Hepatic Stellate Cells Interact with the Immune System: A Bidirectional Crosstalk Network Driving Liver Fibrosis

Hepatic Stellate Cells Interact with the Immune System: A Bidirectional Crosstalk Network Driving Liver Fibrosis
Chu-Ning Zhang 1,2 Guan-Yue Shan 1,3 Hui Wan 1,2 Yu-Xin Zhang 1,2 Zhi-Cheng Gao 1,2 Hai-Jun Li 1,2,4,*
Received: 28 January 2026 Revised: 24 February 2026 Accepted: 31 March 2026 Published: 10 April 2026

© 2026 The authors. This is an open access article under the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/).
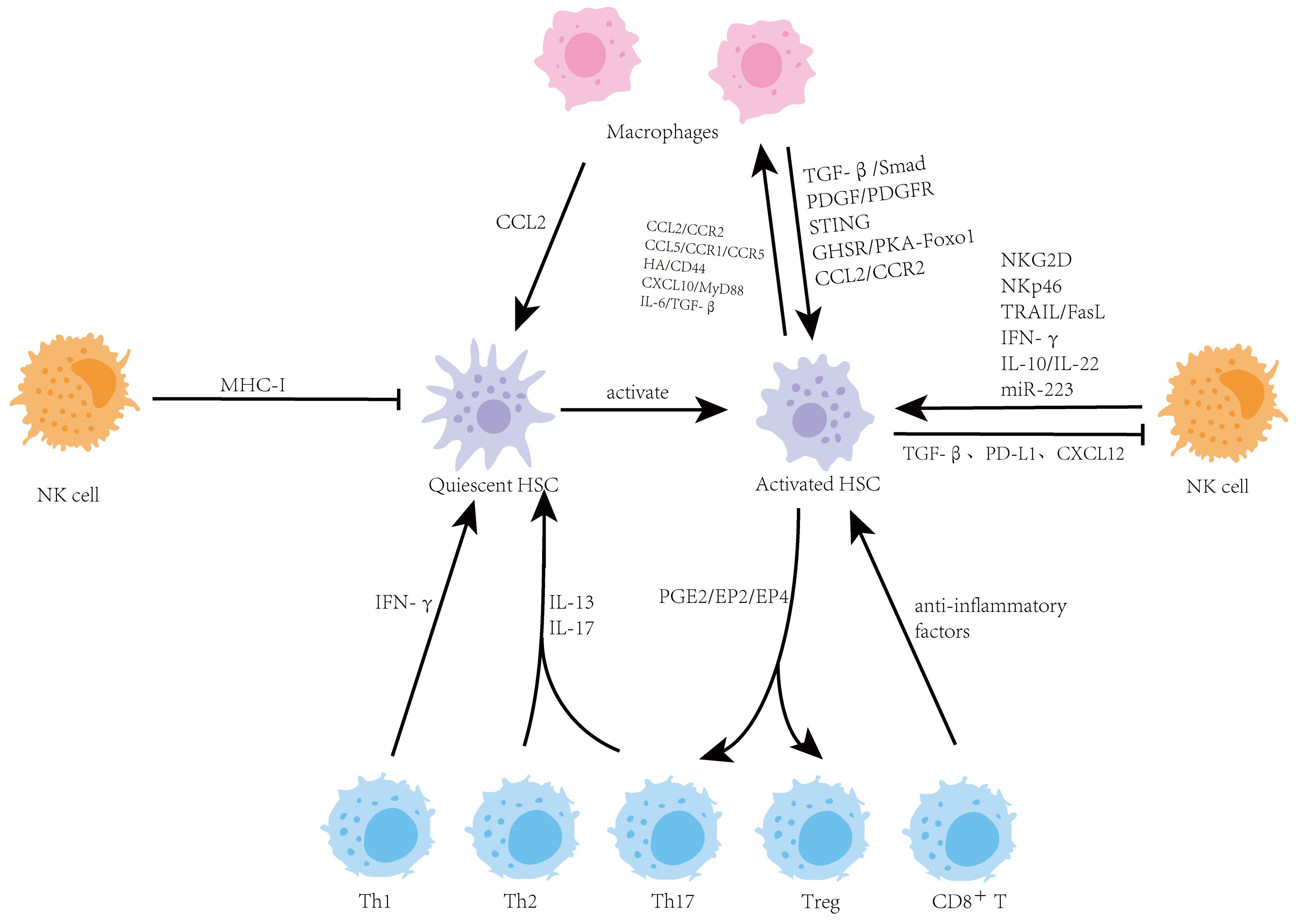
Graphical Abstract
1. Introduction
The liver, due to its unique dual blood supply (portal vein and hepatic artery), forms a highly specialized immune microenvironment. This environment is rich in various immune cell populations, ranging from innate immune cells (such as macrophages and NK cells) to adaptive immune cells (such as T cells and B cells). Under physiological conditions, precise molecular interactions exist between these immune cells and parenchymal cells (hepatocytes, cholangiocytes) as well as non-parenchymal cells (HSCs, liver sinusoidal endothelial cells), maintaining a dynamic balance between immune tolerance and immune surveillance. This effectively defends against pathogens while preventing host tissue damage. However, under the sustained influence of chronic injury factors (such as viruses, alcohol, and metabolic disorders), this delicate balance is disrupted, leading to hepatic fibrosis characterized by HSC activation and excessive ECM deposition. The transformation of HSCs from quiescent lipid-storing cells into activated myofibroblasts represents the “final common pathway” of the fibrotic process. Therefore, deciphering the activation mechanisms of HSCs is central to understanding hepatic fibrosis. However, growing evidence suggests that HSC activation is not an independent event but is deeply embedded within a complex regulatory network composed of various immune cells. For instance, in viral hepatitis, cytotoxic T cells attack infected hepatocytes, releasing a large number of inflammatory cytokines that indirectly activate HSCs; in metabolic dysfunction-associated steatohepatitis (MASH, formerly known as non-alcoholic steatohepatitis, NASH), aberrant polarization of macrophages drives HSC activation and fibrosis progression through the secretion of factors such as transforming growth factor-β (TGF-β).
In addition to the local hepatic immune environment, the spleen has recently been recognized as a crucial extrahepatic regulator of liver fibrosis, forming a functional “spleen-liver axis”. As the largest secondary lymphoid organ, the spleen serves as a major reservoir for monocytes and T cells and plays a central role in systemic immune surveillance. During chronic liver injury, splenic immune cells—particularly macrophages and regulatory T cells—are recruited to the liver via the portal circulation, where they interact with HSCs and amplify the profibrotic response. Splenomegaly, commonly observed in patients with advanced metabolic dysfunction-associated Steatotic liver disease (MASLD, formerly known as non-alcoholic fatty liver disease, NAFLD) or cirrhosis, reflects this systemic immune dysregulation and is associated with increased migration of pro-inflammatory cells to the liver, thereby exacerbating HSC activation and ECM deposition. Conversely, activated HSCs may release soluble mediators into the systemic circulation, modulating the function of splenic immune cells, establishing a bidirectional spleno-hepatic interaction that sustains chronic inflammation. From a clinical perspective, ultrasonographic assessment of the longitudinal spleen diameter has been proposed as a non-invasive biomarker for distinguishing MASH from simple steatosis, with a cutoff value of approximately 116 mm showing high specificity and sensitivity, further supporting the pivotal role of the spleen-liver axis in fibrosis pathogenesis [1]. Therefore, incorporating the spleen-liver axis into the current HSC-centric fibrosis model provides a more systemic perspective on fibrosis pathogenesis.
Recent advances, particularly in single-cell sequencing and spatial transcriptomics, have provided unprecedented insights into the heterogeneity of hepatic immune cells and HSCs, and have precisely mapped their intercellular communication networks. These findings compel us to reassess liver diseases from a more systemic and dynamic perspective. This review will primarily focus on dissecting the bidirectional interactions between HSCs and three key immune cell types central to liver fibrosis: macrophages, NK cells, and T cells [2]. The roles of other immune populations, such as B cells, neutrophils, and innate lymphoid cells (ILCs), will also be briefly discussed, as emerging evidence implicates them in regulating the fibrotic network via antibody-independent mechanisms, neutrophil extracellular traps (NETs) formation, and cytokine secretion, respectively [3,4,5]. By systematically elucidating this core regulatory network of HSC-immune cell interactions, this review aims to provide a theoretical foundation for developing precise therapeutic and preventive strategies targeting key immunoregulatory nodes in liver fibrosis.
2. HSC and Macrophages in Liver Fibrosis
2.1. Overview of the Relationship Between HSC and Macrophages
Macrophages represent the most critical and complex immune cell populations that regulate the fate of HSCs [6]. This complexity is evident not only in the heterogeneity of their cellular origins but also in the dynamic nature of their interactions [7]. Macrophages exhibit high heterogeneity, with distinct subsets precisely regulating HSC activation, proliferation, apoptosis, and clearance through diverse molecular mechanisms [7]. This intricate interaction network has become a central focus of liver fibrosis research [6]. In the liver, macrophages are primarily of two types: embryonically derived, self-renewing resident Kupffer cells, and monocyte-derived macrophages recruited from the bone marrow or peripheral blood during liver injury. These two macrophage populations, together with HSCs, form the core functional unit of the hepatic microenvironment, participating in bidirectional regulatory networks that govern hepatic homeostasis, fibrotic progression, and fibrosis reversal [8].
Under physiological conditions, Kupffer cells maintain hepatic immune tolerance by secreting anti-inflammatory factors and interacting with quiescent HSCs to suppress their aberrant activation, thereby preserving normal liver structure and function [9]. When the liver undergoes chronic injury, this homeostatic balance is disrupted: injury signals first activate Kupffer cells, which subsequently recruit monocyte-derived macrophages. Both macrophage populations regulate HSC activation, proliferation, survival, and phenotypic transition through the secretion of various cytokines, chemokines, and matrix remodelling molecules [10]. Activated HSCs, in turn, influence macrophage recruitment, polarization, and functional reprogramming by secreting chemokines, ECM components, and immunomodulatory molecules, creating a dynamic “cross-talk” network [11]. This interaction is not unidirectional but exhibits remarkable plasticity depending on the degree of liver damage and changes in the inflammatory microenvironment. During fibrosis progression, macrophages display pro-fibrotic synergistic effects, whereas during injury resolution, they transition to pro-reparative functions, thus forming the core regulatory axis that determines the development or regression of liver fibrosis.
2.2. Macrophage-Driven HSC Activation During Fibrosis Progression
The activation of HSCs by macrophages represents a pivotal step in the initiation and progression of liver fibrosis [12]. During fibrogenesis, macrophages with specific functional phenotypes, including M2 macrophages, directly trigger HSC activation programs by secreting various key factors [13]. Meanwhile, macrophages of different origins act synergistically in both temporal and spatial dimensions to collectively amplify fibrogenic effects [14]. TGF-β is the most critical pro-fibrotic factor secreted by macrophages and serves as the core signaling molecule driving HSC activation [9]. TGF-β, primarily secreted by M2 macrophages, binds to receptors on HSC surfaces, activating Smad2/3/4 transcription complexes, which upregulate the expression of pro-fibrotic molecules such as α-smooth muscle actin (α-SMA), type I and III collagen, and hyaluronan synthase 2 (HAS2) [15]. This process promotes the transition of quiescent HSCs to a myofibroblast-like phenotype, enhancing their ECM synthesis capacity. Additionally, macrophages downregulate the endogenous TGF-β receptor inhibitor BAMBI in HSCs via TLR4 signaling, further amplifying TGF-β signaling and exacerbating HSC activation [16].
