Found 36 results
Open Access
Perspective
28 January 2026The Double Face of Exosomes Derived from Mesenchymal Stromal Cells in Fibrotic Lung Diseases: Pathology Contribution or Treatment?
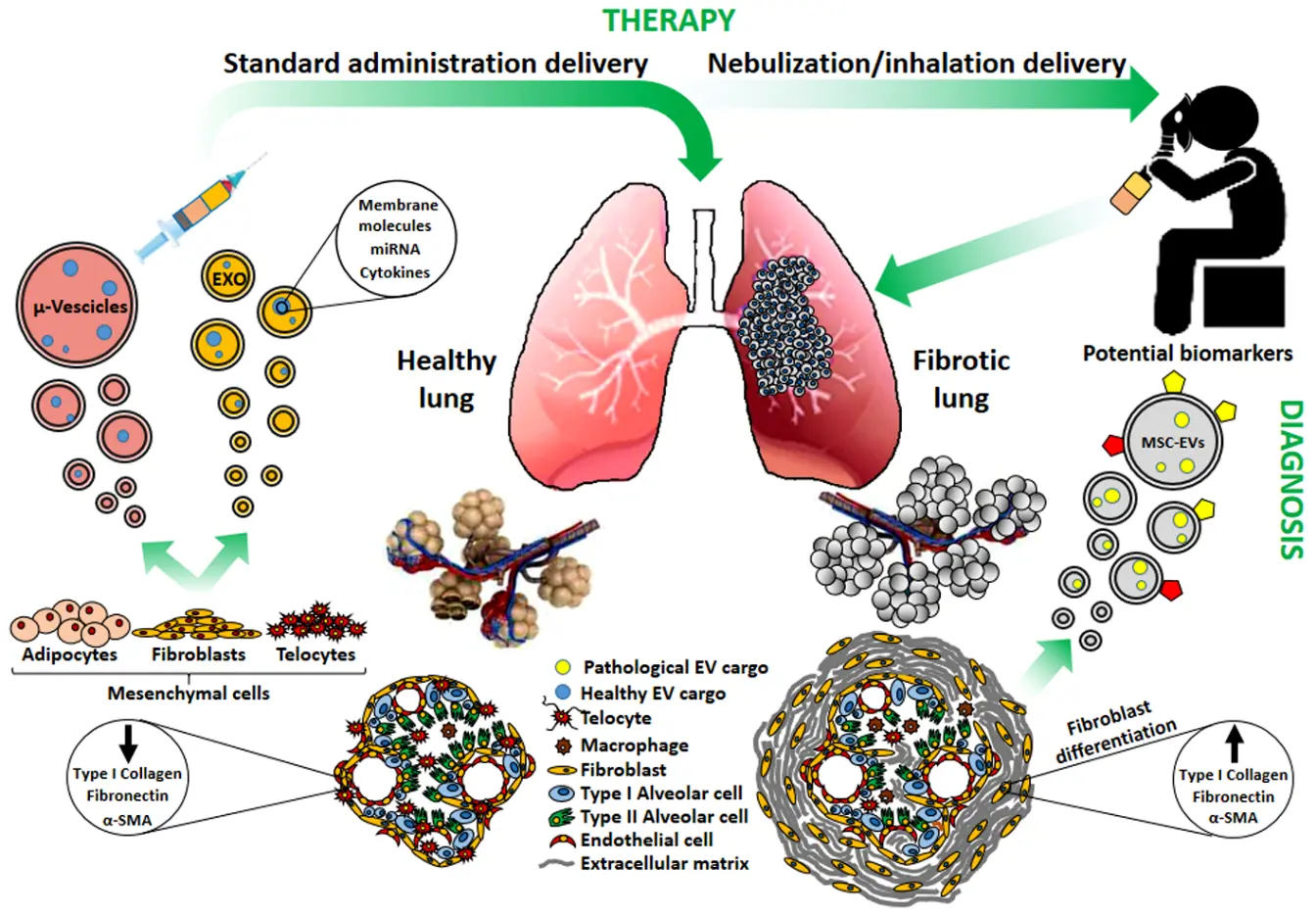
Several studies have attempted to clarify the role of exosomes and/or microvesicles derived from mesenchymal stromal cells (MSCs) (collectively indicated as extracellular vesicles: MSCs-EVs) in pulmonary fibrosis. Depending on their origin and on the micro-environmental context, MSCs-EVs may support or attenuate the fibrotic invasion of the lung, a hallmark of all Interstitial Lung Diseases (ILDs). Indeed, EVs have emerged as pivotal intercellular mediators and their potential diagnostic and therapeutic applications have been suggested. We aim here to elucidate the dual role of MSCs-derived exosomes and microvesicles: the contribution to pulmonary fibrosis progression, exerted by the MSCs-EVs originated from resident MSCs, or the potential therapeutic activity of those generated from healthy MSCs. Actually, MCSs-EVs appear as the frontiers of cell-free therapy and nano-medicine research in a great number of pre-clinical studies, but developments are needed to optimize and standardize their isolation, production and delivery. Interestingly, since the respiratory system directly communicates with the external environment, lung treatment could be approached by MSCs-EVs nebulization as a preferential administration route, integrating targeted pulmonary delivery with an enhanced patient’s compliance. Hence MSCs-EVs may contribute to ILD pathogenesis, display a potential as biomarkers, and still hold promise as therapeutic agents to reduce lung fibrosis. However further researches are needed to validate their clinical application.
