1. Introduction
Chronic Obstructive Pulmonary Disease (COPD) ranks as the third leading cause of morbidity and mortality globally according to the World Health Organization [
1]. The disease primarily manifests as persistent dyspnea that progresses to disability, complicated by acute, life-threatening exacerbations, which are often triggered by infections or tobacco use, particularly in the elderly. Following recent viral pandemics, a significant proportion of survivors developed chronic respiratory syndromes due to impaired lung homeostasis, ineffective repair, or incomplete regeneration [
2,
3,
4].
COPD progression involves robust immune cell infiltration, epithelial/endothelial-mesenchymal transition (EMT/EndMT), and fibroblast-to-myofibroblast transition. These transformations drive airway/vascular remodeling, alveolar bronchiolization, and aberrant alveolar wall changes. Close epithelial-mesenchymal proximity enables critical signaling for stem cell niches, maintaining tissue homeostasis and directing injury repair through mesenchymal-derived signals [
5].
Epithelial and mesenchymal cells constitute the primary lung airways and alveolar structures along with the connective tissues as well as the extracellular matrix (ECM) in maintaining the homeostasis environment. The epithelium lines the internal surfaces of the lungs, while the stroma comprises the connective tissue and vasculature. Disruption of this homeostatic balance by disease can lead to the exhaustion of airway epithelial stem cells and mesenchymal remodeling, contributing to chronic pulmonary diseases like COPD.
Epithelial heterogeneity and lineage functions are well-characterized in lung development and disease. Proximal airways contain distinct cell types, including myoepithelial cells, basal cells, club cells, ciliated cells, goblet cells, and tuft cells [
6,
7,
8]. Basal cells, marked by tumor protein 63 (TP63) and keratin 5 (KRT5), act as stem cells in the trachea, capable of migrating, proliferating, and differentiating into ciliated and club cells to restore lung function after injury [
9,
10,
11]. In the distal regions of the mouse lungs, the epithelial lining is simpler, composed primarily of club and ciliated cells [
10,
12,
13]. Club cells, marked by secretoglobin family 1A member 1 (SCGB1A1, also known as CC10 or CCSP) and cytochrome P450 family 2 subfamily f polypeptide 2 (CYP2F2), exhibit regenerative capacity in response to injury, such as naphthalene-induced damage [
12]. Bronchioalveolar stem cells (BASCs) at the terminal bronchioles and alveolar junctions are marked by both Scgb1a1and surfactant protein C (SP-C) that regenerate bronchiolar/alveolar epithelia [
12,
13,
14,
15,
16,
17]. The alveolar units, responsible for gas exchange, are composed of alveolar Type 2 cells (AT2s) and AT1 cells surrounded by heterogeneous mesenchymal cells. AT2 cells are recognized as potent stem cells in response to injury. Several subpopulations of AT2 cells have been identified, including Axin2
Pos bipotent, Sca1
Pos, Krt8
Pos transitional, Cd44
High, and Pd-l1
HighSp-C
Low cells, which contribute to alveolar repair and regeneration [
18,
19,
20,
21,
22,
23,
24,
25,
26,
27,
28,
29,
30,
31].
Mesenchymal niche studies in COPD are comparatively limited. Recent advances have revealed the lineage hierarchies of mesenchymal cells in lung diseases. Zepp et al., identified a population of platelet derived growth factor receptor alpha positive (Pdgfrα
Pos) mesenchymal alveolar niche cells (MANCs), which are WNT-responsive and critical for maintaining AT2 cells in the mouse distal lung [
32]. Pdgfrα also serves as a marker for lipofibroblasts and supports AT2 function [
6,
33]. Furthermore, recent studies identified another distinct population of Pdgfra
Pos cells, emerging from the proximal airway niche following naphthalene-induced club cell depletion, termed Repair Supportive Mesenchymal Cells (RSMCs) [
30,
31]. Derived from the
Acta2Pos lineage, these cells exhibit enhanced capacity to support club-cell regeneration in vitro and express high levels of Fibroblast growth factor 10 (Fgf10), a key growth factor for branching morphogenesis and lung repair in response to bleomycin-induced injury in mice, human idiopathic pulmonary fibrosis (IPF), and COPD [
34,
35,
36,
37,
38].
Beyond these populations, Axin2
Pos mesenchymal cells contribute to de novo airway smooth muscle cells (ASMC) formation after injury, ASMCs expressing leucine-rich repeat-containing G-protein coupled receptor 6 (Lgr6) promote epithelial repair through a Wnt-Fgf10-signaling axis [
39,
40]. Other notable mesenchymal populations include secreted frizzled related protein 1 positive (Sfrp1
Pos) transitional fibroblasts and collagen triple helix repeat containing 1 positive (Cthrc1
Pos) myofibroblasts, recognized as the primary collagen producers during pulmonary fibrosis [
41,
42,
43]. The spatial distribution between epithelial and mesenchymal cells facilitates direct cell-cell contact and intercellular communications mediated by secreted components such as growth factors, exosomes. Key signaling pathways, including TGF-β, FGF, Hippo, PPARγ, WNT, and SHH, tightly regulate these communications, ensuring tissue homeostasis and promoting repair.
While the intricate epithelial and mesenchymal interplay has been extensively documented as a central driver in the pathogenesis of pulmonary fibrosis, parallel investigations within the COPD context remain remarkably scarce. Although IPF and COPD exhibit distinct core pathological features, they share fundamental dysregulations in key biological processes governed by epithelial-mesenchymal crosstalk. These shared pathological mechanisms include aberrant epithelial-mesenchymal signaling, dysregulated cellular proliferation and differentiation, and impaired stem cell-mediated regeneration [
44]. In this review, we aim to elucidate alterations in epithelial and mesenchymal cell lineages and their interactions that contribute to COPD phenotypes. By synthesizing the current body of knowledge on these cellular transformations, interactions, and regulatory mechanisms, we seek to illuminate potential future research avenues and identify novel COPD therapeutic targets.
2. Epithelial Cell Response in COPD
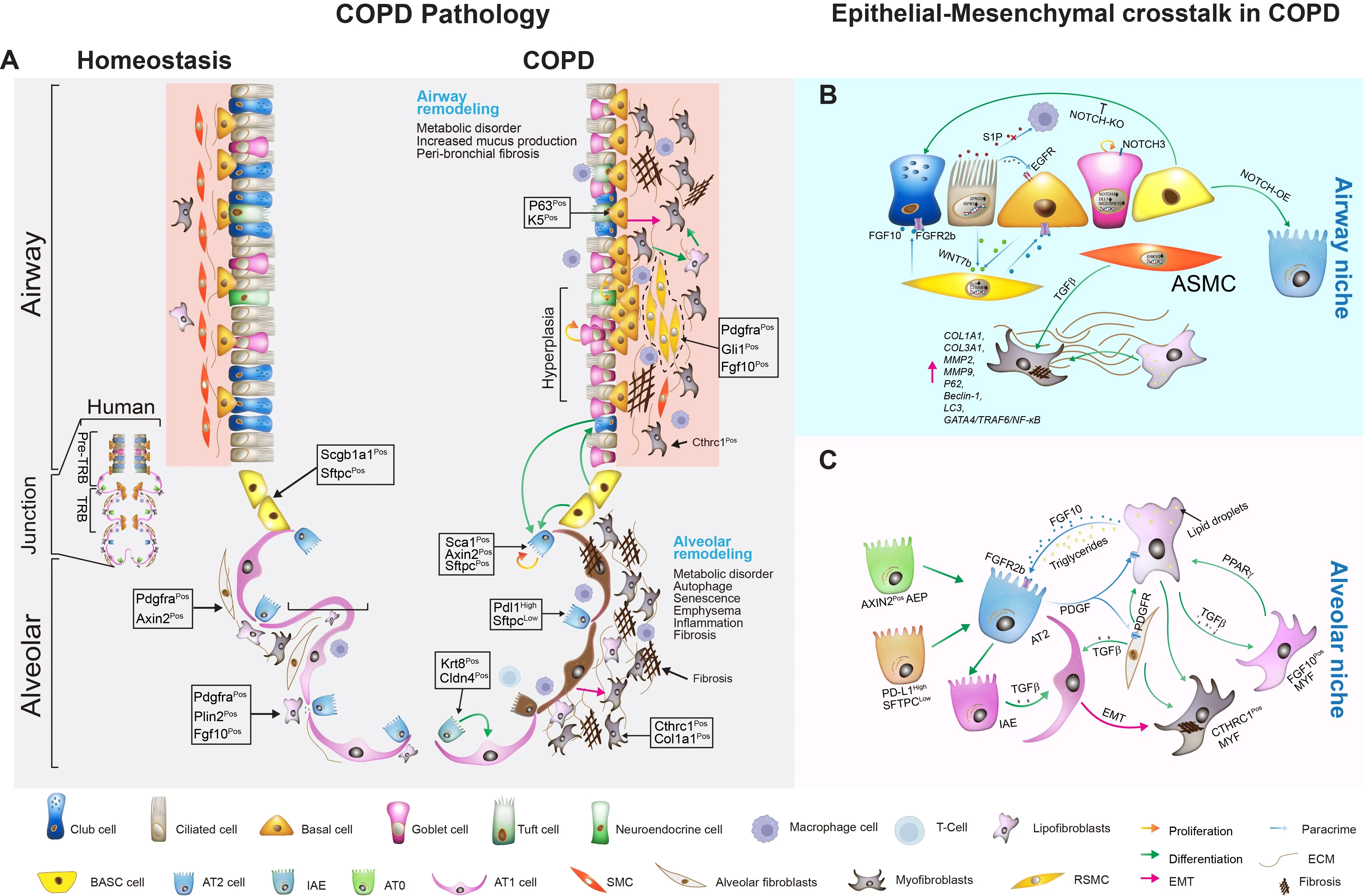
Disruptions in epithelial cell function are a primary cause of COPD progression, driven by processes such as cellular senescence, apoptosis, ferroptosis, and inflammation [
45,
46,
47,
48]. These pathological events lead to epithelial cell damage, exhaustion, and impaired attempts at regeneration. Chronic exposure to cigarette smoke, the leading risk factor for COPD, along with repeated respiratory infections, induces significant alterations in epithelium, including the loss of ciliated cells [
49,
50,
51], an increase in goblet cell numbers, shortened cilia and reduced ciliary beat frequency, all of which compromise the airway’s ability to clear mucus and pathogens [
52,
53,
54]. Epithelial injury triggers a persistent immune response, driving alveolar destruction (emphysema) and airway remodeling, often resulting in peribronchial fibrosis and an increase in airway smooth muscle mass [
50,
55]. These changes also result in excess mucus hyperproduction, a key feature of chronic bronchitis (
A) [
56,
57].
. Cellular dynamics in lung homeostasis and COPD. (<strong>A</strong>) The left panel illustrates key cell types and their spatial distribution in the airway and alveolar regions, including human terminal respiratory bronchioles (TRB) under healthy conditions (Homeostasis), club cells, ciliated cells, basal cells, goblet cells, tuft cells, neuroendocrine cells, BASCs (mouse), AT0 (human), AT2, and AT1 cells. Specific cell markers, adjacent fibroblasts, and the extracellular matrix (ECM) are also depicted. The right panel shows changes in cellular states and marker expression during COPD, highlighting airway remodeling, hyperplasia of goblet and basal cells, flattened ciliated cells, and a fibrotic phenotype. These cellular and signaling alterations are associated with key COPD symptoms and pathological features, including metabolic dysregulation, emphysema, inflammation, and fibrosis. (<strong>B</strong>) The dynamic interactions between ASMCs, RSMCs, and key signaling pathways, including the FGF10-FGFR2b axis, WNT signaling, EGFR activation, and NOTCH signaling in “Airway Niche”. (<strong>C</strong>) The “Alveolar Niche,” focusing on the interactions between AT2 cells and adjacent fibroblasts via FGF10-FGFR2b and PDGFRα signaling. It emphasizes the role of PPARγ-regulated lipid metabolism, showcasing the importance of the lipofibroblast niche in supporting AT2 cells. The involvement of TGF-β signaling in EMT is also shown, linking to fibrosis with downstream effects mediated by Cthrc1<sup>Pos</sup> MYFs.
