1. Introduction
Plant secondary metabolites represent large families of compounds that come from a diverse set of biosynthetic origins. Due to the sessile nature of plants, many of these compounds are involved in host defense mechanisms [
1] and have been repurposed by humans for medicinal use [
2]. Examples of these medicinal compounds include the anti-cancer terpenoid paclitaxel, and the alkaloid analgesic morphine [
3]. Although the biosynthetic precursors for both compounds are different, many of the pathways share similar enzymes, such as the cytochrome P450s responsible for the mono-oxygenation of the carbon backbone precursors in a regio‐ and stereo‐selective manner [
4]. Plant secondary metabolites are often produced and stored in specific tissues in relatively low amounts, making harvesting costly [
5]. The structural complexity of many of these compounds also complicates classical chemical synthesis. Due to prohibitory costs of harvesting and chemical synthesis, and the increasing need to meet green goals such as higher yields and the use of non-arable land, research has thus turned to discovering the biochemical pathways responsible for their synthesis in plants and heterologous expression of these biosynthetic pathways in alternative hosts.
Recently, there has been increasing interest in photosynthetic hosts as chassis for secondary metabolite production and biocatalysis [
6]. Of note are the photosynthetic microbes known as cyanobacteria. Their simple requirement for light, atmospheric CO
2, and inorganic nutrients makes them an attractive alternative to heterotrophic hosts. Although there have been many reports of successful expression of heterologous pathways in cyanobacteria, overall yields have not approached levels seen in traditionally used microbial hosts such as
Escherichia coli or
Saccharomyces cerevisiae. However, recent use of cyanobacteria in whole cell biocatalysis has revealed promising results. In one example, light-driven hydroxylation of testosterone into 15β-hydroxytestosterone was achieved using whole-cells of
Synechocystis sp. PCC 6803 expressing the heterologous P450 monooxygenase, CYP110D1 [
7]. Under optimized reaction conditions a maximum specific activity of 1 U·g·
CDW−1. A value about 2-fold higher than the one achieved using
E. coli. Some remaining challenges includes lack of knowledge about cellular metabolism and the shortage of tools for genetic engineering. However, in cyanobacteria, tools such as promoters, ribosome binding sites and repressors have been characterized previously in efforts to redirect carbon flux to varying degrees of success [
8].
A tool less explored is the partitioning of reducing equivalents towards pathways of interest. This is particularly relevant when engineering P450-dependent metabolism, as these enzymes are electron sinks which require repartitioning of the cell’s reducing power towards the P450s themselves [
6,
9]. While bacterial P450s are soluble, eukaryotic P450s are membrane anchored, normally to the ER-membrane. Eukaryotic P450s also require a dedicated redox partner, usually a membrane bound NADPH P450-oxidoreductase (CPR) [
9]. Membrane localization for heterologously expressed proteins in bacterial systems is notoriously difficult, which presents a challenge for the membrane bound P450s and P450 oxidoreductases. These issues can be circumvented by inserting the P450s into the thylakoid membrane of cyanobacteria or chloroplasts [
10]. We and others have previously shown that heterologously expressed eukaryotic P450s spontaneously insert into the thylakoid membrane and are able to obtain electrons directly from the photosynthetic electron transfer chain via the soluble electron donor ferredoxin [
10,
11,
12,
13,
14,
15,
16]. Flux of photosynthetic reducing equivalents towards P450s is limited due to the presence of competing endogenous electron sinks [
17]. Two of the major sinks include, ferredoxin NADP
+ reductase (FNR), which is responsible for the generation of NADPH, and nitrate assimilation, which accounts for around 80% and 16–21% of the total reducing power generated by photosynthetic apparatus, respectively [
18,
19]. Creation of artificial chimeric P450 has proven useful for the development of efficient biocatalysts. In several promising studies, the P450 catalytic modules fused with reductase modules and multi-domain chimeric P450 enzymes are constructed resulting in more efficient electron transfer, through enhanced catalytic and coupling properties [
20,
21,
22]. We have previously shown that fusing a eukaryotic P450 to a native ferredoxin increases light driven P450 activity in
Nicotiana benthamiana chloroplasts [
23] and that fusing the FMN binding domain (CPR
59–265) from the
Sorghum bicolor Cytochrome P450-oxidoreductase to a eukaryotic P450 increases light driven activity in the presence of competition from FNR [
24]. However, only transient expression in tobacco was used to characterize these fusion enzymes. In the current study we explore the stable expression of these fusion enzymes in
Synechocystis sp. PCC 6803 (hereafter
Synechocystis). We show that the CPR
59–265 fusion improves the light-driven P450 activity of CYP79A1 also in cyanobacteria, but endogenous proteolytic activity may challenge fusion enzyme stability, thus limiting overall yield.
2. Materials and Methods
2.1. Strains and Growth Conditions
E. coli strain NEB 10-β carrying pUC57 or pDF-trc plasmids were grown in standard LB media supplemented with 100 µg/mL carbenicillin or 50 µg/mL spectinomycin, respectively.
E. coli strain HB101 carrying the helper plasmid pRL443 was grown in LB media with 100 µg/mL carbenicillin.
Synechocystis sp. PCC. 6803 wildtype and derived strains were grown on BG5 [
25] plates supplemented with 1.5 g/L HEPES (hereafter BGH5; Sigma-Aldrich, Søborg, Denmark). Plates were kept at 30 °C in continuous light of approximately 40–50 µE without CO
2 supplementation. Liquid cultures were grown in BG11 [
25] media supplemented with 4.75 g/L HEPES (hereafter BGH11). The cultures were grown at 30 °C at approximately 50 µE with constant bubbling of air enriched with 3% CO
2 (
v/
v). Strains carrying pDF-trc were supplemented with 50 µg/mL spectinomycin to their media.
2.2. Construct Design and Cloning
Constructs were subcloned into pUC57 by restriction digest and ligation. Before ligation, backbones were dephosphorylated using Antarctic phosphatase (New England BioLabs; Ipswich, MA, USA). SbCYP79A1 and the FMN binding domain of the
Sorghum cytochrome P450 reductase (denoted CPR
59–265) were obtained in codon optimized form for
Synechocystis from GenScript (Piscataway, NJ, USA). CPR
59–265 was ordered with flanking AvrII and HindIII restriction sites.
Synechocystis Ferredoxin 1 (ssl0020) was obtained by colony PCR. All amplifications were performed using Phusion High-Fidelity DNA Polymerase (New England BioLabs) using primers described in Supplementary Table S1. The QLGGGSGGGGLG linker was purified by restriction digest from previously described constructs [
24]. Assembled fusion constructs were then transferred from pUC57 to pDF-trc [
26] via restriction digest with EcoRI and HindIII and ligation. CYP79A1 and CPR
59–265 were both subcloned directly into pDF-trc to generate two plasmids. The resulting constructs and their abbreviations can be seen in . An empty pDF-trc lasmid, i.e. without any insert was used as an empty vector control.
2.3. Transformation
Transformation of cyanobacteria with empty pDF-trc (empty vector control) and constructs shown in was performed using triparental mating as described [
27]. Spectinomycin at a concentration of 50 µg/mL was used for selection. Plates were incubated at 30 °C and approximately 50 µE of constant light until colonies became visible. Positive clones were confirmed using colony PCR. One transformant for each construct was chosen for further analysis.
2.4. Growth Curve Conditions
Growth curves were carried out in a Multi Cultivator MC 1000-OD (Photon System Instruments; Drásov, Czech Republic). Initially, 20 mL cultures were inoculated from plates and grown in liquid cultures as described above. These were then used to inoculate fresh cultures to an OD730 of ∼0.2 in 80 mL culture vessels using BGH11 as the growth medium supplemented with 50 µg/mL spectinomycin. Expression of the fusion enzymes was induced on day two with 1 mM IPTG. Cultures were continuously bubbled with humidified air supplemented with 3% (
v/
v) CO
2 and kept under 100 µE of constant illumination for 7 days at 30 °C. Samples for LC-MS, OD730, and immunoblot analyses were collected as described specifically below.
2.5. Cell Disruption
Cell pellets were harvested and resuspended in approximately 500 µL of 50 mM Tris-HCl at pH 7.5 containing 2X cOmplete EDTA-free protease inhibitor (Roche; Copenhagen, Denmark). 200 µL of 0.15 mm zirconium oxide beads were added to the cell suspension and disrupted in a NextAdvance Bullet Blender (Troy, NY, USA). Samples were processed twice at power 10 for 5 min each round.
