1. Introduction
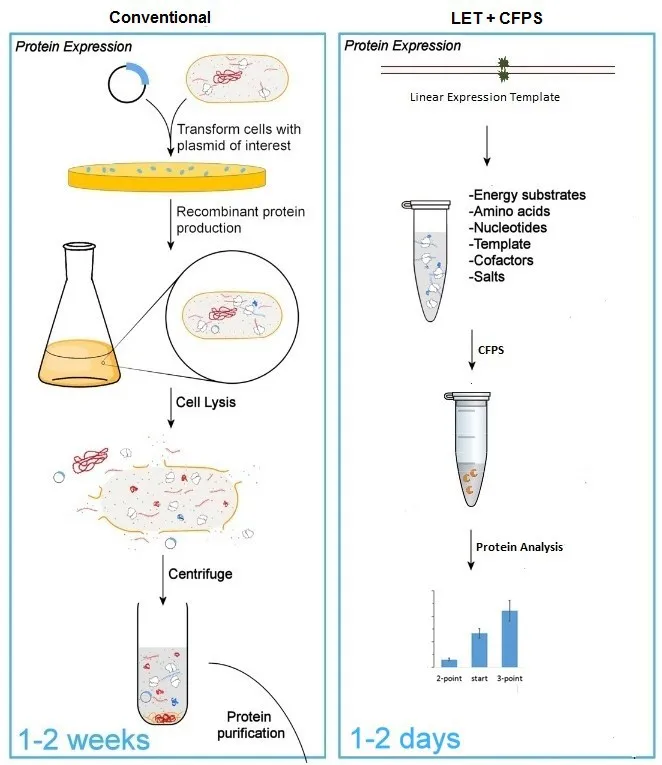
Modification of parts of enzymes’ amino acid sequence by substituting single or few amino acids leads to modified structure and function of the enzymes. In ideal conditions, these modifications will come with improved properties for bioconversion purposes like increased activity or altered substrate specificity. Therefore, numerous methods are developed to screen large number of mutants for beneficial enzyme properties. The combination of mutations positions leads to vast increase of variants that needs to be produced and screened. A conventional protein production and optimization method takes weeks to screen limited number of variants. To overcome this time limitation, libraries can be created using cell-free protein synthesis system (CFPS) based on linear expression template and polymerase chain reaction (PCR) (). In this method multiple variants can be produced and screened within a couple of days. The other key advantages of this PCR-based library screening are associated with open nature of the CFPS platform. Lack of cellular growth means no need for handling of cells and sterile conditions and therefore can be completely automated, thereby reducing the manual labor involved in cloning, protein production and purification steps of protein library screening. This also further reduces the time from production to analysis of proteins for specific mutation point or combinations of them. Fast and flexible manipulation of enzymes and enzyme complexes in an automated enzyme optimization system increases the chances of obtaining non-randomized and accurate results in shorter time.
Synthetic genes can be optimized for expression and constructed for easy mutational manipulation without any regard for the parent gene, thus providing a fast and economically efficient approach [
1]. Advances in the technology for gene synthesis make possible large-scale synthesis of not only one new gene but also whole library of mutants at low cost [
2]. Introduction of point mutations or combinations of point mutations into existing sequences or de novo creation of engineered genes are unique possibility in the in vitro synthesis of large DNA fragments and can be used for bioengineering applications, structural studies, drug development and combinatorial biology [
3]. Various methods have been proposed for in vitro gene synthesis like oligonucleotide ligation [
4], Fok I method [
5], DNA shuffling [
6] and PCR based methods [
7]. Among the PCR based methods, assembly PCR coupled with ordinary PCR is most suitable for large scale gene synthesis starting from short synthetic oligonucleotides [
1]. Hoover and Lubkowski developed an automated software for designing optimized oligonucleotides for PCR based gene synthesis methods called DNAWorks [
1]. This software uses either DNA or protein sequences as input and incase of protein sequences, it designs oligonucleotides based on the codon bias of the chosen host. Rouillard et al developed a web-based tool to design optimized oligonucleotides for in vitro gene assembly [
3]. The specificity and melting temperature uniformity of the oligonucleotides to avoid unspecific assembly and uniform hybridization are ensured by dynamically choosing the length of the oligonucleotides. This tool however only accepts DNA sequence to maximize the versatility.