Unlike the direct pro-fibrotic effect of M2 macrophages, classically activated M1 macrophages primarily promote HSC activation indirectly through the inflammatory microenvironment they establish. In chronic liver injury such as MASLD (previously termed NAFLD), M1 macrophages become polarized in response to lipopolysaccharide (LPS), free fatty acids, or Th1-type cytokines such as interferon-γ (IFN-γ) [17]. These M1 macrophages are the major source of pro-inflammatory cytokines including tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), and reactive oxygen species (ROS) [17]. TNF-α and IL-1β not only act directly on HSCs to activate inflammatory signaling pathways such as NF-κB and JNK, inducing a pro-inflammatory and anti-apoptotic phenotype in HSCs, but also sustain the overall hepatic inflammatory state, providing a favorable microenvironment for persistent HSC activation. Meanwhile, abundant ROS released by M1 macrophages directly induce hepatocyte injury, recruit additional inflammatory cells, and serve as important signaling molecules. ROS enhance growth factor receptor signal transduction (e.g., PDGF receptor) by oxidizing protein tyrosine phosphatases (PTPs) and activates transcription factors, including NF-κB and AP-1 in HSCs, thereby directly or indirectly promoting HSC activation and ECM production [18]. Thus, during fibrosis progression, M1 macrophages establish a critical foundation for HSC activation by shaping an inflammatory and oxidative stress-rich microenvironment.
Members of the platelet-derived growth factor (PDGF) family (PDGF-B, PDGF-C, PDGF-D) represent another critical factor mediating macrophage-induced HSC activation. PDGF secreted by macrophages binds to PDGFR-β on HSC surfaces, activating downstream proliferation and migration signaling pathways that significantly enhance the proliferative capacity of activated HSCs [19]. Experimental data indicate that PDGF-C overexpression can directly induce liver fibrosis and hepatocellular carcinoma, while blocking PDGFR-β signaling effectively inhibits HSC activation and fibrosis progression, underscoring the importance of the PDGF/PDGFR pathway in macrophage-HSC cross-talk [20].
Beyond these classical factors, recent studies have revealed a more diverse set of mechanisms by which macrophages activate HSCs during fibrogenesis. Macrophages can sense increased ECM stiffness in fibrotic livers, activating intracellular STING pathways through calcium influx and mitochondrial damage, subsequently releasing pro-inflammatory cytokines that drive HSC activation. This creates a mechanobiological positive feedback loop of “ECM stiffness-macrophage-HSC” [21]. Experimental research indicates that this activation is triggered by calcium influx mediated by the mechanosensitive channel PIEZO1, leading to mitochondrial damage and mtDNA release, which, in turn, drives the pro-fibrotic macrophage phenotype via the cGAS-STING axis [22]. Clinical evidence further demonstrates that, in human liver tissues from MASH and viral hepatitis-induced cirrhosis, elevated local stiffness significantly correlates with sustained STING pathway activation in macrophages [23]. Moreover, the growth hormone secretagogue receptor (GHSR) in macrophages significantly upregulates pro-fibrotic cytokines such as TNF-α and IL-1β by activating the PKA-Foxo1 signaling axis, indirectly promoting HSC activation [24]. Additionally, chitinase 3-like protein 1 (CHI3L1) from macrophages binds to the IL-13Rα2 receptor on HSCs, activating downstream pro-fibrotic signals, providing new molecular insights into how immune cell secretions directly target HSCs during fibrosis progression [25].
The regulation of HSCs activation by macrophages exhibits remarkable origin specificity, manifesting as distinct regulatory patterns toward quiescent versus activated HSCs, differential molecular interaction modes, and spatiotemporal heterogeneity in signal initiation. This specificity determines that macrophage-HSC crosstalk at different activation stages employs unique ligand-receptor pairs and signaling pathways, ultimately shaping the direction and intensity of fibrotic progression.
In summary, during fibrosis progression, macrophages adopt pro-inflammatory and pro-fibrotic phenotypes—including M1 and M2 subsets—that drive HSC activation through multiple mechanisms: direct stimulation via TGF-β and PDGF, indirect promotion through inflammatory mediators and ROS, and mechanobiological signaling via STING pathway activation. Different macrophage populations exhibit origin-specific functions, with Kupffer cells serving as initiators and monocyte-derived macrophages as sustainers of the fibrotic process.
2.3. HSC-Mediated Macrophage Recruitment and Polarization During Fibrogenesis
Clinical studies have identified a positive association between the number of activated HSCs in cirrhotic liver tissues, CCL2 expression levels, and the degree of macrophage infiltration [26]. Patients with high CCL2 expression exhibit faster fibrosis progression and higher recurrence risks, confirming the critical role of HSC-mediated macrophage recruitment in clinical fibrosis progression [27]. During fibrogenesis, activated HSCs construct a “signal gradient” for macrophage recruitment by actively secreting chemokines and ECM components, recruiting large numbers of monocytes to infiltrate liver injury sites and differentiate into macrophages. This establishes a vicious cycle of “macrophage activation of HSCs—activated HSCs recruiting macrophages”, which serves as a fundamental pathological basis for the continuous progression of liver fibrosis [12].
CCL2 is a core chemokine secreted by activated HSCs during fibrosis progression, forming the key signaling axis mediating monocyte recruitment by binding to the CCR2 receptor on monocytes [27]. It is noteworthy that macrophages regulate HSC activation through distinct temporal mechanisms: Kupffer cells are activated early during injury, secreting CCL2 to recruit monocyte-derived macrophages while directly initiating the initial activation of HSCs [15]. In contrast, monocyte-derived macrophages are recruited in large numbers during the prolonged injury phase, becoming the major source of pro-fibrotic factors (TGF-β, PDGF, etc.) that continuously drive HSC activation and ECM accumulation [28]. Together, they constitute a “dual-driving” mechanism for HSC activation. In chronic liver injury models, activated HSCs express high levels of CCL2, creating a localized high-concentration signal gradient through the portal venous circulation that recruits monocytes from the bone marrow to infiltrate the liver [29]. These monocytes further differentiate into macrophages in the damaged microenvironment, replenishing the hepatic macrophage pool and perpetuating the fibrotic response [30]. Studies have confirmed that blocking the CCL2/CCR2 signaling pathway significantly reduces liver infiltration of monocyte-derived macrophages while inhibiting HSC activation and fibrosis progression [29]. Furthermore, genetic or pharmacological disruption of the CCL2/CCR2 axis effectively attenuates experimental liver fibrosis progression [27]. Pharmacological inhibition of CCL2 accelerates reversal of liver fibrosis by reducing Ly-6C+ macrophage infiltration, and specific CCL2 knockout in HSCs effectively disrupts this vicious fibrotic cycle [31].
In addition to CCL2, activated HSCs secrete CCL5 during fibrogenesis [32], which binds to CCR1 and CCR5 on macrophages, further enhancing their recruitment in fibrotic and inflammatory contexts. A study on spinal cord injury demonstrated that this CCL5/CCR5 axis contributes to macrophage recruitment and their alternative activation [32]. Moreover, hyaluronic acid (HA) synthesized by activated HSCs participates in macrophage recruitment during fibrosis progression. HSCs upregulate the synthesis enzyme HAS2 to produce HA, which then binds to CD44 receptors on macrophages, facilitating their migration and pro-fibrotic function [33].
The impact of activated HSCs on macrophage polarization during fibrogenesis is complex and multifaceted, driving macrophages toward different functional subtypes that collectively exacerbate the fibrotic process. Specifically, in the early stages of inflammation, activated HSCs actively promote macrophage polarization toward the classically activated pro-inflammatory M1 phenotype by secreting specific cytokines and chemokines. For example, in addition to CXCL10, activated HSCs produce large amounts of TNF-α and IL-1β [34]. These cytokines bind to corresponding receptors on macrophage surfaces, activating key transcription pathways such as NF-κB, inducing macrophages to highly express inducible nitric oxide synthase (iNOS) and release large amounts of ROS and pro-inflammatory factors (e.g., IL-6, IL-12) [35], thereby amplifying local inflammatory responses and creating conditions for fibrosis. Simultaneously, during fibrosis progression and persistence, the role of activated HSCs in inducing macrophage polarization toward the alternatively activated M2 phenotype becomes more prominent and critical. Activated HSCs are the primary cellular source of TGF-β in the liver, and TGF-β strongly induces macrophages to express M2 markers such as arginase-1 (Arg-1) and CD206 via autocrine and paracrine mechanisms. Arg-1 consumes L-arginine in the microenvironment, weakening M1 macrophage iNOS-dependent functions while its product ornithine serves as a precursor for polyamine and collagen synthesis, directly supporting the fibrotic process [36]. Additionally, Th2-type cytokines such as IL-13 secreted by HSCs further consolidate the M2 phenotype by activating the STAT6 signaling pathway in macrophages [37]. These M2 macrophages, in turn, secrete large amounts of TGF-β, PDGF, and TIMPs, forming a positive feedback loop during fibrogenesis: HSC activation promotes M2 polarization, and M2 macrophages secrete pro-fibrotic factors that further activate HSCs and inhibit ECM degradation, thereby exacerbating fibrosis [38]. This “recruitment-polarization” coupling mechanism enables recruited macrophages to continually amplify HSC activation effects, driving fibrosis progression.
In summary, activated HSCs during fibrogenesis actively recruit macrophages through chemokines (CCL2, CCL5) and ECM components (HA), while simultaneously directing their polarization toward both M1 and M2 phenotypes. This creates a self-amplifying pro-fibrotic loop wherein recruited macrophages, once polarized, further promote HSC activation and fibrosis progression.
2.4. Macrophage Functions in Fibrosis Resolution and HSC Clearance
After liver injury resolution or therapeutic intervention, liver fibrosis enters a reversal phase during which macrophages undergo functional reprogramming, transitioning from a pro-fibrotic phenotype toward a reparative phenotypic state [7]. This contrasts sharply with their role during fibrotic progression, where macrophages enhance activated HSC survival by secreting cytokines, including IL-1β and TNF-α, thereby maintaining the fibrotic state [39]. During the fibrosis reversal phase, macrophages serve as central mediators facilitating regression through two core pro-resolving functions: inducing HSC apoptosis and degrading pathological ECM [40].
The first key pro-resolving function is the induction of activated HSC apoptosis [41]. During the reversal phase, macrophage phenotypes undergo significant changes, and their secreted cytokine profiles shift from pro-survival to pro-apoptotic [42]. On one hand, macrophages downregulate pro-survival factor expression (TGF-β, PDGF). On the other hand, they trigger apoptotic signaling pathways in activated HSCs through other mechanisms. Experimental evidence indicates that macrophage depletion significantly inhibits activated HSC apoptosis during fibrosis reversal, leading to continued ECM accumulation, while adoptive transfer of reparative macrophages expedites activated HSC clearance and drives liver fibrosis resolution [12]. Additionally, p53-positive senescent HSCs secrete factors that induce macrophage polarization toward the M1 type, which then phagocytose the senescent HSCs, forming a regulatory loop of “senescent HSCs-macrophage clearance” that further promotes fibrosis resolution [43].