Open Access
Article
22 January 2026Therapies Targeting Metabolic Pathways in Lung Fibrosis: Advances and Future Perspectives
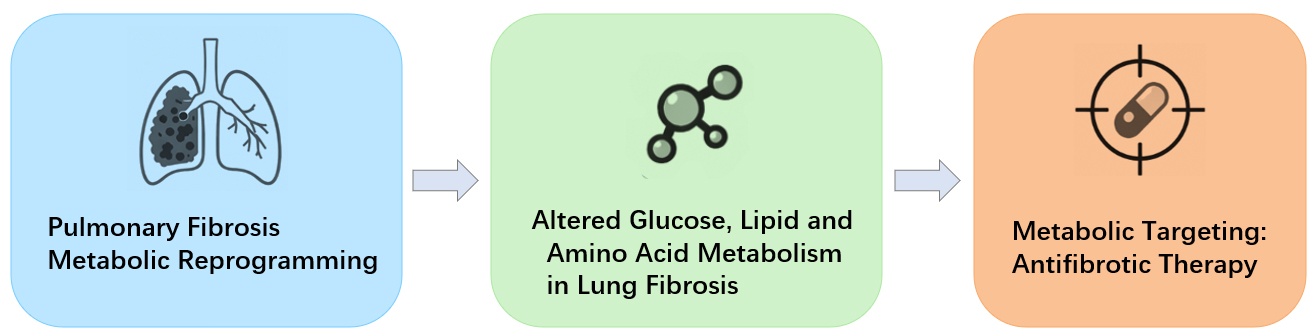
Pulmonary fibrosis is a progressive lung disease associated with high morbidity and mortality. Increasing evidence indicates that metabolic reprogramming is a central driver of fibrogenesis. Multiple cell types in the fibrotic lung, including fibroblasts, alveolar epithelial type II (AEC2) cells, and macrophages, exhibit enhanced glycolysis, dysregulated lipid turnover, and altered amino acid utilization. These metabolic changes promote fibroblast activation, sustain ECM production, and impair epithelial repair. Recent studies have identified key regulatory pathways—such as hypoxia-inducible factor-1α(HIF-1α)-mediated glycolysis, aberrant fatty acid and cholesterol metabolism, and glutamine-dependent anabolic processes—that collectively shape the profibrotic microenvironment. Targeting these metabolic vulnerabilities has shown promising antifibrotic effects in preclinical studies, supporting glycolysis inhibitors, lipid-modulating agents, and amino acid metabolism blockers as potential therapeutic approaches. This review summarizes recent advances in glucose, lipid, and amino acid metabolic reprogramming in pulmonary fibrosis, with IPF discussed as a representative and well-studied subtype, and highlights emerging metabolic-targeted therapeutic strategies. Understanding cell-specific metabolic adaptations may provide new opportunities to develop effective interventions for pulmonary fibrosis, whereas most metabolic mechanisms are shared across fibrotic lung diseases.
