from the corresponding authors upon reasonable request. The RNAseq data used in this study have been deposited in the NCBI Gene Expression Omnibus (GEO) under accession numbers GSE215841 and GSE153880.
Funding
This research was funded by the National Institutes of Health, National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS), grants number K01AR083019 and K24AR060297. The authors also would like to acknowledge the SmartState Center for Inflammation and Fibrosis Research and the Kitty Trask Holt Endowment at the Medical University of South Carolina (MUSC) for financial support.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
-
Abraham DJ, Varga J. Scleroderma: From cell and molecular mechanisms to disease models. Trends Immunol. 2005, 26, 587–595. DOI:10.1016/j.it.2005.09.004 [Google Scholar]
-
Mayes MD, Lacey JV, Jr., Beebe-Dimmer J, Gillespie BW, Cooper B, Laing TJ, et al. Prevalence, incidence, survival, and disease characteristics of systemic sclerosis in a large US population. Arthritis Rheum. 2003, 48, 2246–2255. DOI:10.1002/art.11073 [Google Scholar]
-
Peoples C, Medsger TA, Jr., Lucas M, Rosario BL, Feghali-Bostwick CA. Gender differences in systemic sclerosis: Relationship to clinical features, serologic status and outcomes. J. Scleroderma Relat. Disord. 2016, 1, 204–212. DOI:10.5301/jsrd.5000209 [Google Scholar]
-
Hachulla E, Launay D. Diagnosis and classification of systemic sclerosis. Clin. Rev. Allergy Immunol. 2011, 40, 78–83. DOI:10.1007/s12016-010-8198-y [Google Scholar]
-
Steen VD, Medsger TA, Jr. Severe organ involvement in systemic sclerosis with diffuse scleroderma. Arthritis Rheum. Off. J. Am. Coll. Rheumatol. 2000, 43, 2437–2444. DOI:10.1002/1529-0131(200011)43:11%3C2437::AID-ANR10%3E3.0.CO;2-U [Google Scholar]
-
Medsger TAJ. Systemic sclerosis (scleroderma): Clinical aspects. In Arthritis and Allied Conditions: A Textbook of Rheumatology; Koopman WJ, Ed.; Williams & Wilkins: Baltimore, MD, USA, 1997; pp. 1433–1464. [Google Scholar]
-
Steen V, Medsger T. Systemic sclerosis (scleroderma). In Prognosis in the Rheumatic Diseases; Springer: Berlin/Heidelberg, Germany, 1991; pp. 213–231. [Google Scholar]
-
Ruaro B, Soldano S, Smith V, Paolino S, Contini P, Montagna P, et al. Correlation between circulating fibrocytes and dermal thickness in limited cutaneous systemic sclerosis patients: A pilot study. Rheumatol. Int. 2019, 39, 1369–1376. DOI:10.1007/s00296-019-04315-7 [Google Scholar]
-
Cottin V, Brown KK. Interstitial lung disease associated with systemic sclerosis (SSc-ILD). Respir. Res. 2019, 20, 13. DOI:10.1186/s12931-019-0980-7 [Google Scholar]
-
Steen VD, Medsger TA. Changes in causes of death in systemic sclerosis, 1972–2002. Ann. Rheum. Dis. 2007, 66, 940–944. DOI:10.1136/ard.2006.066068 [Google Scholar]
-
Garrett SM, Baker Frost D, Feghali-Bostwick C. The mighty fibroblast and its utility in scleroderma research. J. Scleroderma Relat. Disord. 2017, 2, 100–107. DOI:10.5301/jsrd.5000240 [Google Scholar]
-
Romano E, Rosa I, Fioretto BS, Matucci-Cerinic M, Manetti M. The role of pro-fibrotic myofibroblasts in systemic sclerosis: From origin to therapeutic targeting. Curr. Mol. Med. 2022, 22, 209–239. DOI:10.2174/0929867328666210325102749 [Google Scholar]
-
Phan SH. Biology of fibroblasts and myofibroblasts. Proc. Am. Thorac. Soc. 2008, 5, 334–337. DOI:10.1513/pats.200708-146DR [Google Scholar]