The second core pro-resolving function is the degradation of excessively deposited ECM [44]. Reparative macrophages secrete large amounts of matrix metalloproteinases (mainly MMP-12 and MMP-13) [45], specifically degrading accumulated type I and III collagen and other ECM components in the fibrotic liver, thus breaking the positive feedback loop between ECM accumulation and HSC activation. Studies have shown that the Ly-6C+ macrophage subset plays a critical role in reversing fibrosis by secreting MMPs [46]. In carbon tetrachloride (CCl4)-induced liver fibrosis models, hepatic macrophages exhibited prominent upregulation of MMP-12 and MMP-13 expression during the fibrosis reversal phase [39]. Conversely, selective depletion of hepatic macrophages led to inadequate MMP synthesis and secretion, thereby abrogating ECM degradation and prolonging established liver fibrosis resolution.
Recently, the role of extracellular vesicle-mediated communication in fibrosis resolution has attracted attention. MiR-411-5p, highly abundant in exosomes released by M2 reparative macrophages, effectively inhibits CAMSAP1 gene expression in HSCs, thereby blocking HSC activation and providing a new epigenetic explanation for fibrosis reversal [47]. Additionally, the TIM4 receptor on macrophages mediates efferocytosis (the clearance of apoptotic cells), which is crucial for maintaining microenvironment homeostasis during fibrosis resolution. During the reversal phase, TIM4-positive macrophages efficiently clear apoptotic HSCs and increase secretion of the anti-inflammatory factor IL-10 through this process, which suppresses remaining HSC activation and promotes fibrous tissue degradation. In contrast, TIM4 dysfunction causes apoptotic cell accumulation and reduced IL-10 secretion, hindering fibrosis resolution [48].
Interactions with HSCs support the pro-resolution functions of macrophages during the reversal phase. Quiescent HSCs secrete anti-inflammatory factors that induce macrophage polarization toward a reparative phenotype [49], while HA synthesized by HSCs promotes macrophage MMP expression and enhances their ECM-degrading capacity via CD44 receptor signaling [47]. Additionally, changes in ECM components (such as decreased type I collagen) feedback to reinforce the reparative macrophage phenotype [50]. This synergy between HSCs and macrophages during the resolution phase constitutes the core molecular mechanism of fibrosis reversal. Clinical studies have shown that the ratio of reparative macrophages in liver tissues from cirrhotic patients correlates positively with the degree of fibrosis reversal, and MMP-12 and MMP-13 expression levels could serve as potential biomarkers for fibrosis resolution, underscoring the clinical significance of macrophage-mediated resolution mechanisms [39].
In summary, during fibrosis resolution, macrophages undergo functional reprogramming toward a reparative phenotype and exert two core pro-resolving functions: inducing activated HSC apoptosis and degrading pathological ECM via MMP secretion. These activities, supported by reciprocal interactions with HSCs, position macrophages as central drivers of fibrosis reversal and regression.
3. Interactions Between HSCs and NK Cells in Liver Fibrosis
3.1. Overview of the Relationship Between HSCs and NK Cells
The activation of HSCs is a pivotal event in the initiation and progression of liver fibrosis. As a major component of the liver’s innate immunity, NK cells play a dual regulatory role in the fibrotic process [51]. The crosstalk between HSCs and NK cells is a dynamic, bidirectional interaction, and its homeostatic balance is a critical determinant of whether the liver microenvironment shifts toward fibrosis progression or regression [52]. Under physiological conditions, quiescent HSCs abundantly express major histocompatibility complex class I (MHC I) molecules, which engage with inhibitory receptors on the NK cell surface and transmit “self-recognition” signals, thereby protecting HSCs from NK cell-mediated cytotoxicity [53]. Upon liver injury, HSCs become activated, downregulate MHC I expression, and upregulate stress-induced ligands such as MICA, which relieve NK cell inhibition and enhance activating signals [54]. At this early stage, NK cells efficiently kill activated HSCs through multiple mechanisms, exerting a potent anti-fibrotic effect that promotes the resolution of early fibrotic changes [55]. For instance, the prostaglandin E2 receptor EP3 is crucial for the effective adhesion and killing of activated HSCs by NK cells, as the EP3 signaling pathway enhances NK cell–HSC contact by regulating integrins such as Itga4. Accordingly, downregulation of EP3 function exacerbates fibrosis by impairing this anti-fibrotic clearance [56].
However, in the context of chronic and persistent injury, this protective interaction becomes unbalanced, driving fibrosis progression. Activated HSCs secrete immunosuppressive molecules such as TGF-β, which actively suppresses NK cell function [57]. The liver lymphocyte population is enriched with NK cells, which play a key role in host defense against viral infection and tumor transformation. Recent evidence from animal models suggests that NK cells also play an important role in inhibiting liver fibrosis by selectively killing early or senescence activated (HSCs) and by producing the anti-fibrotic cytokine IFN-γ. Furthermore, clinical studies have revealed that human NK cells can kill primary human HSCs and that the ability of NK cells from HCV patients to kill HSCs is enhanced and correlates inversely with the stages of liver fibrosis. IFN-α treatment enhances, while other factors (e.g., alcohol, TGF-β) attenuate, the cytotoxicity of NK cells against HSCs, thereby differentially regulating liver fibrogenesis. In addition, the mouse liver lymphocyte population is also enriched for natural killer T (NKT) cells, whereas human liver lymphocytes have a much lower percentage of NKT cells. Many studies suggest that NKT cells promote liver fibrogenesis by producing pro-fibrotic cytokines such as IL-4, IL-13, hedgehog ligands, and osteopontin; however, NKT cells may also attenuate liver fibrosis under certain conditions by killing HSCs and by producing IFN-γ. Finally, the potential for NK and NKT cells to be used as therapeutic targets for anti-fibrotic therapy is discussed. Similarly, in the setting of liver tumor progression or metastasis, the immunosuppressive tumor microenvironment shaped by activated HSCs further drives NK cell exhaustion. As liver disease progresses to advanced stages, NK cells often become functionally exhausted, losing their ability to halt fibrosis progression [58]. Moreover, HSCs can acquire resistance to NK cell-mediated killing by upregulating anti-apoptotic proteins and altering surface ligand expression [59]. This ultimately establishes a vicious cycle of “NK cell dysfunction—persistent HSC activation”, which drives the inexorable progression of fibrosis to end-stage disease [60]. Therefore, therapeutic strategies aimed at restoring NK cell functionality and overcoming HSC resistance mechanisms hold promise for reversing advanced liver fibrosis.
In summary, the balance of HSC–NK cell interactions dictates liver fibrosis outcomes: effective NK cell killing of activated HSCs promotes fibrosis regression, while HSC-induced NK cell suppression and exhaustion drive fibrosis progression.
3.2. NK Cell Cytotoxicity to HSCs
This targeted killing involves multiple mechanisms, which have been reviewed in the context of liver diseases [61]. NK cells exhibit highly selective cytotoxicity against early-activated or senescent HSCs, a process that is part of their broader immunosurveillance against senescent cells [62]. This involves activating receptor-mediated recognition, such as through natural killer group 2 member D (NKG2D); death receptor pathways, such as via TNF-related apoptosis-inducing ligand (TRAIL) and Fas ligand (FasL); and secretion of cytokines, such as IFN-γ. Notably, these anti-fibrotic functions of NK cells are particularly potent in the early stages of liver fibrosis, contributing to fibrosis regression by eliminating activated HSCs.
3.2.1. Activation Receptor-Mediated Cytotoxicity
This is the primary pathway through which NK cells eliminate HSCs. The receptors NKG2D and NKp46 on the surface of NK cells are well-studied core receptors. Activated HSCs upregulate ligands for NKG2D (such as MICA and ULBPs) [63] and ligands for NKp46 (NCR1) [64], which, upon binding to their respective receptors on NK cells, trigger the release of perforin and granzymes, directly inducing apoptosis in HSCs. Emerging activation receptors such as NKp44 [65], killer cell lectin-like receptor G1 (KLRG1) [66], and Toll-like receptor 9 (TLR-9) have also been shown to participate in the recognition and killing of HSCs. Recent studies further reveal that activation of the metabotropic glutamate receptor 5 (mGluR5) on NK cells can enhance their cytotoxicity against activated HSCs, thereby alleviating liver fibrosis [59]. Conversely, lowering the expression of inhibitory receptors (such as KIRs) on NK cells can enhance their killing activity against HSCs, reducing fibrosis. These mechanisms collectively promote fibrosis regression by ensuring efficient clearance of activated HSCs.
3.2.2. Death Receptor-Mediated Apoptotic Pathways
This pathway is an important complement to NK cell killing of HSCs. Activated HSCs specifically upregulate the expression of TNF-related apoptosis-inducing ligand receptor (TRAIL-R) and Fas [67]. NK cells express TRAIL and FasL, which bind to their respective receptors on HSCs, initiating the caspase cascade [68] and inducing programmed cell death in HSCs [67]. Both clinical and animal studies have confirmed that blocking or enhancing this pathway significantly affects HSC survival and the degree of fibrosis, underscoring its role in tipping the balance toward fibrosis regression when active or toward progression when impaired [67].
3.2.3. Cytokine Secretion with Anti-Fibrotic Properties
NK cells exert anti-fibrotic effects indirectly through the secretion of cytokines. IFN-γ is a key molecule that directly induces cell cycle arrest in activated HSCs and inhibits the synthesis of collagen and other ECM components [69]. Notably, in metastatic tumor models, NK cell-derived IFN-γ has also been shown to be crucial for maintaining the dormancy of disseminated tumor cells (DTCs) [70]. Additionally, NK cell-derived interleukin-10 (IL-10) and interleukin-22 (IL-22) can promote HSC senescence by activating the STAT3/p53/p21 signaling pathway [71]. Senescent HSCs not only have reduced capacity to synthesize collagen, but their altered surface markers make them more recognizable and susceptible to clearance by NK cells [72], thereby contributing to fibrosis regression. For instance, curcumin can enhance sensitivity to NK cell killing by inducing HSC senescence [73]. Curcumin induces HSC senescence through multiple molecular pathways, thereby enhancing their susceptibility to NK cell-mediated killing. The core mechanism involves curcumin entering HSCs and inhibiting key pro-survival signaling pathways, such as STAT3 and NF-κB, leading to cell cycle arrest and activation of the senescence-associated secretory phenotype (SASP) [73]. This senescence process is accompanied by significant alterations in cell surface ligands: on one hand, senescent HSCs upregulate ligands for NK cell-activating receptors NKG2D (such as MICA/B) and DNAM-1 (such as PVR/CD155); on the other hand, they may downregulate inhibitory ligands like MHC class I molecules. This shift in ligand profile—characterized by increased activating signals and decreased inhibitory signals—enables NK cells to more effectively recognize and clear senescent HSCs via receptors such as NKG2D and DNAM-1. Research indicates that curcumin triggers this senescence-associated surface molecular reprogramming, transforming HSCs into targets more vulnerable to NK cell attack, thus promoting fibrosis resolution.