Open Access
Review
15 January 2026Renal Aging and Fibrosis in the Elderly: Frontiers in Non-Invasive Assessment
Today’s society has gradually entered an aging phase, and among the elderly population, the risk of chronic kidney disease (CKD) is significantly increased. Renal fibrosis is the key pathological mechanism for the development of chronic kidney disease to end-stage renal disease. With the increase in age, the phenomenon of glomerular sclerosis and interstitial fibrosis in aging kidneys gradually aggravates, and the glomerular filtration rate (GFR) decreases, further affecting renal function. Fibrosis not only accelerates the loss of renal function but also significantly increases the risk of cardiovascular disease, which seriously affects the quality of life and life expectancy of patients. This paper reviews the relevant literature and discusses the characteristics of an aging kidney and the diagnostic methods for renal fibrosis.

Open Access
Review
12 January 2026The Anti-Fibrotic Potential of GLP-1 and GIP Receptor Agonists in Chronic Inflammatory Disorders: Mechanisms and Therapeutic Horizons
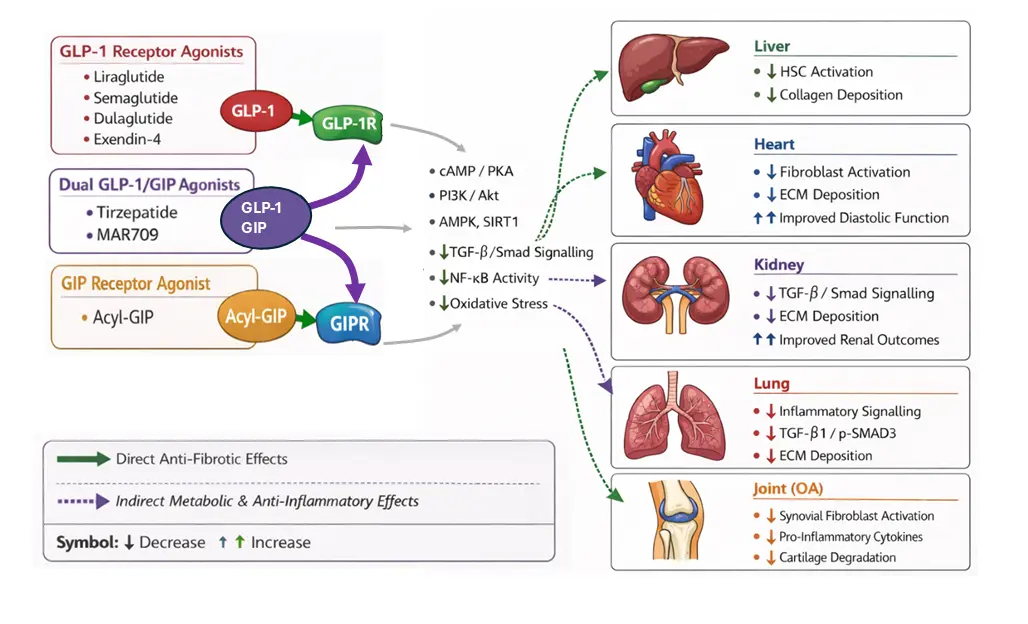
Fibrosis, characterised by the excessive deposition of extracellular matrix via activated fibroblasts, is a pathological feature of several chronic inflammatory disorders, which collectively contribute significantly to global morbidity and mortality. Despite this, current anti-fibrotic therapies are of limited efficacy. However, incretin-based therapies, primarily glucagon-like peptide-1 (GLP-1) receptor agonists, are now emerging as candidate drugs for modulating fibrotic signalling pathways. This review synthesises the growing body of preclinical and clinical evidence that incretin receptor agonists exert direct and indirect anti-fibrotic effects. We detail the molecular mechanisms and survey the promising data across hepatic, cardiac, renal, lung, and joint tissues, which underscore the potential for repurposing of this drug class as a therapeutic strategy for fibro-inflammatory conditions.