2.1.1. Ciliated Cells
Ciliated epithelial cells line the airway and maintain effective mucociliary clearance by moving mucus and trapping pathogens out of the respiratory tract [
58]. In COPD, smoking and aging critically impair the structure and function of these cells, leading to inefficient clearance of mucus [
59,
60]. One of the molecular pathways affected involves Spinster 2 (SPNS2), which regulates sphingosine-1-phosphate (S1P) secretion. Reduced SPNS2 levels in ciliated cells impair macrophage phagocytosis, further compromising lung defense mechanisms [
61]. Additionally, receptor-interacting protein kinase 1 (RIPK1) levels are notably elevated in the ciliated cells of COPD patients. RIPK1 contributes to airway inflammation and remodeling, and blocking RIPK1 has been shown to reduce emphysema and lung function decline (
A) [
62].
2.1.2. Club Cells
Club cells, predominantly found in bronchioles, are important for airway repair, immunomodulation, anti-inflammatory functions, and detoxification [
63]. In COPD, there is a notable reduction in club cells, which correlates with the disease’s severity [
64]. These cells produce SCGB1A1, which modulates immune responses and inhibits harmful enzymes, thereby reducing inflammation and promoting repair mechanisms [
65,
66,
67,
68]. When club cells are ablated in animal models, there is a loss of epithelial regeneration capacity, resulting in squamous metaplasia and fibrosis, changes also observed in the airways of COPD patients [
69]. Notably, CCSP therapy has shown promise in reducing inflammation and supporting lung repair, highlighting its potential as a therapeutic target in COPD [
70,
71,
72]. Furthermore, club cells also act as facultative stem cells, with the ability to regenerate alveolar epithelial cells after injury. Recent studies have demonstrated that club cells can differentiate into AT2 cells, contributing to alveolar repair during emphysema (
A) [
73,
74]. However, club cell-derived AT2 cells exhibit defects in generating alveolar structures in COPD compared to healthy controls, suggesting an impaired regenerative response in the disease state [
74].
2.1.3. Basal Cells
Basal cells serve as stem cells for the airway epithelium, playing a key role in maintaining and regenerating the epithelial lining, which is heavily impacted by smoking [
75]. These cells are responsible for maintaining the stem cell pool through self-renewal and for giving rise to differentiated progenitors that form ciliated and secretory cells. In COPD, basal cell hyperplasia is observed, along with mucous hyperplasia, contributing to chronic airway remodeling [
76]. Basal cells reside in the basal layer of the epithelium, just above the basement membrane, where they can interact with underlying stromal and immune cells. This interaction is likely crucial for the development of airway fibrosis and inflammation [
77,
78]. Basal cells express markers such as KRT5, KRT13, and TP63, which are critical for their differentiation and the formation of the normal airway epithelium [
79,
80,
81]. While basal cells are typically involved in repair processes following injury, their regenerative capacity is diminished, leading to persistent remodeling of the airway in COPD [
79,
82,
83]. Additionally, basal cells express Epidermal Growth Factor Receptor (EGFR), which is activated by EGF produced by ciliated cells in response to cigarette smoke, further driving airway remodeling (
A) [
51,
84].
2.1.4. Goblet Cells
Goblet cell hyperplasia is a prominent characteristic of COPD that contributing to airway obstruction [
85]. Increased mucin production and hypersecretion of mucus are driven by goblet cell proliferation through the NOTCH signaling pathway [
85]. Goblet cells not only act as progenitors to ciliated cells but also play a key role in rapidly escalating mucus production in response to airway insults [
86]. Overproduction of mucus in chronic bronchitis aggravates airflow limitation by blocking small airways, leading to epithelial remodeling and airway collapse [
87]. Despite their clinical significance, the precise mechanisms behind goblet cell hyperplasia in COPD remain poorly understood, and therapeutic options are limited. Recent research on pulsed focused ultrasound offers potential as a novel strategy for selectively reducing goblet cell hyperactivity, providing a promising avenue for COPD treatment [
88].
2.2. Additional Airway Epithelial Cells
In addition to the well-characterized epithelial cell types described above, several less-studied cell types in the airway epithelium—including ionocytes, pulmonary neuroendocrine cells (PNECs), and tuft cells—may significantly contribute to the COPD pathogenesis.
Ionocytes are recently identified epithelial cells that express high levels of CFTR (cystic fibrosis transmembrane conductance regulator) chloride channels, playing a crucial role in maintaining airway surface hydration. Their dysfunction, especially when influenced by cigarette smoke exposure, causes mucus dehydration and impaired mucociliary clearance, exacerbating symptoms in COPD patients [
89]. PNECs are rare epithelial cells that secrete neuropeptides to modulate airway smooth muscle tone and immune responses. In the context of COPD, PNEC hyperplasia and alterations in neuropeptide secretion may exacerbate airway remodeling and chronic inflammation, contributing to disease progression [
90]. Tuft cells, also known as brush cells in the airway, have been found to play roles in innate immune regulation and airway epithelial repair. They release signaling molecules that can modulate inflammation and contribute to the dysregulation of immune responses within the airways of COPD patients [
91].
Therefore, airway epithelial cells are critical in the pathogenesis of COPD, contributing to chronic inflammation, mucus hypersecretion, airway remodeling, and alveolar destruction. A deeper understanding of these cellular responses is crucial for developing targeted therapies that can mitigate disease progression and improve the quality of life for COPD patients.
2.3. Alveolar Epithelial Cells in COPD
Bronchiolization of alveolar regions, a key feature of emphysema, is largely attributed to alveolar epithelial hyperplasia during the repair process following inflammatory damage in COPD. The alveolar epithelium consists mainly of AT1 and AT2 cells. AT2 cells, functioning as resident stem cells, are crucial for alveolar regeneration after injury [
33].
In healthy lungs, AT2 cells tend to proliferate and differentiate into AT1 cells responsible for gas exchange [
92]. However, in COPD Lungs, proportion of AT2 cells lose their ability to regenerate, resulting in impaired alveolar repair [
93]. The interferon-gamma (IFN-γ) and LHX9 signaling axis has been implicated in maintaining a subset of AT2 cells that retain stem cell functionality and resistance to apoptosis under chronic cigarette smoke exposure [
93,
94,
95]. This preservation is also thought to be supported by fatty acid oxidation mechanisms. Additionally, exposure to PM2.5 particles disrupts the differentiation of AT2 cells into AT1 cells, as evidenced by reduced claudin-4 (CLDN4) expression in AT2 cells following exposure [
96]. Recent scRNA-seq analyses of lung tissue from COPD patients have identified a distinct subpopulation of AT2 cells with altered metabolic processes, further contributing to the pathogenesis of emphysema [
97]. Moreover, abnormal T cell proliferation in COPD may inhibit the renewal capabilities of AT2 cells, exacerbating the condition [
98]. In contrast to the mouse lung, the human lung features a distinct airway architecture, including respiratory bronchioles—a transitional region absent in mice. Within these structures lies a unique secretory cell population, respiratory airway secretory (RAS) cells, which serve as unidirectional progenitors for AT2 cells. NOTCH and WNT signaling modulate this process. Notably, RAS cells exhibit transcriptional alterations associated with smoking induced COPD, leading to aberrant AT2 cell states. This human-specific progenitor population plays a vital role in maintaining alveolar homeostasis and is implicated in chronic lung pathology [
99].
AT1 cells tightly adhere to pulmonary venous capillary endothelial cells, covering approximately 95% of the alveolar surface, and are essential for gas exchange. Damage to AT1 cells results in alveolar structure remodeling [
100]. Previous studies in a transgenic mouse model have identified two AT1 cell subpopulations: terminally differentiated Hopx
PosIgfbp2
Pos cells and plastic Hopx
PosIgfbp2
Neg cells. Igfbp2
Neg cells demonstrate
in vitro transdifferentiation potential into AT2 cells and organoid formation (indicating an intermediate differentiation state), Igfbp2 marks terminal AT1 commitment [
101]. However, dual recombinase-mediated lineage tracing studies—which enable specific AT1 labeling—show that AT1 cells do not generate AT2 cells during bleomycin-induced injury
in vivo [
73]. Whether AT1 cells, which align with structurally remodeled capillary endothelia, undergo damage and remodeling remains unknown—a key area for future investigation [
102]. Therefore, strategies targeting AT1 preservation or functional restoration via AT2-to-AT1 differentiation may also mitigate emphysematous destruction in COPD.
3. Activation and Therapeutic Applications of Mesenchymal Cells in COPD
3.1. Mesenchymal Cell Activation in COPD
Beyond the important role of dysfunction in lung epithelia, COPD additionally features progressive remodeling of both airway and alveolar structures, primarily driven by fibroblasts, particularly myofibroblasts. Although mesenchymal niches are known to play critical roles in fibrotic epithelial repair and regeneration, their response, regulation, and signaling mechanisms in COPD are still poorly understood. COPD primarily affects the small conducting airways, where thickening of the airway walls and peribronchiolar fibrosis cause narrowing [
103,
104]. This fibrosis is partly driven by senescent fibroblasts that secrete increased levels of collagen types 1A1 (COL1A1) and 3A1 (COL3A2) and upregulate matrix metalloproteinases (MMP2, MMP9), promoting a profibrotic environment and inducing the differentiation of bronchial smooth muscle cells into myofibroblasts [
105,
106].
ASMCs in COPD exhibit metabolic disturbances, including the accumulation of lactate, glutamine, fatty acids, and amino acids. TGF-β stimulation restores fatty acid oxidation, enhances ribose-5-phosphate production, and promotes nucleotide biosynthesis [
107]. This metabolic reprogramming supports ASMC growth and is linked with altered redox balance and reduced mitochondrial oxidative stress. Inhibiting glycolysis and glutamine depletion reduces ASMC proliferation in COPD [
94]. ASMCs also show increased fibronectin production and upregulation of autophagy markers, potentially accelerating cellular senescence [
108,
109].
Environmental exposures such as CSE (Cigarette smoking exposure) and particulate matter (PM2.5) further exacerbate mesenchymal cell dysfunction. CSE exposure induces pro-proliferative and profibrotic characteristics in fibroblasts through NF-κB activation [
110]. CSE also increases oxidative and ER stress in human lung fibroblasts, promoting their differentiation into myofibroblasts and impairing their ability to support epithelial progenitor function, while activation of the WNT/β-catenin signaling pathway partially restores this impaired function [
111]. PM2.5 exposure triggers a senescence-associated secretory phenotype in ASMCs, enhancing collagen-I and α-SMA synthesis via the GATA4/TRAF6/NF-κB signaling pathway [
112].