Cell free protein synthesis (CFPS) system typically consists of a cell lysate (transcription/translation machinery) and a buffer/energy mix along with template DNA to produce the protein of interest in in-vitro setting [
8]. The cell-free systems were first utilized in 1960’s for defined synthetic RNA translation [
9]. Since then,
Escherichia coli (
E. coli) cell free systems have been used for protein productions though highly limited due to short lived reactions and low yield [
10]. This had led to a number of optimizations in cell lysates [
11] and energy sources [
12]. Due to these optimizations cell-free protein synthesis has become preferred method for protein production, as it offers numerous advantages over the conventional production method [
13]. The open nature of the in-vitro system facilitates modifications of proteins and enables production of difficult proteins like physiologically toxic proteins [
14] and large protein complexes [
15].
. Conventional protein production and purification method vs LET based cell free protein synthesis system. The absence of cloning and purification steps significantly reduces the time from protein expression to analysis of variants with single and mutliple mutations.
In this work, the time efficiency of LET based CFPS protein screening approach is demonstrated using model proteins green fluorescence protein (GFP), aspartokinase III (AKIII) and phosphoserine aminotransferase (serC). A simple monomeric protein like GFP was used to establish the method by achieving fluorescent signal variants like yellow fluorescence protein (YFP) and cyan fluorescence protein (CFP). In case of multimeric proteins like AKIII the up- and down- regulated inhibition and altered substrate specificity of studied variants were used while for serC, variants with altered substrate specificity were selected. AKIII is a bottleneck enzyme in the lysine biosynthetic pathway and one of the three iso-functional aspartokinases. It is naturally inhibited by an allosteric conformation transition caused by the product lysine [
16]. Both of the enzymes have been studied and engineered in our group for metabolic engineering and synthetic biology study of biosynthesis and hence multiple experimental data is available for cross checking the validity of CFPS based results with the conventional based results.
2. Materials and Method
2.1. Linear Expression Template
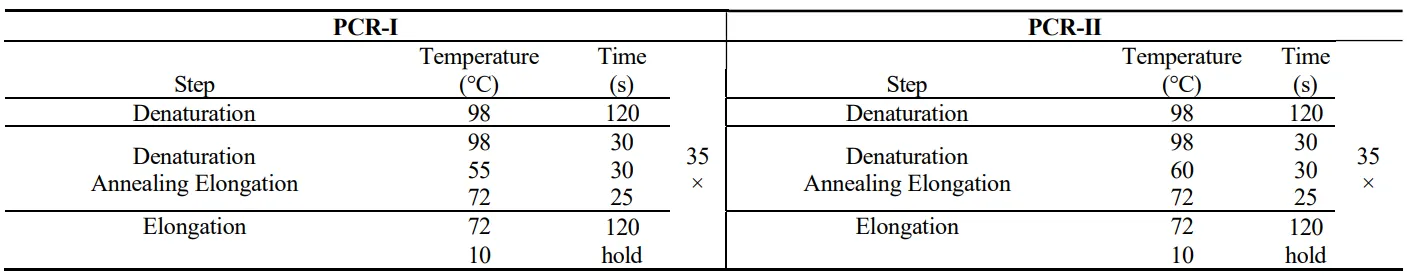
The LETs for the model enzymes were synthesized following a two-step PCR method [
3]. The first PCR was to assemble all the oligonucleotides while the second PCR was used to amplify the final product. The assembly PCR was carried out in a volume of 25 µL containing 12.5 µL of Phusion flash PCR master mix and 25 nM oligonucleotides. The product from assembly PCR was amplified using the outermost oligonucleotides as the forward and reverse primers in 50 µL reaction containing 1 µL of the assembly PCR mix, 25 µL Phusion PCR master mix and 1 µM of each primer ( and ).
