Mitochondrial Fatty Acid Oxidation Dysfunction in Tubulointerstitial Fibrosis: Mechanisms and Therapeutic Advances

Mitochondrial Fatty Acid Oxidation Dysfunction in Tubulointerstitial Fibrosis: Mechanisms and Therapeutic Advances
Jiaojiao Fan
1,2,3
 Siyu Li
1,2,3
Jianjiang Zhang
1,*
Wei Zhou
4,*
Siyu Li
1,2,3
Jianjiang Zhang
1,*
Wei Zhou
4,*
Received: 18 March 2026 Revised: 08 May 2026 Accepted: 22 May 2026 Published: 05 June 2026

© 2026 The authors. This is an open access article under the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/).
1. Introduction
Chronic kidney disease (CKD) represents a growing global health burden and is characterized by a high prevalence and a progressive course toward end-stage kidney disease [1]. Regardless of the nature of the initiating insult, tubulointerstitial fibrosis (TIF) is widely regarded as the final common pathway most closely associated with renal functional decline and poor prognosis [2]. Histologically, TIF is characterized by tubular atrophy, inflammatory cell infiltration, and excessive extracellular matrix accumulation. However, effective therapies capable of halting this process remain elusive. Given the limited success of conventional anti-inflammatory and hemodynamic interventions, there is a pressing need to identify additional pathogenic mechanisms, particularly those involving alterations in cellular metabolism, in order to define new therapeutic targets for preventing fibrotic progression.
The kidney, particularly the proximal tubule, is among the most metabolically active organs and depends heavily on mitochondrial fatty acid oxidation (FAO) to meet the substantial energy demands of solute reabsorption. Under physiological conditions, FAO serves as the principal energy source for proximal tubular epithelial cells and is more efficient than glycolysis in supporting cellular energy production [3,4]. In the fibrotic kidney, however, this metabolic program is profoundly disrupted, as reflected by marked downregulation of key FAO-related enzymes, such as carnitine palmitoyltransferase 1A (CPT1A), and transcriptional regulators, including peroxisome proliferator-activated receptor-α (PPARα) [5]. The resulting impairment of FAO can drive intracellular lipid accumulation, inadequate energy supply, and tubular cell injury, thereby directly promoting fibrotic progression [6]. Accordingly, a focus on FAO impairment provides an important framework for understanding the metabolic basis of TIF.
In view of the central role of FAO impairment in the pathogenesis of TIF, restoration of mitochondrial FAO has emerged as a promising therapeutic strategy. A range of interventions, including PPARα agonists, mitochondrial protectants, and natural compounds, have shown antifibrotic potential in preclinical models [7,8,9]. Nevertheless, a systematic understanding of the pathological manifestations, molecular regulatory networks, and therapeutic strategies related to FAO impairment in TIF remains to be further refined. This review, therefore, aims to systematically examine how mitochondrial FAO dysfunction contributes to TIF and to discuss current therapeutic strategies targeting this metabolic defect, with particular attention to the challenges of clinical translation and future directions.
2. Manifestations of FAO Impairment
TIF is not simply a form of structural scarring, rather, it is a dynamic pathological process accompanied by sustained disruption of energy metabolism in tubular epithelial cells (TECs). Given their strong dependence on mitochondrial FAO for ATP production, TECs are particularly vulnerable to disturbances in FAO, making FAO dysfunction one of the earliest and most prominent metabolic alterations in chronic kidney injury. Evidence from kidney tissues of patients with CKD and from multiple experimental models of renal fibrosis shows that TECs commonly exhibit suppression of FAO-related transcriptional programs, impaired mitochondrial fatty acid utilization, and abnormal lipid accumulation. Together, these abnormalities define a characteristic metabolic phenotype of TIF [10,11]. Overall, impaired mitochondrial FAO in TIF can be summarized into three interrelated manifestations: reduced fatty acid oxidative capacity in renal tubular epithelial cells, mitochondrial dysfunction with defective bioenergetics, and secondary lipid deposition accompanied by lipotoxic stress (Figure 1).
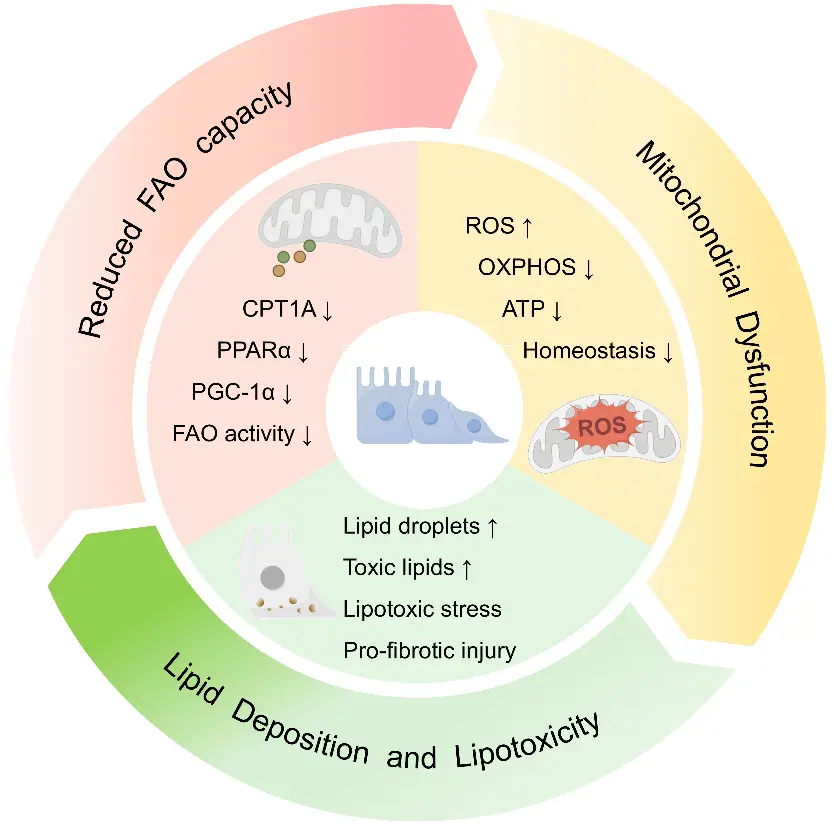
Figure 1. Schematic representation of the manifestations of FAO impairment in tubular epithelial cells during TIF. The central tubular epithelial cell model and circular arrows depict the progression from impaired FAO to mitochondrial dysfunction, lipotoxicity, and subsequent profibrotic tubular injury. Arrows denote increases (↑) or decreases (↓) in the corresponding molecules and biological processes. FAO impairment is associated with downregulation of key FAO regulators and enzymes, including CPT1A, PPARα, and PGC-1α, leading to reduced FAO activity. Concomitantly, mitochondrial dysfunction is characterized by increased ROS production, reduced OXPHOS and ATP generation, and disrupted cellular homeostasis. These changes promote lipid droplet accumulation, toxic lipid deposition, lipotoxic stress, and profibrotic injury.
2.1. Impaired Fatty Acid Oxidation
Under physiological conditions, proximal tubular epithelial cells preferentially use fatty acids as their principal energy substrate. Following cellular uptake and activation, fatty acids are transported into mitochondria via the carnitine shuttle, where they undergo β-oxidation to generate acetyl-CoA, which is subsequently coupled to the tricarboxylic acid cycle and oxidative phosphorylation to meet the high ATP demand of tubular function [12]. This dependence on FAO-dominant oxidative metabolism renders renal tubules particularly sensitive to impaired lipid catabolism and suppression of mitochondrial metabolism.
In fibrotic kidneys, reduced FAO capacity is first reflected by broad downregulation of key FAO-associated enzymes and transcriptional regulators. Seminal studies have shown that, in both human fibrotic kidney biopsy specimens and experimental models including unilateral ureteral obstruction and folic acid-induced kidney injury, tubular epithelial cells exhibit decreased expression of CPT1A, PPARα, peroxisome proliferator-activated receptor gamma coactivator-1α (PGC-1α), and other FAO-related molecules, indicating an overall reduction in mitochondrial fatty acid entry and subsequent oxidative utilization [13,14]. More recent evidence further suggests that this alteration does not reflect an isolated enzymatic defect, but rather coordinated suppression of fatty acid transport, β-oxidation, and the broader mitochondrial oxidative metabolic program [15].
Reduced FAO capacity is also directly manifested as diminished fatty acid oxidative flux and lower efficiency of energy substrate utilization. When fatty acids fail to enter mitochondria efficiently or cannot complete mitochondrial β-oxidation, ATP production in tubular cells becomes constrained, leading to an energetic deficit. Kang et al. [16] demonstrated that selective inhibition of FAO in tubular epithelial cells is sufficient to induce ATP depletion, cell death, dedifferentiation, and lipid accumulation, a phenotype closely resembling the pathological changes observed in fibrotic kidneys. Subsequent studies have reinforced this concept at the level of transcriptional regulation. For example, Kruppel-like factor 15 (KLF15) deficiency aggravates renal injury and fibrosis by suppressing proximal tubular FAO, whereas an imbalance in the Twist1/PGC-1α axis downregulates FAO-related gene expression and promotes the transition of tubular cells toward a profibrotic state [10,17].