FGF10 is crucial for maintaining the alveolar AT2 stem cell niche [
99]. Studies have demonstrated that FGF10 expression is reduced in COPD lungs, disrupting alveolar homeostasis. Remarkably, overexpression of
Fgf10 has been shown to reverse established emphysema in mice, underscoring its therapeutic potential [
36]. Given that Fgf10 is primarily produced by alveolar fibroblasts, an imbalance between fibroblasts and epithelial cells likely contributes to the alveolar remodeling observed in COPD. These intricate biological processes illustrate how mesenchymal cell dysfunction drives the structural changes that exacerbate the obstructive nature of the disease. Although research into FGF10-targeted therapies is still in its early stages, the advancements in cell-based precision therapies targeting this pathway are promising.
3.2. Mesenchymal Stem Cells as Therapies Against COPD
Mesenchymal stem cells (MSCs) have shown significant promise as cell-based therapies in regenerative medicine due to their multipotent differentiation capacity, self-renewal abilities, immunoregulatory properties, and paracrine effects. MSCs have been extensively studied in respiratory diseases such as COPD, asthma, and idiopathic pulmonary fibrosis, demonstrating their potential to regenerate lung tissue [
113,
114,
115,
116,
117,
118,
119,
120,
121]. Transplantation of human umbilical cord-derived MSCs (hUC-MSCs) or their extracellular vesicles (EVs) have shown efficacy in reducing peribronchial and perivascular inflammation, alveolar septal thickening, and goblet cell hyperplasia in COPD rat models. These interventions also reduce alveolar septal loss and downregulate NF-κB subunit P65 levels in affected tissues [
117]. Transplantation of MSCs also alleviates COPD by regulating oxidative stress-related pathways [
122].
Lung resident MSCs (LR-MSCs) are found in comparable numbers in smokers and never-smokers, indicating that the MSC reservoir in COPD patients remains intact [
123]. However, MSCs from current smokers demonstrated diminished capacity to inhibit CD8
Pos T-cell proliferation. This impairment was accentuated by
in vitro exposure to CSE, which reduced the T-cell immunomodulatory capabilities of LR-MSCs [
123]. These results suggest that tobacco smoke-induced oxidative stress can compromise the immunomodulatory and homeostatic functions of LR-MSCs. Furthermore, COPD MSCs show decreased production of Fgf10, essential for effective alveolar repair, and this process can be rescued by activating FGF10/FGFR2b signaling [
36,
124].
During the COVID-19 pandemic, systemic administration of allogeneic MSCs was investigated as a treatment for virus-associated pneumonia. MSCs' anti-inflammatory properties and ability to remodel the extracellular matrix offer a potential strategy to prevent pulmonary fibrosis. Despite promising results in clinical trials, outcomes have been mixed, potentially due to persistent bronchial inflammation, which may attenuate MSC efficacy [
125,
126]. Despite challenges, MSCs offer a promising approach for addressing chronic inflammation and remodeling in COPD ().
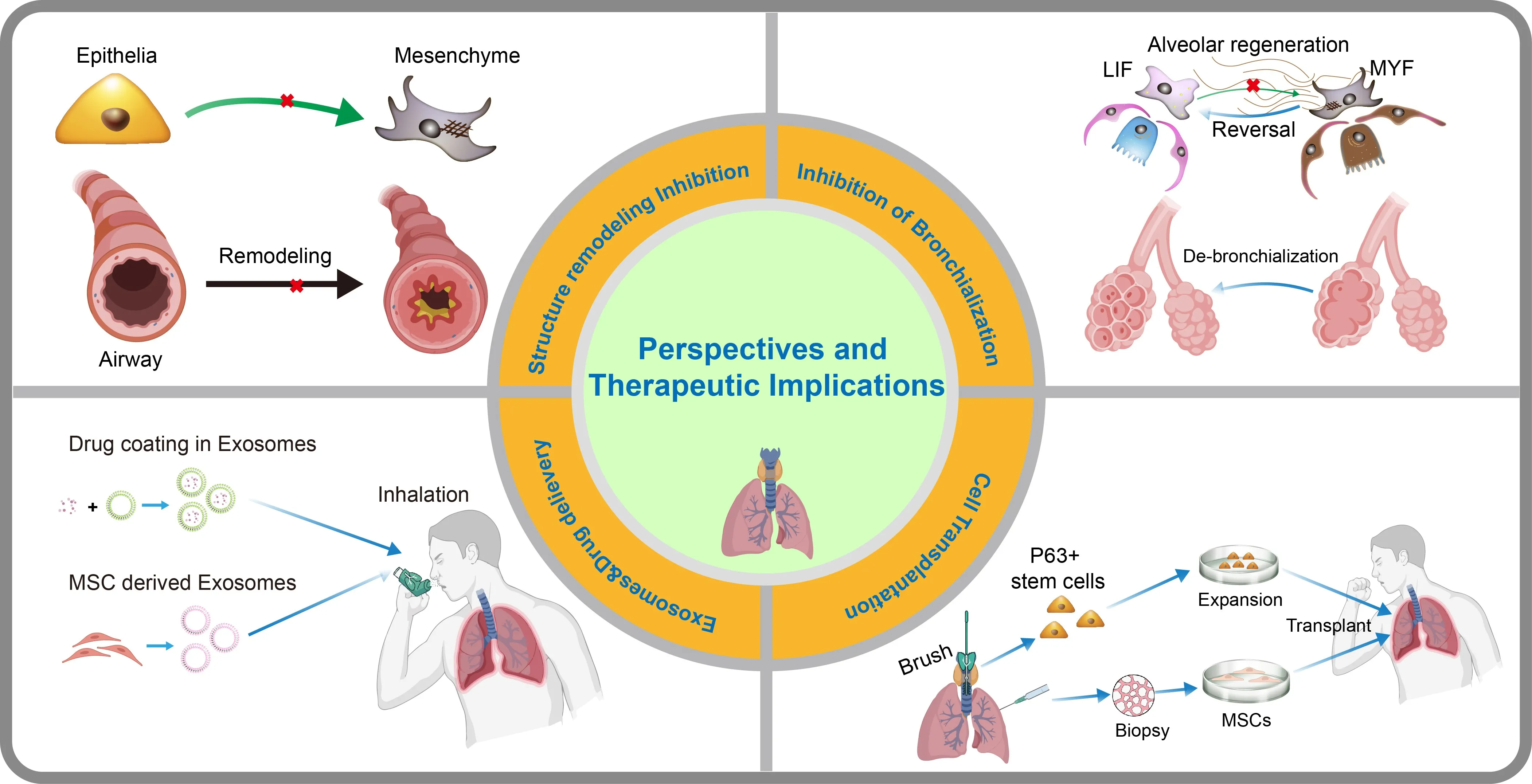
. Perspectives and therapeutic implications of airway and alveolar regeneration in COPD. Based on current insight, several perspectives and therapeutic implications for COPD treatment have been summarized. (<strong>Top Left</strong>): Inhibition of EMT and airway remodeling is critical for preventing pathological transition of epithelial cells into mesenchymal phenotypes, thereby maintaining airway integrity. (<strong>Top Right</strong>): Inhibition of bronchiolization and promotion of de-bronchiolization within the alveolar niche support alveolar regeneration through reversal of pathological changes. (<strong>Bottom Left</strong>): Exosome/Exosome-delivered drugs can be administered via inhalation, enabling targeted delivery to lung tissues for regenerative therapy. (<strong>Bottom Right</strong>): Cell-based therapies involve isolation (biopsy), <em>in vitro</em> expansion, and transplantation of lung epithelial cells to restore respiratory function. This integrative approach aims to advance therapeutic strategies for lung diseases by targeting multiple pathways involved in COPD.
In conclusion, mesenchymal cells, including fibroblasts and MSCs, are critical in the pathogenesis and potential treatment of COPD, despite the fact that fibroblasts drive structural remodeling. Further research is needed to fully understand mesenchymal mobilization and optimize MSC-based strategies for COPD.
4. Cellular Interactions and Key Signaling Pathways in COPD
4.1. Epithelial-to-Mesenchymal Transformation in COPD
The pulmonary epithelial cell lining is a vital barrier against environmental toxins and infections. In COPD, epithelial cells undergo transformation into mobile mesenchymal cells through epithelial-mesenchymal transition (EMT), a process triggered by diseases, infections, or injuries, and mediated by pathways like TGF-β signaling (B) [
127,
128]. EMT is observed not only in lung development but also in respiratory diseases such as COPD, lung cancer, and asthma, where it contributes to excessive collagen and ECM protein deposition [
127,
129,
130].
In COPD, transcription factors like Snail1 are notably upregulated, particularly in cases with α1-antitrypsin deficiency, signifying increased EMT activity [
131]. Additionally, transcription factors like Twist demonstrate increased nuclear translocation in smokers, correlating with greater airflow obstruction [
132]. E-cadherin, an adhesion molecule essential for interepithelial junctions, is significantly decreased in smokers and COPD patients, with its reduction linked to more severe airflow limitations [
133,
134].
Moreover, mesenchymal markers, including N-cadherin, vimentin, α-SMA, type I collagen, and fibronectin, are elevated in COPD patients, promoting increased cell migration, tissue fibrosis, and airflow restriction [
135,
136]. New biomarkers such as β2-microglobulin (β2M) and sphingosine-1-phosphate (S1P) are under investigation for their roles in EMT regulation and their correlation with declining lung function in smoking-induced COPD [
137,
138]. Further research highlights the role of Cullin 4A (CUL4A); silencing this gene inhibits EMT in smoke-exposed lung cells, demonstrating its potential in altering disease progression [
139,
140]. This emerging understanding of EMT biomarkers could contribute to more effective management of smoking-related COPD.
4.2. Key Signaling Pathways Involved in Epithelial-Mesenchymal Interactions
Cellular signaling pathways regulating epithelial-mesenchymal interactions play crucial roles in lung development, homeostasis, and disease. Among these pathways, paracrine-acting FGFs are particularly important in cellular processes such as proliferation, survival, migration, differentiation, and metabolism [
141]. Mesenchyme-derived Fgf10, which binds predominantly to Fgfr2b expressed by epithelial cells, plays a key role in maintaining lung stem/progenitor cell homeostasis, promoting wound healing, and protecting against oxidant-induced DNA damage [
29,
142,
143,
144,
145]. FGF10 haploinsufficiency correlates with impaired lung function and is linked to COPD through single nucleotide polymorphisms (SNPs) near the
Fgf10 locus [
146,
147,
148]. In advanced COPD, decreased Fgf10 expression in the alveolar walls’ been noted. While overexpression of
Fgf10 stimulates AT2 cell proliferation and enhances epithelial progenitor markers via the β-catenin pathway, underscoring its potential in alveolar regeneration (
B) [
20,
36].
Further studies indicate that precise regulation of the Yap-WNT7b-FGF10 pathway is critical for maintaining airway epithelial quiescence and preventing excessive remodeling. Dysregulation of Hippo signaling, seen in COPD, leads to defective repair mechanisms. Elevated nuclear YAP, FGFR2b, and WNT7b levels in the squamous metaplastic regions of the airway epithelium in COPD patients support this conclusion [
144]. Meanwhile, WNT5a and WNT5b, secreted by lung fibroblasts, negatively impact epithelial progenitor proliferation. Specifically, WNT5b inhibits the growth and differentiation of alveolar epithelial progenitors in a WNT/β-catenin-dependent manner, suggesting therapeutic potential in modulating WNT signaling to enhance alveolar repair, particularly in aging lungs affected by COPD [
149]. The role of Fam13a, another key player, has been explored in chronic CSE mouse models.