2.6. Isolation of Thylakoid Membranes
All work was carried out either on ice or at 4 °C in the dark to prevent degradation. Cells were disrupted as described above but, in a buffer containing 400 mM sucrose, 20 mM tricine pH 7.5, 10 mM NaCl, 5 mM MgCl
2 (thylakoid storage buffer) and 2X cOmplete EDTA-free protease inhibitor (Roche). Unbroken cells were removed by centrifugation at 3000×
g for 3 min. The supernatant was collected and spun at 20,000×
g for 20 min. The supernatant was removed, and the resulting pellet was resuspended in 1 mL of thylakoid storage buffer containing an additional 200 mM of NaCl. The sample was spun again at 20,000×
g for 20 min and washed once more with thylakoid storage buffer to remove residual NaCl. Finally, the pellet was resuspended in 30 µL of storage buffer and used subsequently for light driven assays. Chlorophyll a content was quantified by extracting 5 µL of the thylakoid membranes in 96% ethanol, heated for 5 min at 90 °C, and spun down at 20,000×
g for 5 min to pellet the cell debris. Absorbance was measured at 664 nm and chlorophyll a concentration was derived from the following equation [
28]:
2.7. In Vitro Light Driven Assays
Light driven assays of the CYP79A1 fusion enzymes were performed in a total volume of 40 µL containing thylakoids (10 µg chlorophyll equiv.), 20 mM tricine pH 7.5, 100 mM NaCl, 193 µM L-tyrosine, 0.2 µCi·U-
14C-tyrosine (PerkinElmer; Skovlunde, Denmark, 486 mCi·mmol
−1), and 10 mM DTT. If specified, FNR and NADP+ were added to a concentration of 0.6 µM and 1.63 mM, respectively. FNR were purified as previously described [
29,
30,
31]. Assays were performed at 30 °C in a thermomixer at 200 µE white light from a custom-built LED light source. Total assay time was 10 min. The reaction was immediately stopped by extracting twice with 80 µL ethyl acetate. The extracts were concentrated and applied on silica gel 60 F
254 (Merck; Søborg, Denmark) TLC plates. Plates were developed with a toluene:ethyl acetate:methanol (30:8:1
v/
v/
v) mobile phase, dried and stored with storage phosphor screens (Molecular Dynamics; Vallensbæk Strand, Denmark). Visualization was performed using a Typhoon Trio scanner (GE Healthcare; Brøndby, Denmark). U-
14C- labeled
p-hydroxyphenylacetaldoxime was used as a standard.
p-hydroxyphenylacetaldoxime was quantified using GelQuant.NET software (BiochemLab Solutions; http://biochemlabsolutions.com/). Quantification of protein specifically for the light driven assays was performed using immunoblot by loading equal amounts of chl
a onto 12% TGX stain-free gels (Bio-rad; Copenhagen, Denmark). Immunoblot was performed as described below.
2.8. Immunoblotting
10 µg of total protein from whole cell lysate were loaded onto 12% TGX stain-free gels (Bio-Rad) and run at 250V for 30–35 min in Tris-glycine-SDS buffer (Bio-Rad). Proteins were transferred onto a Trans-Blot Turbo PVDF membrane for 7 min at 2.5 A using a Trans-Blot Turbo blotting system (Bio-Rad). Membranes were blocked for 1 h at room temperature in 5% skimmed milk in PBS-t (PBS containing 0.05% Tween-20) and subsequently washed in PBS-t. Primary antibodies were applied for 1 h at room temperature in 2% skimmed milk in PBS-t with either a 1:3000 dilution for anti-SbCYP79A1 or 1:10,000 dilution for anti-SbCPR2b. Membranes were washed in PBS-t and a 1:5000 dilution of swine anti-rabbit HRP conjugated immunoglobulins (Dako; Glostrup, Denmark) was applied for 1 h at room temperature in 2% skim milk in PBS-t. Chemiluminescence was detected using Super Signal West Dura substrate (Thermo Scientific; Roskilde, Denmark) with a ChemiDoc MP imaging system equipped with a cooled CCD camera (Bio-Rad). Quantification was performed using ImageLab 5.2 (Bio-Rad).
2.9. Quantification of p-Hydroxyphenylacetaldoxime
Samples were prepared by harvesting 1 mL of culture supernatant and extracting twice in 500 µL ethyl acetate. The ethyl acetate extract was dried under vacuum using a Scanvac. Residue was resuspended in 100% MeOH and then diluted to 10% MeOH. Samples were filtered through 0.2 µm cellulose acetate filter plates. Dilutions were prepared as needed to fit the standard curve.
p-hydroxyphenylacetaldoxime was quantified on an Advance UHPLC/EVOQTM Elite TripleQuad mass spectrometer essentially as in [
23] (Bruker; Bremen, Germany). Chromatography was performed on a Kinetex Biphenyl column (Phenomenex; Copenhagen, 1.7 µm, 100 Å, 2.1 × 100 mm) at a flow rate of 400 µL/min at an oven temperature of 40 °C. The injection volume was 5 µL. A gradient between A: 2 mM ammonium acetate pH 6.6 and B: 100% MeOH was used as follows: 0–0.3 min isocratic 10% B; 0.3–5 min 10–25% B; 5–5.1 min 25–98% B, 6.1–6.2 min 98–10% B; 6.2–8.5 min isocratic 10% B. Ionization was carried out using negative ESI mode with the following source settings: spray voltage −4500 V; cone temperature 300 °C; cone gas flow 20 psi; heated probe temperature 300 °C; probe gas flow 50 and nebuliser gas flow 60. Nitrogen was used as cone and probe gas and argon as collision gas.
p-Hydroxyphenylacetaldoxime was detected using selected ion monitoring at an
m/z of (−) 150 with E and Z isomers eluting at 3.95 min and 4.45 min, respectively. Standards for (
E/
Z)-
p-hydroxyphenylacetaldoxime were synthesized as previously described [
23]. Standard curves of p-hydroxyphenylacetaldoxime were constructed from 10 to 10,000 ppb.
2.10. Nitrogen Source Feeding Experiment
Strains were grown as described in the growth curve section unless otherwise noted. Triplicates of each strain were grown in BGH11 with either 10 mM NaNO
3 or 10 mM NH
4Cl. Cell numbers were analyzed with a Muse Cell Analyzer (Merck-Millipore; Søborg, Denmark).
2.11. Purification of Soluble CPR59–265 Containing Product
A soluble CPR
59–265 containing product derived from the strain carrying the 79-FMNd construct was purified. Two liters of the 79-FMNd strain were grown in BGH11 with appropriate antibiotics and induced with 1 mM IPTG. Cultures were grown to saturation and harvested. The harvested cells were resuspended in approximately 80 mL of 50 mM Tris-HCl at pH 7.5 with 2X cOmplete EDTA-free protease inhibitor (Roche). The suspension was passed through a Cell Disruptor CF (Constant Systems Ltd.; Daventry, UK). The resulting lysate was spun at 27,500×
g for 1 h. The supernatant was loaded onto 1 mL Q Sepharose FF resin packed into a HR 5/5 column (GE Life Sciences) connected to an ÄKTA start system (GE Life Sciences; Brøndby, Denmark). A linear gradient starting from 50 mM NaCl to 500 mM NaCl in 50 mM Tris-HCl at pH 7.5 was developed over 30 column volumes. The resulting fractions were analyzed by immunoblot. Fractions containing the soluble CPR
59–265 product were combined and concentrated to 2 mL using an Amicon centrifugal filter (Merck) with a 3 kDa cutoff. The sample was further purified by size exclusion chromatography on a HiLoad 26/600 Superdex 200 pg column equilibrated to 50 mM NaCl and 50 mM Tris-HCl at pH 7.5. Fractions were analyzed by immunoblot for soluble CPR
59–265 product and concentrated as before. Protein quantification was roughly estimated using absorbance at 272 nm with the molar extinction coefficient of 38,600 M
−1·cm
−1 [
32].
2.12. Proteomics of Soluble CPR59–265 Product
Lys-C and trypsin partial digests were performed to provide comprehensive protein coverage. Proteomics grade trypsin from porcine pancreas was obtained from Sigma and UHPLC grade solvents were obtained from Merck. Approximately 300 ng of purified soluble CPR
59–265 product was used and reduced using 10 mM DTT and alkylated with 55 mM iodoacetamide. Sequential Lys-C and trypsin digests were performed according to the manufacturer’s protocol. Desalting was performed using Oasis HLB 1cc 10 mg Extraction Cartridges. Coupling of a Dionex Ultimate 3000 series with a timsTOF Pro powered by PASEF mass spectrometer (Bruker) allowed for the identification of the protein. The peptides were separated prior to MS on an Acclaim PepMap column 100, 75 µm × 50 cm nanoViper C18 3 µm 100 A column with the following gradient and solvents: Solvent A: 0.1% formic acid, solvent B: 80% acetonitrile with 0.08% formic acid. The gradient started with isocratic flow for 5 min at 5% B. The concentration of B was increased to 40% B over the next 70 min. A further increase to 80% B was carried out in the following 5 min. This concentration was held isocratically for an additional 5 min before re-equilibration to 5% B for 15 min. The UHPLC was coupled via Captive Spray source to the timsTOF Pro mass spectrometer, and the capillary heated to 180 °C. The timsTOF Pro was acting on a mass range of 100–17,000
m/
z in positive ion mode. Ions were taken with a target intensity threshold and a target intensity of 20,000. The PASEF was run with 10 MS/MS scans and ions with a charge range of 0-5 were allowed. Total cycle time of 1.19 seconds was set and an active exclusion release after 0.4 min. The spectra were searched against the
Synechocystis Uniprot database with 3507 protein entries and the Mascot provided contaminant database using Mascot Daemon 2.6. The full length 79-FMNd protein was also added as a search query as described in Supplementary Table S2. The search was performed with a 15 ppm error for peptide masses, whereas 0.5 Da were allowed for the fragmented products. Carbamidomethylation was set as a fixed modification and oxidation as variable. Cleavage was required on one side of the tryptic peptide. Two missed cleavages were allowed in total. For the peptide IDs, the report from MASCOT has been uploaded as Supplementary data (mascot-reprot.xlsx).