Importantly, the decline in FAO in TIF should not be interpreted simply as the consequence of a single molecular abnormality, but more likely as a manifestation of multilayered disruption within the metabolic network. Although CPT1A has long been regarded as a key rate-limiting node in proximal tubular FAO, and restoration of CPT1A expression can recover FAO and attenuate fibrosis [18], recent findings indicate that tubular CPT1A deletion alone does not necessarily exacerbate fibrosis in some models of chronic injury, suggesting compensation through peroxisomal oxidation or other alternative lipid metabolic pathways [19]. Accordingly, current evidence increasingly supports the view that impaired FAO capacity represents an integrated failure of the tubular oxidative metabolic program rather than an isolated defect in a single enzyme.
Impaired FAO in TECs is a characteristic metabolic abnormality in TIF and contributes to mitochondrial dysfunction, energy deficiency, and fibrotic progression.
2.2. Mitochondrial Dysfunction
With persistent impairment of FAO, renal tubules progressively develop mitochondrial dysfunction, which is mainly characterized by reduced oxidative phosphorylation (OXPHOS), increased production of reactive oxygen species (ROS), and disruption of mitochondrial homeostasis. These alterations do not occur in isolation. Rather, they are mechanistically interconnected and collectively drive tubular epithelial cells from metabolic disequilibrium toward sustained injury and a profibrotic phenotype.
Reduced OXPHOS capacity and insufficient ATP production are direct manifestations of mitochondrial dysfunction. Nicotinamide adenine dinucleotide (NADH) and flavin adenine dinucleotide (FADH2) generated during FAO serve as major electron donors to the electron transport chain (ETC). When FAO is impaired, the ETC is deprived of sufficient reducing equivalents, resulting in inefficient establishment of the transmembrane proton gradient and subsequent suppression of ATP synthase activity [20]. This pathological alteration has been well documented in human CKD kidney tissues and in multiple models of renal fibrosis, in which proximal tubular mitochondria exhibit metabolic derangement characterized by suppressed oxidative metabolism, reduced mitochondrial respiration and ATP production, and a shift toward glycolysis. These findings indicate that, under fibrotic conditions, renal tubules progressively lose the capacity to sustain efficient oxidative metabolism [21,22,23]. The resulting energy deficit not only compromises ATP-dependent ion transport and aggravates cellular injury, but also reflects a broader transition of renal tubules from efficient oxidative energy production to a low-efficiency injury-response state, supporting the view that OXPHOS decline is an important indicator of progressive tubular dysfunction.
Excessive ROS generation and enhanced oxidative stress are major consequences of mitochondrial injury. In dysfunctional mitochondria, electron leakage during electron transport becomes more likely, leading to persistent ROS overproduction that further damages lipids, proteins, and mitochondrial DNA, thereby establishing a self-amplifying cycle [24]. Mitochondrial dysfunction is a major source of oxidative stress in the kidney, and excessive ROS not only directly injures tubular cells but also promotes inflammatory responses, cell death, and activation of profibrotic signaling. Li et al. [25] showed in unilateral ureteral obstruction (UUO) mice and hypoxia-treated HK-2 cells that inhibition of mitophagy impaired the clearance of damaged mitochondria, which in turn led to further accumulation of mitochondrial ROS (mtROS), enhanced activation of the NOD-like receptor family pyrin domain containing 3 (NLRP3) inflammasome, and increased expression of transforming growth factor-β1 (TGF-β1) and α-smooth muscle actin (α-SMA). These changes were partially reversed by the mitochondria-targeted antioxidant MitoTEMPO, suggesting that persistent accumulation of damaged mitochondria can continuously amplify the ROS burden and sustain profibrotic signaling.
Mitochondrial homeostasis is not a static equilibrium, but a dynamic process maintained through the coordinated regulation of mitochondrial biogenesis, quality control, and turnover. Disruption of this homeostatic network further aggravates metabolic decompensation in renal tubules. Kim et al. [26] found in a model of autosomal dominant tubulointerstitial kidney disease that both mitophagy and mitochondrial biogenesis were impaired in renal tubules, leading to persistent accumulation of abnormal mitochondria and activation of the cyclic GMP-AMP synthase (cGAS)-stimulator of interferon genes (STING) pathway. Restoration of mesencephalic astrocyte-derived neurotrophic factor (MANF) expression improved mitochondrial turnover and function and markedly alleviated tubular injury, indicating that disruption of mitochondrial homeostasis itself is sufficient to drive the transition of renal tubules from metabolic adaptation to sustained injury. Yuan et al. [27] further showed in a mouse model of renal fibrosis that suppression of the regulatory program governing mitochondrial biogenesis aggravated mitochondrial dysfunction and renal fibrosis, whereas restoration of this program improved mitochondrial homeostasis through PGC-1α and attenuated tubular injury. Taken together, disruption of mitochondrial homeostasis is not merely a structural abnormality, but a key mechanism driving the progression of renal tubules from reversible metabolic adaptation to irreversible injury.
Taken together, mitochondrial dysfunction promotes tubular injury through bioenergetic failure and oxidative stress, while further impairing fatty acid oxidation and promoting lipid accumulation. These changes may contribute to ongoing fibrotic progression.
2.3. Lipid Deposition and Lipotoxicity
Insufficient FAO also leads to persistent intracellular lipid deposition, accompanied by progressive aggravation of lipotoxicity. Under physiological conditions, fatty acids taken up by TECs are efficiently transported into mitochondria for oxidative utilization, thereby preventing abnormal accumulation of intracellular free fatty acids and their metabolic intermediates. In the fibrotic setting, however, the decline in FAO capacity and impairment of mitochondrial oxidative metabolism disrupt the balance between fatty acid uptake, transport, oxidation, and storage, leading to tubular cells progressively acquiring a metabolic phenotype characterized by abnormal lipid deposition [28,29].
Defective FAO utilization constitutes the pathological basis for abnormal intracellular lipid accumulation. The proximal tubule relies heavily on fatty acids as an energy source, but within the fibrotic microenvironment, blockade of the FAO pathway prevents fatty acids from undergoing mitochondrial β-oxidation. Li et al. [30] observed, across multiple models of kidney injury, that downregulation of PPARα and its downstream FAO-related genes in renal tubules occurred in parallel with lipid deposition and fibrotic changes. In vitro experiments further showed that TGF-β1 treatment at 5 ng/mL for 24 h suppressed oxygen consumption rates in tubular cells and reduced ATP production derived from FAO. Rinaldi et al. [31], using both patient samples and experimental models, further demonstrated that persistent defects in fatty acid metabolism were accompanied by reduced CPT2 expression, lipid accumulation, and amplified lipotoxicity, thereby driving renal tubules into a state of sustained chronic stress. These findings indicate that lipid deposition in TIF does not simply result from excessive fatty acid influx but rather reflects impaired mitochondrial oxidative capacity in renal tubules.
A direct consequence of defective FAO is the abnormal accumulation of metabolic substrates within TECs, which are subsequently converted into lipids with toxic potential and thereby induce cellular injury. Mukhi and colleagues reported that lipid synthetic programs were aberrantly activated in TECs, accompanied by upregulation of lipid droplet-associated markers and inflammatory cell death, suggesting that persistent lipid accumulation in renal tubules can promote renal fibrosis [32]. Similarly, Hou et al. [33] found in diabetic kidney disease models and high glucose-treated HK-2 cells that lipid droplet formation in tubular cells was markedly increased and was accompanied by abnormal accumulation of lipid intermediates such as sphingosine-1-phosphate. SS-31 treatment, administered at 3 mg/kg/day for 10 weeks in db/db mice or at 100 nM for 72 h in high glucose-treated HK-2 cells, restored fatty acid catabolic capacity and conferred renoprotection.
Importantly, such pathological lipid accumulation is not a static metabolic endpoint. Instead, specific accumulated lipid species may act as toxic mediators that actively drive cellular injury. Zhou et al. [34] observed a marked increase in the endogenous lipid mediator 2-arachidonoylglycerol (2-AG) in patient samples. Supplementation with 2-AG in UUO mice further aggravated tubular lipid droplet deposition, FAO decline, and fibrotic changes. Consistently, in HK-2 cells, 2-AG treatment at 100 μM for 24 h reduced ATP production, enhanced Nile Red staining, and increased the expression of fibrosis-related proteins, indicating that abnormal lipid burden itself can directly amplify tubular injury. Liu et al. [35] further showed in diabetic kidney disease that sustained glucagon exposure induced increased lipid deposition in TECs, abnormal mitochondrial morphology, and aggravated injury phenotypes. Conversely, inhibition of tubular glucagon signaling markedly attenuated these changes and delayed the progression of renal fibrosis. Taken together, lipid deposition is not only a consequence of metabolic dysregulation but also a key effector that directly drives tubular injury and fibrotic progression through the toxic actions of specific lipid mediators.
3. Mechanisms of FAO Impairment in TIF
FAO impairment is a central metabolic abnormality underlying the progression of TIF. In TECs, defective FAO not only compromises ATP production but also promotes the accumulation of lipid intermediates, oxidative stress, phenotypic reprogramming, and disturbance of the intrarenal microenvironment. These alterations collectively contribute to tubular injury, interstitial inflammation, fibroblast activation, and abnormal extracellular matrix deposition. The following sections examine how FAO impairment promotes TIF through four interconnected mechanisms: bioenergetic failure, lipotoxicity and oxidative stress, phenotypic alterations and intercellular crosstalk, and dysregulation of profibrotic signaling pathways (Figure 2).