The accuracy of the synthesized genes was then verified using agarose gel electrophoresis (Supplementary Materials Table S1 and Figure S1) and sequence analysis.
. The conditions for PCR-I and PCR-II used to produce GFP LET.
. The conditions for PCR-I and PCR-II used to produce AKIII and serC LET.
A T7 DNA polymerase based lysate was prepared using modified version of the method proposed by Adachi et al [
17]. To prepare the T7 DNA polymerase based cell lysate,
E. coli BL21-DE3 cells were grown at 37 °C overnight in 200 mL of 2YT (yeast extract-tryptone- NaCl) medium shaken at 150 rpm. The overnight culture was added to two separate baffled flasks containing 1 L of 2YT medium to an initial OD
600 higher than 0.1 and incubated at 30 °C with continuous shaking at 150 rpm. When the cell density reaches between OD
600 0.6 and 0.8, one of the two cultures was induced to produce T7 DNA polymerase using IPTG. The cells were then harvested at the mid to late log phase (OD
600 > 3.0).
The harvested cells were then washed three times with S30A buffer (10 mM Tris-acetate buffer (pH 8.0), 14 mM magnesium acetate, 60mM potassium acetate and 2 mM dithiothreitol). The washed cells were then suspended in S30B buffer (same as S30A buffer but with 1 mM dithiothreitol). It was then crushed with silica spheres (Lysing matrix B, MP Biomedicals, USA) using a homogenizer (FastPrep-24, MP Biomedicals, USA) four times at 6.0 m/s for 60 s. For every 1 g of cells 2 mL of S30A buffer and 4 g of the silica spheres were used. The spheres and debris were removed by centrifugation at 17,000×
g for 45 min at 4 °C. The supernatant was then mixed with 0.3 times volume of pre-incubation buffer (295 mM Tris acetate buffer (pH 8.0), 9 mM Mg(OAc)
2, 13.2 mM ATP, 84 mM PEP, 4.5 mM DTT, 40 µM of all 20 amino acids and 6.7 U/mL pyruvate kinase) and incubated at 37 °C for 80 min. The extract was dialysed two times against 50 fold S30B buffer. The first round of dialysis was done for 60 min and the second round was done overnight at 4 °C. The cell extract was centrifuged at 4000 g for 15 min at 4 °C. The supernatant obtained is the cell lysate which is then aliquoted, frozen in liquid nitrogen and stored at −80 °C. The energy solution used during the cell free protein synthesis reaction was prepared as described by Kigawa et al [
18] with the addition of 123 µM of gamS protein. This extract was used to produce model enzymes AKIII and serC and the reaction was performed at 25 °C for 8 h.
The Tx-Tl
E. coli cell extract was prepared by using the protocol published by Sun et al. This extract was used to produce GFP at 29 °C for 4 h.
2.3. Detection of Protein Production
Synthesis of aspartokinase III (AKIII) was performed at 25 °C for 8 h and shaken at 600 rpm. The applicability of T7 DNA polymerase based cell extract for multimeric protein production was tested using AKIII LET containing 6-histidine tag sequence. The quantity of His-tagged protein was then measured using bio-layer interferometry. His-tagged proteins bind to the anti-penta HIS biosensor thereby causing a spectral shift in the reflected white beam. By comparing the intensity of the white beam between the unbound and bound molecules, interferometry is able to quantify the protein in the sample. The Anti-penta-HIS (HIS-1k) biosensor (Pall ForteBio,USA) was used for quantitative measurement since it is pre-immobilized with Penta-HIS antibodies. Since the standard and samples must contain the same background buffer, the storage buffer used to dilute the standard also contains the cell free protein synthesis reaction mixture without the DNA template. The standard protein solutions were prepared using purified aspartokinase III at different concentrations ranging from 0 to 20 µg/mL. The biosensors need to be hydrated in the same storage buffer used to dilute the samples for at least 10 min. The assay was first set using the Data Acquisition 10.0 software from Pall ForteBio in which the biosensors are immersed in the samples for 90 min at 30 °C and shaken at 1000 rpm.