Fam13a deficiency ameliorates the negative effects of CSE on lung epithelial stem cell regeneration via activation of the WNT signaling pathway. Additionally, in human cells and animal models of COPD, CSE has been shown to increase the expression of COPD-related markers, including type I collagen and ERK1/2 activation. This pathway also plays a crucial role in regulating mitochondria fission and fusion, which in turn controls ASMC proliferation (
B) [
150,
151].
The NOTCH signaling pathway is also compromised in COPD, with significant downregulation of NOTCH3, DLL1, and downstream targets such as HES5 and HEY1/2 in COPD patients compared to non-smokers. Despite this global inhibition, NICD1/HEY2 expression is upregulated in regions of mucosal hyperplasia in COPD patients, indicating persistent NOTCH activation, likely triggered by cigarette smoking, which skews differentiation towards the goblet cell lineage (
B) [
152,
153]. Additionally, the involvement of SHH signaling in COPD, pulmonary fibrosis, and fibroblast expansion suggests its role in ECM deposition [
154,
155]. Genome-wide association studies (GWAS
) have identified a locus near HHIP on chromosome 4q31, associated with reduced lung function and altered HHIP gene expression, further implicating this pathway in COPD pathogenesis [
156,
157].
These subpopulation-specific injury-response mechanisms may play a significant role in COPD. Despite the growing prevalence of single-cell RNA sequencing analysis, contemporary research seldom focuses on the responses among these heterogeneous populations. A deeper understanding of epithelial-mesenchymal interactions and key signaling pathways in COPD could provide critical insights for developing targeted therapies to slow disease progression and enhance tissue repair.
5. Implications of Epithelial-Mesenchymal Niche for COPD Therapy
Current management of COPD relies on three principal therapeutic modalities: pharmacological interventions targeting airway smooth muscle contraction and inflammatory pathways; extracellular vesicle-based strategies delivering regulatory biomolecules to modulate fibrotic and immune responses; and cellular transplantation approaches utilizing diverse progenitor sources to enhance tissue repair (). While these interventions provide symptomatic relief and attenuate disease progression, they exhibit limited capacity to restore the fundamental regenerative potential of compromised pulmonary epithelia. This critical gap underscores the imperative to elucidate the mechanisms governing airway and alveolar stem cell dysfunction in COPD. Emerging insights into epithelial-mesenchymal-immune crosstalk now illuminate novel regenerative strategies to reactivate endogenous repair programs, redirect aberrant differentiation trajectories, and reestablish functional niche microenvironments.
.
Therapeutic applications on COPD.
| Treatments |
Sources |
Targets |
References |
| Drug treatments |
Beta-agonists |
β2 receptor on BSMCs |
PMID:22314182
PMID:35311415
|
| LAMA/LABA/ICS |
Glucocorticoid receptor on epithelial cells and
endothelial cells of bronchial vessels |
PMID:37690008
PMID:31305147
PMID:9563367
PMID:37852657
|
Anti-muscarinics,
Anti-cholinergics
|
Muscarinic receptors on ASMCs,
inflammatory cells, and airway epithelial cells |
PMID:27787709 |
| Theophylline |
Adenosine receptor |
PMID:30085566 |
| Extracellular vesicles |
EVs carrying miR-210 |
Myofibroblast |
PMID:33712504 |
| (hUC-MSC)-derived EVs |
Goblet cells, inflammatory cells |
PMID:33436065 |
| EVs carrying miR-21 |
Myofibroblast |
PMID:33712504 |
| EVs carrying miR-181c |
Fibroblast, osteoblast, Endothelial cells
and epithelial cells |
PMID:28806967 |
| Cell transplantation |
P63Pos progenitor cells |
/ |
PMID:38354225 |
| Bone marrow derived mononuclear cells |
/ |
PMID:24255620 |
Adipose tissue-derived
stromal cells |
/ |
PMID:17049053 |
| BM-MSCs |
/ |
PMID:35725505
PMID:20842104
PMID:34974799
|
| iPSC-MSCs |
/ |
PMID:28911970 |
| DASCs |
/ |
PMID:33960563 |
5.1. Airway Stem Cells in Regeneration
Epithelial dysfunction, manifested through cellular damage, senescence, and exhaustion, represents a core pathological feature of COPD. However, the regulatory hierarchy and signaling mechanisms governing epithelial responses remain poorly defined compared to diseases like asthma or IPF. Recent evidence reveals a unique AT2 subpopulation (asATII) co-expressing SP-C and SCGB3A2 within emphysematous alveoli of advanced COPD patients. These cells, potentially derived from migratory airway club cells in experimental models, exhibit critically impaired regenerative capacity in functional assays [
74].
Epithelial-mesenchymal crosstalk is pivotal for lung regeneration. For example, bronchiolar stem cells (BSCs) rely on Fgf10 secreted by mesenchymal cells for their function. Overexpression of
Fgf10 during lung development results in ectopic BSC formation in the airways [
158]. After injury in mice, surviving epithelial cells secrete Wnt7b, which induces Fgf10 expression in ASMCs, promoting secretory cell regeneration [
158,
159]. The HIPPO signaling pathway modulates BSC stability through the Fgf10-Fgfr2b axis [
160,
161]. The BSCs have also been reported to maintain functional intraepithelial airway macrophages [
162], suggesting their significant potential in regulating airway repair. Furthermore, recent studies indicate that RSMCs in non-cartilaginous airways express high levels of Fgf10, which promotes club cell proliferation. These findings highlight that epithelial regeneration is closely modulated by mesenchymal signaling pathways [
145].
These findings underscore a paradoxical phenomenon: while airway-derived progenitors are able to translocate to replenish alveolar compartments, their functional exhaustion ultimately limits repair. Future studies may focus on delineating the crosstalk between proximal and distal epithelial populations, such as BASCs, alongside signaling pathways regulating club cell fate. Such insights may inform cell-based transplantation strategies to reactivate endogenous repair programs () [
163]. It is also important to determine whether RSMCs contribute to basal and club cell repair in COPD, particularly in exhausted epithelia. However, excessive activation of these pathways may lead to hyperplasia, emphasizing the need for a balanced approach to regenerative therapy (B).
5.2. Alveolar Stem Cells and Their Niches in COPD
Alveolar stem cells and their mesenchymal niches play a pivotal role in lung regeneration. A subset of AT2 cells expressing a high level of Cd44 exhibits enhanced proliferation and organoid-forming capabilities. It has been further reported that Gli1
Pos adventitial fibroblasts are a crucial component in supporting the Cd44
High AT2 cells through their production of hyaluronan [
18,
164,
165]. WNT signaling is critical in maintaining AT2 progenitor cells, although there is variability in their dynamics across different studies [
166]. During lung injury repair, intermediate cell populations (e.g., KRT8
High and CLDN4
High alveolar epithelial cells) emerge, influenced by TGFβ and P53 signaling pathways [
167,
168]. In human lungs, a recent study integrated spatial transcriptomics and single-cell RNA sequencing of micro dissected distal airways, identified LGR5
Pos fibroblasts, terminal and respiratory bronchiolar secretory cells (TRB-SCs), and alveolar type-0 (AT0) cells. Organoid and connectome analyses reveal LGR5
Pos fibroblasts as a niche signaling center in contact with epithelia. The AT0 cells represent a transient bipotent state during AT2 regeneration in primates, capable of differentiating into AT1s or TRB-SCs. These findings redefine human lung cell hierarchies and illuminate an epithelial transitional state critical in development, regeneration, and disease, which differs from mouse studies [
169]. Epithelial-immune crosstalk is equally important, the PD-1/PD-L1 inhibitory pathway, known for regulating T-cell activation, critically mediates this interaction. IAAPs, characterized by high PD-L1 and low SP-C expression, contribute significantly to alveolar regeneration (
C) [
29,
170,
171]. These cells warrant further investigation in COPD, with potential for immune-targeted interventions to enhance regeneration. Elucidating how these stem cells respond and adapt within COPD’s chronic inflammatory milieu, and whether their reparative functions are impaired, will be essential [
172]. Therefore, a major goal is to develop strategies that either enhance the growth and transformation potential of these stem cells or modulate niche signals to rebuild functional alveoli, countering structural and functional decline in COPD. Understanding the precise responses and therapeutic potential of defined alveolar epithelial progenitor subpopulations in COPD represents a critical frontier for regenerative approaches.
Pulmonary fibrosis, a hallmark of advanced COPD, is driven by mesenchymal populations like myofibroblasts. PDGFRα-positive alveolar fibroblasts form a supportive niche for AT2 cells, facilitating their function and repair. SFRP1-positive fibroblasts transition into matrix-producing myofibroblasts in response to TGFβ signaling, and contribute to tissue repair post-injury [
173,
174,
175]. Lipofibroblasts, marked by TCF21 and FGF10, support alveolar epithelial regeneration through paracrine signaling and ECM remodeling [
176,
177,
178,
179]. Whether other populations like MANCs and LGR5-positive fibroblasts contribute to the mesenchymal niche in COPD requires further validation [
169,
180].
Analogous to IPF, key therapeutic targets in COPD may include progenitors driving collagen production. For instance, LEPR
Pos alveolar fibroblasts are a key source of pathological CTHRC1
Pos fibroblasts, and targeting Runx2 shows therapeutic potential [
181]. Strategies to deplete these cells from fibrotic tissues, by targeting specific receptors, may prove beneficial. Additional approaches include targeting FGF10, PDGFRα, or TCF21; inhibiting TGFβ-driven SFRP1
Pos fibroblast-to-myofibroblast transition; or conversely activating lipogenic pathways via BMP2/PPARγ agonists—all potentially slowing COPD progression (
C).
6. Conclusions
Both airway and alveolar stem cell niches are vital for lung regeneration, offering significant therapeutic potential for treating COPD. However, balancing regeneration with pathological remodeling presents a challenge. Understanding epithelial-mesenchymal interactions is critical to developing targeted therapies that promote healthy lung regeneration while preventing aberrant repair processes. Further investigation may focus on the origin and development of stem cell lineages that contribute to regeneration in COPD. Translating bench research to bedside applications, such as stem cell implantation, extracellular vesicle based drug treatments or delivery, and cell-based targeted therapies, will be essential for advancing COPD treatment (
).
Author Contributions
Conceptualization: J.-S.Z., J.W., C.C.; Writing—original draft: J.W.; Writing—review and editing: J.-S.Z., W.L., S.B.; Figures and Table creation: J.W., J.-S.Z.; Resource and Funding acquisition: J.-S.Z., W.L., C.C.
Ethics Statement
No ethical statement to declare.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Funding
J.-S.Z. is partially supported by research funds from both the Quzhou Affiliated Hospital and the First Affiliated Hospital of Wenzhou Medical University, the Zhejiang Province Public Welfare Fund Project (LY24H050003). C.C. and W.L. were supported in part by the National Natural Science Foundation of China grants No. 82170017 and No. 82330002, respectively.
Declaration of Competing Interest
The authors declared there is no conflict of interest.
References
-
1.
Dong F, Su R, Ren Y, Yang T. Burden of chronic obstructive pulmonary disease and risk factors in China from 1990 to 2021: Analysis of global burden of disease 2021.
Chin. Med. J. Pulm. Crit. Care Med. 2025,
3, 132–140.
[Google Scholar]
-
2.
Barnes PJ. Chronic obstructive pulmonary disease.
N. Engl. J. Med. 2000,
343, 269–280.
[Google Scholar]
-
3.