3.1. Bioenergetic Dysfunction
FAO impairment disrupts the bioenergetic foundation required for TECs to maintain homeostasis and represents a key mechanism driving TIF. This metabolic defect is not limited to ATP depletion; rather, it reflects a broader decline in mitochondrial oxidative capacity, depriving TECs of the metabolic support required to preserve their differentiated phenotype and sustain physiological function. Han et al. [13] showed that the Notch signaling activation induced by Notch1 intracellular domain (ICN1) overexpression suppresses PGC-1α expression and downregulates FAO-related genes, thereby reducing mitochondrial content and impairing FAO. Conversely, restoration of PGC-1α expression improved FAO defects and attenuated renal fibrosis. These findings suggest that FAO impairment is not merely a secondary change accompanying fibrosis, but may instead represent a critical metabolic link between upstream-injury signals and tubular damage. More recent studies further indicate that, during CKD progression, TECs undergo metabolic reprogramming, shifting from FAO to glycolysis, thereby exacerbating renal fibrosis [36]. This observation supports the view that suppression of FAO is a central biological feature of metabolic remodeling during chronic tubular injury.
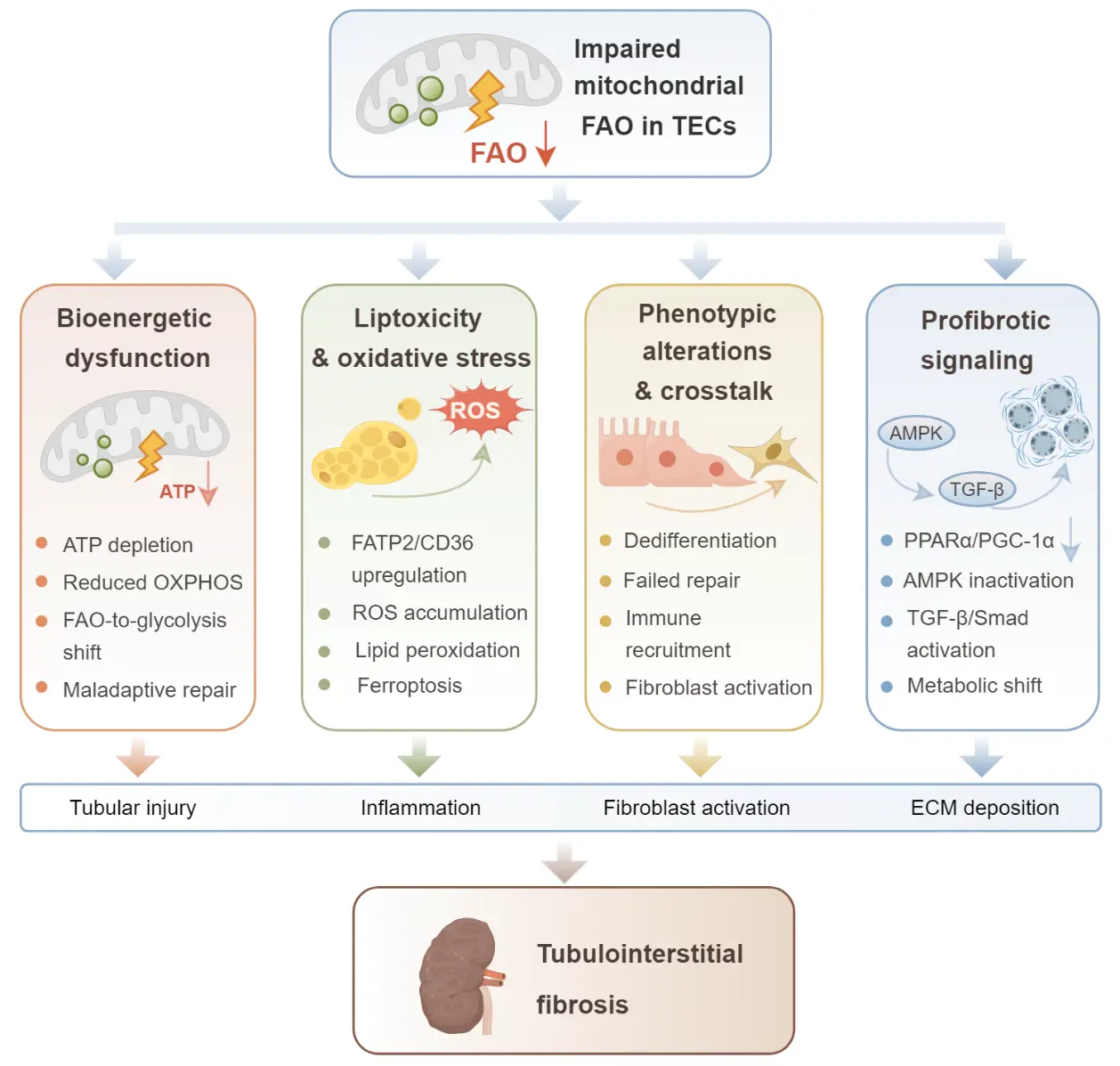
Figure 2. Schematic illustration of the mechanisms linking impaired mitochondrial FAO in TECs to tubulointerstitial fibrosis. Downward arrows denote decreased levels or activities of the molecules and biological processes. Defective FAO leads to ATP depletion and reduced oxidative phosphorylation, promotes lipid accumulation and ROS-associated oxidative damage, induces phenotypic changes and maladaptive intercellular crosstalk, and activates profibrotic signaling pathways including PPARα/PGC-1α suppression, AMPK inactivation, and TGF-β/Smad signaling. These interconnected events contribute to tubular injury, inflammation, fibroblast activation, extracellular matrix deposition, and progression of TIF.
This bioenergetic crisis further reduces the capacity of TECs to mount an appropriate metabolic response to injury. Because TECs perform highly energy-intensive functions, including reabsorption and ion transport, they depend heavily on mitochondrial oxidative metabolism. Once FAO is disrupted, cells not only exhibit reduced ATP supply but also develop decreased oxidative phosphorylation efficiency, diminished respiratory reserve, and disturbed mitochondrial homeostasis. These changes are accompanied by impaired transport function, loss of epithelial integrity, and increased susceptibility to injury. Xu et al. [37] reported that downregulation of GLIS family zinc finger (GLIS1) promotes a metabolic shift from FAO to glycolysis in the kidney, accompanied by increased cellular senescence and aggravated fibrosis. Yang et al. [38] further demonstrated that upregulation of repressor element 1-silencing transcription factor (REST) disrupts FAO in TECs by suppressing molecules involved in mitochondrial energy metabolism, whereas tubule-specific deletion of REST significantly improved mitochondrial bioenergetics and attenuated renal fibrosis. Together, these findings indicate that FAO impairment is not simply a passive response to pre-existing injury, but an active metabolic mechanism that shapes the injury phenotype of TECs.
Persistent FAO impairment not only constrains energy metabolism in TECs but may also influence the outcome of post-injury repair. Recent evidence suggests that disruption of mitochondrial homeostasis can drive maladaptive repair in TECs and promote the transition from acute kidney injury (AKI) to CKD, indicating that metabolic disequilibrium is not merely a consequence of functional impairment, but also a determinant of repair trajectory after injury [39]. Single-cell transcriptomic and multi-omics atlases further suggest that the critical event underlying the progression from tubular injury to chronic fibrosis is not the acute insult itself, but the failure of a subset of injured TECs to reacquire a mature epithelial phenotype. Instead, these cells remain in a state of maladaptive repair characterized by persistent dedifferentiation, upregulation of inflammatory mediators, and sustained activation of profibrotic signaling [40]. Persistence of this maladaptive repair state may therefore constitute an important mechanism linking tubular injury to chronic fibrotic progression. Accordingly, FAO impairment may promote TIF by disrupting metabolic homeostasis and reparative capacity in TECs, thereby shifting them from reversible injury toward a sustained maladaptive repair state.
Collectively, FAO impairment disrupts bioenergetic homeostasis in TECs, compromises their physiological function and injury response, and promotes maladaptive repair, thereby contributing to TIF progression.
3.2. Lipotoxicity and Oxidative Stress
Lipotoxicity resulting from abnormal lipid accumulation is another important mechanism driving the progression of TIF, in part through persistent induction of oxidative stress and amplification of profibrotic responses. Chen et al. [29] reported that the fatty acid transport protein 2 (FATP2) is markedly upregulated in fibrotic kidney tissue and injured TECs. Increased FATP2 expression promotes abnormal fatty acid uptake and reprograms lipid metabolism, thereby disrupting the balance between fatty acid influx and oxidative utilization. This imbalance aggravates lipid deposition while inducing oxidative injury. Pharmacological inhibition of FATP2 with FATP2i restored FAO activity and attenuated renal interstitial fibrosis. Studies in diabetic kidney disease further suggest that upregulation of the fatty acid transport receptor cluster of differentiation 36 (CD36) also exacerbates mitochondrial dysfunction and FAO impairment in TECs, indicating a close association between defective oxidative utilization, increased ROS accumulation, and aggravated mitochondrial injury. Conversely, CD36 deletion helps relieve mitochondrial stress and attenuate tubular injury [41].