2.4. Screening Assays
Aspartokinase III (AKIII) activity was measured using the ATP sensor ‘QUEEN’ (quantitative evaluator of cellular energy) [
19] and hydroxymate method [
20]. The assay was performed in a 200 µL reaction solution containing 200 mMTris-HCl (pH 8.2), 10 mM MgSO
4·6H
2O, 10 mM aspartate, 2 mM ATP, 160 mM NH2OH (neutralized with KOH), 0.007 µM ATP sensor and appropriate amounts of enzyme [
21]. After incubation at 30 °C for 60 min, the enzyme activity was stopped by incubation at 4 °C for 5 min. The fluorescent signals from the ATP sensor were measured using the flow cytometryCytoFLEX (Beckman Coulter, Germany). Since the reaction mixture does not contain any cells, 0.909 µm polystyrene beads are used to trigger the events Wurm et al [
22]. The polystyrene beads were added to the mixture to the final concentration of 1:20000 dissolved in 0.01% polyethylene glycol solution. Each sample was measured with 50,000 events at a flow rate of 30 µL/min and the gain was adjusted to 1000 FITC and 27 KO525 while the threshold of FSH-H was manually set to 10,000.
Phosphoserine aminotransferase (serC) was screened using a serC-glutamate dehydrogenase coupled assay [
23]. During the oxidative deamination of L-glutamate, product of our enzymatic reaction the cofactor NAD
+ is reduced to NADH resulting in absorbance at 340 nm (). The reaction conditions were optimized to ensure the transamination reaction is the rate limiting step. This can be guaranteed by adding excess GDH compared to serC and its mutants. It was performed in 200 µL reaction solution containing 200 mM potassium buffer (pH 8.2), 5 mM α-ketoglutarate, 15 mM homoserine, 6 mM NAD
+ and 16 U of glutamate dehydrogenase (bovine liver). The samples containing the reaction mixture were pre- incubated at 30 °C for 5 min and then the reaction was initiated by adding 18 µg of enzyme. The enzymatic reaction was monitored by measuring the absorbance of NAD
+ at 340 nm (ε = 6220 M
−1 cm
−1) every 20 s for 30 min.
. serC-GDH coupled enzyme assay. The transamination reaction catalyzed by serC produces L-glutamate which is in turn oxidatively deaminated by GDH in the presence of NAD+. The reduction of NAD+ can be observed at 340 nm.
3. Results and Discussion
3.1. Wavelength Shift of GFP Variants
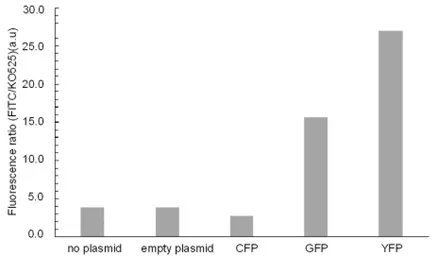
The wavelength shifts of GFP to yellow (YFP) and blue (CFP) were measured using different fluorescent channels of CytoFLEX. While GFP and YFP can be measured using the FITC channel (488 nm excitation and 525 nm emission), Krome Orange 525 (KO525) (405 nm excitation and 525 nm emission) can be used to measure GFP and CFP. A 2-point mutation to obtain YFP and a 3-point mutation to obtain CFP led to the successful production of GFP variants. The ratio of FITC to KO channel signals for YFP is the highest while CFP is the lowest (). This corresponds well with the emission wavelength of GFP and its variants produced. Since CFP can be measured only by Krome Orange the ratio was the lowest while YFP can be measured only by FITC and therefore the highest. GFP can be measured by both the channels and so was in between these two values. This trend was stable and well pronounced and the variants were significantly distinguishable. This whole process from obtaining LETs to measuring the fluorescence using the flow cytometer took less than six hours and was fully automated in a liquid handling system. This proof of concept for a fast and automated protein screening workflow with the model monomeric protein GFP is applied for the other multimeric proteins studied in this work.
. Ratio of fluorescence signals in FITC to KO channels for GFP and its variants CFP and YFP. YFP contains two mutations – S65T and T203Y while CFP contains three mutations—N146I, S175G and H231L.
