Deadline for manuscript submissions: 31 August 2026.



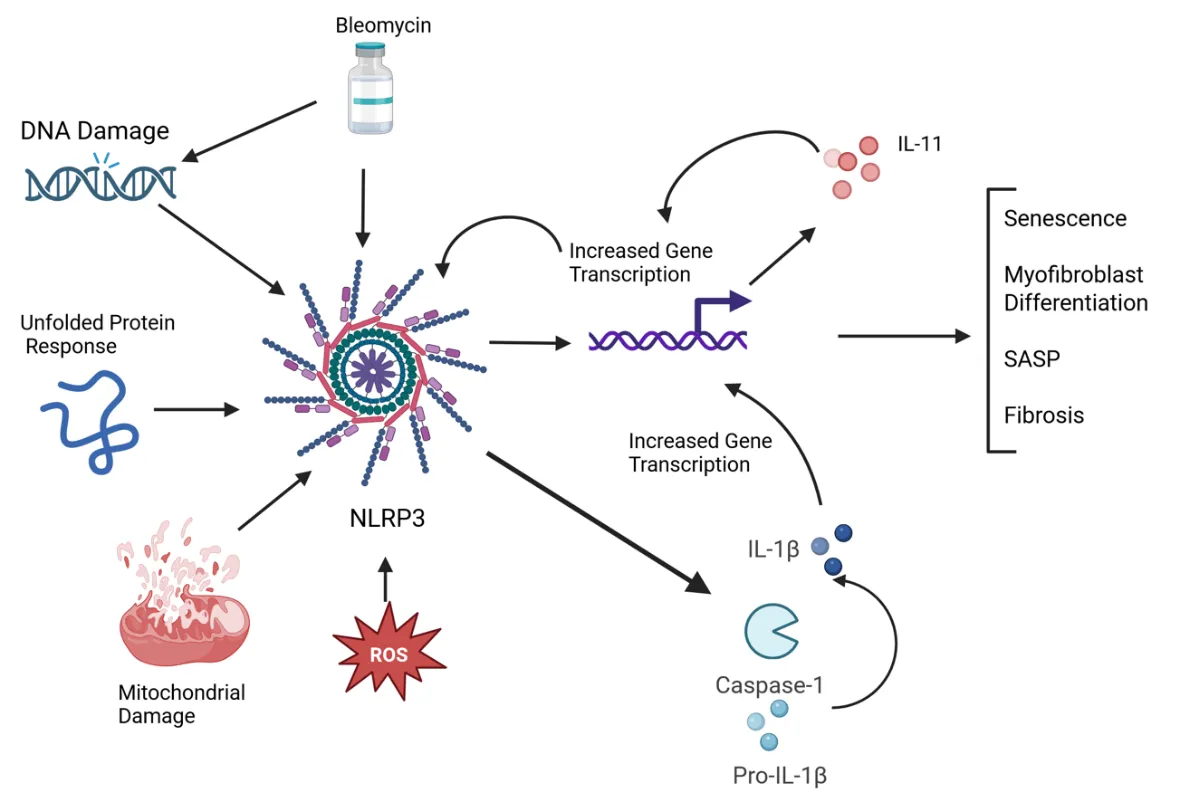
Systemic sclerosis (SSc) is an autoimmune disease characterized by widespread fibrosis affecting multiple organ systems. There is clinical heterogeneity among patients with SSc in terms of the organs affected. However, the pathophysiology of the disease remains elusive. The NLRP3 inflammasome is upregulated in SSc and exerts its fibrotic effects through activation of caspase-1, which in turn activates a fibrotic signaling cascade, resulting in increased collagen deposition and myofibroblast transition. Recently, IL-11 has been shown to be elevated in disease and has been shown to participate in downstream signaling via the NLRP3 inflammasome. A significant number of patients with SSc will develop pulmonary involvement, termed interstitial lung disease (SSc-ILD). Though this type of pulmonary involvement is distinct from other types of pulmonary fibrosis (such as idiopathic pulmonary fibrosis), it may be a valuable model to study mechanisms of fibrosis that could apply to other fibrotic diseases. Here, we discuss recent advances in understanding the mechanisms of the NLRP3 inflammasome and IL-11 in SSc pulmonary fibroblasts. We tie together some of the recent findings, such as senescence, the unfolded protein response, and reactive oxygen species, that contribute to fibrotic pathology via modulating NLRP3 activation, possibly leading to IL-11 expression.