Christenson SA, Smith BM, Bafadhel M, Putcha N. Chronic obstructive pulmonary disease.
Lancet 2022,
399, 2227–2242.
[Google Scholar]
-
4.
Stolz D, Mkorombindo T, Schumann DM, Agusti A, Ash SY, Bafadhel M, et al. Towards the elimination of chronic obstructive pulmonary disease: a Lancet Commission.
Lancet 2022,
400, 921–972.
[Google Scholar]
-
5.
Abohalaka R. Bronchial epithelial and airway smooth muscle cell interactions in health and disease.
Heliyon 2023,
9, e19976.
[Google Scholar]
-
6.
Zepp JA, Morrisey EE. Cellular crosstalk in the development and regeneration of the respiratory system.
Nat. Rev. Mol. Cell. Biol. 2019,
20, 551–566.
[Google Scholar]
-
7.
Hogan B, Tata PR. Cellular organization and biology of the respiratory system.
Nat. Cell Biol. 2019, doi:10.1038/s41556-019-0357-7.
[Google Scholar]
-
8.
Pardo-Saganta A, Law BM, Gonzalez-Celeiro M, Vinarsky V, Rajagopal J. Ciliated cells of pseudostratified airway epithelium do not become mucous cells after ovalbumin challenge.
Am. J. Respir. Cell. Mol. Biol. 2013,
48, 364–373.
[Google Scholar]
-
9.
Kotton DN, Morrisey EE. Lung regeneration: mechanisms, applications and emerging stem cell populations.
Nat. Med. 2014,
20, 822–832.
[Google Scholar]
-
10.
Reynolds SD, Reynolds PR, Pryhuber GS, Finder JD, Stripp BR. Secretoglobins SCGB3A1 and SCGB3A2 define secretory cell subsets in mouse and human airways.
Am. J. Respir. Crit. Care Med. 2002,
166, 1498–509.
[Google Scholar]
-
11.
Zhang JS, Dhlamini Q, Guo Q, Quan M, Wu J, Lyu H, et al. Progress and Gaps in Respiratory Disease Research and Treatment: Highlights of the IRM 2024 in Shanghai.
J. Respir. Biol. Transl. Med. 2024,
1, 10021.
[Google Scholar]
-
12.
Giangreco A, Reynolds SD, Stripp BR. Terminal bronchioles harbor a unique airway stem cell population that localizes to the bronchoalveolar duct junction.
Am. J. Pathol. 2002,
161, 173–182.
[Google Scholar]
-
13.
Reynolds SD, Hong KU, Giangreco A, Mango GW, Guron C, Morimoto Y, et al. Conditional clara cell ablation reveals a self-renewing progenitor function of pulmonary neuroendocrine cells.
Am. J. Physiol. Lung Cell. Mol. Physiol. 2000,
278, L1256–1263.
[Google Scholar]
-
14.
Jones-Freeman B, Starkey MR. Bronchioalveolar stem cells in lung repair, regeneration and disease.
J. Pathol. 2020,
252, 219–226.
[Google Scholar]
-
15.
Hong KU, Reynolds SD, Giangreco A, Hurley CM, Stripp BR. Clara cell secretory protein-expressing cells of the airway neuroepithelial body microenvironment include a label-retaining subset and are critical for epithelial renewal after progenitor cell depletion.
Am. J. Respir. Cell. Mol. Biol. 2001,
24, 671–681.
[Google Scholar]
-
16.
Peake JL, Reynolds SD, Stripp BR, Stephens KE, Pinkerton KE. Alteration of pulmonary neuroendocrine cells during epithelial repair of naphthalene-induced airway injury.
Am. J. Pathol. 2000,
156, 279–286.
[Google Scholar]
-
17.
Reynolds SD, Giangreco A, Power JH, Stripp BR. Neuroepithelial bodies of pulmonary airways serve as a reservoir of progenitor cells capable of epithelial regeneration.
Am. J. Pathol. 2000,
156, 269–278.
[Google Scholar]
-
18.
Chen Q, Suresh Kumar V, Finn J, Jiang D, Liang J, Zhao YY, et al. CD44(high) alveolar type II cells show stem cell properties during steady-state alveolar homeostasis.
Am. J. Physiol. Lung Cell. Mol. Physiol. 2017,
313, L41–L51.
[Google Scholar]
-
19.
Zacharias WJ, Frank DB, Zepp JA, Morley MP, Alkhaleel FA, Kong J, et al. Regeneration of the lung alveolus by an evolutionarily conserved epithelial progenitor.
Nature 2018,
555, 251–255.
[Google Scholar]
-
20.
Nabhan AN, Brownfield DG, Harbury PB, Krasnow MA, Desai TJ. Single-cell Wnt signaling niches maintain stemness of alveolar type 2 cells.
Science 2018,
359, 1118–1123.
[Google Scholar]
-
21.
Travaglini KJ, Nabhan AN, Penland L, Sinha R, Gillich A, Sit RV, et al. A molecular cell atlas of the human lung from single-cell RNA sequencing.
Nature 2020,
587, 619–625.
[Google Scholar]
-
22.
Liu Y, Kumar VS, Zhang W, Rehman J, Malik AB. Activation of type II cells into regenerative stem cell antigen-1(+) cells during alveolar repair.
Am. J. Respir. Cell. Mol. Biol. 2015,
53, 113–124.
[Google Scholar]
-
23.
Choi J, Park JE, Tsagkogeorga G, Yanagita M, Koo BK, Han N, et al. Inflammatory Signals Induce AT2 Cell-Derived Damage-Associated Transient Progenitors that Mediate Alveolar Regeneration.
Cell Stem Cell 2020,
27, 366–382.e7.
[Google Scholar]
-
24.
Kobayashi Y, Tata A, Konkimalla A, Katsura H, Lee RF, Ou J, et al. Persistence of a regeneration-associated, transitional alveolar epithelial cell state in pulmonary fibrosis.
Nat. Cell Biol. 2020,
22, 934–946.
[Google Scholar]
-
25.
Riemondy KA, Jansing NL, Jiang P, Redente EF, Gillen AE, Fu R, et al. Single cell RNA sequencing identifies TGFβ as a key regenerative cue following LPS-induced lung injury.
JCI Insight 2019,
5, e123637.
[Google Scholar]
-
26.
Strunz M, Simon LM, Ansari M, Kathiriya JJ, Angelidis I, Mayr CH, et al. Alveolar regeneration through a Krt8+ transitional stem cell state that persists in human lung fibrosis.
Nat. Commun. 2020,
11, 3559.
[Google Scholar]
-
27.
Verheyden JM, Sun X. A transitional stem cell state in the lung.
Nat. Cell Biol. 2020,
22, 1025–1026.
[Google Scholar]
-
28.
Wu H, Yu Y, Huang H, Hu Y, Fu S, Wang Z, et al. Progressive pulmonary fibrosis is caused by elevated mechanical tension on alveolar stem cells.
Cell 2021,
184, 845–846.
[Google Scholar]
-
29.
Ahmadvand N, Khosravi F, Lingampally A, Wasnick R, Vazquez-Armendariz AI, Carraro G, et al. Identification of a novel subset of alveolar type 2 cells enriched in PD-L1 and expanded following pneumonectomy.
Eur. Respir. J. 2021,
58, 2004168.
[Google Scholar]
-
30.
Kathiriya JJ, Wang C, Zhou M, Brumwell A, Cassandras M, Le Saux CJ, et al. Human alveolar type 2 epithelium transdifferentiates into metaplastic KRT5(+) basal cells.
Nat. Cell Biol. 2022, 24, 10–23.
[Google Scholar]
-
31.
Quan M, Guo Q, Yan X, Yu C, Yang L, Zhang Y, et al. Parkin deficiency aggravates inflammation-induced acute lung injury by promoting necroptosis in alveolar type II cells.
Chin. Med. J. Pulm. Crit. Care Med. 2024,
2, 265–278.
[Google Scholar]
-
32.
Zepp JA, Zacharias WJ, Frank DB, Cavanaugh CA, Zhou S, Morley MP, et al. Distinct Mesenchymal Lineages and Niches Promote Epithelial Self-Renewal and Myofibrogenesis in the Lung.
Cell 2017,
170, 1134–1148.e10.
[Google Scholar]
-
33.
Barkauskas CE, Cronce MJ, Rackley CR, Bowie EJ, Keene DR, Stripp BR, et al. Type 2 alveolar cells are stem cells in adult lung.
J. Clin. Invest. 2013,
123, 3025–3036.
[Google Scholar]
-
34.
Morrisey EE, Hogan BL. Preparing for the first breath: genetic and cellular mechanisms in lung development.
Dev. Cell 2010,
18, 8–23.
[Google Scholar]
-
35.
Bellusci S, Grindley J, Emoto H, Itoh N, Hogan BL. Fibroblast growth factor 10 (FGF10) and branching morphogenesis in the embryonic mouse lung.
Development 1997,
124, 4867–4878.
[Google Scholar]
-
36.
Hadzic S, Wu CY, Gredic M, Pak O, Loku E, Kraut S, et al. Fibroblast growth factor 10 reverses cigarette smoke- and elastase-induced emphysema and pulmonary hypertension in mice.
Eur. Respir. J. 2023,
62, 2201606.
[Google Scholar]
-
37.
El Agha E, Kramann R, Schneider RK, Li X, Seeger W, Humphreys BD, et al. Mesenchymal Stem Cells in Fibrotic Disease.
Cell Stem Cell 2017,
21, 166–177.
[Google Scholar]
-
38.
El Agha E, Moiseenko A, Kheirollahi V, De Langhe S, Crnkovic S, Kwapiszewska G, et al. Two-Way Conversion between Lipogenic and Myogenic Fibroblastic Phenotypes Marks the Progression and Resolution of Lung Fibrosis.
Cell Stem Cell 2017,
20, 261–273.e3.
[Google Scholar]
-
39.
Lee JH, Tammela T, Hofree M, Choi J, Marjanovic ND, Han S, et al. Anatomically and Functionally Distinct Lung Mesenchymal Populations Marked by Lgr5 and Lgr6.
Cell 2017,
170, 1149–1163.e12.
[Google Scholar]
-
40.
Liu X, Zhang X, Liang J, Noble PW, Jiang D. The concept of Sfrp1(+) transitional fibroblasts: the key to dissociating lineage heterogeneity and fate of invasive fibroblasts in pulmonary fibrosis?
Eur. Respir. J. 2024,
63, 2400498.
[Google Scholar]
-
41.
Mukhatayev Z, Adilbayeva A, Kunz J. CTHRC1: An Emerging Hallmark of Pathogenic Fibroblasts in Lung Fibrosis.
Cells 2024,
13, 946.
[Google Scholar]
-
42.
Feng Y, Hu J, Liu F, Shang Y. Collagen Triple Helix Repeat Containing 1 Deficiency Protects Against Airway Remodeling and Inflammation in Asthma Models In Vivo and In Vitro.
Inflammation 2023,
46, 925–940.
[Google Scholar]
-
43.
Tsukui T, Sun KH, Wetter JB, Wilson-Kanamori JR, Hazelwood LA, Henderson NC, et al. Collagen-producing lung cell atlas identifies multiple subsets with distinct localization and relevance to fibrosis.
Nat. Commun. 2020,
11, 1920.
[Google Scholar]
-
44.
Liu G, Philp AM, Corte T, Travis MA, Schilter H, Hansbro NG, et al. Therapeutic targets in lung tissue remodelling and fibrosis.
Pharmacol. Ther. 2021,
225, 107839.