While applying the previously proven workflow for other model enzymes AKIII and serC, a roadblock was encountered when the Tx-Tl extract used for GFP production was not able to produce these proteins at high enough concentrations. To overcome this problem, another extract dependent on T7 DNA polymerase was used. The viability of this particular extract to produce large multimeric proteins was tested. Two main variables for the CFPS system were temperature and time of the production process. Lowering the temperature of the reaction can improve the productivity by lowering the rate at which the linear DNA template is degraded by exonucleases [
24]. The cell-free protein synthesis reactions were reduced from 29 °C to 27 °C and 25 °C. When reduced to 27 °C the protein production only increased by 2 µg/mL, however when reduced to 25 °C the production increased by 8 µg/mL compared to 29 °C and 6 µg/mL compared to 27 °C (a). Then, the effect of time of the reaction was tested to see if the yield can be further increased by extending the reaction. However, there was no substantial increase in the amount of protein produced after 8 h and 10 h (b). Therefore 25 °C for 8 h was chosen as the conditions for cell-free protein synthesis. The LET based CFPS system was also compared to the plasmid based CFPS system to check if there is any difference between a linear and circular DNA template. There was no any significant difference when the DNA template was changed. This might be due to the addition of gamS protein and lowering of the temperature. Therefore by using linear expression template from the PCR method, no compromise is being made in terms of the yield of the protein.
. The concentration of protein produced in cell-free system measured using biolayer-interferometry. (a) The protein production at different temperatures was carried out to find the optimal temperature. These reactions were also carried out for 8 h. (b) The protein production was tested for different times at 25 °C. (The error bars indicate 1% confidence interval n = 3). pAKIII refers to plasmid based CFPS samples; AKIII refers to LET based CFPS samples.
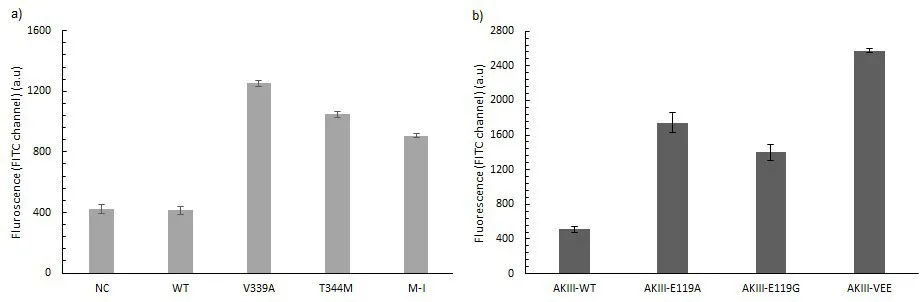
Using the conditions obtained in the previous section, the model enzymes aspartokinase III and phosphoserine aminotransferase (serC) and their variants were produced in cell free environment from LET in a fully automated liquid handling system. Since this is only to establish a new and fully automated protein screening method, the variants of both the enzymes were already tested through conventional methods. First for AKIII, V339A [
21] showed high activity even at the highest lysine concentration tested and therefore was used as positive control. Mutants T344M [
25] and M-I were then tested against V339A and the wild type to check if the method can differentiate mutants of varying levels of activity. The fluorescence signal of V339A was considerably higher than those of the other samples (a). The fluorescent signal was then subjected to significance analysis using ANOVA and Tukey test. Since small changes in the signals were not readily noticeable, these significance tests were necessary. The mutants with moderate level of activity were identified using the significance analysis (b). Mutant T344M was not only different from the wild type but also other mutants. Even though mutant M-I was not shown to be significantly different at a
p value of 0.01, it was different at a
p value of 0.05.