2.13. Molecular Modeling of the FMN Binding Domain
The FMN domain of
Sorghum bicolor CPR2b residues 59-265 was truncated based on a model presented previously [
33] of the full-length protein and visualized and aligned against the secondary structure of
Synechococcus elongatus PCC 7942 flavodoxin (PDB entry 1CZN) using Pymol Open Source (Schrödinger LLC; New York, NY, USA).
3. Results
3.1. Engineered Strains
To investigate the effects of P450-redox partner fusions stably transformed into a cyanobacterial host, we initially designed 3 constructs: a construct comprising just SbCYP79A1 (denoted 79), a construct where CYP79A1 was fused with the major
Synechocystis ferredoxin, Fd1 construct (denoted 79-Fd1), and a construct where CYP79A1 was fused with the CPR
59–265 from the P450 oxidoreductase found in
S. bicolor (denoted 79-FMNd) (). The fusions were facilitated by a flexible 12 amino acid glycine-serine linker. Fd1 is the major ferredoxin involved in photosynthesis [
34] in
Synechocystis and CPR
59–265 is responsible for native electron transfer by CPR to CYP79A1 in
S. bicolor. All coding sequences were codon optimized to ensure proper expression in
Synechocystis and placed under the strong and inducible
trc promoter on the broad host range vector pDF-trc. The constructed vectors were transformed into
Synechocystis cells and the resulting colonies were genotyped and screened for expression and activity using immunoblot analysis and LC-MS quantification of produced compounds.
. Constructs used in this study. Constructs depicted are located on a pDF-trc backbone under the IPTG inducible trc promoter. Construct abbreviations are given to the right of each construct. A pDF-trc backbone without any DNA insert is used as empty vector control.
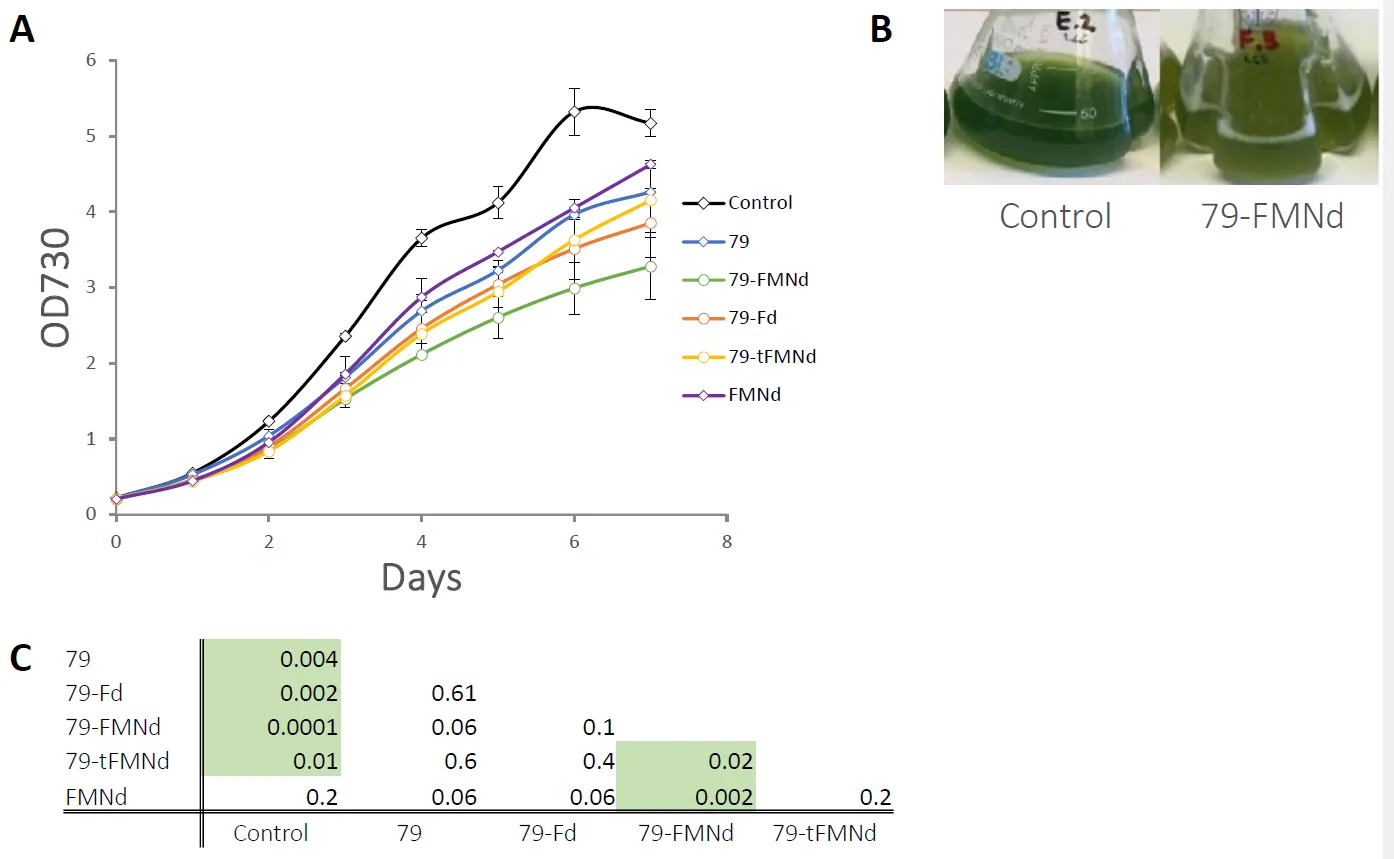
Growth curve experiments were carried out to investigate for any growth defects in 79, 79-Fd1 and 79-FMNd strains after induction on day 2 with 1 mM IPTG. All strains overexpressing a CYP79A1 variant reached a maximum OD
730 after 7 days that was significantly lower than the empty control (A), demonstrating that expression of CYP79A1 alone was detrimental to growth. Both fusion strains reached a maximum OD
730 lower than the unfused 79 strain, with the 79-FMNd strain performing marginally worse than the 79-Fd strain (A). Although these trends were consistent between repeat experiments, the differences between the strains carrying the different CYP79A1 constructs are not statistically significant.
. Growth characteristics of analysed strains. (A) Strains were grown photoautotrophically for seven days in triplicate and optical density was measured at 730 nm daily (OD730). (B) Photo of the empty vector control to the left and 79-FMNd to the right. The empty control displays a color indicative of normal growth and the 79-FMNd strain displays a color indicative of chlorosis. (C) The resulting density on the last day was calculated for pairwise significance on the last day (Holms-Sidak). p-values are given in the table below the graph with significant pairs (p < 0.05) highlighted in green.
When compared to the empty vector control strain the 79-FMNd strain showed a chlorotic-like phenotype which manifests as a visible color difference that is characteristic of nitrogen deficiency due to the degradation of phycobilliproteins [
35] (B). This yellow phenotype was consistent among several independent transformants obtained from transformation with the 79-FMNd construct (data not shown), indicating that the phenotype is a direct consequence of the vector and not a spurious mutation.