Lipotoxicity-associated oxidative stress not only directly injures TECs but also activates multiple signaling pathways involved in inflammation and fibrosis, thereby aggravating the imbalance in the local microenvironment. Excessive ROS and lipid peroxidation products can activate key signaling networks such as NF-κB and TGF-β/Smad, which, in turn, induce the production and release of monocyte chemoattractant protein-1 (MCP-1), tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6), and other chemokines and profibrotic mediators. These events enhance immune cell recruitment, fibroblast activation, and extracellular matrix deposition, ultimately promoting TIF progression [42,43,44,45].
More recent studies on ferroptosis further suggest that lipid peroxidation serves as a critical link between inflammatory amplification and fibrotic progression [46,47]. In chronic kidney injury, ferroptosis contributes to tubular epithelial cell injury and is characterized by the accumulation of lipid peroxidation products, suppression of the antioxidant defense axis formed by glutathione peroxidase 4 (GPX4) and xCT (solute carrier family 7 member 11, SLC7A11), and disruption of redox homeostasis. Inhibition of ferroptosis can partially ameliorate these abnormalities and attenuate renal injury [48]. Zhang et al. [49] similarly found in the UUO model that liproxstatin-1 treatment at 10 mg/kg/day for 14 consecutive days after surgery reduced renal iron deposition, lipid peroxidation, and tubular epithelial cell death, while significantly attenuating collagen deposition and renal fibrosis. Their in vitro experiments further showed that HK-2 cells undergoing ferroptosis-like changes could promote activation of neighboring fibroblasts through paracrine mechanisms. In a study of diabetic kidney disease, Gan et al. found that canagliflozin enhanced FAO, increased ATP production and CPT1A expression, and attenuated tubular ferroptosis in db/db mice and high glucose-treated HK-2 cells [50]. Consistently, Dai et al. reported in UUO and folic acid-induced chronic kidney disease models that inhibition of acyl-CoA synthetase long-chain family member 4 (ACSL4) reduced lipid peroxide accumulation, alleviated tubular ferroptosis, and attenuated tubulointerstitial fibrosis [51]. These findings suggest that FAO dysfunction may induce lipid peroxide accumulation, trigger ferroptosis in TECs, and promote fibrotic progression.
Overall, lipotoxicity driven by abnormal lipid accumulation promotes oxidative stress, profibrotic signaling, and microenvironmental imbalance, thereby contributing to sustained fibrotic responses in renal tubules.
3.3. Tubulointerstitial Crosstalk
Under conditions of FAO impairment and persistent injury-related stress, TECs develop not only intracellular metabolic and structural abnormalities, but also sustained aberrant repair-associated phenotypes. Recent single-cell transcriptomic and spatial omics studies have shown that these abnormal tubular epithelial cells are often distributed adjacent to fibroblasts, immune cells, and other interstitial components, together forming a fibrosis-associated pathological cellular ecosystem. Notably, the spatial architecture of this fibrotic microenvironment is closely associated with declining renal function and disease progression [52]. These findings indicate a close link between injured TECs and the local fibrotic microenvironment.
Functional communication between abnormal TECs and interstitial fibroblasts is a major manifestation of tubulointerstitial crosstalk. Using single-cell multimodal approaches, Aggarwal et al. [53] found that proximal tubular cells that failed to complete normal repair after injury persistently maintained an abnormal SRY-box transcription factor 9 (SOX9) program, accompanied by activation of Wnt signaling. These cells were associated with fibroproliferative responses in neighboring fibroblasts, suggesting that maladaptively repaired TECs may contribute to the maintenance and expansion of the local profibrotic microenvironment. This study provides relatively direct evidence that injured tubular epithelial cells can influence the state of interstitial cells through paracrine mechanisms.
In addition to fibroblasts, abnormal TECs can further amplify profibrotic responses by recruiting immune cells and facilitating intercellular communication. Doke et al. [54] identified a population of aberrant tubular cells with marked proinflammatory and profibrotic features and showed that these cells secrete C-X-C motif chemokine ligand 1 (CXCL1) to recruit basophils expressing C-X-C motif chemokine receptor 2 (CXCR2), which subsequently serve as an important source of IL-6, thereby promoting renal fibrotic progression. In addition, Melchinger et al. [55] reported that, during the transition from AKI to CKD, a subset of failed-repair proximal tubular cells newly expresses vascular cell adhesion molecule-1 (VCAM-1). This change enhances their adhesion to and interaction with immune cells, thereby sustaining the local inflammatory microenvironment and aggravating subsequent nephron loss. In recent years, extracellular vesicle-mediated epithelial-interstitial communication has also been recognized as an important component of this process. Liu et al. [56] showed that exosomes derived from stressed or injured renal tubules can regulate the survival and fate of interstitial fibroblasts, thereby influencing the progression of renal fibrosis. Collectively, these findings indicate that abnormal TECs can convert localized epithelial injury into sustained interstitial activation through multiple mechanisms, including chemokines, adhesion molecules, and extracellular vesicles.
Genetic fate-tracing studies have shown that although cultured proximal epithelial cells can express myofibroblast-specific markers in vitro, they remain within the renal tubules in vivo and fail to migrate into the interstitium or undergo complete phenotypic transformation [57]. Another study analyzed the cellular composition of the human kidney using single-cell RNA sequencing and also confirmed that the main cellular sources of scar-forming cells during renal fibrosis are pericytes and fibroblasts [58]. In fact, experimental studies on TECs phenotypic transition further showed that these partial epithelial-to-mesenchymal transition (EMT) cells, which acquire mesenchymal features but do not leave the basement membrane, remain arrested in the G2 phase of the cell cycle, lose their repair capacity, and may indirectly regulate the interstitial microenvironment through paracrine signaling, leading to parenchymal cell injury and activation of profibrotic signaling pathways [59,60].
Overall, FAO dysfunction may drive renal TECs into a maladaptive repair state. Consequently, maladaptive tubular epithelial cells may enhance sustained tubulointerstitial crosstalk with fibroblasts through paracrine signaling, immune cell recruitment, and adhesion molecule-mediated cellular interactions, thereby promoting the formation of a local profibrotic microenvironment and extracellular matrix deposition and ultimately contributing to TIF progression.
3.4. Profibrotic Signaling Pathways
FAO impairment and the resulting progression of renal fibrosis are not driven by a single molecular event, but are jointly regulated by multiple signaling pathways involved in metabolic control and injury responses. Among these, PPARα and PGC-1α are key regulators of tubular fatty acid oxidation and mitochondrial metabolic homeostasis. On the one hand, proximal tubule-specific overexpression of PPARα attenuates renal fibrosis and inflammation in the UUO model and reduces the expression of fibrosis-related molecules, including TGF-β1, collagen, and α-SMA [61]. On the other hand, PGC-1α, a central regulator of mitochondrial biogenesis and oxidative metabolism, is closely associated with abnormal mitochondrial dynamics, enhanced oxidative stress, and aggravated fibrosis when downregulated in tubular cells. Nam et al. further showed that restoration of PGC-1α activity improves mitochondrial homeostasis and suppresses activation of the NLR family pyrin domain containing 3 (NLRP3) inflammasome, thereby alleviating renal injury and fibrosis [62]. More recent work also suggests that signal transducer and activator of transcription 6 (STAT6) promotes tubular lipid accumulation and fibrotic progression by transcriptionally repressing PPARα and its downstream FAO-related genes [30].
AMP-activated protein kinase (AMPK) primarily functions in energy sensing and metabolic reprogramming and represents a key node linking cellular energy status to FAO activity. AMPK activation promotes fatty acid oxidation, preserves mitochondrial function, and suppresses abnormal matrix deposition, whereas AMPK inactivation predisposes renal tubules to a metabolic shift from oxidative phosphorylation to glycolysis. Liu et al. [63] found in a model of chronic allograft fibrosis that disruption of the liver kinase B1 (LKB1)-AMPK pathway in tubular epithelial cells impaired glucose and fatty acid metabolic homeostasis and was accompanied by increased collagen secretion, extracellular matrix remodeling, and aggravated fibrosis. Pharmacological activation of AMPK, particularly with metformin, ameliorated these metabolic abnormalities and attenuated renal fibrosis. Another study further showed in UUO model and TGF-β1 induced tubular cells that enhanced AMPK phosphorylation improves protein kinase B (AKT)-PGC-1α activity, restores mitochondrial function, and reduces lipid accumulation, thereby attenuating profibrotic changes [64]. These findings indicate that AMPK is not only a critical sensor of cellular energy stress but also an important coordinator of FAO, mitochondrial homeostasis, and fibrotic outcomes.
Compared with the metabolic regulatory axes described above, the TGF-β/Smad pathway serves as a major signaling hub linking metabolic imbalance to profibrotic responses. At the level of metabolic regulation, TGF-β1 stimulation suppresses fatty acid oxidation in tubular cells and is accompanied by reduced CPT1A activity, ATP depletion, and lipid deposition, thereby connecting canonical profibrotic signaling with FAO impairment [65]. Further studies have shown that activation of Smad3, a downstream effector of TGF-β, contributes to the downregulation of PGC-1α expression in tubular cells, whereas blockade of Smad3-related signaling helps preserve its expression [66]. More recently, TGF-β-activated Smad3 has also been shown to further weaken the PGC-1α-associated mitochondrial protective program by repressing PR/SET domain 16 (PRDM16) transcription, thereby leading to mitochondrial injury, lipid deposition, and aggravated fibrosis in renal tubules [27]. These findings suggest that the TGF-β/Smad pathway not only participates in canonical profibrotic responses but may also promote metabolic reprogramming and fibrotic progression by disrupting tubular fatty acid oxidation and mitochondrial metabolic homeostasis.