3.2.4. Exosome-Mediated Novel Regulatory Mechanisms
Recent studies have revealed that NK cells can regulate HSCs not only through direct contact but also via the release of exosomes, providing a novel non-contact regulatory perspective for understanding complex intercellular communication within the liver microenvironment. Exosomes derived from NK cells have been confirmed to directly inhibit HSC activation. Research demonstrates that exosomes isolated from NK cells can be effectively taken up by HSCs, significantly suppressing HSC proliferation, reducing the expression of key activation markers (such as α-SMA), and alleviating the progression of liver fibrosis [74]. This confirms that NK cells can exert important immunoregulatory functions through paracrine exosome release, contributing to fibrosis regression. At the molecular level, this inhibitory effect is primarily achieved by delivering specific microRNAs via exosomes. Among them, miR-223 is a key effector molecule. Exosomes originating from NK cells efficiently transport miR-223 into HSCs. Inside HSCs, miR-223 targets and inhibits the mRNA of autophagy-related gene 7 (ATG7), thereby significantly suppressing the cellular autophagy process. The inhibition of autophagy flux hinders the transformation of HSCs from a quiescent state to an activated myofibroblast-like phenotype, ultimately achieving the effect of inhibiting their activation [75]. This “miR-223-ATG7-autophagy” axis clearly elucidates a precise post-transcriptional regulatory pathway that promotes fibrosis regression by preventing HSC activation [76]. In summary, existing evidence indicates that NK cell regulation of HSCs via the exosomal pathway is a multi-layered process: on one hand, the exosomes possess an overall function of inhibiting HSC activation [74]; on the other hand, the miR-223 they carry accomplishes this function through the specific molecular mechanism of targeting ATG7 and interfering with autophagy [75]. This exosome-mediated non-contact intercellular communication enriches the understanding of NK cell immunoregulatory functions and offers new potential therapeutic targets for promoting fibrosis resolution.
3.3. Mechanisms of HSC-Induced Suppression of NK Cell Function
In advanced liver fibrosis, continuously activated HSCs create an immunosuppressive microenvironment that diminishes the anti-fibrotic capacity of NK cells, a key reason why fibrosis becomes difficult to reverse and progresses to end-stage disease. The mechanisms by which HSCs suppress NK cells involve core pathways common to chronic liver injury, as well as etiology-specific modulations prominent in infection or cancer settings.
3.3.1. Core Immunosuppressive Mechanisms Mediated by HSCs
Activated HSCs are the primary source of TGF-β in the fibrotic liver. TGF-β suppresses NK cell function at multiple levels: (1) it mediates the engulfment of NK cells through Rac1 and Cdc42 signaling pathways (emperipolesis) [77], directly leading to NK cell apoptosis and a reduction in their numbers; (2) it significantly downregulates the expression of NK cell activation receptors (such as NKG2D and NKp46) and inhibits the production of effector molecules like IFN-γ and TRAIL. Both preclinical and clinical studies have shown that levels of NKG2D and IFN-γ are significantly decreased in NK cells during late fibrosis, while blocking TGF-β signaling can partially restore NK cell function [77]. Moreover, activated HSCs upregulate programmed death-ligand 1 (PD-L1), which binds to the programmed death-1 (PD-1) receptor on NK cells, thereby transmitting strong inhibitory signals that directly weaken NK cell cytotoxicity [78]. This is one of the key mechanisms through which HSCs achieve immune evasion, thereby driving fibrosis progression. Additionally, excessive deposition of ECM can create physical barriers that obstruct effective contact between NK cells and HSCs, further limiting the cytotoxic efficacy of NK cells and perpetuating fibrosis progression.
3.3.2. NK Cell Exhaustion in the Context of Chronic Viral Infection
In chronic hepatitis B virus (HBV) infection, the persistent viral antigens and inflammation drive a unique vicious cycle. Activated HSCs not only secrete high levels of TGF-β but may also directly respond to viral signals, further amplifying the suppression of NK cells [79]. The resulting NK cell exhaustion is characterized by profound impairment in cytotoxicity and cytokine production, which compromises both antiviral immunity and the surveillance of activated HSCs, thereby facilitating fibrosis progression.
3.3.3. NK Cell Dysfunction in the Tumor Microenvironment
Within the liver tumour microenvironment, HSCs interact with cancer cells, exacerbating NK cell suppression and promoting tumor progression and fibrosis. A key mechanism is the secretion of CXCL12 by activated HSCs. CXCL12 significantly diminishes the capacity of NK cells to produce IFN-γ, critically impairing their immunosurveillance function, which has been demonstrated as a pivotal regulatory axis in models of breast cancer liver metastasis [70]. Furthermore, tumor cells themselves often express high levels of PD-L1 and other immune checkpoint ligands, creating a synergistic inhibitory milieu with HSCs that drives NK cells toward a deeply exhausted phenotype, thereby accelerating disease progression.
3.3.4. Therapeutic Implications
These mechanisms collectively lead to an “exhausted” phenotype in NK cells during advanced liver fibrosis, characterized by diminished degranulation activity, proliferative capacity, and effector functions. Thus, therapies solely aimed at activating NK cells may have limited efficacy. Future treatment strategies may need to adopt combination therapies, such as targeting activated HSCs (for instance, using nanoparticle delivery systems to silence their CXCL12 secretion) while also activating NK cell functions (such as using EP3 [56] or mGluR5 [59] agonists) to comprehensively address the immunosuppressive microenvironment and restore immunological surveillance.
In essence, the transition from fibrosis regression to progression is governed by the dynamic interplay between NK cell-mediated killing of HSCs and HSC-induced suppression of NK cells; restoring the balance in favor of NK cell function is key to reversing advanced fibrosis.
4. HSCs and T Cells in Liver Fibrosis
4.1. Overview of the Relationship Between HSCs and T Cells
T cells, as core components of adaptive immunity, exhibit a complex and often contradictory functional spectrum in liver fibrosis [80]. This duality is evident not only in the distinct functional roles of different T cell subsets [81] but also in the divergent outcomes observed across clinical and experimental studies. The liver’s unique immune microenvironment underpins this functional diversity: the ratio of CD8+ to CD4+ T cells is significantly higher in the liver than in peripheral blood [82], and the phenotype and function of resident lymphocytes differ markedly from their circulating counterparts [83]. This peculiarity directly influences how T cells regulate the progression or resolution of fibrosis.
In terms of functional differentiation, T cell subsets can be broadly categorized as pro-fibrotic (promoting fibrosis progression), anti-fibrotic (promoting fibrosis resolution), or dual-functional. For instance, Th2 and Th17 cells drive fibrosis progression by activating HSCs and sustaining inflammation via cytokines such as IL-13 and IL-17. In contrast, Th1 cells support fibrosis resolution by secreting IFN-γ, which inhibits HSC activation and promotes their apoptosis [84]. γδ T cells, which comprise 15–25% of hepatic T cells [85], exhibit a dual role: they can contribute to fibrosis resolution by inducing HSC apoptosis via the Fas/FasL pathway, but also promote fibrosis progression through IL-17 secretion [80]. Notably, γδ T cells that adopt a tissue-resident memory phenotype (TRM), such as those expressing CD69 and CXCR6, have been shown to actively drive fibrosis resolution in animal models.
Clinical observations further illustrate this complexity. In chronic hepatitis C virus (HCV)-infected renal transplant recipients, those not receiving immunosuppressive therapy show significantly slower fibrosis progression compared to those who do [86], suggesting that broad T cell suppression may inadvertently block protective responses. Conversely, in HIV/HCV co-infected individuals [87], the loss of CD4+ T cells accelerates fibrosis progression [88], highlighting the protective role of specific subsets. These seemingly contradictory findings underscore the functional heterogeneity of T cells: non-specific immunosuppression differs fundamentally from the selective depletion of particular subsets in its impact on fibrosis dynamics.
4.2. Differential Regulation of HSCs by CD4+ T Cells
CD4+ T helper cells differentiate into distinct functional subsets that differentially regulate liver fibrosis through cytokine secretion. The balance between these subsets—such as Th1/Th2 and Th17/Treg—is critical in determining whether fibrosis progresses or resolves [89].
4.2.1. Th1 Cells (Pro-Resolution): Inhibition of HSC Activation via IFN-γ
Th1 cells are a classic pro-resolution T cell subset. Their core anti-fibrotic function is mediated by IFN-γ, which exerts multi-level inhibitory effects on HSCs [90]. IFN-γ directly impairs HSC activity, upregulates Smad7 in a STAT1-dependent manner, and suppresses TGF-β1-mediated Smad2/Smad3 phosphorylation and nuclear translocation, thereby abrogating ECM synthesis. It also promotes apoptosis of activated HSCs and inhibits Th2 cells from secreting pro-fibrotic factors like IL-13. In animal models, Th1-polarized C57BL/6 mice exhibit significantly less severe fibrosis compared to Th2-polarized BALB/c mice, confirming the crucial role of Th1 cells in driving fibrosis resolution [91].
4.2.2. Th2 and Th17 Cells (Pro-Fibrotic): Promotion of HSC Activation and Inflammation
Both Th2 and Th17 cells are key drivers of fibrosis progression, though through distinct mechanisms. Th2 cells promote fibrosis primarily by secreting IL-13, which stimulates TGF-β1 synthesis and enhances HSC-mediated ECM production. Th17 cells play an even more critical role in fibrosis progression [92]. Their signature cytokine, IL-17, activates HSCs via ERK1/2 and p38 signaling and induces type I collagen synthesis through the STAT3 pathway [93]. IL-17 also upregulates TGF-β1 receptors on HSCs in a JNK-dependent manner and recruits neutrophils and monocytes to sustain the pro-inflammatory microenvironment required for fibrosis progression [44].
IL-22, a cytokine produced by Th17 cells, plays a dual role in liver fibrosis, with opposing effects dependent on the disease stage. It promotes fibrosis during sustained injury and active disease progression. For instance, in advanced HBV-related fibrosis, IL-22 enhances TGF-β1 signaling via the p38 MAPK pathway [94], driving HSC activation and excessive ECM deposition. Conversely, it exerts anti-fibrotic effects primarily in the context of acute or early-stage injury. In these settings, IL-22 can induce HSC senescence via the STAT3/SOCS3/p53/p21 axis [95], thereby halting HSC proliferation and facilitating fibrosis resolution. Thus, the temporal context and molecular microenvironment critically determine whether IL-22 drives fibrogenesis or supports repair.
4.2.3. Regulatory T Cells in Liver Fibrosis
Regulatory T cells (Tregs), defined by FOXP3 expression, display marked functional duality in liver fibrosis. On one hand, they can support fibrosis resolution by suppressing pro-fibrotic CD8+ and Th17 cells via IL-10 [96], thereby dampening intrahepatic inflammation. In vitro, Tregs inhibit HSC activation in a dose-dependent manner, and in animal models of biliary fibrosis [97], Treg depletion exacerbates disease. On the other hand, Tregs secrete TGF-β1, a potent activator of HSCs that promotes ECM synthesis and inhibits degradation via TIMP upregulation, thereby driving fibrosis progression [98]. Thus, the net effect of Tregs depends on the balance of cytokines in the local microenvironment. The Th17/Treg ratio is increasingly recognized as a key determinant of fibrosis outcome: an elevated ratio in HBV patients correlates with accelerated progression and poor prognosis [97].