Open Access
Commentary
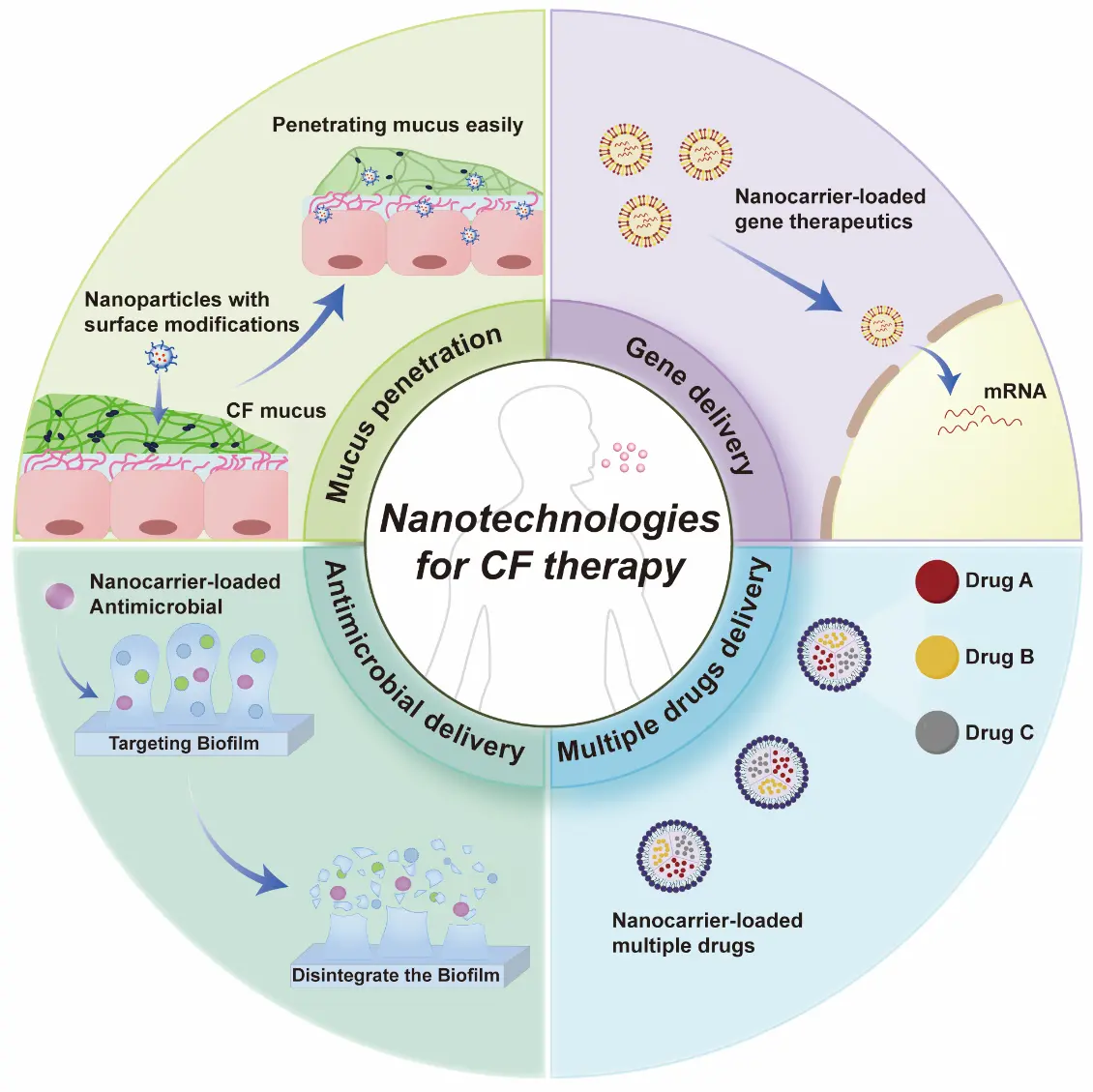
29 December 2025Breath and Life: Emerging Nanotechnologies for Cystic Fibrosis Therapy
The treatment of cystic fibrosis (CF) remains challenging due to formidable biological barriers in the lungs, including thick mucus and resilient biofilms that severely limit the efficacy of conventional therapies. Nanotechnology, engineered to overcome these barriers, is emerging as a transformative approach for CF therapy. This opinion highlighted the most recent and advanced nanotechnologies, categorizing them into four strategic frontiers: (1) nanocarriers that achieve mucus penetration through surface modifications; (2) nanoplatforms for efficient delivery of genetic therapeutics; (3) nanocarriers for antimicrobial delivery to cure infections associated with CF; and (4) combinatorial nanomedicines for synchronized delivery of multiple drugs. We concluded that, with the help of these nanotechnologies, therapies for CF will now undergo a paradigm shift, moving CF from a fatal disease to a treatable and potentially curable one. Although the clinical transition is challenging, it holds immense promise for revolutionizing CF management.
Open Access
Commentary
20 October 2025Sulfatide Inhibits Growth of Fibroblasts and Is a Potential Treatment against Fibrosis