PPARα/PGC-1α, AMPK, and TGF-β/Smad jointly promote FAO impairment and TIF progression. Targeting these metabolic networks may help delay renal fibrosis.
4. Therapeutic Targets in FAO Impairment
Given the central role of FAO impairment in tubular injury and fibrotic progression, targeting this metabolic defect has become an important focus of recent research aimed at delaying renal fibrosis. Current evidence indicates that relevant interventions extend beyond simply restoring fatty acid oxidation. Increasingly, these strategies also seek to improve mitochondrial function, attenuate lipotoxicity, and suppress profibrotic responses. The following sections summarize recent advances in interventions targeting FAO impairment, including approaches designed to restore FAO, improve mitochondrial function, alleviate lipotoxicity and profibrotic responses, and address current challenges and translational prospects (Table 1).
Table 1. Representative therapeutic strategies targeting FAO dysfunction in TIF.
|
Category |
Intervention |
Target/Pathway |
Experimental Model |
Treatment Regimen |
Main Effects |
Ref. |
|---|---|---|---|---|---|---|
|
FAO restoration |
Fenofibrate |
NF-κB; TGF-β1/Smad3 |
Type 2 diabetic rats |
150 mg/kg/day, 10 weeks |
Reduced inflammation and TIF |
[67] |
|
FAO restoration |
Fenofibrate |
Lipid metabolism; MCP-1/PAI-1 signaling |
Lipotoxic kidney injury models; palmitate-treated proximal TECs |
0.05% wt/wt diet, 12 weeks; 10-50 μM in vitro |
Reduced inflammation and TIF |
[7] |
|
FAO restoration |
ZLN005 |
PGC-1α/TFAM |
UUO mice; TGF-β1-treated TECs |
40 mg/kg, pre-UUO to day 7; 10 μM, 48 h |
Attenuated lipotoxic renal injury |
[68] |
|
FAO restoration |
IL-37 |
CPT1A |
DKD model; HG-treated HK-2 cells |
IL-37tg mice; recIL-37 300 ng/mL, 2 h pretreatment + HG 48 h |
Restored mitochondrial homeostasis; reduced fibrosis |
[69] |
|
Mitochondrial protection |
Metformin |
HIF-1α/MIOX; PINK1/Parkin mitophagy |
DKD mice; HG-treated HK-2 cells |
200 mg/kg/day, 8 weeks; 500 μM, 48 h |
Restored FAO; reduced lipid deposition and fibrosis |
[70] |
|
Mitochondrial protection |
Sulforaphane |
Mitochondrial biogenesis; β-oxidation; mitochondrial dynamics |
RUUO rats |
1 mg/kg/day, post-UUO days 2–6 |
Reduced lipid accumulation and fibrosis |
[71] |
|
Mitochondrial protection |
Piericidin analogue S14 |
LKB1/autophagy |
ADR, UUO, UIRI, 5/6NX; TGF-β1-treated TECs |
0.5 or 1.0 mg/kg/day in vivo; 0.5 μM in vitro |
Preserved mitochondrial homeostasis; reduced fibrosis |
[72] |
|
Lipotoxicity reduction |
Astragaloside IV |
FATP2-mediated fatty acid uptake |
DKD rats; PA-BSA-treated NRK-52E cells |
AS-IV 10/20 mg/kg/day, 8 weeks; 5–20 μM in vitro; lipofermata 5 mg/kg/day, 2 weeks |
Reduced tubular lipotoxicity and inflammation |
[73] |
|
Lipotoxicity reduction |
Liproxstatin-1 |
Ferroptosis |
UUO mice; ferroptotic HK-2 cells; renal fibroblasts |
10 mg/kg/day, 14 days after surgery |
Reduced ferroptosis and TIF |
[49] |
|
Lipotoxicity reduction |
Ferrostatin-1 |
SLC7A11/GPX4-related ferroptosis |
HN mice; UA-treated HK-2 cells |
2 mg/kg/day, 17 d; 1 μM, 24 h |
Increased UA excretion; reduced ferroptosis and fibrosis |
[74] |
4.1. Restoration of FAO
Restoring FAO is the most direct strategy for correcting FAO impairment. PPARα agonists, represented by fenofibrate, are among the classic approaches used to enhance FAO. Li et al. [67] found that, in a diabetic rat model, fenofibrate administered by intragastric gavage at 150 mg/kg/day for 10 weeks reduced renal inflammatory responses and tubulointerstitial fibrosis. These effects were associated with suppression of NF-κB activity and TGF-β1/Smad3 signaling. Tanaka et al. [7] further showed that fenofibrate, administered as a 0.05% wt/wt dietary supplement for 12 weeks in HFD-fed mice and used at 10–50 μmol/L in palmitate-stimulated proximal tubular cells, attenuated lipotoxicity-induced glomerular and tubulointerstitial injury, enhanced the expression of renal lipid catabolism-related enzymes, and suppressed the production of inflammatory and profibrotic mediators such as MCP-1 and plasminogen activator inhibitor-1 (PAI-1) in proximal tubular cells.
PGC-1α, a key regulator of mitochondrial biogenesis and oxidative metabolism, contributes to the restoration of tubular metabolic homeostasis when activated. In the UUO model and in TGF-β1-stimulated tubular cells, the PGC-1α activator ZLN005 at 40 mg/kg in UUO mice and 10 μM for 48 h in tubular cells restored the expression of mitochondrial transcription factor A (TFAM) and other molecules involved in mitochondrial homeostasis and FAO. This effect improved mitochondrial biogenesis and bioenergetic balance, suppressed the expression of fibrotic markers, and ultimately attenuated renal interstitial fibrosis [68].
In addition to modulating upstream metabolic regulators such as PPARα and PGC-1α, targeting key enzymes within the FAO pathway is also an important strategy for restoring fatty acid oxidative capacity. Among these enzymes, CPT1A, the rate-limiting enzyme responsible for mitochondrial entry of fatty acids for β-oxidation, is regarded as a critical point of impairment in tubular FAO. A study in diabetic kidney disease showed that interleukin-37 (IL-37), an anti-inflammatory cytokine of the interleukin-1 family, ameliorated the decline in FAO in tubular cells by upregulating CPT1A and attenuated proteinuria, lipid deposition, and in renal fibrosis in IL-37 transgenic DKD mice and high glucose-treated HK-2 cells [69]. In the setting of chronic allograft fibrosis, promotion of lipid utilization and enhancement of CPT1A-mediated FAO were likewise shown to alleviate renal fibrosis [75].
4.2. Improvement of Mitochondrial Function
Improving mitochondrial function represents an important strategy for correcting FAO impairment. Wu et al. [70] found in a model of diabetic kidney disease that the biguanide antidiabetic agent metformin at 200 mg/kg/day for 8 weeks in db/db mice and 500 μM for 48 h in high glucose-treated HK-2 cells suppresses the hypoxia-inducible factor-1α (HIF-1α)/myo-inositol oxygenase (MIOX) axis, promotes PTEN-induced kinase 1 (PINK1)/Parkin-dependent mitophagy, restores mitochondrial membrane potential in tubular cells, and attenuates lipid peroxidation and fibrosis. These findings suggest that improving mitochondrial quality control may help slow the progression of chronic kidney injury. In parallel, interventions targeting mitochondrial oxidative stress and bioenergetic imbalance have also shown renoprotective effects. Aranda-Rivera et al. [71] reported in ureteral obstruction-related models that the natural isothiocyanate sulforaphane administered intraperitoneally at 1 mg/kg/day from day 2 to day 6 after UUO enhances mitochondrial biogenesis, restores mitochondrial oxygen consumption and β-oxidation, and improves mitochondrial dynamics and autophagic flux, thereby reducing lipid accumulation and renal interstitial fibrosis. In addition, Liu et al. [72] showed that the LKB1 activator piericidin analogue S14 at 1 mg/kg/day in mouse models and 0.5 μM in TGF-β1-stimulated TECs alleviates mitochondrial dysfunction and renal fibrosis by promoting autophagy and preserving mitochondrial homeostasis in tubular epithelial cells. Together, these findings further support the importance of improving mitochondrial quality control for delaying the progression of chronic kidney injury.
4.3. Attenuation of Lipotoxicity
Attenuating lipotoxicity and its secondary profibrotic consequences represents an important complementary strategy for targeting FAO impairment. Such approaches may reduce lipid accumulation while simultaneously suppressing oxidative stress, inflammatory amplification, and interstitial activation. Among these strategies, targeting lipid influx to inhibit abnormal fatty acid uptake provides a direct means of reducing intracellular lipid accumulation. For example, Astragaloside IV at 10 or 20 mg/kg/day for 8 weeks in DKD rats and 5–20 μM in BSA-PA-treated NRK-52E cells alleviates lipid accumulation in tubular epithelial cells in diabetic kidney disease by inhibiting FATP2-mediated fatty acid transport, while also improving mitochondrial dysfunction and inflammatory responses. Further validation using lipofermata at 5 mg/kg/day for 2 weeks or Fatp2 siRNA confirmed a critical role for FATP2 in this protective effect, suggesting that inhibition of FATP2-mediated fatty acid transport may be an effective strategy for alleviating tubular lipotoxicity [73].