-
Renaud L, Waldrep KM, da Silveira WA, Pilewski JM, Feghali-Bostwick CA. First Characterization of the Transcriptome of Lung Fibroblasts of SSc Patients and Healthy Donors of African Ancestry. Int. J. Mol. Sci. 2023, 24, 3645. DOI:10.3390/ijms24043645 [Google Scholar]
-
Malaab M, Renaud L, Takamura N, Zimmerman KD, da Silveira WA, Ramos PS, et al. Anti-fibrotic factor KLF4 is repressed by the miR-10/TFAP2A/TBX5 axis in dermal fibroblasts: Insights from twins discordant for systemic sclerosis. Ann. Rheum. Dis. 2022, 81, 268–277. DOI:10.1136/annrheumdis-2021-221050 [Google Scholar]
-
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. DOI:10.1186/s13059-014-0550-8 [Google Scholar]
-
Roach KM, Duffy SM, Coward W, Feghali-Bostwick C, Wulff H, Bradding P. The K+ channel KCa3.1 as a novel target for idiopathic pulmonary fibrosis. PLoS ONE 2013, 8, e85244. DOI:10.1371/journal.pone.0085244 [Google Scholar]
-
Wu C-Y, Hsu W-L, Tsai M-H, Chai C-Y, Yen C-J, Chen C-H, et al. A potential new approach for treating systemic sclerosis: Dedifferentiation of SSc fibroblasts and change in the microenvironment by blocking store-operated Ca2+ entry. PLoS ONE 2019, 14, e0213400. DOI:10.1371/journal.pone.0213400 [Google Scholar]
-
Kittana N. Calcium signaling in cardiac fibroblasts: Roles in fibrosis and therapeutic implications. Cardiovasc. Drugs Ther. 2026, 40, 393–410. DOI:10.1007/s10557-025-07699-w [Google Scholar]
-
Atanelishvili I, Akter T, Noguchi A, Vuyiv O, Wollin L, Silver RM, et al. Anti-fibrotic efficacy of nintedanib in a cellular model of systemic sclerosis-associated interstitial lung disease. Clin. Exp. Rheumatol. 2019, 37, 115–124. [Google Scholar]
-
Roach KM, Wulff H, Feghali-Bostwick C, Amrani Y, Bradding P. Increased constitutive αSMA and Smad2/3 expression in idiopathic pulmonary fibrosis myofibroblasts is KCa3.1-dependent. Respir. Res. 2014, 15, 155. DOI:10.1186/s12931-014-0155-5 [Google Scholar]
-
Xu D, Li T, Wang R, Mu R. Expression and pathogenic analysis of integrin family genes in systemic sclerosis. Front. Med. 2021, 8, 674523. DOI:10.3389/fmed.2021.674523 [Google Scholar]
-
Paine R, Ward PA. Cell adhesion molecules and pulmonary fibrosis. Am. J. Med. 1999, 107, 268–279. DOI:10.1016/S0002-9343(99)00226-0 [Google Scholar]
-
Dufour AM, Alvarez M, Russo B, Chizzolini C. Interleukin-6 and type-I collagen production by systemic sclerosis fibroblasts are differentially regulated by interleukin-17A in the presence of transforming growth factor-beta 1. Front. Immunol. 2018, 9, 1865. DOI:10.3389/fimmu.2018.01865 [Google Scholar]
-
Feghali CA, Bost KL, Boulware DW, Levy LS. Control of IL-6 expression and response in fibroblasts from patients with systemic sclerosis. Autoimmunity 1994, 17, 309–318. DOI:10.3109/08916939409010671 [Google Scholar]
-
Shima Y. Cytokines involved in the pathogenesis of SSc and problems in the development of anti-cytokine therapy. Cells 2021, 10, 1104. DOI:10.3390/cells10051104 [Google Scholar]
-
Forte E, Ramialison M, Nim HT, Mara M, Li JY, Cohn R, et al. Adult mouse fibroblasts retain organ-specific transcriptomic identity. Elife 2022, 11, e71008. DOI:10.7554/eLife.71008 [Google Scholar]
-