Fibrosis of vital organs such as the lungs, liver, and kidneys is a serious condition without effective causal treatment. Here, we suggest the use of the sphingolipid sulfatide and its isoform C16, which we have found to inhibit the growth of fibroblasts. In the lungs, sulfatide can be easily administered via an inhalation spray. Alternatively, fenofibrate, an anti-cholesterol drug with no major side effects, may be used, as it enhances the body’s own production of sulfatide.

Open Access
Article
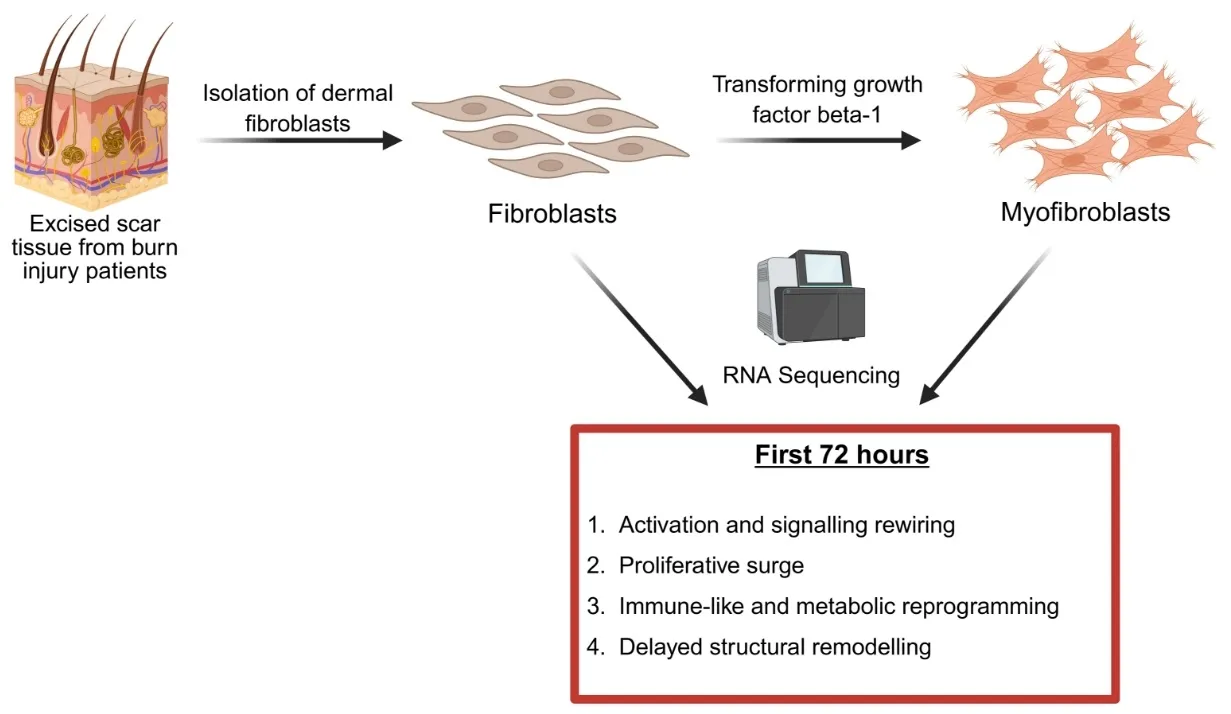
09 October 2025Identification of Pathways That Drive Myofibroblast Transformation in Hypertrophic Scars
Hypertrophic scars (HTS) are a common complication of burn injuries and are characterized by excessive dermal fibrosis driven by the transformation of resident dermal fibroblasts to profibrotic myofibroblasts. Although single cell and bulk RNA transcriptomics analysis of HTS and normal skin tissue samples were performed previously, transcriptomics of the transformation of fibroblasts to myofibroblasts has not been studied. Here, we report the data obtained from RNA sequencing of fibroblasts before and after exposure to transforming growth factor beta 1 (TGF-β1) and highlight the pathways that are up- and down-regulated during myofibroblast transformation. Our results suggest increased cellular signalling and rewiring, proliferative surge, immune-like and metabolic reprogramming, and delayed structural remodelling as four groups of events during the transformation of human primary dermal fibroblasts to myofibroblasts.