Fibrotic diseases are driven by the excessive accumulation of extracellular matrix (ECM), particularly collagens, leading to progressive tissue stiffness and organ dysfunction. While many factors contribute to fibrosis—including cytokine signaling, integrin-mediated mechanotransduction, and altered ECM degradation—the synthesis and secretion of collagen remain central bottlenecks. Collagen biosynthesis is a complex process involving extensive post-translational modification and intracellular trafficking. The export of procollagen from the endoplasmic reticulum (ER) requires Transport and Golgi Organisation 1 (TANGO1), a transmembrane organizer of ER exit sites that coordinates cargo selection, membrane remodeling, and connectivity between the ER and the ER-Golgi-Intermediate-Comaprtment (ERGIC). By assembling into ring-like structures at ER exit sites, TANGO1 builds a secretory route for bulky cargoes that bypasses conventional vesicle constraints. Loss of TANGO1 disrupts collagen secretion and causes developmental defects across various species. In fibrotic tissues, TANGO1 expression is upregulated, linking secretory machinery to pathological matrix deposition. Recent work has identified specific interfaces within the complex of TANGO1 with its vertebrate paralogue Cutaneous T-cell lymphoma-associated antigen 5 (cTAGE5) as targets for cell-permeant peptide inhibitors. Inhibitors that selectively and specifically block TANGO1 complex formation reduce collagen secretion in fibroblasts and scar formation in vivo, offering a new strategy to modulate fibrotic processes.

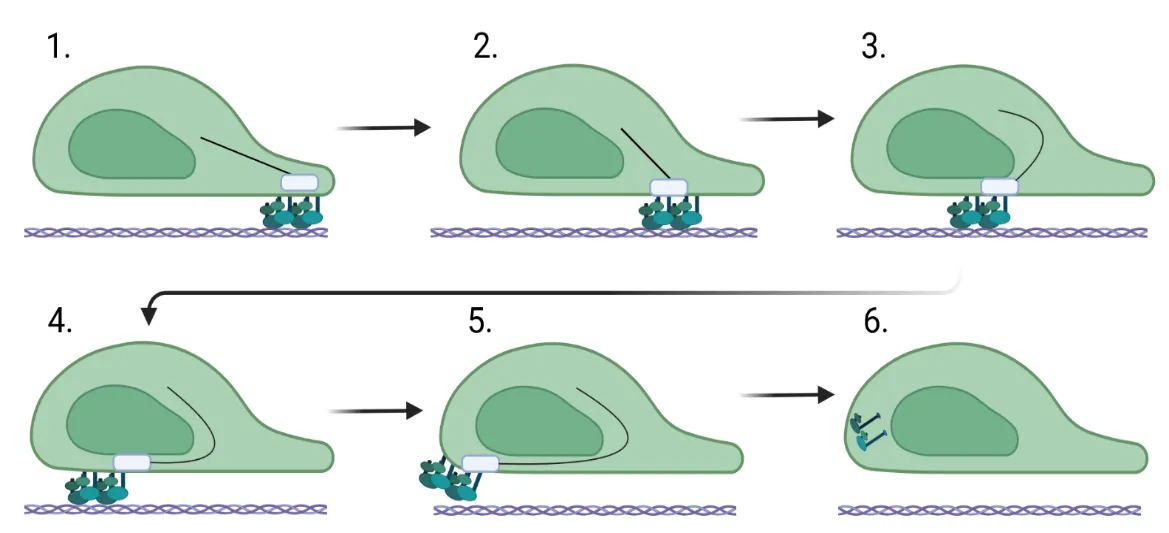
Fibroblast migration is a critical factor in wound healing, but also plays a fundamental role in fibrosis. For a fibroblast to migrate, the cell must be able to assemble factors that help it crawl across the extracellular matrix. Most of this movement is facilitated through the assembly and stability of the cytoskeleton that connects focal adhesion engagement with the extracellular matrix to intracellular stress fibers that wrap around the nucleus. These intracellular stress fibers help to polarize the fibroblast and orient the nucleus in the direction it is traveling. Changes in intracellular signaling for the fibroblast to move are also required, and this is necessitated by downstream signaling mediated by sonic hedgehog, WNT/β-catenin, ROCK/Rho, and PI3K/AKT. These changes regulate the stability of the cytoskeleton and, in addition, increase the expression of genes involved in cell migration. This review assimilates what is known about the function of the cytoskeleton in migration and the role of intracellular signaling pathways in fibrosis.
Cellular communication network factor 2 (CCN2, formerly known as ‘connective tissue growth factor’ or ‘CTGF’) was the subject of anti-fibrotic drug development programs, largely in FibroGen, starting in the mid-1990s. This led to the development of FG-3019 (pamrevlumab) as a lead drug that was used initially to target diabetic nephropathy and subsequently pancreatic cancer, pulmonary fibrosis and Duchenne’s muscular dystrophy. All these programs failed clinically; diabetes in early development, and the others at Phase III. Could these failures have been anticipated? Is ‘CTGF’ dead as an anti-fibrotic target? What might have been done differently or could be done differently in the future? This personal commentary—based on years of experience first at FibroGen working on the ‘CTGF’ program and then as an independent academic researcher---aims to address at least some of these issues.