In case of serC, the screening method in this automated system is the same as the one of the conventionally available method of detection for aminotransferase enzymes. This is due to the fact that the GDH coupled assay is capable of detecting aminotransferase in crude environment instead of needing pure enzyme. This made it easier to compare the activity from both the ways since the only difference was the method used to produce the protein. Four different mutants (two 2-point mutations mutants and two 3-point mutations mutants) along with the wildtype were produced using cell free protein synthesis by LETs and tested. The wildtype shows very low activity towards homoserine and therefore used as negative control since the goal was to increase the affinity towards homoserine. The activity of mutant WW towards homoserine was more than 4 times higher than the wildtype. The increase in the activity of other mutants ranged from 2 to 3 times higher than the wildtype. These results were well corroborated by the work done by Zhang et al. [
26]. The results showed that the LET based CFPS system was able to produce the mutants with the desired change in enzyme properties as the conventional protein production but in a faster and more flexible manner. Using LETs as DNA template for CFPS via PCR approach allowed for easier gene manipulation wherein three point mutations were introduced by just exchanging the oligonucleotides without changing the established set up. This method offers the flexibility to create mutants with multiple point mutations without any need for cellular cloning procedures.
. (
a) The fluorescent emission of the ATP sensor of wildtype and different mutants of AKIII at lysine concentration of 200 mM (error bars indicate 1% confidence interval,
n = 50,000). (
b) The significant difference between various mutants of AKIII from the QUEEN fluorescent signal. There is significant difference between the wildtype and the mutants V339A [
21] and T344M [
25] (
p < 0.01) (
p cut off is the red line). There was also mild difference between the wildtype and mutant M-I. (
c) The activity of serC mutants measured using the serC-GDH coupled assay. The mutants were produced by using the LET based CFPS method performed at 25 °C for 8 h. (
d) Activity of serC mutants produced using LET based CFPS method (black) compared to the same mutants produced conventionally (grey). Activities are measured using the same serC-GDH coupled assay in both cases.
As shown in the previous sections, this fully automated protein production and screening method is able to produce the DNA template and corresponding protein and to screen the produced proteins within one day. This is mainly due to lack of the cloning and protein purification steps. This was further demonstrated when two different aspartokinase III modifications were tested; the lysine feedback resistant AKIII mutants (same variants as in the previous experiments) and AKIII mutants with varied substrate specificity (malate instead of aspartate) [
27]. For the change in substrate specificity, three mutants from Walther et al. [
27] were selected and tested. All eight samples were produced and tested in a single run. Even though changes in two different enzyme properties are tested, these samples can be processed simultaneously. Since the mutants with malate as substrate does not produce aspartyl hydroxymate, the hydroxymate method by Black and Wright cannot be used to screen them. However, the ATP sensor does not have this drawback. In both cases, the FITC channel results were able to differentiate not only the mutants from wildtype but also various mutants (). The mutants V339A and VEE had the highest fluorescence signal in their respective cases and the trend of the FITC signal for these mutants corroborates well with previous results and results of Walther et al. [
27].
. (a) Fluorescent emission of ATP sensor of wildtype AKIII in comparison with feedback resistant mutant AKIII produced in fully automated system at lysine concentration of 200 mM (error bars indicate 1% confidence interval n = 50000). (b) Fluorescent signal of wildtype AKIII in comparison with AKIII mutants with different substrate specificity (malate instead of aspartate) (same error bars).
This overall workflow from LET to screening using the ATP sensor took 15 h to complete. Time consuming step in this process is the cell free protein synthesis. This took 8 h to produce the amount of protein required for the screening method. If the amount of protein required for the screening method can be lowered or if the concentration of protein produced can be increased the time taken for workflow can be further reduced. For instance, the time for the workflow in case of GFP was only six hours since the protein was produced in four hours. This was also possible because GFP did not require any screening assay.
4. Conclusions
We introduced an efficient production and screening method using fully automated liquid handling system. Currently, the overall workflow includes linear expression template production, cell free protein synthesis system and protein screening. Wavelength shift of GFP was predicted, tested and detected while different mutants of AKIII and serC were produced and tested using this workflow. It was shown that the conventional screening methods can be adapted and modified to be used in an automated system. This whole process took from 6 to 15 h to complete depending on the protein while the conventional method would take about a week to finish. The flexibility and reliability of this process was shown by introducing multiple point mutations with ease in all the model enzymes. This workflow can be further integrated with computational prediction to create new mutants, thus achieving feedback guided enzyme optimization.