Open Access
Review
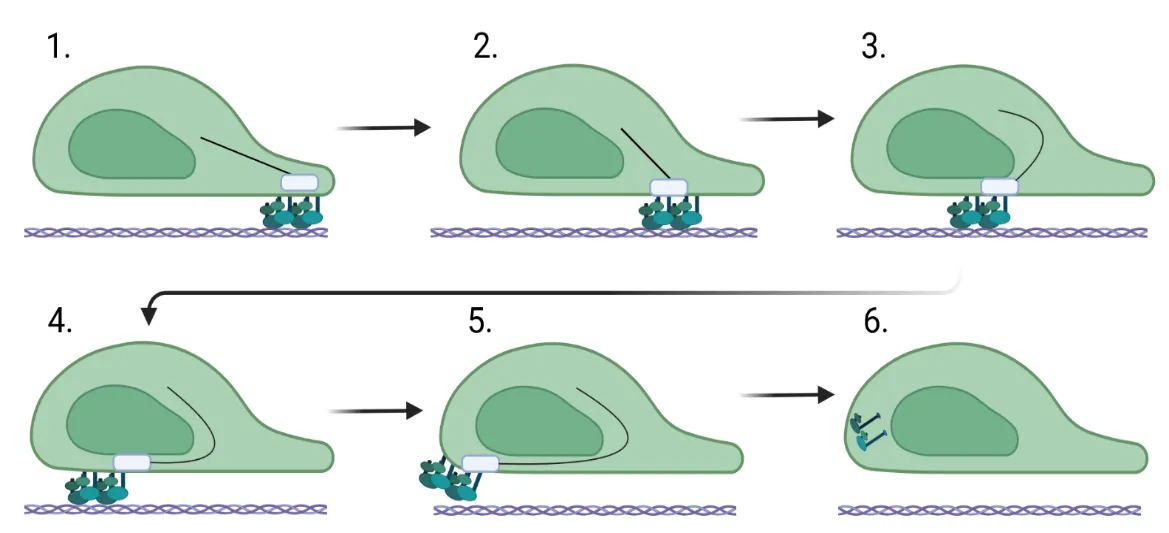
27 June 2025Fibroblast Migration in Fibrosis
Fibroblast migration is a critical factor in wound healing, but also plays a fundamental role in fibrosis. For a fibroblast to migrate, the cell must be able to assemble factors that help it crawl across the extracellular matrix. Most of this movement is facilitated through the assembly and stability of the cytoskeleton that connects focal adhesion engagement with the extracellular matrix to intracellular stress fibers that wrap around the nucleus. These intracellular stress fibers help to polarize the fibroblast and orient the nucleus in the direction it is traveling. Changes in intracellular signaling for the fibroblast to move are also required, and this is necessitated by downstream signaling mediated by sonic hedgehog, WNT/β-catenin, ROCK/Rho, and PI3K/AKT. These changes regulate the stability of the cytoskeleton and, in addition, increase the expression of genes involved in cell migration. This review assimilates what is known about the function of the cytoskeleton in migration and the role of intracellular signaling pathways in fibrosis.
Open Access
Review
27 June 2025Targeting Collagen Secretion as a Potential Therapeutic Strategy to Modulate Fibrosis