In MASH (previously termed NASH), Tregs exhibit an additional pro-fibrotic mechanism [99]. A distinct Treg subset producing amphiregulin (Areg) accumulates in the liver and directly activates HSCs via the EGFR pathway, bypassing TGF-β signaling entirely [100]. This promotes both fibrosis progression and insulin resistance, revealing a novel pathway through which Tregs contribute to disease progression in metabolic liver injury.
4.3. Indirect Pro-Fibrotic Role of CD8+ Cytotoxic T Cells in Liver Fibrosis
CD8+ T cells were once considered a homogeneous population, but recent studies reveal significant functional heterogeneity in their regulation of liver fibrosis [101]. In CCl4- or TAA-induced fibrosis models, adoptive transfer of spleen-derived CD8+ T cells exacerbates fibrosis progression [102], an effect blocked by IL-10. However, CD8+ T cell deficiency does not alter fibrosis outcomes in MDR2 knockout mice, suggesting that their role is context-dependent and mediated by specific subsets [2].
The pro-fibrotic effect of CD8+ T cells is largely indirect, involving the secretion of inflammatory cytokines that sustain a microenvironment conducive to HSC activation [103]. A newly identified CD8+ Treg subset adds further complexity, though its role in fibrosis remains to be fully elucidated. Notably, a breakthrough study revealed that liver-resident CD8+ TRM can directly recognize and induce apoptosis of activated HSCs, actively driving fibrosis resolution in animal models. This challenges the traditional view that CD8+ T cells promote fibrosis and highlights their potential as therapeutic targets for resolution [101].
4.4. HSCs as Antigen-Presenting Cells for Reverse Regulation of T Cells
HSCs are not merely passive targets of T cell regulation; they actively shape the immune microenvironment to favor fibrosis progression. Activated HSCs express MHC I constitutively and can be induced to express MHC II under inflammatory signals, enabling antigen presentation to CD8+ and CD4+ T cells [104]. Although direct evidence for cross-presentation in HSCs remains limited, their expression of MHC I and ability to acquire exogenous antigens from the fibrotic milieu suggest a potential, albeit unproven, role in cross-priming CD8+ T cells against hepatocyte-derived or stromal antigens. However, their low expression of co-stimulatory molecules (e.g., CD80/CD86) and high expression of co-inhibitory molecules (e.g., PD-L1) render them “defective” antigen-presenting cells. This typically induces T cell anergy, exhaustion, or apoptosis, promoting immune tolerance and supporting fibrosis progression. In this context, the impact on naïve versus memory T cells likely diverges: defective antigen presentation by HSCs preferentially tolerizes naïve T cells during primary activation due to inadequate co-stimulation, whereas their engagement with memory T cells—which have lower activation thresholds—may more directly trigger inhibitory pathways such as PD-1/PD-L1 to suppress recall responses.
HSCs also directly modulate T cell function through multiple pathways. PD-L1 on HSCs engages PD-1 on T cells, inducing apoptosis [105]. Secreted TGF-β1 suppresses T cell activation and promotes Treg differentiation. In HBV-related fibrosis, HSCs upregulate Th17 and Treg numbers via PGE2/EP2/EP4 signaling [106]. In early CCl4-induced fibrosis, HSCs collaborate with γδ T cells to amplify IL-17 production, establishing a pro-fibrotic inflammatory loop. Conversely, in liver transplant recipients with recurrent hepatitis, LPS-stimulated HSCs expand allogeneic Tregs via an IDO1-AHR pathway, reinforcing immune suppression.
Beyond direct effects, HSCs indirectly suppress T cells by shaping myeloid cell phenotypes. In hepatocellular carcinoma, HSCs secrete CX3CL1 to recruit CX3CR1+ macrophages, which upregulate arginase 1 and deplete arginine, thereby suppressing CD8+ T cell function [107]. Although identified in cancer, this “HSC–macrophage–CD8+ T cell” axis likely operates in fibrotic microenvironments as well, further entrenching immunosuppression and promoting fibrosis progression.
In summary, HSCs establish a multidimensional immunosuppressive network—through defective antigen presentation, direct T cell modulation, and myeloid cell remodeling—that ensures their persistent activation and drives fibrosis progression.
4.5. Chapter Summary: A Cytokine-Mediated Bidirectional Regulatory Network
This chapter has detailed the cytokine-mediated bidirectional regulatory network between HSCs and T cells, a central mechanism governing liver fibrosis progression or resolution [108]. T cells exert their effects through subset-specific cytokine profiles: Th1 cells promote resolution via IFN-γ; Th2 and Th17 cells drive progression via IL-13 and IL-17; Tregs play dual roles depending on context; and CD8+ T cells can either promote progression through inflammation or actively drive resolution via direct HSC killing [47].
Conversely, HSCs shape the immune environment to favor progression by inducing T cell apoptosis via PD-L1, suppressing T cell function via TGF-β, and modulating Th17/Treg balance [109]. This bidirectional regulation forms a complex network centered on key cytokines such as TGF-β1, IL-17, IFN-γ, and IL-10. The Th17/Treg and Th1/Th2 balances are critical determinants of fibrosis outcome.
A deeper understanding of this network—particularly the molecular mechanisms governing key cytokines and signaling pathways—offers new opportunities for targeted therapy. Advances in single-cell sequencing have revealed previously unrecognized heterogeneity within both T cell and HSC populations, paving the way for more precise interventions. Future strategies may include enhancing protective subsets, such as γδ TRM or CD8+ TRM, to promote resolution; selectively blocking pathogenic Treg functions (e.g., Areg secretion) without compromising immunosuppression; and disrupting HSC-mediated immunosuppressive axes (e.g., CX3CL1-CX3CR1) to reverse the microenvironment that sustains fibrosis progression. By targeting specific cell subsets and pathways, it may be possible to rebalance this bidirectional network and open new avenues for the treatment of liver fibrosis.
5. Summary and Outlook
Liver fibrosis represents the core pathological process in the progression of various chronic liver diseases. This review systematically clarifies how the complex interaction network composed of HSCs, macrophages, NK cells, and T cells dynamically regulates the progression and reversal of fibrosis. As summarized in Table 1, the bidirectional crosstalk between HSCs and these immune cells plays a central role in this process.
Table 1. Summary of the bidirectional crosstalk between HSCs and major immune cells in liver fibrosis.
|
Immune Cell Type |
Key Molecules/Pathways |
Main Effects |
Functional Significance in Liver Fibrosis |
|---|---|---|---|
|
Macrophages |
TGF-β, PDGF, TNF-α, IL-1β, ROS, CCL2/CCR2, MMPs, TIM4, Exosomes |
Activate HSCs & promote ECM deposition; Induce HSC apoptosis & degrade ECM |
Drive fibrotic progression; Promote fibrosis resolution |
|
NK Cells |
NKG2D, TRAIL, IFN-γ, TGF-β, PD-L1 |
Kill activated HSCs; Get suppressed by activated HSCs |
Anti-fibrotic in early stages; Functional exhaustion in late stages |
|
T Cells |
IFN-γ (Th1), IL-13/IL-17 (Th2/Th17), TGF-β (Tregs), PD-L1 |
Inhibit or activate HSCs; Their differentiation/function is regulated by HSCs |
Determine fibrotic direction; Subject to HSC-mediated immunosuppression |
This review focuses on the core of the immune regulatory network in liver fibrosis, namely the bidirectional interactions among macrophages, NK cells, T cells, and HSCs.
5.1. Macrophages: The Microenvironment Dominator and a Plastic Target
Macrophages are central regulators of the hepatic immune microenvironment. By secreting key factors such as TGF-β and PDGF, they potently drive the activation of HSCs into myofibroblasts, acting as the core engine of fibrosis. Their therapeutic potential stems from their high functional plasticity—they can polarize to pro-inflammatory (M1) or pro-fibrotic/repair (M2-like) phenotypes under different signals, making the reprogramming of macrophages a direct strategy for reversing fibrosis.
5.2. NK Cells: Early Defenders and Chronic Phase Silencers
NK cells play a crucial role in anti-fibrotic innate immunity. In the early stages of injury, they inhibit HSC activation by secreting IFN-γ and induce apoptosis of activated HSCs through pathways such as FasL and TRAIL, forming an important physiological defense barrier. However, during chronic persistent injury, their cytotoxicity and IFN-γ production capacity are often exhausted or inhibited, leading to the failure of this protective mechanism.
5.3. T Cells: A Sophisticated Regulatory System for Dynamic Balance
Different T cell subsets constitute a dynamically balanced regulatory network: Th1 cells (secreting IFN-γ) exert anti-fibrotic effects, while Th2 cells (secreting IL-4, IL-13) and Th17 cells (secreting IL-17) promote inflammation and fibrosis progression. The roles of regulatory T cells (Tregs) and cytotoxic T cells (CD8+ T cells) are particularly complex, possessing dual characteristics that can both inhibit immune-mediated damage and, under certain conditions, promote fibrosis, with their net effect highly dependent on the specific microenvironmental context.
Other immune cell populations also play significant roles in the process of liver fibrosis. B cells and plasma cells, besides forming immune complexes through antibody production that exacerbate inflammatory damage, can also secrete cytokines such as IL-10 to exert immunomodulatory effects; certain immunoglobulins they produce (e.g., IgG) may also directly activate HSCs. Neutrophils, as the first immune cells recruited during acute injury, cause direct hepatocyte damage and activate HSCs by releasing myeloperoxidase (MPO) and neutrophil elastase (NE), and by forming NETs, acting as key players in initiating the the early fibrogenic events. ILCs, as tissue-resident innate immune cells, have different subsets (ILC1s, ILC2s, and ILC3s) that mirror the functions of Th1, Th2, and Th17 cells, respectively, regulating the balance between inflammation and fibrosis at early stages before adaptive T-cell immune responses are fully established.
The immune regulation of liver fibrosis is a dynamic process centered on the bidirectional interaction network between HSCs and macrophages, NK cells, and T cells. Targeting the functional status of these three core immune cell types or their communication network with HSCs, and leveraging their intrinsic plasticity to shift the immune balance towards an anti-fibrotic direction, represents the most promising strategy for future immunotherapy.
Author Contributions
Conceptualization, C.-N.Z., G.-Y.S. and H.-J.L. Methodology, C.-N.Z., G.-Y.S. and H.-J.L.; Investigation, C.-N.Z., G.-Y.S. and H.W.; Data Curation, C.-N.Z., Y.-X.Z. and Z.-C.G.; Writing—Original Draft, C.-N.Z.; Writing—Review & Editing, H.-J.L., G.-Y.S., H.W. and Y.-X.Z.; Visualization, Z.-C.G. and Y.-X.Z.; Supervision, H.-J.L.; Project Administration, H.-J.L.; Funding Acquisition, H.-J.L.
Ethics Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All data used in the present study are available from the corresponding author upon reasonable request.
Funding
This research was funded by the Natural Science Foundation of Jilin Province (YDZJ202401218ZYTS to HL).