Hung CF, Rohani MG, Lee S-S, Chen P, Schnapp LM. Role of IGF-1 pathway in lung fibroblast activation. Respir. Res. 2013, 14, 102. DOI:10.1186/1465-9921-14-102 [Google Scholar]
-
Pilewski JM, Liu L, Henry AC, Knauer AV, Feghali-Bostwick CA. Insulin-like growth factor binding proteins 3 and 5 are overexpressed in idiopathic pulmonary fibrosis and contribute to extracellular matrix deposition. Am. J. Pathol. 2005, 166, 399–407. DOI:10.1016/S0002-9440(10)62263-8 [Google Scholar]
-
Veraldi KL, Feghali-Bostwick CA. Insulin-like growth factor binding proteins-3 and-5: Central mediators of fibrosis and promising new therapeutic targets. Open Rheumatol. J. 2012, 6, 140–145. DOI:10.2174/1874312901206010140 [Google Scholar]
-
Yasuoka H, Yamaguchi Y, Feghali-Bostwick CA. The pro-fibrotic factor IGFBP-5 induces lung fibroblast and mononuclear cell migration. Am. J. Respir. Cell Mol. Biol. 2009, 41, 179–188. DOI:10.1165/rcmb.2008-0211OC [Google Scholar]
-
Nguyen X-X, Nishimoto T, Takihara T, Mlakar L, Bradshaw AD, Feghali-Bostwick C. Lysyl oxidase directly contributes to extracellular matrix production and fibrosis in systemic sclerosis. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2021, 320, L29–L40. DOI:10.1152/ajplung.00173.2020 [Google Scholar]
-
Sun T, Huang Z, Liang W-C, Yin J, Lin WY, Wu J, et al. TGFβ2 and TGFβ3 isoforms drive fibrotic disease pathogenesis. Sci. Transl. Med. 2021, 13, eabe0407. DOI:10.1126/scitranslmed.abe0407 [Google Scholar]
-
Garrett SM, Hsu E, Thomas JM, Pilewski JM, Feghali-Bostwick C. Insulin-like growth factor (IGF)-II-mediated fibrosis in pathogenic lung conditions. PLoS ONE 2019, 14, e0225422. DOI:10.1371/journal.pone.0225422 [Google Scholar]
-
Sun T, Vander Heiden JA, Gao X, Yin J, Uttarwar S, Liang W-C, et al. Isoform-selective TGF-β3 inhibition for systemic sclerosis. Med 2024, 5, 132–147.e7. DOI:10.1016/j.medj.2023.12.011 [Google Scholar]
-
Allanore Y. Limited cutaneous systemic sclerosis: The unfairly neglected subset. J. Scleroderma Relat. Disord. 2016, 1, 241–246. DOI:10.5301/jsrd.5000216 [Google Scholar]
-
Yamaguchi Y, Ono J, Masuoka M, Ohta S, Izuhara K, Ikezawa Z, et al. Serum periostin levels are correlated with progressive skin sclerosis in patients with systemic sclerosis. Br. J. Dermatol. 2013, 168, 717–725. DOI:10.1111/bjd.12117 [Google Scholar]
-
Yoshihara T, Nanri Y, Nunomura S, Yamaguchi Y, Feghali-Bostwick C, Ajito K, et al. Periostin plays a critical role in the cell cycle in lung fibroblasts. Respir. Res. 2020, 21, 38. DOI:10.1186/s12931-020-1299-0 [Google Scholar]
-
Maldonado M, Salgado-Aguayo A, Herrera I, Cabrera S, Ortíz-Quintero B, Staab-Weijnitz CA, et al. Upregulation and nuclear location of MMP28 in alveolar epithelium of idiopathic pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2018, 59, 77–86. DOI:10.1165/rcmb.2017-0223OC [Google Scholar]
-
Ferreras M, Felbor U, Lenhard T, Olsen BR, Delaissé J-M. Generation and degradation of human endostatin proteins by various proteinases. FEBS Lett. 2000, 486, 247–251. DOI:10.1016/S0014-5793(00)02249-3 [Google Scholar]
-
Mouawad JE, Sharma S, Renaud L, Pilewski JM, Nadig SN, Feghali-Bostwick C. Reduced Cathepsin L expression and secretion into the extracellular milieu contribute to lung fibrosis in systemic sclerosis. Rheumatology 2023, 62, 1306–1316. DOI:10.1093/rheumatology/keac411 [Google Scholar]
-