Fibrotic diseases are driven by the excessive accumulation of extracellular matrix (ECM), particularly collagens, leading to progressive tissue stiffness and organ dysfunction. While many factors contribute to fibrosis—including cytokine signaling, integrin-mediated mechanotransduction, and altered ECM degradation—the synthesis and secretion of collagen remain central bottlenecks. Collagen biosynthesis is a complex process involving extensive post-translational modification and intracellular trafficking. The export of procollagen from the endoplasmic reticulum (ER) requires Transport and Golgi Organisation 1 (TANGO1), a transmembrane organizer of ER exit sites that coordinates cargo selection, membrane remodeling, and connectivity between the ER and the ER-Golgi-Intermediate-Comaprtment (ERGIC). By assembling into ring-like structures at ER exit sites, TANGO1 builds a secretory route for bulky cargoes that bypasses conventional vesicle constraints. Loss of TANGO1 disrupts collagen secretion and causes developmental defects across various species. In fibrotic tissues, TANGO1 expression is upregulated, linking secretory machinery to pathological matrix deposition. Recent work has identified specific interfaces within the complex of TANGO1 with its vertebrate paralogue Cutaneous T-cell lymphoma-associated antigen 5 (cTAGE5) as targets for cell-permeant peptide inhibitors. Inhibitors that selectively and specifically block TANGO1 complex formation reduce collagen secretion in fibroblasts and scar formation in vivo, offering a new strategy to modulate fibrotic processes.