Supplementary Materials
The supporting information can be found at: https://www.sciepublish.com/article/pii/97.
Acknowledgments
This work was finally supported by the National Key R&D Program of China (2022YFA0912000), by the Westlake University Center for Synthetic Biology and Integrated Bioengineering and by the Westlake Education Foundation.
Author Contributions
Conceptualization, A.-P.Z. and U.J.; Methodology, S.R.S., U.J, S.I. and A.-P.Z.; Investigation, S.R.S., and S.I.; Resources, A.-P.Z.; Data Curation, S.S. and S.I.; Writing—Original Draft Preparation, S.S. and S.I.; Writing—Review & Editing, U.J. and A.-P.Z.; Supervision, A.-P.Z.; Funding Acquisition, A.-P.Z.
Funding
This research was funded by the National Key R&D Program of China (2022YFA0912000), the Westlake University Center for Synthetic Biology and Integrated Bioengineering, the Westlake Education Foundation and the Hamburg University of Technology.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
-
1.
Hoover DM, Lubkowski J. DNAWorks: an automated method for designing oligonucleotides for PCR-based gene synthesis.
Nucleic Acids Res. 2002,
30, e43.
[Google Scholar]
-
2.
Gao X, LeProust E, Zhang H, Srivannavit O, Gulari E, Yu P, et al. A flexible light-directed DNA chip synthesis gated by deprotection using solution photogenerated acids.
Nucleic Acids Res. 2001,
29, 4744–4450.
[Google Scholar]
-
3.
Rouillard JM, Lee W, Truan G, Gao X, Zhou X, Gulari E. Gene2Oligo: oligonucleotide design for in vitro gene synthesis.
Nucleic Acids Res. 2004,
32, W176–W180.
[Google Scholar]
-
4.
Goeddel DV, Kleid DG, Bolivar F, Heyneker HL, Yansura DG, Crea R, et al. Expression in
Escherichia coli of chemically synthesized genes for human insulin.
Proc. Natl. Acad. Sci. USA 1979,
76, 106–110.
[Google Scholar]
-
5.
Mandecki W, Boiling TJ. FokI method of gene synthesis.
Gene 1988,
68, 101–107.
[Google Scholar]
-
6.
Crameri A, Stemmer WC. Combinatorial multiple cassette mutagenesis creates all the permutations of mutant and wild-type sequences.
BioTechniques 1995,
18, 194–196.
[Google Scholar]
-
7.
Dillon PJ, Rosen CA. A rapid method for the construction of synthetic genes using the polymerase chain reaction.
BioTechniques 1990,
9, 298–300.
[Google Scholar]
-
8.
Takahashi MK, Hayes CA, Chappell J, Sun ZZ, Murray RM, Noireaux V, et al. Characterizing and prototyping genetic networks with cell-free transcription–translation reactions.
Methods 2015,
86, 60–72.
[Google Scholar]
-
9.
Noireaux V, Bar-Ziv R, Libchaber A. Principles of cell-free genetic circuit assembly.
Proc. Natl. Acad. Sci. USA 2003,
100, 12672–12677.
[Google Scholar]
-
10.
Katzen F, Chang G, Kudlicki W. The past, present and future of cell-free protein synthesis.
Trends Biotechnol. 2005,
23, 150–156.
[Google Scholar]
-
11.
Kigawa T, Yabuki T, Yoshida Y, Tsutsui M, Ito Y, Shibata T, et al. Cell‐free production and stable‐isotope labeling of milligram quantities of proteins.
FEBS Lett. 1999,
442, 15–19.
[Google Scholar]
-
12.
Kim DM, Swartz JR. Regeneration of adenosine triphosphate from glycolytic intermediates for cell‐free protein synthesis.
Biotechnol. Bioeng. 2001,
74, 309–316.
[Google Scholar]
-
13.