In addition to abnormal fatty acid uptake, lipid peroxidation and ferroptosis represent another important pathological axis through which lipotoxicity promotes fibrotic progression. Zhang et al. found in the UUO model that the ferroptosis inhibitor liproxstatin-1 reduced iron deposition, lipid peroxidation, and cell death in tubular epithelial cells, while significantly attenuating collagen deposition and renal interstitial fibrosis. Further in vitro experiments showed that liproxstatin-1 also suppressed the secretion of profibrotic factors from tubular cells undergoing ferroptosis, thereby weakening their ability to promote the proliferation and activation of neighboring fibroblasts [49]. Consistent with these findings, Li et al. [74] reported in a model of hyperuricemic nephropathy that the ferroptosis inhibitor ferrostatin-1 administered at 2 mg/kg/day intraperitoneally for 17 days increased uric acid excretion and reduced tubular dilation and collagen deposition. At the same time, ferrostatin-1 partially restored the expression of ferroptosis-related molecules such as solute carrier family 7 member 11 (SLC7A11) and GPX4, thereby improving renal function and attenuating fibrosis.
5. Limitations and Perspectives
Although therapeutic strategies targeting FAO impairment have shown promise in improving tubular metabolic imbalance, alleviating mitochondrial dysfunction, and attenuating lipotoxic and profibrotic responses, the available evidence still derives predominantly from animal models and cell-based experiments. Clinical translational research remains limited, particularly due to a lack of large-scale, prospective human studies. Meanwhile, most candidate interventions exert pleiotropic effects, and the extent to which their renoprotective actions depend on direct modulation of the FAO pathway remains unclear. In addition, the safety, effective dosing, target populations, and long-term benefits of these strategies still require systematic clinical validation.
Notably, the therapeutic efficacy of FAO-targeted interventions may be highly dependent on disease stage. During early or potentially reversible tubular injury, restoring FAO may help preserve metabolic homeostasis in viable TECs, reduce lipid peroxidation, and attenuate aberrant epithelial-interstitial crosstalk. However, when fibrosis has progressed to extensive tubular epithelial cell loss and marked extracellular matrix deposition, restoration of FAO alone may be insufficient to reverse the established fibrotic architecture. This suggests that FAO-targeted therapy for renal fibrosis requires consideration of the early intervention window.
Future work should further define the key pathological events underlying FAO impairment and clarify their interactions with inflammation, oxidative stress, and disruption of mitochondrial homeostasis. On this basis, more specific and clinically translatable therapeutic targets should be identified, while efforts to advance clinically relevant studies and biomarker discovery should be strengthened to facilitate the clinical application of FAO-targeted strategies in the prevention and treatment of tubulointerstitial fibrosis.
6. Summary
Mitochondrial FAO impairment is increasingly recognized as an important metabolic basis of tubulointerstitial fibrosis. By disrupting bioenergetic homeostasis, promoting lipotoxicity, amplifying oxidative stress, and activating profibrotic signaling, FAO impairment contributes to disease progression through multiple interconnected mechanisms. Therefore, restoration of mitochondrial FAO represents a promising therapeutic strategy for delaying renal fibrotic progression. Nevertheless, given the complexity of the underlying regulatory network, the precise mechanisms linking FAO dysregulation to fibrosis, as well as its translational potential in clinical settings, require further investigation.
Statement of the Use of Generative AI and AI-Assisted Technologies in the Writing Process
During the preparation of this manuscript, the authors utilized the generative AI tool ChatGPT to refine the language of the entire text, aiming to enhance the fluency of English expression and adherence to academic standards. The AI tool was not involved in the study design, the derivation of conclusions, or the creation of figures, tables, and references. All text generated or modified by the AI has been carefully reviewed, revised, and approved by the authors sentence by sentence. The authors assume full responsibility for the final content of the published article.
Acknowledgments
We thank the Hub Laboratory of Precision Diagnosis and Treatment for Chronic Kidney Disease in Henan Province for providing technical and platform support for this study.
Author Contributions
Conceptualization, J.F. and W.Z.; Writing-Original Draft Preparation, S.L.; Writing-Review & Editing, J.F. and J.Z.; Supervision, J.Z. and W.Z.; Project Administration, J.Z. and W.Z.
Ethics Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No data were generated or analyzed in this review.
Funding
This study was supported by the National Natural Science Foundation of China (grant number 82400854; 82500910); China Postdoctoral Science Foundation (grant number 2024M763008); and the Henan Provincial Medical Science and Technology Research Project (grant number LHGJ20240189).
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
-
Francis A, Harhay MN, Ong ACM, Tummalapalli SL, Ortiz A, Fogo AB, et al. Chronic kidney disease and the global public health agenda: An international consensus. Nat. Rev. Nephrol. 2024, 20, 473–485. DOI:10.1038/s41581-024-00820-6 [Google Scholar]
-
Huang R, Fu P, Ma L. Kidney fibrosis: From mechanisms to therapeutic medicines. Signal Transduct. Target. Ther. 2023, 8, 129. DOI:10.1038/s41392-023-01379-7 [Google Scholar]
-
Lee LE, Doke T, Mukhi D, Susztak K. The key role of altered tubule cell lipid metabolism in kidney disease development. Kidney Int. 2024, 106, 24–34. DOI:10.1016/j.kint.2024.02.025 [Google Scholar]
-
Gewin LS. Sugar or fat? Renal tubular metabolism reviewed in health and disease. Nutrients 2021, 13, 1580. DOI:10.3390/nu13051580 [Google Scholar]
-
Jao TM, Nangaku M, Wu CH, Sugahara M, Saito H, Maekawa H, et al. ATF6α downregulation of PPARα promotes lipotoxicity-induced tubulointerstitial fibrosis. Kidney Int. 2019, 95, 577–589. DOI:10.1016/j.kint.2018.09.023 [Google Scholar]
-
Han SH, Malaga-Dieguez L, Chinga F, Kang HM, Tao J, Reidy K, et al. Deletion of LKB1 in renal tubular epithelial cells leads to CKD by altering metabolism. J. Am. Soc. Nephrol. 2016, 27, 439–453. DOI:10.1681/ASN.2014121181 [Google Scholar]
-
Tanaka Y, Kume S, Araki S, Isshiki K, Chin-Kanasaki M, Sakaguchi M, et al. Fenofibrate, a PPARα agonist, has renoprotective effects in mice by enhancing renal lipolysis. Kidney Int. 2011, 79, 871–882. DOI:10.1038/ki.2010.530 [Google Scholar]
-
Szeto HH, Liu S, Soong Y, Alam N, Prusky GT, Seshan SV. Protection of mitochondria prevents high-fat diet-induced glomerulopathy and proximal tubular injury. Kidney Int. 2016, 90, 997–1011. DOI:10.1016/j.kint.2016.06.013 [Google Scholar]
-
Hu H, Li W, Hao Y, Peng Z, Zou Z, Liang W. Baicalin ameliorates renal fibrosis by upregulating CPT1α-mediated fatty acid oxidation in diabetic kidney disease. Phytomedicine 2024, 122, 155162. DOI:10.1016/j.phymed.2023.155162 [Google Scholar]
-
Piret SE, Attallah AA, Gu X, Guo Y, Gujarati NA, Henein J, et al. Loss of proximal tubular transcription factor kruppel-like factor 15 exacerbates kidney injury through loss of fatty acid oxidation. Kidney Int. 2021, 100, 1250–1267. DOI:10.1016/j.kint.2021.08.031 [Google Scholar]
-