[Google Scholar]
-
45.
Tang X, Li Z, Yu Z, Li J, Zhang J, Wan N, et al. Effect of curcumin on lung epithelial injury and ferroptosis induced by cigarette smoke.
Hum. Exp. Toxicol. 2021,
40, S753–S762.
[Google Scholar]
-
46.
Wei T, Wang X, Lang K, Song Y, Luo J, Gu Z, et al. Peroxiredoxin 6 Protects Pulmonary Epithelial Cells From Cigarette-related Ferroptosis in Chronic Obstructive Pulmonary Disease.
Inflammation 2024,
48, 662–675.
[Google Scholar]
-
47.
Peng K, Yao YX, Lu X, Wang WJ, Zhang YH, Zhao H, et al. Mitochondrial dysfunction-associated alveolar epithelial senescence is involved in CdCl(2)-induced COPD-like lung injury.
J. Hazard. Mater. 2024,
476, 135103.
[Google Scholar]
-
48.
Liu W, Liang W, Zhang C, Liu H, Li H, Zhou L, et al. LncR-GAS5 decrease in adenine phosphoribosyltransferase expresssion via binding TAF1 to increase kidney damage created by CIH.
Heliyon 2024,
10, e33084.
[Google Scholar]
-
49.
Kurie JM, Shin HJ, Lee JS, Morice RC, Ro JY, Lippman SM, et al. Increased epidermal growth factor receptor expression in metaplastic bronchial epithelium.
Clin. Cancer Res. 1996,
2, 1787–1793.
[Google Scholar]
-
50.
Shaykhiev R, Zuo WL, Chao I, Fukui T, Witover B, Brekman A, et al. EGF shifts human airway basal cell fate toward a smoking-associated airway epithelial phenotype.
Proc. Natl. Acad. Sci. USA 2013,
110, 12102–12107.
[Google Scholar]
-
51.
Bals R, Hiemstra PS. Innate immunity in the lung: how epithelial cells fight against respiratory pathogens.
Eur. Respir. J. 2004,
23, 327–333.
[Google Scholar]
-
52.
Rogers DF. The airway goblet cell.
Int. J. Biochem. Cell Biol. 2003,
35, 1–6.
[Google Scholar]
-
53.
Leopold PL, O'Mahony MJ, Lian XJ, Tilley AE, Harvey BG, Crystal RG. Smoking is associated with shortened airway cilia.
PLoS ONE 2009,
4, e8157.
[Google Scholar]
-
54.
Ramos EM, De Toledo AC, Xavier RF, Fosco LC, Vieira RP, Ramos D, et al. Reversibility of impaired nasal mucociliary clearance in smokers following a smoking cessation programme.
Respirology 2011,
16, 849–855.
[Google Scholar]
-
55.
Carlier FM, de Fays C, Pilette C. Epithelial Barrier Dysfunction in Chronic Respiratory Diseases.
Front. Physiol. 2021,
12, 691227.
[Google Scholar]
-
56.
Coyne CB, Vanhook MK, Gambling TM, Carson JL, Boucher RC, Johnson LG. Regulation of airway tight junctions by proinflammatory cytokines.
Mol. Biol. Cell 2002,
13, 3218–334.
[Google Scholar]
-
57.
Chen R, Cui Y, Ip MS, Mak JC. Cigarette smoke induces endoplasmic reticulum stress-associated mucus hypersecretion via orosomucoid 1-like protein 3 in airway epithelia.
Free Radic. Res. 2025,
59, 342–355.
[Google Scholar]
-
58.
Dong X, Ding M, Zhang J, Ogülür I, Pat Y, Akdis M, et al. Involvement and therapeutic implications of airway epithelial barrier dysfunction in type 2 inflammation of asthma.
Chin. Med. J. 2022,
135, 519–531.
[Google Scholar]
-
59.
Bailey KL. Aging Diminishes Mucociliary Clearance of the Lung.
Adv. Geriatr. Med. Res. 2022,
4, e220005.
[Google Scholar]
-
60.
Alter P, Baker JR, Dauletbaev N, Donnelly LE, Pistenmaa C, Schmeck B, et al. Update in Chronic Obstructive Pulmonary Disease 2019.
Am. J. Respir. Crit. Care Med. 2020,
202, 348–355.
[Google Scholar]
-
61.
Tran HB, Jersmann H, Truong TT, Hamon R, Roscioli E, Ween M, et al. Disrupted epithelial/macrophage crosstalk via Spinster homologue 2-mediated S1P signaling may drive defective macrophage phagocytic function in COPD.
PLoS ONE 2017,
12, e0179577.
[Google Scholar]
-
62.
Van Eeckhoutte HP, Donovan C, Kim RY, Conlon TM, Ansari M, Khan H, et al. RIPK1 kinase-dependent inflammation and cell death contribute to the pathogenesis of COPD.
Eur. Respir. J. 2023,
61, 2201506.
[Google Scholar]
-
63.
Hiemstra PS, Bourdin A. Club cells, CC10 and self-control at the epithelial surface.
Eur. Respir. J. 2014,
44, 831–832.
[Google Scholar]
-
64.
Gamez AS, Gras D, Petit A, Knabe L, Molinari N, Vachier I, et al. Supplementing defect in club cell secretory protein attenuates airway inflammation in COPD.
Chest 2015,
147, 1467–1476.
[Google Scholar]
-
65.
Snyder JC, Reynolds SD, Hollingsworth JW, Li Z, Kaminski N, Stripp BR. Clara cells attenuate the inflammatory response through regulation of macrophage behavior.
Am. J. Respir. Cell. Mol. Biol. 2010,
42, 161–171.
[Google Scholar]
-
66.
Katavolos P, Ackerley CA, Clark ME, Bienzle D. Clara cell secretory protein increases phagocytic and decreases oxidative activity of neutrophils.
Vet. Immunol. Immunopathol. 2011,
139, 1–9.
[Google Scholar]
-
67.
Tokita E, Tanabe T, Asano K, Suzaki H, Rubin BK. Club cell 10-kDa protein attenuates airway mucus hypersecretion and inflammation.
Eur. Respir. J. 2014,
44, 1002–1010.
[Google Scholar]
-
68.
Pilette C, Godding V, Kiss R, Delos M, Verbeken E, Decaestecker C, et al. Club cell protein 16 and disease progression in chronic obstructive pulmonary disease.
Am. J. Respir. Crit. Care Med. 2013,
188, 1413–1419.
[Google Scholar]
-
69.
Perl AK, Riethmacher D, Whitsett JA. Conditional depletion of airway progenitor cells induces peribronchiolar fibrosis.
Am. J. Respir. Crit. Care Med. 2011,
183, 511–521.
[Google Scholar]
-
70.
Pilette C, Godding V, Kiss R, Delos M, Verbeken E, Decaestecker C, et al. Reduced epithelial expression of secretory component in small airways correlates with airflow obstruction in chronic obstructive pulmonary disease.
Am. J. Respir. Crit. Care Med. 2001, 163, 185–194.
[Google Scholar]
-
71.
Shijubo N, Itoh Y, Yamaguchi T, Imada A, Hirasawa M, Yamada T, et al. Clara cell protein-positive epithelial cells are reduced in small airways of asthmatics.
Am. J. Respir. Crit. Care Med. 1999,
160, 930–933.
[Google Scholar]
-
72.
Kelly FL, Kennedy VE, Jain R, Sindhwani NS, Copeland CF, Snyder LD, et al. Epithelial clara cell injury occurs in bronchiolitis obliterans syndrome after human lung transplantation.
Am. J. Transplant. 2012,
12, 3076–3084.
[Google Scholar]
-
73.
Liu K, Meng X, Liu Z, Tang M, Lv Z, Huang X, et al. Tracing the origin of alveolar stem cells in lung repair and regeneration.
Cell 2024,
187, 2428–2445.e20.
[Google Scholar]
-
74.
Hu Y, Hu Q, Ansari M, Riemondy K, Pineda R, Sembrat J, et al. Airway derived emphysema-specific alveolar type II cells exhibit impaired regenerative potential in COPD.
Eur. Respir. J. 2024,
64, 2302071.
[Google Scholar]
-
75.
Rock JR, Onaitis MW, Rawlins EL, Lu Y, Clark CP, Xue Y, et al. Basal cells as stem cells of the mouse trachea and human airway epithelium.
Proc. Natl. Acad. Sci. USA 2009,
106, 12771–12775.
[Google Scholar]
-
76.
Shaykhiev R. Airway Epithelial Progenitors and the Natural History of Chronic Obstructive Pulmonary Disease.
Am. J. Respir. Crit. Care Med. 2018,
197, 847–849.
[Google Scholar]
-
77.
Araya J, Cambier S, Markovics JA, Wolters P, Jablons D, Hill A, et al. Squamous metaplasia amplifies pathologic epithelial-mesenchymal interactions in COPD patients.
J. Clin. Invest. 2007,
117, 3551–3562.
[Google Scholar]
-
78.
Rao W, Wang S, Duleba M, Niroula S, Goller K, Xie J, et al. Regenerative Metaplastic Clones in COPD Lung Drive Inflammation and Fibrosis.
Cell 2020,
181, 848–864.e18.
[Google Scholar]
-
79.
Whetstone CE, Ranjbar M, Omer H, Cusack RP, Gauvreau GM. The Role of Airway Epithelial Cell Alarmins in Asthma.
Cells 2022,
11, 1105.
[Google Scholar]
-
80.
Préfontaine D, Hamid Q. Airway epithelial cells in asthma.
J. Allergy Clin. Immunol. 2007,
120, 1475–1478.
[Google Scholar]
-
81.
Jones DL, Morley MP, Li X, Ying Y, Zhao G, Schaefer SE, et al. An injury-induced mesenchymal-epithelial cell niche coordinates regenerative responses in the lung.
Science 2024,
386, eado5561.
[Google Scholar]
-
82.
Warner SM, Hackett TL, Shaheen F, Hallstrand TS, Kicic A, Stick SM, et al. Transcription factor p63 regulates key genes and wound repair in human airway epithelial basal cells.
Am. J. Respir. Cell. Mol. Biol. 2013,
49, 978–988.
[Google Scholar]
-
83.
Stevens PT, Kicic A, Sutanto EN, Knight DA, Stick SM. Dysregulated repair in asthmatic paediatric airway epithelial cells: the role of plasminogen activator inhibitor-1.
Clin. Exp. Allergy 2008,
38, 1901–1910.
[Google Scholar]
-
84.
Nadel JA, Burgel PR. The role of epidermal growth factor in mucus production.
Curr. Opin. Pharmacol. 2001,
1, 254–258.
[Google Scholar]
-
85.
Jing Y, Gimenes JA, Mishra R, Pham D, Comstock AT, Yu D, et al. NOTCH3 contributes to rhinovirus-induced goblet cell hyperplasia in COPD airway epithelial cells.
Thorax 2019,
74, 18–32.
[Google Scholar]
-
86.
Rogers DF. Airway goblet cells: responsive and adaptable front-line defenders.
Eur. Respir. J. 1994,
7, 1690–1706.
[Google Scholar]
-
87.
Kim V, Criner GJ. Chronic bronchitis and chronic obstructive pulmonary disease.
Am. J. Respir. Crit. Care Med. 2013,
187, 228–237.
[Google Scholar]
-
88.
Zhu H, Leng J, Ju R, Qu S, Tian J, Leng H, et al. Advantages of pulsed electric field ablation for COPD: Excellent killing effect on goblet cells.