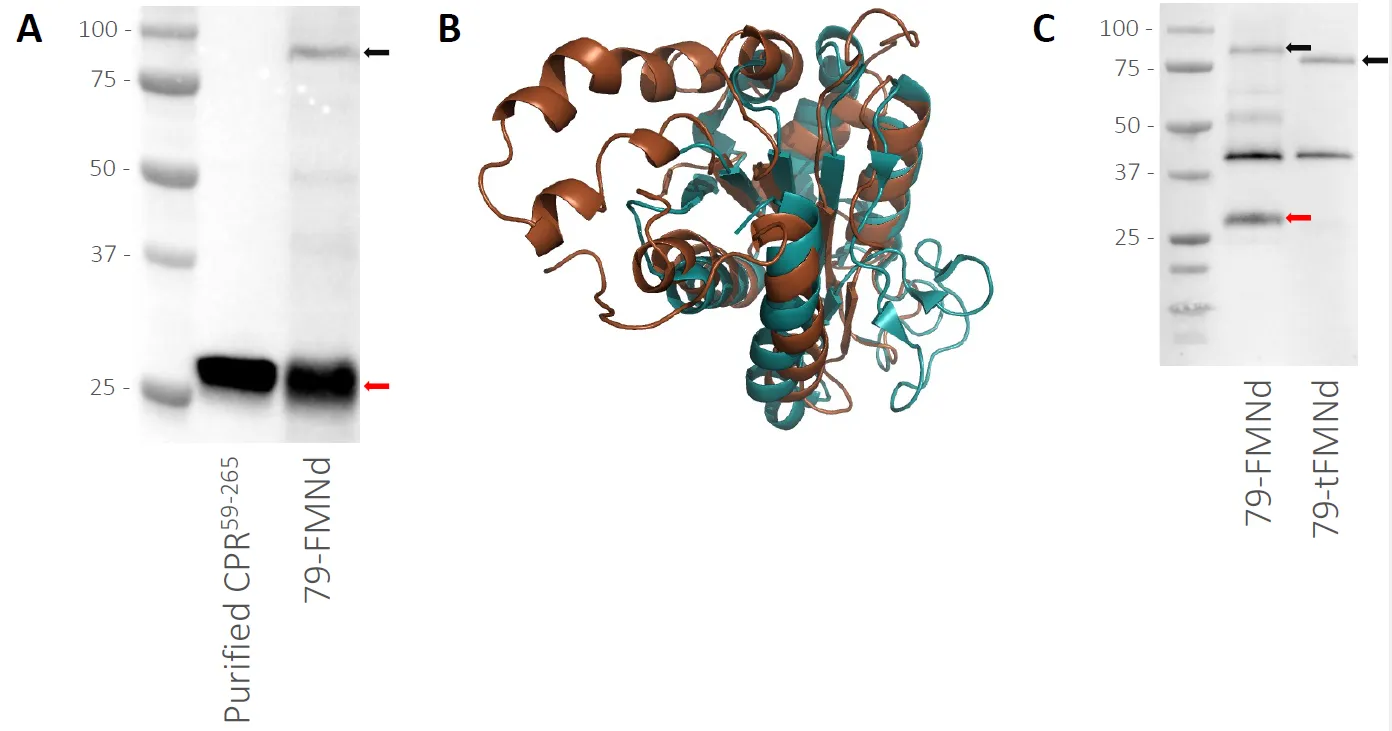
3.3. The 79-FMNd Strain Produces a Soluble CPR59–265 Containing Product
We analyzed the full cell lysate of the 79-FMNd strain by immunoblot using antibodies against cytochrome P450 reductase (CPR) from
S. bicolor (A). The full-length fusion protein was found at the expected size but surprisingly, the lysate also contained an additional band around 25 kDa, approximately the same size as CPR
59–265. We suspected that the additional band was the result of proteolytic degradation near or around the linker region of the fusion enzyme. A molecular model of CPR
59–265 was built and aligned to a flavodoxin from
Synechococcus elongatus (PDB: 1CZN) (B). The flavodoxin, shown in teal, and the FMN binding domain, shown in brown, align with an RMSD of 3.46 Å and display similar overall folds. The model, however, revealed two additional alpha helices that apparently do not constitute the core flavodoxin fold. We hypothesized that this region would be primarily required to provide physical connection to the transmembrane domain in the full-length CPR enzyme, a function dispensable for electron donor activity, and that this region could be the target of proteolytic cleavage
in vivo. We therefore generated a new vector and strain dubbed 79-tFMNd, wherein the 42 amino acid N-terminal stretch of the CPR
59–265 corresponding to the two extra alpha helices was truncated (). An additional strain expressing just the FMN binding domain (FMNd, ) was generated to investigate the effects of the overexpression of the soluble electron donor by itself. Growth characterization of the 79-tFMNd strain showed that it reached a maximal OD730 that was significantly below the empty vector control strain but also significantly higher than the 79-FMNd strain (A). The strain overexpressing just CPR
59–265 (FMNd) did not reach a maximal OD
730 that was statistically significant from the empty vector control. Immunoblot analysis of the resulting strain 79-tFMNd revealed the disappearance of the soluble CPR
59–265 containing product (C).
. Soluble CPR59–265 containing product. (A) Immunoblot of the 79-FMNd strain (right) vs. purified CPR59–265 (left). The fusion enzyme can be seen at the top. (B) Structural alignment of CPR59–265 (brown) to Synechococcus elongatus flavodoxin (teal, PDB 1CZN). (C) Immunoblot of the 79-FMNd and 79-tFMNd strains. Red arrows denote the soluble CPR59–265 containing product and black arrows denote the full-length fusion protein.
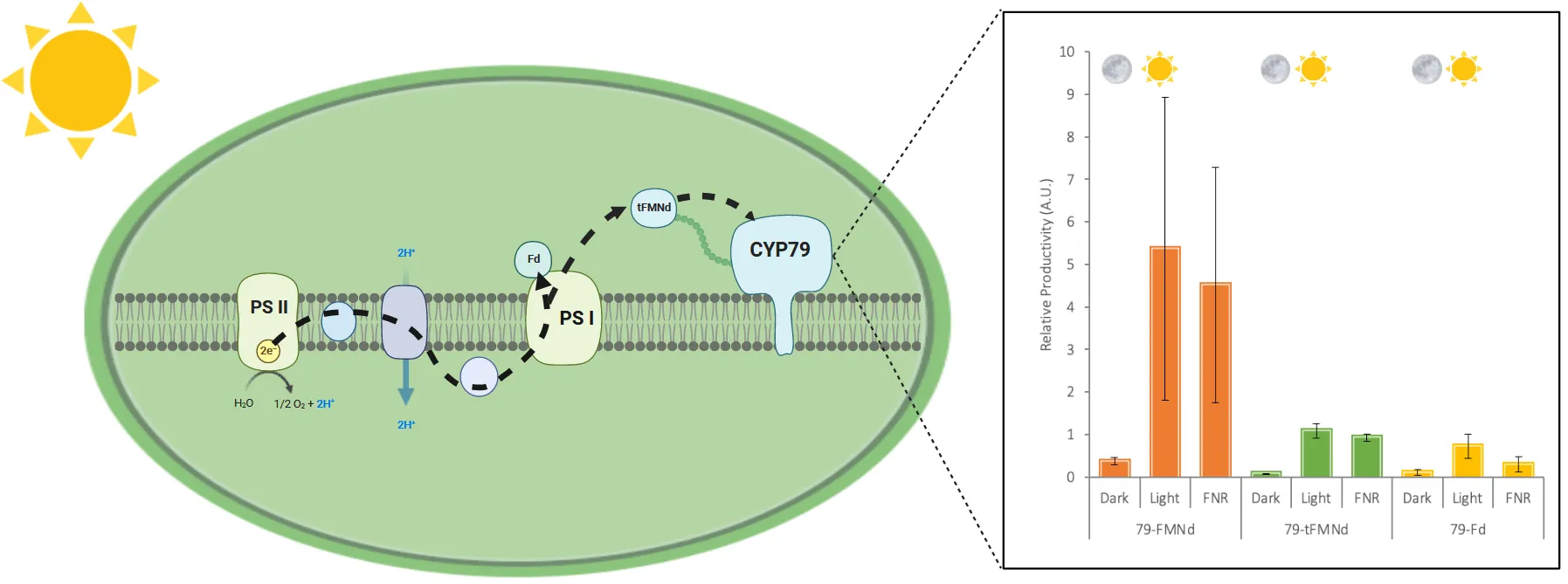
To investigate the electron transfer activity of the redox partner fusion enzymes in the context of cyanobacterial thylakoids, we performed
in vitro light-driven assays (). For this we isolated thylakoid membranes from cultures of the following fusion constructs: 79-Fd1, 79-FMNd and 79-tFMNd. The membranes were assayed in the dark to control for background activity, in the light to characterize productivity of the enzymes without a competing electron sink, and with added FNR and NADP
+ to characterize the productivity of the enzymes in the presence of the main competing electron sink
in vivo [
18]. Absolute enzyme activity was normalized to protein abundance of CYP79A1 determined by immunoblot analysis carried out on a portion of the isolated thylakoid membranes. Whereas the difference in activity in the presence or absence of FNR for the 79-Fd strain was around 2-fold, the difference in activity in the presence or absence of FNR was negligible in thylakoids isolated from the 79-FMNd and the 79-tFMNd strain (). However, there was a large decrease in activity in the 79-tFMNd thylakoids, suggesting an important role in the truncated portion for activity. Although the differences between the light and FNR in the 79-Fd strain were not statistically significant, they recapitulate a trend seen in transiently expressed fusion enzymes in
N. benthamiana [
24]. Altogether, the 79-FMNd fusion enzyme had the highest relative enzyme activity.
. In vitro light driven assays. Thylakoids extracted from strains expressing fusion constructs were assayed p-hydroxyphenylacetaldoxime formation in the dark, light, and in the presence of the competing electron sink FNR and NADP+. p-hydroxyphenylacetaldoxime was normalized to relative protein abundance in the thylakoid membranes.