Yamaguchi Y, Takihara T, Chambers RA, Veraldi KL, Larregina AT, Feghali-Bostwick CA. A peptide derived from endostatin ameliorates organ fibrosis. Sci. Transl. Med. 2012, 4, ra171–ra136. DOI:10.1126/scitranslmed.3003421 [Google Scholar]
-
Sharma S, Watanabe T, Nishimoto T, Takihara T, Mlakar L, Nguyen X-X, et al. E4 engages uPAR and enolase-1 and activates urokinase to exert anti-fibrotic effects. JCI Insight 2021, 6, e144935. DOI:10.1172/jci.insight.144935 [Google Scholar]
-
Ota C, Ng-Blichfeldt J-P, Korfei M, Alsafadi HN, Lehmann M, Skronska-Wasek W, et al. Dynamic expression of HOPX in alveolar epithelial cells reflects injury and repair during the progression of pulmonary fibrosis. Sci. Rep. 2018, 8, 12983. DOI:10.1038/s41598-018-31214-x [Google Scholar]
-
Feghali CA. Regulation of Fibroblast Function in Systemic Sclerosis; Tulane University: New Orleans, LA, USA, 1992. [Google Scholar]
-
Fang D, Yuan X, Yi J, Li Z, Li X, Guo W, et al. Targeting PTGDS inhibits pro-inflammatory fibroblasts associated with skin fibrosis in systemic sclerosis. Rheumatology 2025, 64, 5551–5561. DOI:10.1093/rheumatology/keaf276 [Google Scholar]
-
Ünlü B, Türsen Ü, Rajabi Z, Jabalameli N, Rajabi F. The immunogenetics of systemic sclerosis. In The Immunogenetics of Dermatologic Diseases; Springer: Berlin/Heidelberg, Germany, 2022; pp. 259–298. [Google Scholar]
-
Ye RD, Sun L. Emerging functions of serum amyloid A in inflammation. J. Leucoc. Biol. 2015, 98, 923–929. DOI:10.1189/jlb.3VMR0315-080R [Google Scholar]
-
Tsukui T, Wolters PJ, Sheppard D. Alveolar fibroblast lineage orchestrates lung inflammation and fibrosis. Nature 2024, 631, 627–634. DOI:10.1038/s41586-024-07660-1 [Google Scholar]
-
Brandenberger R, Schmidt A, Linton J, Wang D, Backus C, Denda S, et al. Identification and characterization of a novel extracellular matrix protein nephronectin that is associated with integrin α8β1 in the embryonic kidney. J. Cell Biol. 2001, 154, 447–458. DOI:10.1083/jcb.200103069 [Google Scholar]
-
Drabek K, van de Peppel J, Eijken M, van Leeuwen JP. GPM6B regulates osteoblast function and induction of mineralization by controlling cytoskeleton and matrix vesicle release. J. Bone Miner. Res. 2011, 26, 2045–2051. DOI:10.1002/jbmr.435 [Google Scholar]
-
Murata H, Zhou L, Ochoa S, Hasan A, Badiavas E, Falanga V. TGF-β3 stimulates and regulates collagen synthesis through TGF-β1-dependent and independent mechanisms. J. Investig. Dermatol. 1997, 108, 258–262. DOI:10.1111/1523-1747.ep12286451 [Google Scholar]
-
Je Y-J, Choi D-K, Sohn K-C, Kim H-R, Im M, Lee Y, et al. Inhibitory role of Id1 on TGF-β-induced collagen expression in human dermal fibroblasts. Biochem. Biophys. Res. Commun. 2014, 444, 81–85. DOI:10.1016/j.bbrc.2014.01.010 [Google Scholar]
-
Li X, Ma T-K, Wang P, Shi H, Hai S, Qin Y, et al. HOXD10 attenuates renal fibrosis by inhibiting NOX4-induced ferroptosis. Cell Death Dis. 2024, 15, 398. DOI:10.1038/s41419-024-06780-w [Google Scholar]
-
Li L, Ran J, Li L, Chen G, Zhang S, Wang Y. Gli3 is a novel downstream target of miR-200a with an anti-fibrotic role for progression of liver fibrosis in vivo and in vitro. Mol. Med. Rep. 2020, 21, 1861–1871. DOI:10.3892/mmr.2020.10997 [Google Scholar]
-