Bioelectrochemistry 2024,
158, 108726.
[Google Scholar]
-
89.
Montoro DT, Haber AL, Biton M, Vinarsky V, Lin B, Birket SE, et al. A revised airway epithelial hierarchy includes CFTR-expressing ionocytes.
Nature 2018,
560, 319–324.
[Google Scholar]
-
90.
Noguchi M, Furukawa KT, Morimoto M. Pulmonary neuroendocrine cells: physiology, tissue homeostasis and disease.
Dis. Model Mech. 2020,
13, dmm046920.
[Google Scholar]
-
91.
Strine MS, Wilen CB. Tuft cells are key mediators of interkingdom interactions at mucosal barrier surfaces.
PLoS Pathog. 2022,
18, e1010318.
[Google Scholar]
-
92.
Desai TJ, Brownfield DG, Krasnow MA. Alveolar progenitor and stem cells in lung development, renewal and cancer.
Nature 2014,
507, 190–194.
[Google Scholar]
-
93.
Okutomo K, Fujino N, Yamada M, Saito T, Ono Y, Okada Y, et al. Increased LHX9 expression in alveolar epithelial type 2 cells of patients with chronic obstructive pulmonary disease.
Respir. Investig. 2022,
60, 119–128.
[Google Scholar]
-
94.
Tsutsumi A, Ozaki M, Chubachi S, Irie H, Sato M, Kameyama N, et al. Exposure to Cigarette Smoke Enhances the Stemness of Alveolar Type 2 Cells.
Am. J. Respir. Cell. Mol. Biol. 2020,
63, 293–305.
[Google Scholar]
-
95.
Irie H, Ozaki M, Chubachi S, Hegab AE, Tsutsumi A, Kameyama N, et al. Short-term intermittent cigarette smoke exposure enhances alveolar type 2 cell stemness via fatty acid oxidation.
Respir. Res. 2022,
23, 41.
[Google Scholar]
-
96.
Yu H, Lin Y, Zhong Y, Guo X, Lin Y, Yang S, et al. Impaired AT2 to AT1 cell transition in PM2.5-induced mouse model of chronic obstructive pulmonary disease.
Respir. Res. 2022,
23, 70.
[Google Scholar]
-
97.
Sauler M, McDonough JE, Adams TS, Kothapalli N, Barnthaler T, Werder RB, et al. Characterization of the COPD alveolar niche using single-cell RNA sequencing.
Nat. Commun. 2022,
13, 494.
[Google Scholar]
-
98.
Wang C, Hyams B, Allen NC, Cautivo K, Monahan K, Zhou M, et al. Dysregulated lung stroma drives emphysema exacerbation by potentiating resident lymphocytes to suppress an epithelial stem cell reservoir.
Immunity 2023,
56, 576–591.e10.
[Google Scholar]
-
99.
Basil MC, Cardenas-Diaz FL, Kathiriya JJ, Morley MP, Carl J, Brumwell AN, et al. Human distal airways contain a multipotent secretory cell that can regenerate alveoli.
Nature 2022,
604, 120–126.
[Google Scholar]
-
100.
Thompson BT, Chambers RC, Liu KD. Acute Respiratory Distress Syndrome.
N. Engl. J. Med. 2017,
377, 562–572.
[Google Scholar]
-
101.
Wang Y, Tang Z, Huang H, Li J, Wang Z, Yu Y, et al. Pulmonary alveolar type I cell population consists of two distinct subtypes that differ in cell fate.
Proc. Natl. Acad. Sci. USA 2018,
115, 2407–2412.
[Google Scholar]
-
102.
Bhattarai P, Lu W, Hardikar A, Dey S, Gaikwad AV, Shahzad AM, et al. Endothelial to mesenchymal transition is an active process in smokers and patients with early COPD contributing to pulmonary arterial pathology.
ERJ Open Res. 2024,
10, 00767-2023.
[Google Scholar]
-
103.
Hogg JC. Pathophysiology of airflow limitation in chronic obstructive pulmonary disease.
Lancet 2004,
364, 709–721.
[Google Scholar]
-
104.
McDonough JE, Yuan R, Suzuki M, Seyednejad N, Elliott WM, Sanchez PG, et al. Small-airway obstruction and emphysema in chronic obstructive pulmonary disease.
N. Engl. J. Med. 2011,
365, 1567–1575.
[Google Scholar]
-
105.
Barnes PJ. Small airway fibrosis in COPD.
Int. J. Biochem. Cell Biol. 2019,
116, 105598.
[Google Scholar]
-
106.
Eapen MS, Lu W, Hackett TL, Singhera GK, Mahmood MQ, Hardikar A, et al. Increased myofibroblasts in the small airways, and relationship to remodelling and functional changes in smokers and COPD patients: potential role of epithelial-mesenchymal transition.
ERJ Open Res. 2021,
7, 00876-2020.
[Google Scholar]
-
107.
Michaeloudes C, Kuo CH, Haji G, Finch DK, Halayko AJ, Kirkham P, et al. Metabolic re-patterning in COPD airway smooth muscle cells.
Eur. Respir. J. 2017,
50, 1700202.
[Google Scholar]
-
108.
Araya J, Tsubouchi K, Sato N, Ito S, Minagawa S, Hara H, et al. PRKN-regulated mitophagy and cellular senescence during COPD pathogenesis.
Autophagy 2019,
15, 510–526.
[Google Scholar]
-
109.
Fang L, Zhang M, Li J, Zhou L, Tamm M, Roth M. Airway Smooth Muscle Cell Mitochondria Damage and Mitophagy in COPD via ERK1/2 MAPK.
Int. J. Mol. Sci. 2022,
23, 13987.
[Google Scholar]
-
110.
Krimmer DI, Burgess JK, Wooi TK, Black JL, Oliver BG. Matrix proteins from smoke-exposed fibroblasts are pro-proliferative.
Am. J. Respir. Cell. Mol. Biol. 2012,
46, 34–39.
[Google Scholar]
-
111.
Khedoe PP, van Schadewijk WA, Schwiening M, Ng-Blichfeldt JP, Marciniak SJ, Stolk J, et al. Cigarette smoke restricts the ability of mesenchymal cells to support lung epithelial organoid formation.
Front. Cell Dev. Biol. 2023,
11, 1165581.
[Google Scholar]
-
112.
Cheng PP, Yu F, Chen SJ, Feng X, Jia ZH, Hu SH, et al. PM2.5 exposure-induced senescence-associated secretory phenotype in airway smooth muscle cells contributes to airway remodeling.
Environ. Pollut. 2024,
347, 123674.
[Google Scholar]
-
113.
El Omar R, Beroud J, Stoltz JF, Menu P, Velot E, Decot V. Umbilical cord mesenchymal stem cells: the new gold standard for mesenchymal stem cell-based therapies?
Tissue Eng. Part B Rev. 2014,
20, 523–544.
[Google Scholar]
-
114.
Abbaszadeh H, Ghorbani F, Derakhshani M, Movassaghpour AA, Yousefi M, Talebi M, et al. Regenerative potential of Wharton's jelly-derived mesenchymal stem cells: A new horizon of stem cell therapy.
J. Cell Physiol. 2020,
235, 9230–9240.
[Google Scholar]
-
115.
Wang P, Cui Y, Wang J, Liu D, Tian Y, Liu K, et al. Mesenchymal stem cells protect against acetaminophen hepatotoxicity by secreting regenerative cytokine hepatocyte growth factor.
Stem Cell Res. Ther. 2022,
13, 94.
[Google Scholar]
-
116.
Chen Y, Huang H, Li G, Yu J, Fang F, Qiu W. Dental-derived mesenchymal stem cell sheets: a prospective tissue engineering for regenerative medicine.
Stem Cell Res. Ther. 2022,
13, 38.
[Google Scholar]
-
117.
Ridzuan N, Zakaria N, Widera D, Sheard J, Morimoto M, Kiyokawa H, et al. Human umbilical cord mesenchymal stem cell-derived extracellular vesicles ameliorate airway inflammation in a rat model of chronic obstructive pulmonary disease (COPD).
Stem Cell Res. Ther. 2021,
12, 54.
[Google Scholar]
-
118.
Antunes MA, Lapa e Silva JR, Rocco PR. Mesenchymal stromal cell therapy in COPD: from bench to bedside.
Int. J. Chron. Obstruct. Pulmon. Dis. 2017,
12, 3017–3027.
[Google Scholar]
-
119.
Yen BL, Yen ML, Wang LT, Liu KJ, Sytwu HK. Current status of mesenchymal stem cell therapy for immune/inflammatory lung disorders: Gleaning insights for possible use in COVID-19.
Stem Cells Transl. Med. 2020,
9, 1163–1173.
[Google Scholar]
-
120.
Feng B, Zhu J, Xu Y, Chen W, Sheng X, Feng X, et al. Immunosuppressive effects of mesenchymal stem cells on lung B cell gene expression in LPS-induced acute lung injury.
Stem Cell Res. Ther. 2020,
11, 418.
[Google Scholar]
-
121.
Wang YY, Li XZ, Wang LB. Therapeutic implications of mesenchymal stem cells in acute lung injury/acute respiratory distress syndrome.
Stem Cell Res. Ther. 2013,
4, 45.
[Google Scholar]
-
122.
Zhou Y, Zhou W, Li Y, Zhang J. MSCs regulate oxidative stress through the Nrf2 pathway to treat chronic obstructive pulmonary disease.
BMC Pulm. Med. 2025,
25, 304.
[Google Scholar]
-
123.
Cruz T, Lopez-Giraldo A, Noell G, Guirao A, Casas-Recasens S, Garcia T, et al. Smoking Impairs the Immunomodulatory Capacity of Lung-Resident Mesenchymal Stem Cells in Chronic Obstructive Pulmonary Disease.
Am. J. Respir. Cell. Mol. Biol. 2019,
61, 575–583.
[Google Scholar]
-
124.
Kruk DM, Wisman M, Noordhoek JA, Nizamoglu M, Jonker MR, de Bruin HG, et al. Paracrine Regulation of Alveolar Epithelial Damage and Repair Responses by Human Lung-Resident Mesenchymal Stromal Cells.
Cells 2021,
10, 2860.
[Google Scholar]
-
125.
Najar M, Martel-Pelletier J, Pelletier JP, Fahmi H. Novel insights for improving the therapeutic safety and efficiency of mesenchymal stromal cells.
World J. Stem Cells 2020,
12, 1474–1491.
[Google Scholar]
-
126.
Paris GC, Azevedo AA, Ferreira AL, Azevedo YM, Rainho MA, Oliveira GP, et al. Therapeutic potential of mesenchymal stem cells in multiple organs affected by COVID-19.
Life Sci. 2021,
278, 119510.
[Google Scholar]
-
127.
Jolly MK, Ward C, Eapen MS, Myers S, Hallgren O, Levine H, et al. Epithelial-mesenchymal transition, a spectrum of states: Role in lung development, homeostasis, and disease.
Dev. Dyn. 2018,
247, 346–358.
[Google Scholar]
-
128.
Dongre A, Weinberg RA. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer.
Nat. Rev. Mol. Cell Biol. 2019,
20, 69–84.
[Google Scholar]
-
129.
Kim DH, Xing T, Yang Z, Dudek R, Lu Q, Chen YH. Epithelial Mesenchymal Transition in Embryonic Development, Tissue Repair and Cancer: A Comprehensive Overview.
J. Clin. Med. 2017,
7, doi:10.3390/jcm7010001..
[Google Scholar]
-
130.