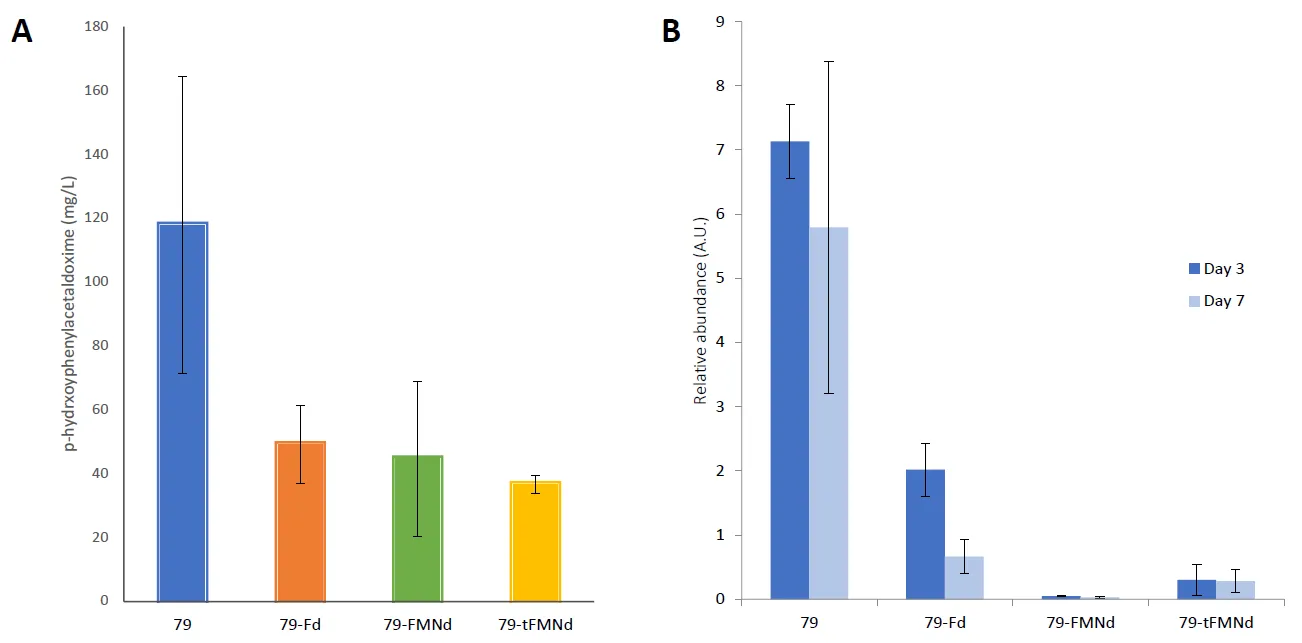
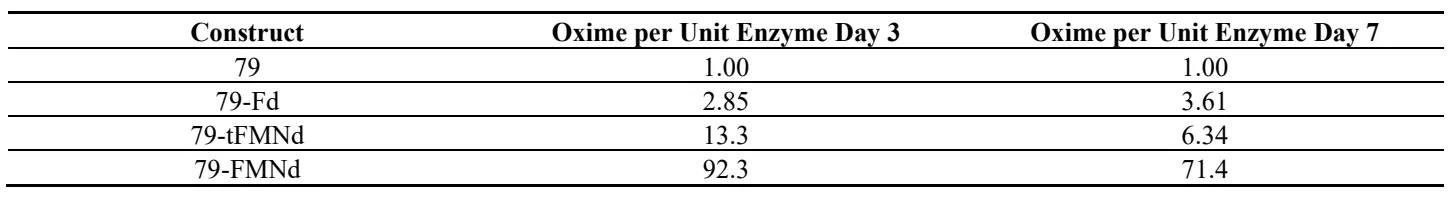
The yield of
p-hydroxyphenylacetaldoxime, i.e., the
in vivo yield, was also quantified from cultures of each cyanobacterial strain by LC-MS. The 79 strain produced the most
p-hydroxyphenylacetaldoxime at approximately 118 mg/L, with the 79-Fd, 79-FMNd, and 79-tFMNd producing between 36.5 to 49 mg/L (A). Protein expression was induced on day 2 and relative protein abundance quantified on day 3 and day 7 (B). Fusion enzymes had lower abundance when compared to the unfused 79 enzyme. The 79-Fd, 79-tFMNd and 79-FMNd strains had approximately 3.5-fold, 23-fold, and 142-fold lower enzyme levels when compared to unfused 79 enzyme. Despite differences in enzyme activity shown in
in vitro experiments, yields recovered from growth experiments did not correlate with these differences in activity. The steady state enzyme levels likely explain the disconnect between enzyme activity and absolute yield, with the 79 strain producing the most
p-hydroxyphenylacetaldoxime and the remaining enzyme fusions producing similar amounts. Day 7 abundance of enzyme by immunoblot analysis, using an antibody against CYP79A1, shows a drop in enzyme levels across all strains. When absolute yield is normalized to relative protein abundance to either day, the productivity trends seen in the
in vitro light driven assay are reproduced
in vivo ().
Figure 5. In vivo oxime yield and enzyme abundance. (A) Absolute p-hydroxyphenylacetaldoxime levels were measured by LC-MS at the end of a 7-day fermentation. (B) Relative protein abundance levels were measured on day 3 and day 7 of the fermentation.
Table 1. In vivo oxime levels normalized to relative protein abundance. Yields were quantified from growth curve experiments and normalized to protein abundance. The resulting activities were then normalized to the 79 activity of the same day (same column).
Because the 79-FMNd strain had large amounts of soluble CPR
59–265 containing product and it showed a chlorotic phenotype, we hypothesized nitrate assimilation may have been compromised. To investigate this possibility, we fed the empty vector control, 79-Fd, 79-FMNd, 79-tFMNd, and FMNd strain with either 10 mM NaNO
3 or 10 mM NH
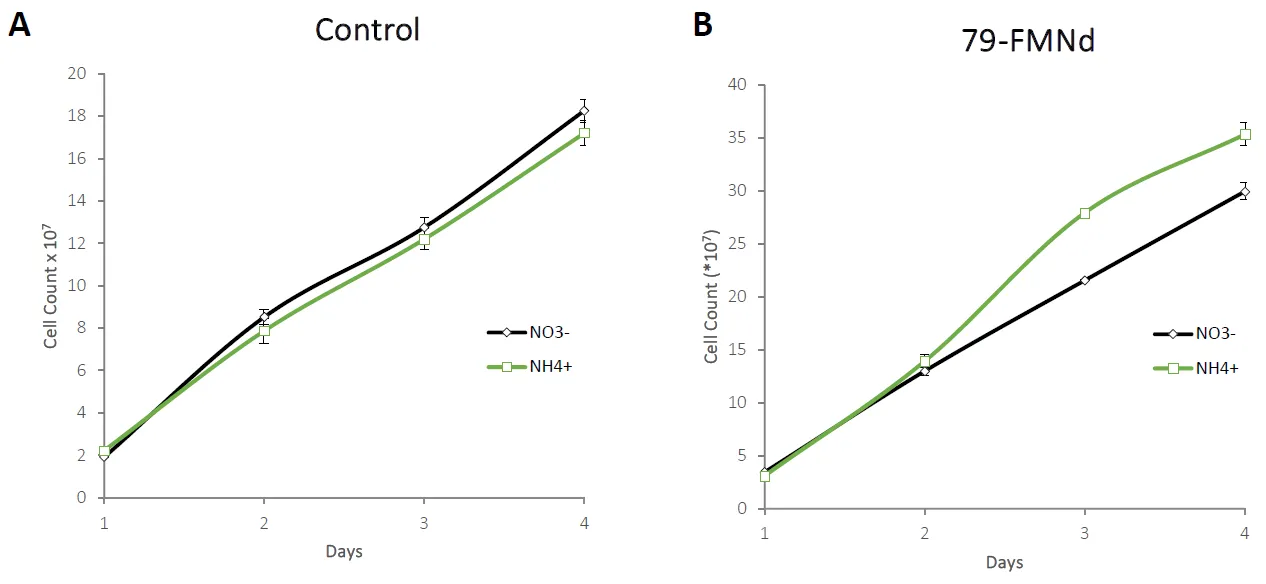
4Cl. Due to differences in optical scattering properties at 730 nm when strains were grown with different nitrogen sources, we opted to use cell count as a measure of growth difference. The empty vector control strain showed no difference in growth between nitrogen sources (A), while the 79-FMNd strain showed increased growth with NH
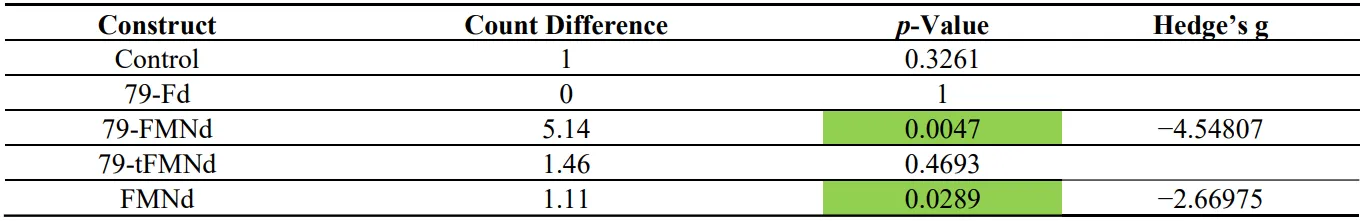
4Cl as the nitrogen source up to day 4 (B). After day 4, the difference in cell count diminishes, presumably due to a different factor limiting growth, e.g. light. All strains were analyzed for cell count differences on day 4, where only the 79-FMNd and FMNd strain showed significant differences in growth with the two different nitrogen sources ().