Clark AJ, Parikh SM. Targeting energy pathways in kidney disease: The roles of sirtuins, AMPK, and PGC1α. Kidney Int. 2021, 99, 828–840. DOI:10.1016/j.kint.2020.09.037 [Google Scholar]
-
Bhargava P, Schnellmann RG. Mitochondrial energetics in the kidney. Nat. Rev. Nephrol. 2017, 13, 629–646. DOI:10.1038/nrneph.2017.107 [Google Scholar]
-
Han SH, Wu MY, Nam BY, Park JT, Yoo TH, Kang SW, et al. PGC-1α protects from notch-induced kidney fibrosis development. J. Am. Soc. Nephrol. 2017, 28, 3312–3322. DOI:10.1681/ASN.2017020130 [Google Scholar]
-
Chen S, Li J, Liang Y, Zhang M, Qiu Z, Liu S, et al. β-catenin-inhibited sumoylation modification of LKB1 and fatty acid metabolism is critical in renal fibrosis. Cell Death Dis. 2024, 15, 769. DOI:10.1038/s41419-024-07154-y [Google Scholar]
-
Chen YY, Chen XG, Zhang S. Druggability of lipid metabolism modulation against renal fibrosis. Acta Pharmacol. Sin. 2022, 43, 505–519. DOI:10.1038/s41401-021-00660-1 [Google Scholar]
-
Kang HM, Ahn SH, Choi P, Ko YA, Han SH, Chinga F, et al. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat. Med. 2015, 21, 37–46. DOI:10.1038/nm.3762 [Google Scholar]
-
Liu L, Ning X, Wei L, Zhou Y, Zhao L, Ma F, et al. Twist1 downregulation of PGC-1α decreases fatty acid oxidation in tubular epithelial cells, leading to kidney fibrosis. Theranostics 2022, 12, 3758–3775. DOI:10.7150/thno.71722 [Google Scholar]
-
Miguel V, Tituana J, Herrero JI, Herrero L, Serra D, Cuevas P, et al. Renal tubule CPT1A overexpression protects from kidney fibrosis by restoring mitochondrial homeostasis. J. Clin. Investig. 2021, 131, e140695. DOI:10.1172/JCI140695 [Google Scholar]
-
Hammoud S, Ivanova A, Osaki Y, Funk S, Yang H, Viquez O, et al. Tubular CPT1A deletion minimally affects aging and chronic kidney injury. JCI Insight 2024, 9, e171961. DOI:10.1172/jci.insight.171961 [Google Scholar]
-
Bang S, Choi SH, Jeong SM. Beyond bioenergetics: Emerging roles of mitochondrial fatty acid oxidation in stress response and aging. Cells 2025, 14, 1956. DOI:10.3390/cells14241956 [Google Scholar]
-
Verissimo T, Faivre A, Rinaldi A, Lindenmeyer M, Delitsikou V, Veyrat-Durebex C, et al. Decreased renal gluconeogenesis is a hallmark of chronic kidney disease. J. Am. Soc. Nephrol. 2022, 33, 810–827. DOI:10.1681/ASN.2021050680 [Google Scholar]
-
Wang Y, Li H, Jiang S, Fu D, Lu X, Lu M, et al. The glycolytic enzyme PFKFB3 drives kidney fibrosis through promoting histone lactylation-mediated NF-κB family activation. Kidney Int. 2024, 106, 226–240. DOI:10.1016/j.kint.2024.04.016 [Google Scholar]
-
Yuan Q, Lv Y, Ding H, Ke Q, Shi C, Luo J, et al. CPT1α maintains phenotype of tubules via mitochondrial respiration during kidney injury and repair. Cell Death Dis. 2021, 12, 792. DOI:10.1038/s41419-021-04085-w [Google Scholar]
-
Doke T, Susztak K. The multifaceted role of kidney tubule mitochondrial dysfunction in kidney disease development. Trends Cell Biol. 2022, 32, 841–853. DOI:10.1016/j.tcb.2022.03.012 [Google Scholar]
-
Li J, Lin Q, Shao X, Li S, Zhu X, Wu J, et al. HIF1α-BNIP3-mediated mitophagy protects against renal fibrosis by decreasing ros and inhibiting activation of the NLRP3 inflammasome. Cell Death Dis. 2023, 14, 200. DOI:10.1038/s41419-023-05587-5 [Google Scholar]
-
Kim Y, Li C, Gu C, Fang Y, Tycksen E, Puri A, et al. MANF stimulates autophagy and restores mitochondrial homeostasis to treat autosomal dominant tubulointerstitial kidney disease in mice. Nat. Commun. 2023, 14, 6493. DOI:10.1038/s41467-023-42154-0 [Google Scholar]
-
Yuan Q, Tang B, Zhu Y, Wan C, Xie Y, Xie Y, et al. PRDM16 acts as a therapeutic downstream target of TGF-β signaling in chronic kidney disease. JCI Insight 2025, 10, e191458. DOI:10.1172/jci.insight.191458 [Google Scholar]
-
Mitrofanova A, Merscher S, Fornoni A. Kidney lipid dysmetabolism and lipid droplet accumulation in chronic kidney disease. Nat. Rev. Nephrol. 2023, 19, 629–645. DOI:10.1038/s41581-023-00741-w [Google Scholar]
-
Chen Y, Yan Q, Lv M, Song K, Dai Y, Huang Y, et al. Involvement of FATP2-mediated tubular lipid metabolic reprogramming in renal fibrogenesis. Cell Death Dis. 2020, 11, 994. DOI:10.1038/s41419-020-03199-x [Google Scholar]
-
Li J, Yang Y, Li Q, Wei S, Zhou Y, Yu W, et al. STAT6 contributes to renal fibrosis by modulating PPARα-mediated tubular fatty acid oxidation. Cell Death Dis. 2022, 13, 66. DOI:10.1038/s41419-022-04515-3 [Google Scholar]
-
Rinaldi A, Lazareth H, Poindessous V, Nemazanyy I, Sampaio JL, Malpetti D, et al. Impaired fatty acid metabolism perpetuates lipotoxicity along the transition to chronic kidney injury. JCI Insight 2022, 7, e161783. DOI:10.1172/jci.insight.161783 [Google Scholar]
-
Mukhi D, Li L, Liu H, Doke T, Kolligundla LP, Ha E, et al. ACSS2 gene variants determine kidney disease risk by controlling de novo lipogenesis in kidney tubules. J. Clin. Investig. 2023, 134, e172963. DOI:10.1172/JCI172963 [Google Scholar]
-
Hou Y, Tan E, Shi H, Ren X, Wan X, Wu W, et al. Mitochondrial oxidative damage reprograms lipid metabolism of renal tubular epithelial cells in the diabetic kidney. Cell. Mol. Life Sci. 2024, 81, 23. DOI:10.1007/s00018-023-05078-y [Google Scholar]
-
Zhou S, Ling X, Zhu J, Liang Y, Feng Q, Xie C, et al. MAGL protects against renal fibrosis through inhibiting tubular cell lipotoxicity. Theranostics 2024, 14, 1583–1601. DOI:10.7150/thno.92848 [Google Scholar]
-
Liu X, Chen J, Gu S, Chen Y, Zhang R, Zang Q, et al. Prolonged glucagon exposure rewires lipid oxidation and drives diabetic kidney disease progression. Nat. Commun. 2025, 16, 8561. DOI:10.1038/s41467-025-63529-5 [Google Scholar]
-
Zhang L, Tian M, Zhang M, Li C, Wang X, Long Y, et al. Forkhead box protein K1 promotes chronic kidney disease by driving glycolysis in tubular epithelial cells. Adv. Sci. 2024, 11, e2405325. DOI:10.1002/advs.202405325 [Google Scholar]
-
Xu L, Chen S, Fan Q, Zhu Y, Mei H, Wang J, et al. N6-methyladenosine regulates metabolic remodeling in kidney aging through transcriptional regulator GLIS1. BMC Biol. 2024, 22, 302. DOI:10.1186/s12915-024-02100-y [Google Scholar]
-
Yang Y, Gong S, Zhou C, Xin W, Qin S, Yao M, et al. Rest contributes to renal fibrosis through inducing mitochondrial energy metabolism imbalance in tubular epithelial cells. Cell Commun. Signal. 2025, 23, 176. DOI:10.1186/s12964-025-02166-3 [Google Scholar]
-
Liu X, Zhang Y, Wang Y, Yang Y, Qiao Z, Zhan P, et al. Tubular mydgf slows progression of chronic kidney disease by maintaining mitochondrial homeostasis. Adv. Sci. 2025, 12, e2409756. DOI:10.1002/advs.202409756 [Google Scholar]
-
Ledru N, Wilson PC, Muto Y, Yoshimura Y, Wu H, Li D, et al. Predicting proximal tubule failed repair drivers through regularized regression analysis of single cell multiomic sequencing. Nat. Commun. 2024, 15, 1291. DOI:10.1038/s41467-024-45706-0 [Google Scholar]
-
Niu H, Ren X, Tan E, Wan X, Wang Y, Shi H, et al. CD36 deletion ameliorates diabetic kidney disease by restoring fatty acid oxidation and improving mitochondrial function. Ren. Fail. 2023, 45, 2292753. DOI:10.1080/0886022X.2023.2292753 [Google Scholar]
-
Patera F, Gatticchi L, Cellini B, Chiasserini D, Reboldi G. Kidney fibrosis and oxidative stress: From molecular pathways to new pharmacological opportunities. Biomolecules 2024, 14, 137. DOI:10.3390/biom14010137 [Google Scholar]
-
Tang PM, Nikolic-Paterson DJ, Lan HY. Macrophages: Versatile players in renal inflammation and fibrosis. Nat. Rev. Nephrol. 2019, 15, 144–158. DOI:10.1038/s41581-019-0110-2 [Google Scholar]
-
Liu Y. Renal fibrosis: New insights into the pathogenesis and therapeutics. Kidney Int. 2006, 69, 213–217. DOI:10.1038/sj.ki.5000054 [Google Scholar]
-
Stenvinkel P, Chertow GM, Devarajan P, Levin A, Andreoli SP, Bangalore S, et al. Chronic inflammation in chronic kidney disease progression: Role of NRF2. Kidney Int. Rep. 2021, 6, 1775–1787. DOI:10.1016/j.ekir.2021.04.023 [Google Scholar]