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
-
Tarantino G, Citro V, Balsano C. Liver-spleen axis in nonalcoholic fatty liver disease. Expert Rev. Gastroenterol. Hepatol. 2021, 15, 759–769. DOI:10.1080/17474124.2021.1914587 [Google Scholar]
-
Gilgenkrantz H, Sayegh RA, Lotersztajn S. Immunoregulation of Liver Fibrosis: New Opportunities for Antifibrotic Therapy. Annu. Rev. Pharmacol. Toxicol. 2025, 65, 281–299. DOI:10.1146/annurev-pharmtox-020524-012013 [Google Scholar]
-
Novobrantseva TI, Majeau GR, Amatucci A, Kogan S, Brenner I, Casola S, et al. Attenuated liver fibrosis in the absence of B cells. J. Clin. Investig. 2005, 115, 3072–3082. DOI:10.1172/JCI24798 [Google Scholar]
-
Shen J, Huang S, Wang Y, Wang Q, Lin S, Guan W, et al. PAD4+ neutrophils promote hepatic stellate cell activation and accelerate MASH fibrosis progression viaNET-DNA/TAOK1/MAPK pathways. JCI Insight 2026, 11, e191479. DOI:10.1172/jci.insight.191479 [Google Scholar]
-
Mathä L, Romera-Hernández M, Steer CA, Yin YH, Orangi M, Shim H, et al. Migration of Lung Resident Group 2 Innate Lymphoid Cells Link Allergic Lung Inflammation and Liver Immunity. Front. Immunol. 2021, 12, 679509. DOI:10.3389/fimmu.2021.679509 [Google Scholar]
-
Kisseleva T, Brenner D. Molecular and cellular mechanisms of liver fibrosis and its regression. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 151–166. DOI:10.1038/s41575-020-00372-7 [Google Scholar]
-
Ma X, Qiu J, Zou S, Tan L, Miao T. The role of macrophages in liver fibrosis: Composition, heterogeneity, and therapeutic strategies. Front. Immunol. 2024, 15, 1494250. DOI:10.3389/fimmu.2024.1494250 [Google Scholar]
-
Ouyang R, Li X, Hao J, Lin J, Lan H, Peng J, et al. Decoding the heterogeneity of liver-resident macrophages in chronic liver diseases: Therapeutic responses to immunomodulatory strategies. Front. Pharmacol. 2025, 16, 1708240. DOI:10.3389/fphar.2025.1708240 [Google Scholar]
-
Wen Y, Lambrecht J, Ju C, Tacke F. Hepatic macrophages in liver homeostasis and diseases-diversity, plasticity and therapeutic opportunities. Cell Mol. Immunol. 2021, 18, 45–56. DOI:10.1038/s41423-020-00558-8 [Google Scholar]
-
Li W, Chen L, Zhou Q, Huang T, Zheng W, Luo F, et al. Liver macrophage-derived exosomal miRNA-342-3p promotes liver fibrosis by inhibiting HPCAL1 in stellate cells. Hum. Genom. 2025, 19, 9. DOI:10.1186/s40246-025-00722-z [Google Scholar]
-
Zhong H, Liu C, Huang Z, Tan P, Chen H, Fu W. Crosstalk between Hepatic Stellate Cells and Hepatic Macrophages in Metabolic Dysfunction-Associated Steatohepatitis. Am. J. Pathol. 2025, 195, 1040–1056. DOI:10.1016/j.ajpath.2025.02.003 [Google Scholar]
-
Ran J, Yin S, Issa R, Zhao Q, Zhu G, Zhang H, et al. Key role of macrophages in the progression of hepatic fibrosis. Hepatol. Commun. 2025, 9, e0602. DOI:10.1097/HC9.0000000000000602 [Google Scholar]
-
Yin X, Liu M, Xu Y, Zhong H, Zhang J, Xie H, et al. Macrophage-derived DLL4 promotes liver fibrosis by activating the Notch pathway in hepatic stellate cells. Free Radic. Biol. Med. 2026, 242, 431–443. DOI:10.1016/j.freeradbiomed.2025.10.275 [Google Scholar]
-
Feng D, Guan Y, Wang Y, Maccioni L, Mackowiak B, Gao B. Characterisation of macrophages in healthy and diseased livers in mice: Identification of necrotic lesion-associated macrophages. eGastroenterology 2025, 3, e100189. DOI:10.1136/egastro-2025-100189 [Google Scholar]
-
Li J, Zhao YR, Tian Z. Roles of hepatic stellate cells in acute liver failure: From the perspective of inflammation and fibrosis. World J. Hepatol. 2019, 11, 412–420. DOI:10.4254/wjh.v11.i5.412 [Google Scholar]
-
Seki E, De Minicis S, Österreicher CH, Kluwe J, Osawa Y, Brenner DA, et al. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat. Med. 2007, 13, 1324–1332. DOI:10.1038/nm1663 [Google Scholar]
-
Li Z, Ying W, Zhuang T, Xie Y, Kim J. Exercise-mediated macrophage polarization modulates the targeted therapeutic effect of NAFLD: A review. Phys. Act. Nutr. 2023, 27, 10–16. DOI:10.20463/pan.2023.0023 [Google Scholar]
-
Zou X, Ke Y, Shao Y, Liu S, Shi T. Liver Fibrosis: Interactions Between Cells and Microenvironments. Turk. J. Gastroenterol. 2025, 36, 711–722. DOI:10.5152/tjg.2025.25313 [Google Scholar]
-
Campbell JS, Hughes SD, Gilbertson DG, Palmer TE, Holdren MS, Haran AC, et al. Platelet-derived growth factor C induces liver fibrosis, steatosis, and hepatocellular carcinoma. Proc. Natl. Acad. Sci. USA 2005, 102, 3389–3394. DOI:10.1073/pnas.0409722102 [Google Scholar]
-
Kocabayoglu P, Lade A, Lee YA, Dragomir AC, Sun X, Fiel MI, et al. β-PDGF receptor expressed by hepatic stellate cells regulates fibrosis in murine liver injury, but not carcinogenesis. J. Hepatol. 2015, 63, 141–147. DOI:10.1016/j.jhep.2015.01.036 [Google Scholar]
-
Han H, Yu D, Liu Y, Jia H, Liu W. Matrix mechanical remodeled carrier-free nanosystem for programmable closed-loop reversal of liver fibrosis via STING alkylation. Sci. Adv. 2025, 11, eadz4126. DOI:10.1126/sciadv.adz4126 [Google Scholar]
-
Sun L, Wang Y, Kan T, Wang H, Cui J, Wang L, et al. Elevated expression of Piezo1 activates the cGAS-STING pathway in chondrocytes by releasing mitochondrial DNA. Osteoarthr. Cartil. 2025, 33, 601–615. DOI:10.1016/j.joca.2025.02.778 [Google Scholar]
-
Wang X, Rao H, Zhao J, Wee A, Li X, Fei R, et al. STING expression in monocyte-derived macrophages is associated with the progression of liver inflammation and fibrosis in patients with nonalcoholic fatty liver disease. Lab. Investig. 2020, 100, 542–552. DOI:10.1038/s41374-019-0342-6 [Google Scholar]
-
Kim DM, Pan Q, Liu Z, Ai W, Han HW, Banu SK, et al. GHSR-Foxo1 Signaling in Macrophages Promotes Liver Fibrosis via Inflammatory Response and Hepatic Stellate Cell Activation. Adv. Sci. 2025, 12, e04223. DOI:10.1002/advs.202504223 [Google Scholar]
-
Zheng Q, Cao Y, Jiang X, Wang X, Wang X, He Y, et al. Decoding the CHI3L1/IL-13Rα2 signaling nexus in MASH-fibrosis pathogenesis. Sci. Adv. 2025, 11, eadz3223. DOI:10.1126/sciadv.adz3223 [Google Scholar]
-
Li X, Liu S, Li T, Yang Q, Chen Y, Meng Y, et al. Inflammatory factor CCL2 enhances the interaction between monocyte-macrophage cells and liver parenchymal cells to promote liver inflammation and fibrosis in biliary atresia. BMC Pediatr. 2025, 25, 643. DOI:10.1186/s12887-025-05984-z [Google Scholar]
-
Guo S, Zhang Q, Guo Y, Yin X, Zhang P, Mao T, et al. The role and therapeutic targeting of the CCL2/CCR2 signaling axis in inflammatory and fibrotic diseases. Front. Immunol. 2024, 15, 1497026. DOI:10.3389/fimmu.2024.1497026 [Google Scholar]
-
Cheng D, Chai J, Wang H, Fu L, Peng S, Ni X. Hepatic macrophages: Key players in the development and progression of liver fibrosis. Liver Int. 2021, 41, 2279–2294. DOI:10.1111/liv.14940 [Google Scholar]
-
Geervliet E, Karkdijk E, Bansal R. Inhibition of intrahepatic monocyte recruitment by Cenicriviroc and extracellular matrix degradation by MMP1 synergistically attenuate liver inflammation and fibrogenesis in vivo. Sci. Rep. 2024, 14, 16897. DOI:10.1038/s41598-024-67926-6 [Google Scholar]
-
Xi S, Zheng X, Li X, Jiang Y, Wu Y, Gong J, et al. Activated Hepatic Stellate Cells Induce Infiltration and Formation of CD163+ Macrophages via CCL2/CCR2 Pathway. Front. Med. 2021, 8, 627927. DOI:10.3389/fmed.2021.627927 [Google Scholar]
-
Baeck C, Wei X, Bartneck M, Fech V, Heymann F, Gassler N, et al. Pharmacological inhibition of the chemokine C-C motif chemokine ligand 2 (monocyte chemoattractant protein 1) accelerates liver fibrosis regression by suppressing Ly-6C (+) macrophage infiltration in mice. Hepatology 2014, 59, 1060–1072. DOI:10.1002/hep.26783 [Google Scholar]
-
Yagura K, Ohtaki H, Tsumuraya T, Sato A, Miyamoto K, Kawada N, et al. The enhancement of CCL2 and CCL5 by human bone marrow-derived mesenchymal stem/stromal cells might contribute to inflammatory suppression and axonal extension after spinal cord injury. PLoS ONE 2020, 15, e0230080. DOI:10.1371/journal.pone.0230080 [Google Scholar]
-
Meran S, Martin J, Luo DD, Steadman R, Phillips A. Interleukin-1β induces hyaluronan and CD44-dependent cell protrusions that facilitate fibroblast-monocyte binding. Am. J. Pathol. 2013, 182, 2223–2240. DOI:10.1016/j.ajpath.2013.02.038 [Google Scholar]
-
Sipka T, Peroceschi R, Hassan-Abdi R, Groß M, Ellett F, Begon-Pescia C, et al. Damage-Induced Calcium Signaling and Reactive Oxygen Species Mediate Macrophage Activation in Zebrafish. Front. Immunol. 2021, 12, 636585. DOI:10.3389/fimmu.2021.636585 [Google Scholar]
-
Li W, Luo F, Wu X, Fan B, Yang M, Zhong W, et al. Anti-Inflammatory Effects and Mechanisms of Dandelion in RAW264.7 Macrophages and Zebrafish Larvae. Front. Pharmacol. 2022, 13, 906927. DOI:10.3389/fphar.2022.906927 [Google Scholar]