Figure 6. Nitrogen source feeding experiment. Strains were characterized in both 10 mM NaNO3 and 10 mM NH4Cl were compared using cell counting over the course of six days. The left (A) depicts the empty vector control strain, and the right (B) depicts growth of 79-FMNd.
Table 2. Day 4 nitrogen source feeding experiments. Difference in cell count, p-value, and effect size. Cell count differences between cultures grown with nitrate or ammonia were calculated and normalized to the cell count difference of the empty control strain. Strains displaying a significant difference in cell count (p < 0.05) are highlighted in green.
To investigate the possibility of
in vivo protease activity responsible for the generation of the CPR
59–265 containing product, we purified the CPR
59–265 containing product from 79-FMNd using ion exchange and size exclusion chromatography. Purified protein samples were digested partially using Lys-C and trypsin and subjected to timsTOF Pro-MS. Peptides from CPR
59–265 were identified to 93% coverage, while only one peptide was found from CYP79A1. Two non-tryptic peptides were identified at the N-terminus of CPR
59–265, one of which comprised the C-terminus of CYP79A1, while the second comprised the linker domain minus 2 residues at its N-terminus (). The predicted fragment in between the first two identified fragments was not identified, although some precursor masses were found manually which could fit the missing fragment. Protease cleavage site prediction using PROSPER [
36], did not identify any known cleavage sites at these two fragments sites.
. N-terminal proteomics of the soluble CPR59–265 product in 79-FMNd. Small black arrows denote the two putative cleavage sites.
4. Discussion
4.1. P450-CPR59–265 Fusion Activity Is Limited by Abundance
Although the activity of the P450-CPR
59–265 fusion is greater than all the other fusions tested both
in vivo and
in vitro, this characteristic does not manifest into greater yields
in vivo. Fusion proteins had low abundance in comparison to unfused CYP79A1, with the 79-FMNd having the lowest abundance. Here,
in vitro data showed less reduction in CYP79A1 activity in the presence of FNR (2-fold) compared to data shown in
N.benthamiana, where CYP79A1 fused to various
A. thaliana ferredoxins experienced greater than 10-fold reduction in activity in the presence of FNR [
23]. This could be due to the use of plant FNR in our
in vitro assays, whose affinity to cyanobacterial ferredoxin may be lower than the affinity to plant ferredoxin. Stable expression of the fusion enzymes resulted in strains with comparable yields, largely explained by the differences in steady state enzyme levels. In general, however, trends seen previously in
N. benthamiana using transient expression [
24], have largely been recapitulated in an
in vivo setting by expressing the fusion constructs in
Synechocystis.
4.2. Soluble CPR59–265 Interferes with Nitrate Assimilation
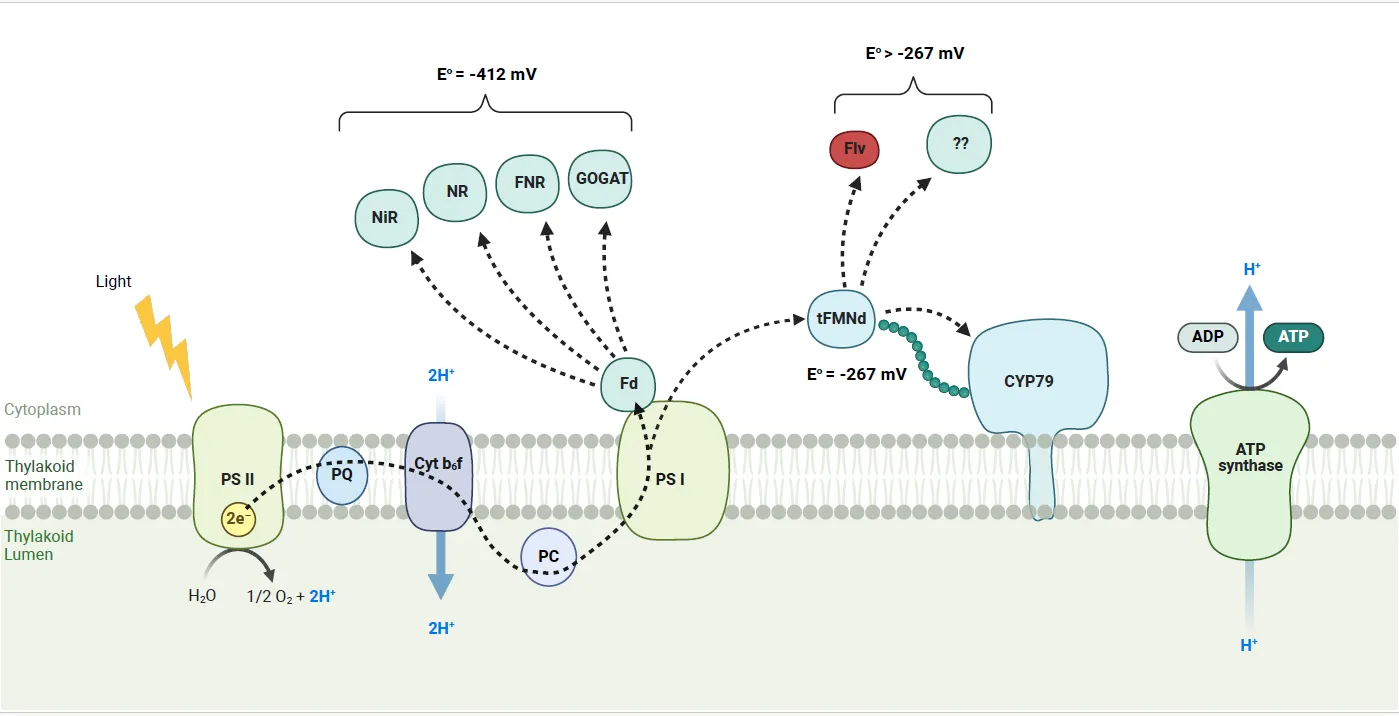
Nitrate assimilation in cyanobacteria is an energy intensive process, which uses 8 reducing equivalents to reduce nitrate to ammonia [
37]. In cyanobacteria, both nitrate reductase and nitrite reductase are ferredoxin dependent. Isolated thylakoids have shown light driven reduction of nitrate to nitrite and ammonium in the presence of ferredoxin [
38]. We find that using a more reduced nitrogen source improves growth of the strains 79-FMNd and FMNd. This suggests that expression of soluble CPR
59–265 (i.e., FMNd) interferes with nitrate assimilation, since both these strains contain the soluble CPR
59–265. CPR
59–265 has a midpoint reduction potential of −267 mV [
24], drastically higher than the native ferredoxin which has a midpoint reduction potential of −412 mV [
34]. It is thus conceivable that transfer of electrons to nitrate and nitrite reductase from CPR
59–265 is impaired by the more positive redox potential of CPR
59–265 (see also ). It was previously shown that specific residues, and not just electrostatic steering, are important for the interaction between cyanobacterial nitrate reductase and ferredoxin [
39]. Therefore, the affinity of CPR
59–265 to nitrate reductase is likely lower than that of the native ferredoxin and thus explains at least in part the drop in nitrate assimilation. The size of the effect of soluble CPR
59–265 in 79-FMNd compared to the FMNd strain is potentially explained by the presence of the P450. Electrons normally used in the formation of biomass can instead be redirected to formation of
p-hydroxyphenylacetaldoxime or participate in a P450 mediated ROS generation [
40]. The destination of electrons in the FMNd strain, however, is unknown. A potential sink could be the flavodiiron proteins that reduce oxygen into water using reducing power from ferredoxin [
41].
. Schematic illustration of light driven P450 activity. Shown is the photosynthetic membrane with photosystem I and II (PSI and PSII). Electrons are derived from water oxidation at PSII and transferred to ferredoxin (Fd) or FMN at PSI in a light-dependent manner. Fd or FMN, in this case the CPR59–265 domain, can then reduce the P450 or the natural Fd-requiring reductases like Rubisco, NR and NIR. The CPR59–265 domain has a midpoint reduction potential of −267 mV, less negative than the −412 mV for native cyanobacterial Fd. This means that photo-reduced CPR59–265 is less likely to donate electrons to the endogenous enzymes and more dedicated to donate electrons towards the P450 enzyme. Indicated are also putative electron sinks photo-reduced CPR59–265 like flavodiiron proteins and unknown enzymes (Created with Biorender.com).