Wu K, Ma F, Shen J, Zhang H, Wan Y, He X, et al. LncRNA FPASL suppresses fibroblast proliferation through its DNA methylation via DNMT3b in hypertrophic scar: DNMT3b-mediated lncRNA FPASL in fibroblast proliferation. Acta Biochim. et Biophys. Sin. 2022, 54, 1854–1862. DOI:10.3724/abbs.2022181 [Google Scholar]
-
Takamura N, Renaud L, da Silveira WA, Feghali-Bostwick C. PDGF promotes dermal fibroblast activation via a novel mechanism mediated by signaling through MCHR1. Front. Immunol. 2021, 12, 745308. DOI:10.3389/fimmu.2021.745308 [Google Scholar]
-
Hsu E, Feghali-Bostwick CA. Insulin-like growth factor-II is increased in systemic sclerosis-associated pulmonary fibrosis and contributes to the fibrotic process via Jun N-terminal kinase-and phosphatidylinositol-3 kinase-dependent pathways. Am. J. Pathol. 2008, 172, 1580–1590. DOI:10.2353/ajpath.2008.071021 [Google Scholar]
-
Waldrep KM, Rodgers JI, Garrett SM, Wolf BJ, Feghali-Bostwick CA. The role of SOX9 in IGF-II-mediated pulmonary fibrosis. Int. J. Mol. Sci. 2023, 24, 11234. DOI:10.3390/ijms241411234 [Google Scholar]
-
Salamito M, Nauroy P, Ruggiero F. The collagen superfamily: Everything you always wanted to know. In The Collagen Superfamily and Collagenopathies; Springer: Berlin/Heidelberg, Germany, 2021; pp. 1–22. [Google Scholar]
-
Watanabe T, Baker Frost D, Mlakar L, Heywood J, da Silveira WA, Hardiman G, et al. A human skin model recapitulates systemic sclerosis dermal fibrosis and identifies COL22A1 as a TGFβ early response gene that mediates fibroblast to myofibroblast transition. Genes 2019, 10, 75. DOI:10.3390/genes10020075 [Google Scholar]
-
Marčetić M, Bufan B, Drobac M, Antić Stanković J, Arsenović Ranin N, Milenković MT, et al. Multifaceted biological properties of verbascoside/acteoside: Antimicrobial, cytotoxic, anti-inflammatory, and immunomodulatory effects. Antibiotics 2025, 14, 697. DOI:10.3390/antibiotics14070697 [Google Scholar]
-
Xiao Y, Ren Q, Wu L. The pharmacokinetic property and pharmacological activity of acteoside: A review. Biomed. Pharmacother. 2022, 153, 113296. DOI:10.1016/j.biopha.2022.113296 [Google Scholar]
-
Chen C-Y, Tung H-Y, Tseng Y-F, Huang J-S, Shi L-S, Ye Y-L. Verbascoside and isoverbascoside ameliorate transforming growth factor β1-induced collagen expression by lung fibroblasts through Smad/non-Smad signaling pathways. Life Sci. 2022, 308, 120950. DOI:10.1016/j.lfs.2022.120950 [Google Scholar]
-
Zhou M, Zhang S, Bai X, Cai Y, Zhang Z, Zhang P, et al. Acteoside delays the fibrosis process of diabetic nephropathy by anti-oxidation and regulating the autophagy-lysosome pathway. Eur. J. Pharmacol. 2024, 978, 176715. DOI:10.1016/j.ejphar.2024.176715 [Google Scholar]
-
Roth M, Eickelberg O, Kohler E, Erne P, Block LH. Ca2+ channel blockers modulate metabolism of collagens within the extracellular matrix. Proc. Natl. Acad. Sci. USA 1996, 93, 5478–5482. DOI:10.1073/pnas.93.11.5478 [Google Scholar]
-
Liao C-T, Lin Y-C, Huang H-J, Liu W-C, Kan W-C, Chiu H-W. Docking and database screening identify manidipine as a potential modulator of matrix metalloproteinase-7 in chronic kidney disease. Biomed. Pharmacother. 2025, 193, 118748. DOI:10.1016/j.biopha.2025.118748 [Google Scholar]
-
Baker Frost D, Savchenko A, Takamura N, Wolf B, Fierkens R, King K, et al. A positive feedback loop exists between estradiol and IL-6 and contributes to dermal fibrosis. Int. J. Mol. Sci. 2024, 25, 7227. DOI:10.3390/ijms25137227 [Google Scholar]