-
Través PG, Luque A, Hortelano S. Macrophages, inflammation, and tumor suppressors: ARF, a new player in the game. Mediat. Inflamm. 2012, 2012, 568783. DOI:10.1155/2012/568783 [Google Scholar]
-
Li B, Dong B, Xie L, Li Y. Exploring Advances in Natural Plant Molecules for Allergic Rhinitis Immunomodulation In Vivo and In Vitro. Int. J. Gen. Med. 2025, 18, 529–565. DOI:10.2147/IJGM.S493021 [Google Scholar]
-
Ge Z, Chen Y, Ma L, Hu F, Xie L. Macrophage polarization and its impact on idiopathic pulmonary fibrosis. Front. Immunol. 2024, 15, 1444964. DOI:10.3389/fimmu.2024.1444964 [Google Scholar]
-
Weng SY, Wang X, Vijayan S, Tang Y, Kim YO, Padberg K, et al. IL-4 Receptor Alpha Signaling through Macrophages Differentially Regulates Liver Fibrosis Progression and Reversal. EBioMedicine 2018, 29, 92–103. DOI:10.1016/j.ebiom.2018.01.028 [Google Scholar]
-
Wang L, Yang K, Xie X, Wang S, Gan H, Wang X, et al. Macrophages as Multifaceted Orchestrators of Tissue Repair: Bridging Inflammation, Regeneration, and Therapeutic Innovation. J. Inflamm. Res. 2025, 18, 8945–8959. DOI:10.2147/JIR.S527764 [Google Scholar]
-
Friedman SL. Mac the knife? Macrophages- the double-edged sword of hepatic fibrosis. J. Clin. Investig. 2005, 115, 29–32. DOI:10.1172/JCI23928 [Google Scholar]
-
Duffield JS, Forbes SJ, Constandinou CM, Clay S, Partolina M, Vuthoori S, et al. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J. Clin. Investig. 2005, 115, 56–65. DOI:10.1172/JCI22675 [Google Scholar]
-
Yu S, Ji G, Zhang L. The role of p53 in liver fibrosis. Front. Pharmacol. 2022, 13, 1057829. DOI:10.3389/fphar.2022.1057829 [Google Scholar]
-
Xiao Y, Wu ZN, Lu J, Wang YD. Role of metabolic reprogramming-mediated hepatic stellate cell activation in the pathogenesis of hepatic fibrosis. Zhonghua Gan Zang Bing Za Zhi 2024, 32, 1053–1056. DOI:10.3760/cma.j.cn501113-20240223-00088 [Google Scholar]
-
Lagente V, Le Quement C, Boichot E. Macrophage metalloelastase (MMP-12) as a target for inflammatory respiratory diseases. Expert. Opin. Ther. Targets 2009, 13, 287–295. DOI:10.1517/14728220902751632 [Google Scholar]
-
Zhang S, Wan D, Zhu M, Wang G, Zhang X, Huang N, et al. CD11b+ CD43hi Ly6Clo splenocyte-derived macrophages exacerbate liver fibrosis via spleen-liver axis. Hepatology 2023, 77, 1612–1629. DOI:10.1002/hep.32782 [Google Scholar]
-
Sun Y, Zhao M, Cheng L, He X, Shen S, Lv J, et al. Reduction of alternative polarization of macrophages by short-term activated hepatic stellate cell-derived small extracellular vesicles. J. Exp. Clin. Cancer Res. 2025, 44, 117. DOI:10.1186/s13046-025-03380-0 [Google Scholar]
-
Shi H, Wang X, Sloas C, Gerlach B, Yurdagul A, Moore MP, et al. Impaired TIM4-mediated efferocytosis by liver macrophages contributes to fibrosis in metabolic dysfunction-associated steatohepatitis. Sci. Transl. Med. 2025, 17, eadv2106. DOI:10.1126/scitranslmed.adv2106 [Google Scholar]
-
Seyfried AN, Maloney JM, MacNamara KC. Macrophages Orchestrate Hematopoietic Programs and Regulate HSC Function During Inflammatory Stress. Front. Immunol. 2020, 11, 1499. DOI:10.3389/fimmu.2020.01499 [Google Scholar]
-
Huang X, Li Z, Huang Y, Zhang Q, Cui Y, Shi X, et al. Vimentin intermediate filaments coordinate actin stress fibers and podosomes to determine the extracellular matrix degradation by macrophages. Dev. Cell 2025, 60, 1669–1685.e6. DOI:10.1016/j.devcel.2025.01.016 [Google Scholar]
-
Zheng M, Tian Z. Liver-Mediated Adaptive Immune Tolerance. Front. Immunol. 2019, 10, 2525. DOI:10.3389/fimmu.2019.02525 [Google Scholar]
-
Zhang Y, Wu Y, Shen W, Wang B, Yuan X. Crosstalk between NK cells and hepatic stellate cells in liver fibrosis (Review). Mol. Med. Rep. 2022, 25, 208. DOI:10.3892/mmr.2022.12724 [Google Scholar]
-
Fink T, Ebbesen P, Koppelhus U, Zachar V. Natural killer cell-mediated basal and interferon-enhanced cytotoxicity against liver cancer cells is significantly impaired under in vivo oxygen conditions. Scand. J. Immunol. 2003, 58, 607–612. DOI:10.1111/j.1365-3083.2003.01347.x [Google Scholar]
-
Kahraman A, Fingas CD, Syn WK, Gerken G, Canbay A. Role of stress-induced NKG2D ligands in liver diseases. Liver Int. 2012, 32, 370–382. DOI:10.1111/j.1478-3231.2011.02608.x [Google Scholar]
-
Muhanna N, Tair LA, Doron S, Amer J, Azzeh M, Mahamid M, et al. Amelioration of hepatic fibrosis by NK cell activation. Gut 2011, 60, 90–98. DOI:10.1136/gut.2010.211136 [Google Scholar]
-
Tao X, Zhang R, Du R, Yu T, Yang H, Li J, et al. EP3 enhances adhesion and cytotoxicity of NK cells toward hepatic stellate cells in a murine liver fibrosis model. J. Exp. Med. 2022, 219, e20212414. DOI:10.1084/jem.20212414 [Google Scholar]
-
Gao B, Radaeva S. Natural killer and natural killer T cells in liver fibrosis. Biochim. Biophys. Acta 2013, 1832, 1061–1069. DOI:10.1016/j.bbadis.2012.09.008 [Google Scholar]
-
Zhang Q, Bi J, Zheng X, Chen Y, Wang H, Wu W, et al. Blockade of the checkpoint receptor TIGIT prevents NK cell exhaustion and elicits potent anti-tumor immunity. Nat. Immunol. 2018, 19, 723–732. DOI:10.1038/s41590-018-0132-0 [Google Scholar]
-
Choi W, Ryu T, Lee J, Shim Y, Kim M, Kim H, et al. Metabotropic Glutamate Receptor 5 in Natural Killer Cells Attenuates Liver Fibrosis by Exerting Cytotoxicity to Activated Stellate Cells. Hepatology 2021, 74, 2170–2185. DOI:10.1002/hep.31875 [Google Scholar]
-
Jeong WI, Park O, Suh YG, Byun JS, Park SY, Choi E, et al. Suppression of innate immunity (natural killer cell/interferon-γ) in the advanced stages of liver fibrosis in mice. Hepatology 2011, 53, 1342–1351. DOI:10.1002/hep.24190 [Google Scholar]
-
Zheng M, Sun H, Tian Z. Natural killer cells in liver diseases. Front. Med. 2018, 12, 269–279. DOI:10.1007/s11684-018-0621-4 [Google Scholar]
-
Gergues M, Bari R, Koppisetti S, Gosiewska A, Kang L, Hariri RJ. Senescence, NK cells, and cancer: Navigating the crossroads of aging and disease. Front. Immunol. 2025, 16, 1565278. DOI:10.3389/fimmu.2025.1565278 [Google Scholar]
-
Cadoux M, Caruso S, Pham S, Gougelet A, Pophillat C, Riou R, et al. Expression of NKG2D ligands is downregulated by β-catenin signalling and associates with HCC aggressiveness. J. Hepatol. 2021, 74, 1386–1397. DOI:10.1016/j.jhep.2021.01.017 [Google Scholar]
-
Gur C, Doron S, Kfir-Erenfeld S, Horwitz E, Abu-tair L, Safadi R, et al. NKp46-mediated killing of human and mouse hepatic stellate cells attenuates liver fibrosis. Gut 2012, 61, 885–893. DOI:10.1136/gutjnl-2011-301400 [Google Scholar]
-
Gong Y, Jin L, Duan L, Xiao J, Li Y, Wang H, et al. Extracellular Vesicles Derived From Streptococcus anginosus Aggravate Lupus Nephritis by Triggering TLR2-MyD88-NF-κB Signalling in NK Cells. J. Extracell. Vesicles 2025, 14, e70134. DOI:10.1002/jev2.70134 [Google Scholar]
-
Rodriguez-Sevilla JJ, Ganan-Gomez I, Kumar B, Thongon N, Ma F, Chien KS, et al. Natural killer cells’ functional impairment drives the immune escape of pre-malignant clones in early-stage myelodysplastic syndromes. Nat. Commun. 2025, 16, 3450. DOI:10.1038/s41467-025-58662-0 [Google Scholar]
-
Li R, Li Z, Feng Y, Yang H, Shi Q, Tao Z, et al. PDGFRβ-targeted TRAIL specifically induces apoptosis of activated hepatic stellate cells and ameliorates liver fibrosis. Apoptosis 2020, 25, 105–119. DOI:10.1007/s10495-019-01583-3 [Google Scholar]
-
Jang JH, Kim H, Jun H, Park CY, Kim JY, Yeo M, et al. Targeting RBM39 with Tasisulam enhances TRAIL-induced apoptosis through DR5 upregulation and Bcl-2 downregulation in renal cell carcinoma. Biochem. Pharmacol. 2025, 236, 116877. DOI:10.1016/j.bcp.2025.116877 [Google Scholar]
-
Gao B, Radaeva S, Jeong WI. Activation of natural killer cells inhibits liver fibrosis: A novel strategy to treat liver fibrosis. Expert Rev. Gastroenterol. Hepatol. 2007, 1, 173–180. DOI:10.1586/17474124.1.1.173 [Google Scholar]
-
Correia AL, Guimaraes JC, Auf der Maur P, De Silva D, Trefny MP, Okamoto R, et al. Hepatic stellate cells suppress NK cell-sustained breast cancer dormancy. Nature 2021, 594, 566–571. DOI:10.1038/s41586-021-03614-z [Google Scholar]
-
Huang Y, Chen M, Guo Q, Chen Y, Zhang L, Chen Z, et al. Interleukin-10 promotes primary rat hepatic stellate cell senescence by upregulating the expression levels of p53 and p21. Mol. Med. Rep. 2018, 17, 5700–5707. DOI:10.3892/mmr.2018.8592 [Google Scholar]
-
Gao Y, Chi Y, Chen Y, Wang W, Li H, Zheng W, et al. Multi-omics analysis of human mesenchymal stem cells shows cell aging that alters immunomodulatory activity through the downregulation of PD-L1. Nat. Commun. 2023, 14, 4373. DOI:10.1038/s41467-023-39958-5 [Google Scholar]
-