The expression level of the fusion enzymes suggests that the presence of the linker and the redox partner affects the stability of the proteins or the secondary structure of their mRNAs. We have shown that the truncation of the 79-FMNd fusion to create the 79-tFMNd fusion abrogates the accumulation of soluble CPR
59–265 product. Data from proteomics points to two putative N-terminal sites, both non-tryptic and one which potentially suggests internal translation. Although the absence of detected peptides is not conclusive evidence of the identity of the N-terminus, both peptides place the resulting protein near the expected size according to western blot. In the case of internal translation in cyanobacteria, ribosome binding can proceed by either a Shine-Dalgarno dependent pathway or a Shine-Dalgarno independent pathway [
42]. The Shine-Dalgarno independent mechanism is correlated with a decrease in secondary mRNA structure. There are no apparent Shine-Dalgarno sequences preceding the putative N-terminal sites. Because the truncation occurs after the putative N-terminal sites, abrogation of CPR
59–265 must occur by the formation of strong secondary structure which occludes the internal ribosome binding site, assuming the product is formed by internal translation. However, it is known that active protein translation disfavors secondary structure, as the ribosome unwinds the secondary structure of the mRNA [
43]. Instead, we hypothesize that the CPR
59–265 containing protein is the result of proteolysis. Because the truncation of the CPR domain is not located at the putative cleavage sites, it seems unlikely the accumulation of CPR
59–265 containing protein is the result of a specific interaction with an endoprotease. Instead, the portion truncated could potentially make the enzyme more accessible to a non-specific cellular protease. For example, it is known that FtsH in
E. coli can degrade substrates by access to an internal loop [
44]. Similarly, the portion truncated could extend the flexible linker such that an orthologous FtsH, which sits on the thylakoid membrane in
Synechocystis, has access to initiate proteolysis. Previously, we have shown that the C-terminal fusions of two different P450s and a glycosyltransferase to integral membrane proteins from the Twin-arginine translocation pathway resulted in only soluble P450 and glycosyltransferase being detected in
Synechocystis [
45]. This data supports the non-specific protease hypothesis.
4.4. Soluble CPR59–265 as a Tool to Partition Electrons to Complex P450 Containing Pathways
The CPR
59–265 has a midpoint reduction potential of −267 mV [
24], less negative than the −412 mV for native cyanobacterial ferredoxin [
34]. This means that CPR
59–265 reduced by photosystem I is less likely to donate electrons to endogenous enzymes involved in processes like CO
2 fixation and nitrate assimilation in the cyanobacterial cell and in principle more dedicated to directing electrons towards the CYP79 (). This is supported by the observation that redox partner fusion enzymes show superior productivities by relative abundance. However, the ability to express these enzymes at high levels remains an issue. Another potential limitation is the scalability of this strategy. In biosynthetic pathways with multiple P450s, such as forskolin or taxol [
46,
47], fusing a redox partner onto all P450s involved could potentially incur issues of steric crowding. The thylakoid membrane in its native state is densely packed to the extent that protein diffusion is significantly affected [
48]. Because membrane or near-membrane dynamics are important for various processes such as non-photochemical quenching [
49] or the transition between different supercomplexes of PSI, PSII, and phycobilisomes [
48], the presence of multiple integral membrane proteins with tethered domains could potentially interfere in these processes. An attractive strategy would be to use the soluble CPR
59–265 as an electron flux valve. For example, when a culture is grown to a certain density or a precursor pool reaches a desired level, the soluble CPR
59–265 could be expressed to redirect flux towards product formation. This could potentially be used to implement growth decoupled production or prevent futile cycling of the P450s by ensuring substrate availability for the P450s. However, it must be borne in mind that recent studies suggest that the redox partners not only function as auxiliary electron transfer proteins but are also important P450 function modulators affecting reactivity and selectivity [
9].
5. Conclusions
We have shown that stable transformation of redox fusion partners in Synechocystis reproduce trends seen in transiently expressed N. benthiana. Fusion with CPR59–265 results in an enzyme with the highest productivity. Fusion enzyme stability remains a limiting factor for the production of p-hydroxyphenylacetaldoxime and other potential products. Surprisingly, we discovered either a degradation product or cryptic translation of soluble CPR59–265 protein. Characterization of strains containing soluble CPR59–265 showed impaired growth due to interference with nitrate assimilation.
Supplementary Materials
The supporting information can be found at: https://www.sciepublish.com/article/pii/184.
Acknowledgments
We would like to thank Lisbeth Mikkelsen for maintaining the cyanobacteria strains developed for this work.
Author Contributions
Experiments were conceived and designed by L.S., S.B.M., A.Z.N., N.D. and P.E.J. The majority of the experimental work was carried out by L.S. A.M. performed the proteomic analysis and M.B. performed the metabolite analysis. The manuscript was written by L.S. with input from all co-authors.
Ethics Statement
Not applicable.
Informed Consent Statement
Not applicable.
Funding
The authors acknowledge financial support from the Copenhagen Plant Science Centre, the VILLUM Foundation “Light-driven biosynthesis: Improving photosynthesis by designing and exploring novel electron transfer pathways” (13363), the Novo Nordisk Foundation project “Harnessing the Energy of the Sun for Biomass Conversion” (NNF16OC0021832), a Novo Nordisk Foundation postdoctoral fellowship for S. B. M. “Integrating approaches to develop Nicotiana tabacum as a small molecule production platform” (NNF18OC0032350), the Novo Nordisk Foundation project “Photosynthetic cell factories for production of phenylpropanoids (PhotoPro)” (NNF19OC0057634), and the Carlsberg Foundation (CF17-0657). The authors would also like to thank the Danish National Research Foundation for support of the DynaMo Center (DNRF grant99) and the DynaMo Metabolomics Facility for LC-MS analysis.
Declaration of Competing Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
-
1.
Wink M. Plant breeding: importance of plant secondary metabolites for protection against pathogens and herbivores.
Theor. Appl. Genet. 1988,
75, 225–233.
[Google Scholar]
-
2.
Wang Y, Pan H, Wang F, Shen C. Microbial P450 repertoire (P450ome) and its application feasibility in pharmaceutical industry, chemical industry, and environmental protection.
Biotechnol. Bioeng. 2024,
121, 7–25.
[Google Scholar]
-
3.
Prakash B, Kujur A, Yadav A. Drug synthesis from natural products: a historical overview and future perspective. In Synthesis of Medicinal Agents from Plants; Elsevier: Amsterdam, The Netherlands; 2018; pp. 25–46.
-
4.
Guengerich FP. Mechanisms of Cytochrome P450-Catalyzed Oxidations.
ACS Catal. 2018,
8, 10964–10976.
[Google Scholar]
-
5.
Verpoorte R, Contin A, Memelink J. Biotechnology for the production of plant secondary metabolites.
Phytochem. Rev. 2002,
1, 13–25.
[Google Scholar]
-
6.
Agustinus B, Gillam EMJ. Solar-powered P450 catalysis: Engineering electron transfer pathways from photosynthesis to P450s.
J. Inorg. Biochem. 2023,
245, 112242.
[Google Scholar]
-
7.
Mascia F, Pereira SB, Pacheco CC, Oliveira P, Solarczek J, Schallmey A, et al. Light-driven hydroxylation of testosterone by
Synechocystis sp. PCC 6803 expressing the heterologous CYP450 monooxygenase CYP110D1.
Green Chem. 2022,
24, 6156–6167.
[Google Scholar]
-
8.
Santos-Merino M, Singh AK, Ducat DC. New applications of synthetic biology tools for cyanobacterial metabolic engineering.
Front. Bioeng. Biotechnol. 2019,
7, 33.
[Google Scholar]
-
9.
Li S, Du L, Bernhardt R. Redox Partners: Function Modulators of Bacterial P450 Enzymes.
Trends Microbiol. 2022,
28, 445–454.
[Google Scholar]
-
10.
Nielsen AZ, Mellor SB, Vavitsas K, Wlodarczyk AJ, Gnanasekaran T, de Jesus MPRH, et al. Extending the biosynthetic repertoires of cyanobacteria and chloroplasts.
Plant J. 2016,
87, 87–102.
[Google Scholar]
-
11.
Berepiki A, Gittins JR, Moore CM, and Bibby TS. Rational engineering of photosynthetic electron flux enhances light-powered cytochrome P450 activity.
Synth. Biol. 2018,
3, ysy009.
[Google Scholar]
-
12.
Jensen K, Jensen PE, Møller BL. Light-Driven Cytochrome P450 Hydroxylations.
ACS Chem. Biol. 2011,
6, 533–539.
[Google Scholar]
-
13.
Nielsen AZ, Ziersen B, Jensen K, Lassen LM, Olsen CE, Møller BL, et al. Redirecting Photosynthetic Reducing Power toward Bioactive Natural Product Synthesis.
ACS Synth. Biol. 2013,
2, 308–315.
[Google Scholar]
-
14.
Santos-Merino M, Torrado A, Davis GA, Röttig A, Bibby TS, Kramer DM, et al. Improved photosynthetic capacity and photoprotection via heterologous metabolism engineering in cyanobacteria.
Proc. Natl. Acad. Sci. USA 2020,
118, e2021523118.
[Google Scholar]
-
15.
Tüllinghoff A, Toepel J, Bühler B. Enlighting Electron Routes in Oxyfunctionalizing
Synechocystis sp. PCC 6803.
ChemBioChem 2024,
25, e202300475.
[Google Scholar]
-
16.
Wlodarczyk A, Gnanasekaran T, Nielsen AZ, Zulu NN, Mellor SB, Luckner M, et al. Metabolic engineering of light-driven cytochrome P450 dependent pathways into
Synechocystis sp. PCC 6803.