-
Sanz AB, Sanchez-Nino MD, Ramos AM, Ortiz A. Regulated cell death pathways in kidney disease. Nat. Rev. Nephrol. 2023, 19, 281–299. DOI:10.1038/s41581-023-00694-0 [Google Scholar]
-
Carney EF. Ferroptotic stress promotes the AKI to CKD transition. Nat. Rev. Nephrol. 2021, 17, 633. DOI:10.1038/s41581-021-00482-8 [Google Scholar]
-
Kim S, Kang SW, Joo J, Han SH, Shin H, Nam BY, et al. Characterization of ferroptosis in kidney tubular cell death under diabetic conditions. Cell Death Dis. 2021, 12, 160. DOI:10.1038/s41419-021-03452-x [Google Scholar]
-
Zhang B, Chen X, Ru F, Gan Y, Li B, Xia W, et al. Liproxstatin-1 attenuates unilateral ureteral obstruction-induced renal fibrosis by inhibiting renal tubular epithelial cells ferroptosis. Cell Death Dis. 2021, 12, 843. DOI:10.1038/s41419-021-04137-1 [Google Scholar]
-
Gan T, Wang Q, Song Y, Shao M, Zhao Y, Guo F, et al. Canagliflozin improves fatty acid oxidation and ferroptosis of renal tubular epithelial cells via FOXA1-CPT1A axis in diabetic kidney disease. Mol. Cell Endocrinol. 2024, 582, 112139. DOI:10.1016/j.mce.2023.112139 [Google Scholar]
-
Dai Y, Chen Y, Mo D, Jin R, Huang Y, Zhang L, et al. Inhibition of ACSL4 ameliorates tubular ferroptotic cell death and protects against fibrotic kidney disease. Commun. Biol. 2023, 6, 907. DOI:10.1038/s42003-023-05272-5 [Google Scholar]
-
Abedini A, Levinsohn J, Klotzer KA, Dumoulin B, Ma Z, Frederick J, et al. Single-cell multi-omic and spatial profiling of human kidneys implicates the fibrotic microenvironment in kidney disease progression. Nat. Genet. 2024, 56, 1712–1724. DOI:10.1038/s41588-024-01802-x [Google Scholar]
-
Aggarwal S, Wang Z, Rincon Fernandez Pacheco D, Rinaldi A, Rajewski A, Callemeyn J, et al. SOX9 switch links regeneration to fibrosis at the single-cell level in mammalian kidneys. Science 2024, 383, eadd6371. DOI:10.1126/science.add6371 [Google Scholar]
-
Doke T, Abedini A, Aldridge DL, Yang YW, Park J, Hernandez CM, et al. Single-cell analysis identifies the interaction of altered renal tubules with basophils orchestrating kidney fibrosis. Nat. Immunol. 2022, 23, 947–959. DOI:10.1038/s41590-022-01200-7 [Google Scholar]
-
Melchinger I, Guo K, Li X, Guo J, Cantley LG, Xu L. VCAM-1 mediates proximal tubule-immune cell cross talk in failed tubule recovery during AKI-to-CKD transition. Am. J. Physiol. Renal Physiol. 2024, 327, F610–F622. DOI:10.1152/ajprenal.00076.2024 [Google Scholar]
-
Liu X, Liu Z, Wang C, Miao J, Zhou S, Ren Q, et al. Kidney tubular epithelial cells control interstitial fibroblast fate by releasing tnfaip8-encapsulated exosomes. Cell Death Dis. 2023, 14, 672. DOI:10.1038/s41419-023-06209-w [Google Scholar]
-
Humphreys BD, Lin SL, Kobayashi A, Hudson TE, Nowlin BT, Bonventre JV, et al. Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am. J. Pathol. 2010, 176, 85–97. DOI:10.2353/ajpath.2010.090517 [Google Scholar]
-
Kuppe C, Ibrahim MM, Kranz J, Zhang X, Ziegler S, Perales-Paton J, et al. Decoding myofibroblast origins in human kidney fibrosis. Nature 2021, 589, 281–286. DOI:10.1038/s41586-020-2941-1 [Google Scholar]
-
Lovisa S, LeBleu VS, Tampe B, Sugimoto H, Vadnagara K, Carstens JL, et al. Epithelial-to-mesenchymal transition induces cell cycle arrest and parenchymal damage in renal fibrosis. Nat. Med. 2015, 21, 998–1009. DOI:10.1038/nm.3902 [Google Scholar]
-
Grande MT, Sanchez-Laorden B, Lopez-Blau C, De Frutos CA, Boutet A, Arevalo M, et al. Snail1-induced partial epithelial-to-mesenchymal transition drives renal fibrosis in mice and can be targeted to reverse established disease. Nat. Med. 2015, 21, 989–997. DOI:10.1038/nm.3901 [Google Scholar]
-
Li S, Mariappan N, Megyesi J, Shank B, Kannan K, Theus S, et al. Proximal tubule PPARα attenuates renal fibrosis and inflammation caused by unilateral ureteral obstruction. Am. J. Physiol. Renal Physiol. 2013, 305, F618–F627. DOI:10.1152/ajprenal.00309.2013 [Google Scholar]
-
Nam BY, Jhee JH, Park J, Kim S, Kim G, Park JT, et al. PGC-1α inhibits the NLRP3 inflammasome via preserving mitochondrial viability to protect kidney fibrosis. Cell Death Dis. 2022, 13, 31. DOI:10.1038/s41419-021-04480-3 [Google Scholar]
-
Liu B, Zhang Y, Wang Y, Meng Q, Zhang D, Yang H, et al. Pharmacological targeting of AMPK to restore glucose and fatty acid metabolism homeostasis attenuates transplanted kidney fibrosis. Biochim. Biophys. Acta Mol. Basis Dis. 2025, 1871, 167510. DOI:10.1016/j.bbadis.2024.167510 [Google Scholar]
-
Li X, Zhuge Z, Carvalho L, Braga VA, Lucena RB, Li S, et al. Inorganic nitrate and nitrite ameliorate kidney fibrosis by restoring lipid metabolism via dual regulation of amp-activated protein kinase and the AKT-PGC1α pathway. Redox Biol. 2022, 51, 102266. DOI:10.1016/j.redox.2022.102266 [Google Scholar]
-
Song X, Du Z, Yao Z, Tang X, Zhang M. Rhein improves renal fibrosis by restoring CPT1A-mediated fatty acid oxidation through SirT1/STAT3/twist1 pathway. Molecules 2022, 27, 2344. DOI:10.3390/molecules27072344 [Google Scholar]
-
Jiang M, Fan J, Qu X, Li S, Nilsson SK, Sun YBY, et al. Combined blockade of Smad3 and JNK pathways ameliorates progressive fibrosis in folic acid nephropathy. Front. Pharmacol. 2019, 10, 880. DOI:10.3389/fphar.2019.00880 [Google Scholar]
-
Li L, Emmett N, Mann D, Zhao X. Fenofibrate attenuates tubulointerstitial fibrosis and inflammation through suppression of nuclear factor-κB and transforming growth factor-β1/Smad3 in diabetic nephropathy. Exp. Biol. Med. 2010, 235, 383–391. DOI:10.1258/ebm.2009.009218 [Google Scholar]
-
Zhu P, Ma H, Cui S, Zhou X, Xu W, Yu J, et al. ZLN005 alleviates in vivo and in vitro renal fibrosis via PGC-1α-mediated mitochondrial homeostasis. Pharmaceuticals 2022, 15, 434. DOI:10.3390/ph15040434 [Google Scholar]
-
Xiong L, He T, Liu C, Qin S, Xiao T, Xin W, et al. IL-37 ameliorates renal fibrosis by restoring CPT1A-mediated fatty acid oxidation in diabetic kidney disease. Kidney Dis. 2023, 9, 104–117. DOI:10.1159/000529460 [Google Scholar]
-
Wu Q, Zhao Y, Huang F. Metformin alleviates renal tubular injury in diabetic kidney disease by activating mitophagy and inhibiting ferroptosis via HIF-1α/MIOX axis. J. Pharm. Anal. 2025, 15, 101284. DOI:10.1016/j.jpha.2025.101284 [Google Scholar]
-
Aranda-Rivera AK, Amador-Martinez I, Aparicio-Trejo OE, Leon-Contreras JC, Hernandez-Pando R, Saavedra E, et al. Sulforaphane restores mitochondrial β-oxidation and reduces renal lipid accumulation in a model of releasing unilateral ureteral obstruction. Antioxidants 2025, 14, 288. DOI:10.3390/antiox14030288 [Google Scholar]
-
Liu C, Wang X, Wang X, Zhang Y, Min W, Yu P, et al. A new LKB1 activator, piericidin analogue S14, retards renal fibrosis through promoting autophagy and mitochondrial homeostasis in renal tubular epithelial cells. Theranostics 2022, 12, 7158–7179. DOI:10.7150/thno.78376 [Google Scholar]
-
Wang J, Wang L, Feng X, Xu Y, Zhou L, Wang C, et al. Astragaloside IV attenuates fatty acid-induced renal tubular injury in diabetic kidney disease by inhibiting fatty acid transport protein-2. Phytomedicine 2024, 134, 155991. DOI:10.1016/j.phymed.2024.155991 [Google Scholar]
-
Li Y, Zheng F, Zhong S, Zhao K, Liao H, Liang J, et al. Protecting against ferroptosis in hyperuricemic nephropathy: The potential of ferrostatin-1 and its inhibitory effect on URAT1. Eur. J. Pharmacol. 2024, 971, 176528. DOI:10.1016/j.ejphar.2024.176528 [Google Scholar]
-
Peng L, Wang C, Yu S, Li Q, Wu G, Lai W, et al. Dysregulated lipid metabolism is associated with kidney allograft fibrosis. Lipids Health Dis. 2024, 23, 37. DOI:10.1186/s12944-024-02021-3 [Google Scholar]