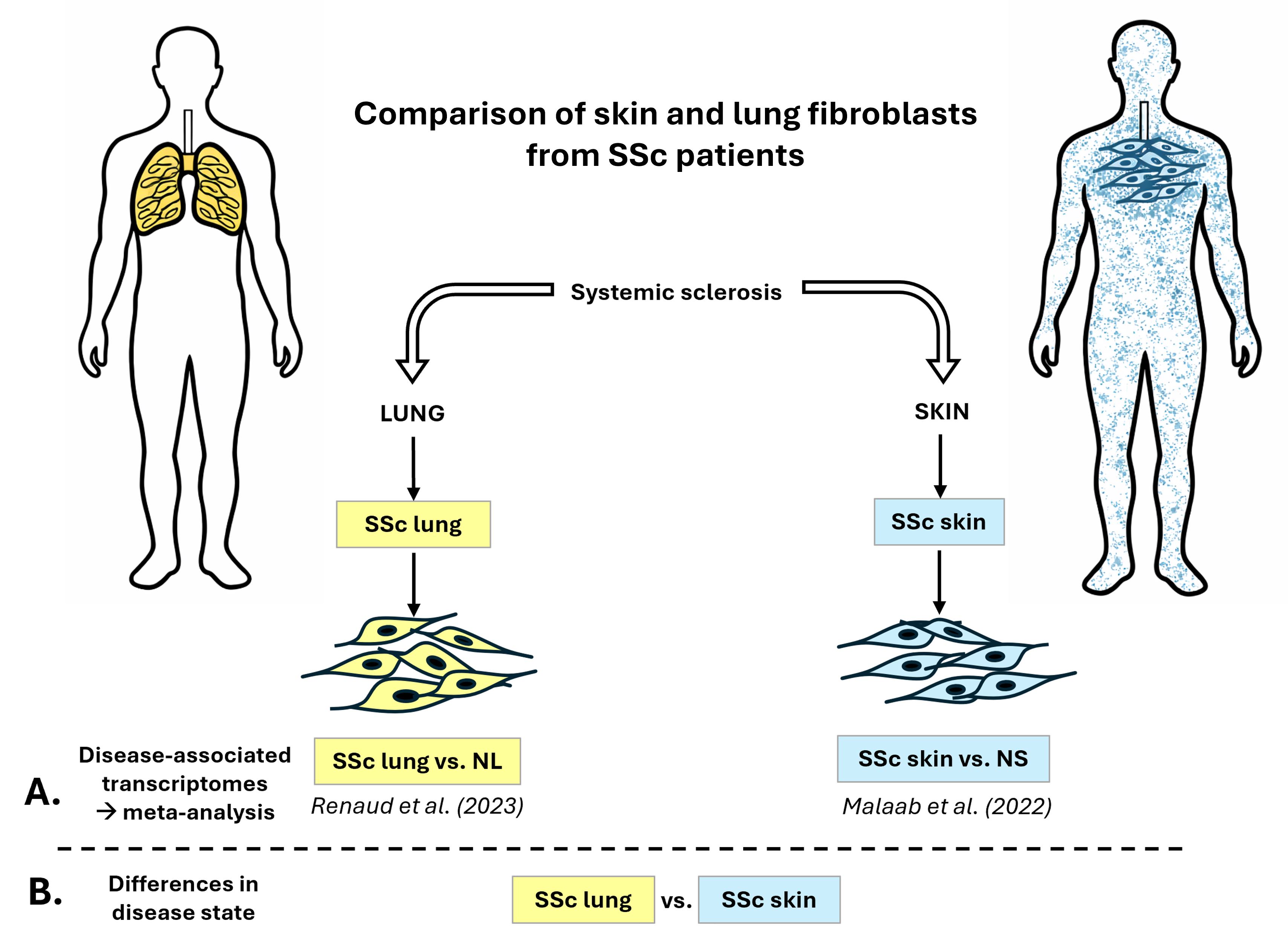
Systemic Sclerosis (SSc) is a chronic autoimmune disease characterized by fibrosis in connective tissues. Fibroblasts are the effector cells of fibrosis since they contribute to the production of collagen and other extracellular matrix components. The goal of this study is to compare the transcriptomic profiles of primary human SSc skin and SSc lung fibroblasts. First, we conducted a meta-analysis of differentially expressed (DE) genes from two previously published differential analyses (SSc vs. normal) using skin and lung fibroblasts, observing 8.7% overlap in DE genes and 30% overlap in impacted pathways. Next, we characterized the signature of several genes of interest from the pro- and anti-fibrotic programs within the unique and overlap groups and explored overlapping drugs that are predicted to revert DE genes to “normal expression”. Finally, we identified 3760 DE genes between SSc lung and SSc skin fibroblasts, highlighting that fibroblasts in the disease state carry a tissue-specific signature that should be taken into consideration for therapeutic development. We also identified core genes that can serve as common targets for both skin and lung in SSc. To our knowledge, this is the first study to describe overlapping genes and pathways in primary human skin and lung fibroblasts from SSc patients.