Open Access
Commentary
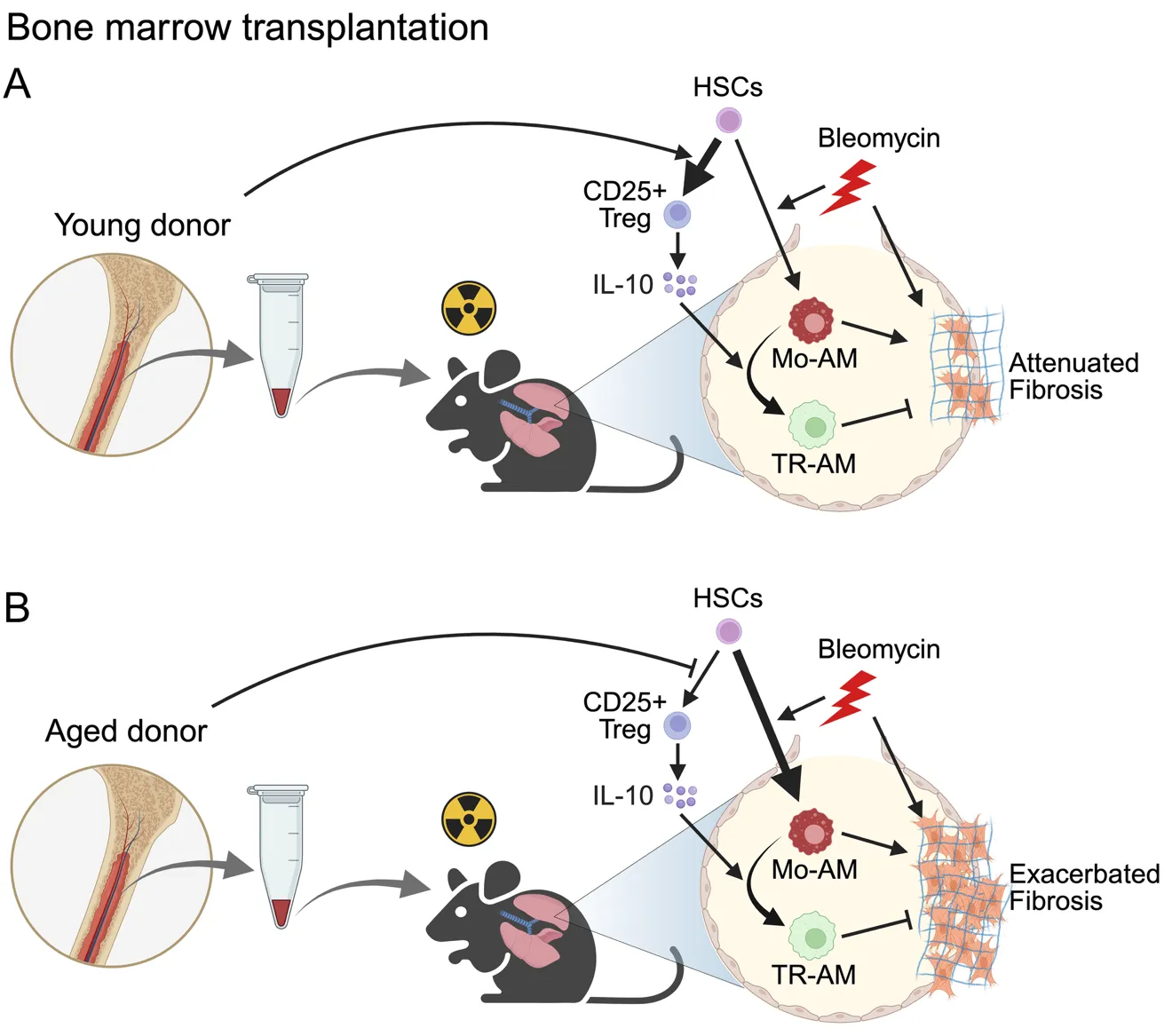
12 June 2025Bone Marrow Transplantation as a Future Therapeutic Strategy for Idiopathic Pulmonary Fibrosis: Lessons from Hematopoietic Aging
Idiopathic pulmonary fibrosis (IPF) is a fatal disease with limited therapeutic options. Lung transplantation is the only curative treatment, but it is rarely available due to a lack of suitable donors. In a recent publication in Science Immunology, Farhat et al. demonstrated that bone marrow transplantation from young donors alleviates fibrosis by restoring immune resolution in aged hosts in animal models. Aged hematopoietic cells exacerbate fibrosis through the persistence of inflammatory macrophages and impaired Treg-derived IL-10, highlighting bone marrow rejuvenation as a potential treatment strategy for IPF.