Metab. Eng. 2016,
33, 1–11.
[Google Scholar]
-
17.
Mellor SB, Vavitsas K, Nielsen AZ, Jensen PE. Photosynthetic fuel for heterologous enzymes: the role of electron carrier proteins.
Photosynth. Res. 2017,
134, 329–342.
[Google Scholar]
-
18.
Backhausen JE, Kitzmann C, Horton P, Scheibe R. Electron acceptors in isolated intact spinach chloroplasts act hierarchically to prevent over-reduction and competition for electrons.
Photosynth. Res. 2000,
64, 1–13.
[Google Scholar]
-
19.
Grund M, Jakob T, Wilhelm C, Bühler B, Schmid A. Electron balancing under different sink conditions reveals positive effects on photon efficiency and metabolic activity of
Synechocystis sp. PCC 6803.
Biotechnol. Biofuels 2019,
12, 43.
[Google Scholar]
-
20.
Sadeghi SJ, Gilardi G. Chimeric P450 enzymes: Activity of artificial redox fusions driven by different reductases for biotechnological applications.
Biotechnol. Appl. Biochem. 2013,
60, 102–110.
[Google Scholar]
-
21.
Degregorio D, D’Avino S, Castrignanò S, Di Nardo G, Sadeghi SJ, Catucci G, et al. Human Cytochrome P450 3A4 as a Biocatalyst: Effects of the Engineered Linker in Modulation of Coupling Efficiency in 3A4-BMR Chimeras.
Front. Pharmacol. 2017,
8, 121.
[Google Scholar]
-
22.
Castrignanò S, D’Avino S, Di Nardo G, Catucci G, Sadeghi SJ, Gilardi G. Modulation of the interaction between human P450 3A4 and B. megaterium reductase via engineered loops.
Biochim. Biophys. Acta Proteins Proteom. 2018,
1866, 116–125.
[Google Scholar]
-
23.
Mellor SB, Nielsen AZ, Burow M, Motawia MS, Jakubauskas D, Møller BL, et al. Fusion of Ferredoxin and Cytochrome P450 Enables Direct Light-Driven Biosynthesis.
ACS Chem. Biol. 2016,
11, 1862–1869.
[Google Scholar]
-
24.
Mellor SB, Vinde MH, Nielsen AZ, Hanke GT, Abdiaziz K, Roessler MM, et al. Defining optimal electron transfer partners for light-driven cytochrome P450 reactions.
Metab. Eng. 2019,
55, 33–43.
[Google Scholar]
-
25.
Stanier RY, Kunisawa R, Mandel M, Cohen-Bazire G. Purification and properties of unicellular blue-green algae (order Chroococcales).
Bacteriol. Rev. 1971,
35, 171–205.
[Google Scholar]
-
26.
Guerrero F, Carbonell V, Cossu M, Correddu D, Jones PR. Ethylene Synthesis and Regulated Expression of Recombinant Protein in
Synechocystis sp. PCC 6803.
PLoS ONE 2012,
7, e50470.
[Google Scholar]
-
27.
Huang H-H, Camsund D, Lindblad P, Heidorn T. Design and characterization of molecular tools for a synthetic biology approach towards developing cyanobacterial biotechnology.
Nucleic Acids Res. 2010,
38, 2577–2593.
[Google Scholar]
-
28.
Lichtenthaler HK. Chlorophylls and Carotenoids: Pigments of Photosynthetic Biomembranes.
Methods Enzymol. 1987,
148, 350–382.
[Google Scholar]
-
29.
Buchanan BB, Arnon DI. Ferredoxins from Photosynthetic Bacteria, Algae, and Higher Plants.
Methods Enzymol. 1971,
23, 413–440.
[Google Scholar]
-
30.
Serrano A, Rivas J. Purification of ferredoxin-NADP+ oxidoreductase from cyanobacteria by affinity chromatography on 2′,5′-ADP-Sepharose 4B.
Anal. Biochem. 1982,
126, 109–115.
[Google Scholar]
-
31.
Zygadlo A, Jensen PE, Leister D, Scheller HV. Photosystem I lacking the PSI-G subunit has a higher affinity for plastocyanin and is sensitive to photodamage.
Biochim. Biophys. Acta Bioenerg. 2005,
1708, 154–163.
[Google Scholar]
-
32.
Ragsdale SW, Ljungdahl LG. Characterization of ferredoxin, flavodoxin, and rubredoxin from Clostridium formicoaceticum grown in media with high and low iron contents.
J. Bacteriol. 1984,
157, 1–6.
[Google Scholar]
-
33.
Jensen K, Osmani SA, Hamann T, Naur P, Møller BL. Homology modeling of the three membrane proteins of the dhurrin metabolon: Catalytic sites, membrane surface association and protein-protein interactions.
Phytochemistry 2011,
72, 2113–2123.
[Google Scholar]
-
34.
Bottin H, Lagoutte B. Ferredoxin and flavodoxin from the cyanobacterium
Synechocystis sp PCC 6803.
Biochim. Biophys. Acta 1992,
1101, 48–56.
[Google Scholar]
-
35.
Baier A, Winkler W, Korte T, Lockau W, Karradt A. Degradation of phycobilisomes in synechocystis sp. pcc6803 evidence for essential formation of an nbla1/nbla2 heterodimer and its codegradation by a clp protease complex.
J. Biol. Chem. 2014,
289, 11755–11766.
[Google Scholar]
-
36.
Song J, Tan H, Perry AJ, Akutsu T, Webb GI, Whisstock JC, et al. PROSPER: An Integrated Feature-Based Tool for Predicting Protease Substrate Cleavage Sites.
PLoS ONE 2012,
7, e50300.
[Google Scholar]
-
37.
Flores E, Frías JE, Rubio LM, Herrero A. Photosynthetic nitrate assimilation in cyanobacteria.
Photosynth. Res. 2005,
83, 117–133.
[Google Scholar]
-
38.
Manzano C, Candau P, Gomez-Moreno C, Relimpio AM, Losada M. Ferredoxin-dependent photosynthetic reduction of nitrate and nitrite by particles of anacystis nidulans.
Mol. Cell. Biochem. 1976,
10, 161–169.
[Google Scholar]
-
39.
Srivastava AP, Hardy EP, Allen JP, Vaccaro BJ, Johnson MK, Knaff DB. Identification of the Ferredoxin-Binding Site of a Ferredoxin-Dependent Cyanobacterial Nitrate Reductase.
Biochemistry 2017,
56, 5582–5592.
[Google Scholar]
-
40.
Johnston WA, Hunter DJB, Noble CJ, Hanson GR, Stok JE, Hayes MA, et al. Cytochrome P450 is present in both ferrous and ferric forms in the resting state within intact Escherichia coli and hepatocytes.
J. Biol. Chem. 2011,
286, 40750–40759.
[Google Scholar]
-
41.
Santana-Sanchez A, Solymosi D, Mustila H, Bersanini L, Aro EM, Allahverdiyeva Y. Flavodiiron proteins 1–to-4 function in versatile combinations in O
2 photoreduction in cyanobacteria.
Elife 2019,
8, 45766.
[Google Scholar]
-
42.
Wei Y, Xia X. Unique Shine–Dalgarno Sequences in Cyanobacteria and Chloroplasts Reveal Evolutionary Differences in Their Translation Initiation.
Genome Biol. Evol. 2019,
11, 3194–3206.
[Google Scholar]
-
43.
Mustoe AM, Busan S, Rice GM, Hajdin CE, Peterson BK, Ruda VM, et al. Pervasive Regulatory Functions of mRNA Structure Revealed by High-Resolution SHAPE Probing.
Cell 2018,
173, 181–195.e18.
[Google Scholar]
-
44.
Okuno T, Yamanaka K, Ogura T. An AAA protease FtsH can initiate proteolysis from internal sites of a model substrate, apo-flavodoxin.
Genes Cells 2006,
11, 261–268.
[Google Scholar]
-
45.
Henriques de Jesus MPR. Novel use of Tat components to increase metabolic flux in light-driven biosynthesis. PhD Theses; Copenhagen University, Copenhagen, Denmark, 2017.
-
46.
Pateraki I, Andersen-Ranberg J, Jensen NB, Wubshet SG, Heskes AM, Forman V, et al. Total biosynthesis of the cyclic AMP booster forskolin from
Coleus forskohlii.
eLife 2017,
6, e23001.
[Google Scholar]
-
47.
Croteau R, Ketchum REB, Long RM, Kaspera R, Wildung MR. Taxol biosynthesis and molecular genetics.
Phytochem. Rev. 2006,
5, 75–97.
[Google Scholar]
-
48.
Casella S, Huang F, Mason D, Zhao G-Y, Johnson GN, Mullineaux CW, et al. Dissecting the Native Architecture and Dynamics of Cyanobacterial Photosynthetic Machinery.
Mol. Plant 2017,
10, 1434–1448.
[Google Scholar]
-
49.
Bailey S, Grossman A. Photoprotection in Cyanobacteria: Regulation of Light Harvesting.
Photochem. Photobiol. 2008,
84, 1410–1420.
[Google Scholar]