Jin H, Jia Y, Yao Z, Huang J, Hao M, Yao S, et al. Hepatic stellate cell interferes with NK cell regulation of fibrogenesis via curcumin induced senescence of hepatic stellate cell. Cell. Signal. 2017, 33, 79–85. DOI:10.1016/j.cellsig.2017.02.006 [Google Scholar]
-
Wang L, Wang Y, Quan J. Exosomes derived from natural killer cells inhibit hepatic stellate cell activation and liver fibrosis. Hum. Cell 2020, 33, 582–589. DOI:10.1007/s13577-020-00371-5 [Google Scholar]
-
Wang L, Wang Y, Quan J. Exosomal miR-223 derived from natural killer cells inhibits hepatic stellate cell activation by suppressing autophagy. Mol. Med. 2020, 26, 81. DOI:10.1186/s10020-020-00207-w [Google Scholar]
-
Li QY, Gong T, Huang YK, Kang L, Warner CA, Xie H, et al. Role of noncoding RNAs in liver fibrosis. World J. Gastroenterol. 2023, 29, 1446–1459. DOI:10.3748/wjg.v29.i9.1446 [Google Scholar]
-
Market M, Tennakoon G, Scaffidi M, Cook DP, Angka L, Ng J, et al. Preventing Surgery-Induced NK Cell Dysfunction Using Anti-TGF-β Immunotherapeutics. Int. J. Mol. Sci. 2022, 23, 14608. DOI:10.3390/ijms232314608 [Google Scholar]
-
Huang Y, Chen Z, Jang JH, Baig MS, Bertolet G, Schroeder C, et al. PD-1 blocks lytic granule polarization with concomitant impairment of integrin outside-in signaling in the natural killer cell immunological synapse. J. Allergy Clin. Immunol. 2018, 142, 1311–1321.e8. DOI:10.1016/j.jaci.2018.02.050 [Google Scholar]
-
Fugier E, Marche H, Thélu MA, Macek Jílková Z, Van Campenhout N, Dufeu-Duchesne T, et al. Functions of liver natural killer cells are dependent on the severity of liver inflammation and fibrosis in chronic hepatitis C. PLoS ONE 2014, 9, e95614. DOI:10.1371/journal.pone.0095614 [Google Scholar]
-
Wang X, Lin R, Li D, Ye W, Yang Z, Wu N, et al. Modulation of γδ T cells by USF3: Implications for liver fibrosis and immune regulation. Int. Immunopharmacol. 2025, 148, 114100. DOI:10.1016/j.intimp.2025.114100 [Google Scholar]
-
Cardon A, Guinebretière T, Dong C, Gil L, Ado S, Gavlovsky PJ, et al. Single cell profiling of circulating autoreactive CD4 T cells from patients with autoimmune liver diseases suggests tissue imprinting. Nat. Commun. 2025, 16, 1161. DOI:10.1038/s41467-025-56363-2 [Google Scholar]
-
Martin OP, Wallace MS, Oetheimer C, Patel HB, Butler MD, Wong LP, et al. Single-cell atlas of human liver and blood immune cells across fatty liver disease stages reveals distinct signatures linked to liver dysfunction and fibrogenesis. Nat. Immunol. 2025, 26, 1596–1611. DOI:10.1038/s41590-025-02255-y [Google Scholar]
-
Burtis AEC, DeNicola DMC, Ferguson ME, Santos RG, Pinilla C, Kriss MS, et al. Ag-driven CD8+ T cell clonal expansion is a prominent feature of MASH in humans and mice. Hepatology 2025, 81, 591–608. DOI:10.1097/HEP.0000000000000971 [Google Scholar]
-
Zhan C, Peng C, Wei H, Wei K, Ou Y, Zhang Z. Diverse Subsets of γδT Cells and Their Specific Functions Across Liver Diseases. Int. J. Mol. Sci. 2025, 26, 2778. DOI:10.3390/ijms26062778 [Google Scholar]
-
Zhou QH, Wu FT, Pang LT, Zhang TB, Chen Z. Role of γδT cells in liver diseases and its relationship with intestinal microbiota. World J. Gastroenterol. 2020, 26, 2559–2569. DOI:10.3748/wjg.v26.i20.2559 [Google Scholar]
-
Chamroonkul N, Bansal MB. HIV and the liver. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 1–2. DOI:10.1038/s41575-018-0085-7 [Google Scholar]
-
Garcia-Broncano P, Medrano LM, Berenguer J, González-García J, Jiménez-Sousa MÁ, Carrero A, et al. Dysregulation of the Immune System in HIV/HCV-Coinfected Patients According to Liver Stiffness Status. Cells 2018, 7, 196. DOI:10.3390/cells7110196 [Google Scholar]
-
Kim AY, Chung RT. Coinfection with HIV-1 and HCV—A one-two punch. Gastroenterology 2009, 137, 795–814. DOI:10.1053/j.gastro.2009.06.040 [Google Scholar]
-
Basset L, Chevalier S, Danger Y, Arshad MI, Piquet-Pellorce C, Gascan H, et al. Interleukin-27 and IFNγ regulate the expression of CXCL9, CXCL10, and CXCL11 in hepatitis. J. Mol. Med. 2015, 93, 1355–1367. DOI:10.1007/s00109-015-1319-6 [Google Scholar]
-
Ji J, Chen Z, He W, Chen Y, Ye D, Zhong Y, et al. CXCR6 defines a distinct resident state in γδ T cells for protecting from liver cirrhosis. Hepatology 2025, 10–1097. DOI:10.1097/HEP.0000000000001643 [Google Scholar]
-
Knight B, Akhurst B, Matthews VB, Ruddell RG, Ramm GA, Abraham LJ, et al. Attenuated liver progenitor (oval) cell and fibrogenic responses to the choline deficient, ethionine supplemented diet in the BALB/c inbred strain of mice. J. Hepatol. 2007, 46, 134–141. DOI:10.1016/j.jhep.2006.08.015 [Google Scholar]
-
Hadian Y, Bagood MD, Dahle SE, Sood A, Isseroff RR. Interleukin-17: Potential Target for Chronic Wounds. Mediat. Inflamm. 2019, 2019, 1297675. DOI:10.1155/2019/1297675 [Google Scholar]
-
Kong X, Feng D, Wang H, Hong F, Bertola A, Wang FS, et al. Interleukin-22 induces hepatic stellate cell senescence and restricts liver fibrosis in mice. Hepatology 2012, 56, 1150–1159. DOI:10.1002/hep.25744 [Google Scholar]
-
Yoon JS, Lee CW. Protein phosphatases regulate the liver microenvironment in the development of hepatocellular carcinoma. Exp. Mol. Med. 2022, 54, 1799–1813. DOI:10.1038/s12276-022-00883-0 [Google Scholar]
-
Seki E, Schwabe RF. Hepatic inflammation and fibrosis: Functional links and key pathways. Hepatology 2015, 61, 1066–1079. DOI:10.1002/hep.27332 [Google Scholar]
-
Roh YS, Park S, Lim CW, Kim B. Depletion of Foxp3+ Regulatory T Cells Promotes Profibrogenic Milieu of Cholestasis-Induced Liver Injury. Dig. Dis. Sci. 2015, 60, 2009–2018. DOI:10.1007/s10620-014-3438-2 [Google Scholar]
-
Li J, Qiu SJ, She WM, Wang FP, Gao H, Li L, et al. Significance of the balance between regulatory T (Treg) and T helper 17 (Th17) cells during hepatitis B virus related liver fibrosis. PLoS ONE 2012, 7, e39307. DOI:10.1371/journal.pone.0039307 [Google Scholar]
-
Wu KJ, Qian QF, Zhou JR, Sun DL, Duan YF, Zhu X, et al. Regulatory T cells (Tregs) in liver fibrosis. Cell Death Discov. 2023, 9, 53. DOI:10.1038/s41420-023-01347-8 [Google Scholar]
-
Savage TM, Fortson KT, de los Santos-Alexis K, Oliveras-Alsina A, Rouanne M, Rae SS, et al. Amphiregulin from regulatory T cells promotes liver fibrosis and insulin resistance in non-alcoholic steatohepatitis. Immunity 2024, 57, 303–318.e6. DOI:10.1016/j.immuni.2024.01.009 [Google Scholar]
-
Ajith A, Merimi M, Arki MK, Hossein-khannazer N, Najar M, Vosough M, et al. Immune regulation and therapeutic application of T regulatory cells in liver diseases. Front. Immunol. 2024, 15, 1371089. DOI:10.3389/fimmu.2024.1371089 [Google Scholar]
-
Koda Y, Teratani T, Chu PS, Hagihara Y, Mikami Y, Harada Y, et al. CD8+ tissue-resident memory T cells promote liver fibrosis resolution by inducing apoptosis of hepatic stellate cells. Nat. Commun. 2021, 12, 4474. DOI:10.1038/s41467-021-24734-0 [Google Scholar]
-
Wang W, Green M, Choi JE, Gijón M, Kennedy PD, Johnson JK, et al. CD8+ T cells regulate tumour ferroptosis during cancer immunotherapy. Nature 2019, 569, 270–274. DOI:10.1038/s41586-019-1170-y [Google Scholar]
-
Wang H, Tsung A, Mishra L, Huang H. Regulatory T cell: A double-edged sword from metabolic-dysfunction-associated steatohepatitis to hepatocellular carcinoma. eBioMedicine 2024, 101, 105031. DOI:10.1016/j.ebiom.2024.105031 [Google Scholar]
-
Winau F, Quack C, Darmoise A, Kaufmann SHE. Starring stellate cells in liver immunology. Curr. Opin. Immunol. 2008, 20, 68–74. DOI:10.1016/j.coi.2007.10.006 [Google Scholar]
-
Zhao W, Su W, Kuang P, Zhang L, Liu J, Yin Z, et al. The role of hepatic stellate cells in the regulation of T-cell function and the promotion of hepatocellular carcinoma. Int. J. Oncol. 2012, 41, 457–464. DOI:10.3892/ijo.2012.1497 [Google Scholar]
-
Li X, Su Y, Hua X, Xie C, Liu J, Huang Y, et al. Levels of hepatic Th17 cells and regulatory T cells upregulated by hepatic stellate cells in advanced HBV-related liver fibrosis. J. Transl. Med. 2017, 15, 75. DOI:10.1186/s12967-017-1167-y [Google Scholar]
-
Aoyama T, Inokuchi S, Brenner DA, Seki E. CX3CL1-CX3CR1 interaction prevents carbon tetrachloride-induced liver inflammation and fibrosis in mice. Hepatology 2010, 52, 1390–1400. DOI:10.1002/hep.23795 [Google Scholar]
-
Tao J, Wu Z, Liang Y, Wang J, Tang M, Huang S, et al. Lhx2 specifically expressed in HSCs promotes liver regeneration and inhibits liver fibrosis. Hepatology 2025, 82, 683–701. DOI:10.1097/HEP.0000000000001201 [Google Scholar]
-
Ma F, Liu X, Zhang Y, Tao Y, Zhao L, Abusalamah H, et al. Tumor extracellular vesicle-derived PD-L1 promotes T cell senescence through lipid metabolism reprogramming. Sci. Transl. Med. 2025, 17, eadm7269. DOI:10.1126/scitranslmed.adm7269 [Google Scholar]