Terada T, Kusano S, Matsuda T, Shirouzu M, Yokoyama S. Cell-Free Protein Production for Structural Biology. In Advanced Methods in Structural Biology; Springer: Tokyo, Japan, 2016, pp. 83–102.
-
14.
Carlson ED, Gan R, Hodgman CE, Jewett MC. Cell-free protein synthesis: applications come of age.
Biotechnol. Adv. 2012,
30, 1185–1194.
[Google Scholar]
-
15.
Shinoda T, Shinya N, Ito K, Ishizuka-Katsura Y, Ohsawa N, Terada T, et al. Cell-free methods to produce structurally intact mammalian membrane proteins.
Sci. Rep. 2016,
6, 30442.
[Google Scholar]
-
16.
Stadtman ER, Cohen GN, LeBras G, de Robichon-Szulmajster H. Feed-back inhibition and repression of aspartokinase activity in
Escherichia coli and
Saccharomyces cerevisiae.
J. Biol. Chem. 1961,
236, 2033–2038.
[Google Scholar]
-
17.
Adachi J, Katsura K, Seki E, Takemoto C, Shirouzu M, Terada T, et al. Cell-free protein synthesis using S30 extracts from
Escherichia coli RFzero strains for efficient incorporation of non-natural amino acids into proteins.
Int. J. Mol. Sci. 2019,
20, 492.
[Google Scholar]
-
18.
Kigawa T, Yabuki T, Matsuda N, Matsuda T, Nakajima R, Tanaka A, et al. Preparation of
Escherichia coli cell extract for highly productive cell-free protein expression.
J. Struct. Funct. Genom. 2004,
5, 63–68.
[Google Scholar]
-
19.
Yaginuma H, Kawai S, Tabata KV, Tomiyama K, Kakizuka A, Komatsuzaki T, et al. Diversity in ATP concentrations in a single bacterial cell population revealed by quantitative single-cell imaging.
Sci. Rep. 2014,
4, 6522.
[Google Scholar]
-
20.
Black S, Wright NG. β-Aspartokinase and β-aspartyl phosphate.
J. Biol. Chem. 1955,
213, 27–38.
[Google Scholar]
-
21.
Chen Z, Rappert S, Sun J, Zeng AP. Integrating molecular dynamics and co-evolutionary analysis for reliable target prediction and deregulation of the allosteric inhibition of aspartokinase for amino acid production.
J. Biotechnol. 2011,
154, 248–254.
[Google Scholar]
-
22.
Wurm M, Ilhan S, Jandt U, Zeng AP. Direct and highly sensitive measurement of fluorescent molecules in bulk solutions using flow cytometry.
Anal. Biochem. 2019,
570, 32–42.
[Google Scholar]
-
23.
Walton CJ, Chica RA. A high-throughput assay for screening L-or D-amino acid specific aminotransferase mutant libraries.
Anal. Biochem. 2013,
441, 190–198.
[Google Scholar]
-
24.
Seki E, Matsuda N, Yokoyama S, Kigawa T. Cell-free protein synthesis system from Escherichia coli cells cultured at decreased temperatures improves productivity by decreasing DNA template degradation.
Anal. Biochem. 2008,
377, 156–161.
[Google Scholar]
-
25.
Kikuchi Y, Kojima H, Tanaka T. Mutational analysis of the feedback sites of lysine-sensitive aspartokinase of
Escherichia coli.
FEMS Microbiol. Lett. 1999,
173, 211–215.
[Google Scholar]
-
26.
Zhang Y, Ma C, Dischert W, Soucaille P, Zeng A-P. Engineering of phosphoserine aminotransferase for L-homoserine conversion to 4-hydroxy-2-ketobutyrate in a glycerol-independent pathway of 1,3-propanediol production from glucose.
Biotechnol. J. 2019,
14, 1900003.
[Google Scholar]
-
27.
Walther T, Topham CM, Irague R, Auriol C, Baylac A, Cordier H, et al. Construction of a synthetic metabolic pathway for biosynthesis of the non-natural methionine precursor 2,4-dihydroxybutyric acid.
Nat. Commun. 2017,
8, 15828.
[Google Scholar]