Knight DA, Grainge CL, Stick SM, Kicic A, Schuliga M. Epithelial Mesenchymal Transition in Respiratory Disease: Fact or Fiction.
Chest 2020,
157, 1591–1596.
[Google Scholar]
-
131.
Koczulla AR, Jonigk D, Wolf T, Herr C, Noeske S, Klepetko W, et al. Krüppel-like zinc finger proteins in end-stage COPD lungs with and without severe alpha1-antitrypsin deficiency.
Orphanet. J. Rare Dis. 2012,
7, 29.
[Google Scholar]
-
132.
Mahmood MQ, Walters EH, Shukla SD, Weston S, Muller HK, Ward C, et al. β-catenin, Twist and Snail: Transcriptional regulation of EMT in smokers and COPD, and relation to airflow obstruction.
Sci. Rep. 2017,
7, 10832.
[Google Scholar]
-
133.
Zheng L, Jiang YL, Fei J, Cao P, Zhang C, Xie GF, et al. Circulatory cadmium positively correlates with epithelial-mesenchymal transition in patients with chronic obstructive pulmonary disease.
Ecotoxicol. Environ. Saf. 2021,
215, 112164.
[Google Scholar]
-
134.
Shirahata T, Nakamura H, Nakajima T, Nakamura M, Chubachi S, Yoshida S, et al. Plasma sE-cadherin and the plasma sE-cadherin/sVE-cadherin ratio are potential biomarkers for chronic obstructive pulmonary disease.
Biomarkers 2018,
23, 414–421.
[Google Scholar]
-
135.
Mahmood MQ, Sohal SS, Shukla SD, Ward C, Hardikar A, Noor WD, et al. Epithelial mesenchymal transition in smokers: large versus small airways and relation to airflow obstruction.
Int. J. Chron. Obstruct. Pulmon. Dis. 2015,
10, 1515–1524.
[Google Scholar]
-
136.
Milara J, Peiró T, Serrano A, Cortijo J. Epithelial to mesenchymal transition is increased in patients with COPD and induced by cigarette smoke.
Thorax 2013,
68, 410–420.
[Google Scholar]
-
137.
Nomura T, Huang WC, Zhau HE, Josson S, Mimata H, Chung LW. β2-Microglobulin-mediated signaling as a target for cancer therapy.
Anticancer Agents Med. Chem. 2014,
14, 343–352.
[Google Scholar]
-
138.
De Cunto G, Brancaleone V, Riemma MA, Cerqua I, Vellecco V, Spaziano G, et al. Functional contribution of sphingosine-1-phosphate to airway pathology in cigarette smoke-exposed mice.
Br. J. Pharmacol. 2020,
177, 267–281.
[Google Scholar]
-
139.
Sharma P, Nag A. CUL4A ubiquitin ligase: a promising drug target for cancer and other human diseases.
Open Biol. 2014,
4, 130217.
[Google Scholar]
-
140.
Ren Y, Zhang Y, Fan L, Jiao Q, Wang Y, Wang Q. The cullin4A is up-regulated in chronic obstructive pulmonary disease patient and contributes to epithelial-mesenchymal transition in small airway epithelium.
Respir. Res. 2019,
20, 84.
[Google Scholar]
-
141.
Ornitz DM, Itoh N. The Fibroblast Growth Factor signaling pathway.
Wiley Interdiscip. Rev. Dev. Biol. 2015,
4, 215–266.
[Google Scholar]
-
142.
Yuan T, Volckaert T, Redente EF, Hopkins S, Klinkhammer K, Wasnick R, et al. FGF10-FGFR2B Signaling Generates Basal Cells and Drives Alveolar Epithelial Regeneration by Bronchial Epithelial Stem Cells after Lung Injury.
Stem Cell Rep. 2019,
12, 1041–1055.
[Google Scholar]
-
143.
Gupte VV, Ramasamy SK, Reddy R, Lee J, Weinreb PH, Violette SM, et al. Overexpression of fibroblast growth factor-10 during both inflammatory and fibrotic phases attenuates bleomycin-induced pulmonary fibrosis in mice.
Am. J. Respir. Crit. Care Med. 2009,
180, 424–436.
[Google Scholar]
-
144.
Volckaert T, Yuan T, Chao CM, Bell H, Sitaula A, Szimmtenings L, et al. Fgf10-Hippo Epithelial-Mesenchymal Crosstalk Maintains and Recruits Lung Basal Stem Cells.
Dev. Cell 2017,
43, 48–59.e5.
[Google Scholar]
-
145.
Moiseenko A, Vazquez-Armendariz AI, Kheirollahi V, Chu X, Tata A, Rivetti S, et al. Identification of a Repair-Supportive Mesenchymal Cell Population during Airway Epithelial Regeneration.
Cell Rep. 2020,
33, 108549.
[Google Scholar]
-
146.
Hadzic S, Wu CY, Avdeev S, Weissmann N, Schermuly RT, Kosanovic D. Lung epithelium damage in COPD - An unstoppable pathological event?
Cell Signal 2020,
68, 109540.
[Google Scholar]
-
147.
Kruk DM, Wisman M, Bruin HG, Lodewijk ME, Hof DJ, Borghuis T, et al. Abnormalities in reparative function of lung-derived mesenchymal stromal cells in emphysema.
Am. J. Physiol. Lung Cell. Mol. Physiol. 2021,
320, L832–L844.
[Google Scholar]
-
148.
Klar J, Blomstrand P, Brunmark C, Badhai J, Håkansson HF, Brange CS, et al. Fibroblast growth factor 10 haploinsufficiency causes chronic obstructive pulmonary disease.
J. Med. Genet. 2011,
48, 705–709.
[Google Scholar]
-
149.
Wu X, van Dijk EM, Ng-Blichfeldt JP, Bos IS, Ciminieri C, Königshoff M, et al. Mesenchymal WNT-5A/5B Signaling Represses Lung Alveolar Epithelial Progenitors.
Cells 2019,
8, 1147.
[Google Scholar]
-
150.
Su Y, Han W, Kovacs-Kasa A, Verin AD, Kovacs L. HDAC6 Activates ERK in Airway and Pulmonary Vascular Remodeling of Chronic Obstructive Pulmonary Disease.
Am. J. Respir. Cell. Mol. Biol. 2021,
65, 603–614.
[Google Scholar]
-
151.
Aravamudan B, Thompson M, Sieck GC, Vassallo R, Pabelick CM, Prakash YS. Functional Effects of Cigarette Smoke-Induced Changes in Airway Smooth Muscle Mitochondrial Morphology.
J. Cell Physiol. 2017,
232, 1053–1068.
[Google Scholar]
-
152.
Boucherat O, Chakir J, Jeannotte L. The loss of Hoxa5 function promotes Notch-dependent goblet cell metaplasia in lung airways.
Biol. Open 2012, 1, 677–691.
[Google Scholar]
-
153.
Danahay H, Pessotti AD, Coote J, Montgomery BE, Xia D, Wilson A, et al. Notch2 is required for inflammatory cytokine-driven goblet cell metaplasia in the lung.
Cell Rep. 2015,
10, 239–252.
[Google Scholar]
-
154.
Elias JA, Lee CG, Zheng T, Ma B, Homer RJ, Zhu Z. New insights into the pathogenesis of asthma.
J. Clin. Invest. 2003,
111, 291–297.
[Google Scholar]
-
155.
Hogg JC, Timens W. The pathology of chronic obstructive pulmonary disease.
Annu. Rev. Pathol. 2009,
4, 435–459.
[Google Scholar]
-
156.
Wilk JB, Chen TH, Gottlieb DJ, Walter RE, Nagle MW, Brandler BJ, et al. A genome-wide association study of pulmonary function measures in the Framingham Heart Study.
PLoS Genet. 2009,
5, e1000429.
[Google Scholar]
-
157.
Zhou X, Baron RM, Hardin M, Cho MH, Zielinski J, Hawrylkiewicz I, et al. Identification of a chronic obstructive pulmonary disease genetic determinant that regulates HHIP.
Hum. Mol. Genet. 2012,
21, 1325–1335.
[Google Scholar]
-
158.
Volckaert T, Campbell A, Dill E, Li C, Minoo P, De Langhe S. Localized Fgf10 expression is not required for lung branching morphogenesis but prevents differentiation of epithelial progenitors.
Development 2013,
140, 3731–3742.
[Google Scholar]
-
159.
Volckaert T, Dill E, Campbell A, Tiozzo C, Majka S, Bellusci S, et al. Parabronchial smooth muscle constitutes an airway epithelial stem cell niche in the mouse lung after injury.
J. Clin. Invest. 2011,
121, 4409–4419.
[Google Scholar]
-
160.
Tata PR, Mou H, Pardo-Saganta A, Zhao R, Prabhu M, Law BM, et al. Dedifferentiation of committed epithelial cells into stem cells in vivo.
Nature 2013,
503, 218–223.
[Google Scholar]
-
161.
Zhao R, Fallon TR, Saladi SV, Pardo-Saganta A, Villoria J, Mou H, et al. Yap tunes airway epithelial size and architecture by regulating the identity, maintenance, and self-renewal of stem cells.
Dev. Cell 2014,
30, 151–165.
[Google Scholar]
-
162.
Kooistra T, Saez B, Roche M, Egea-Zorrilla A, Li D, Anketell D, et al. Airway basal stem cells are necessary for the maintenance of functional intraepithelial airway macrophages.
Cell Rep. 2025,
44, 115860.
[Google Scholar]
-
163.
Wang Y, Meng Z, Liu M, Zhou Y, Chen D, Zhao Y, et al. Autologous transplantation of P63(+) lung progenitor cells for chronic obstructive pulmonary disease therapy.
Sci. Transl. Med. 2024,
16, eadi3360.
[Google Scholar]
-
164.
Chen Q, Hirai H, Chan M, Zhang J, Cho M, Randell SH, et al. Characterization of perivascular alveolar epithelial stem cells and their niche in lung homeostasis and cancer.
Stem Cell Rep. 2024,
19, 890–905.
[Google Scholar]
-
165.
Zhang J, Liu Y. Epithelial stem cells and niches in lung alveolar regeneration and diseases.
Chin. Med. J. Pulm. Crit. Care Med. 2024, 2, 17–26.
[Google Scholar]
-
166.
Hogan BL, Barkauskas CE, Chapman HA, Epstein JA, Jain R, Hsia CC, et al. Repair and regeneration of the respiratory system: complexity, plasticity, and mechanisms of lung stem cell function.
Cell Stem Cell 2014,
15, 123–138.
[Google Scholar]
-
167.
Adams TS, Schupp JC, Poli S, Ayaub EA, Neumark N, Ahangari F, et al. Single-cell RNA-seq reveals ectopic and aberrant lung-resident cell populations in idiopathic pulmonary fibrosis.
Sci. Adv. 2020,
6, eaba1983.
[Google Scholar]
-
168.
Habermann AC, Gutierrez AJ, Bui LT, Yahn SL, Winters NI, Calvi CL, et al. Single-cell RNA sequencing reveals profibrotic roles of distinct epithelial and mesenchymal lineages in pulmonary fibrosis.
Sci. Adv. 2020,
6, eaba1972.
[Google Scholar]
-
169.
Kadur Lakshminarasimha Murthy P, Sontake V, Tata A, Kobayashi Y, Macadlo L, Okuda K, et al. Human distal lung maps and lineage hierarchies reveal a bipotent progenitor.
Nature 2022,
604, 111–119.
[Google Scholar]
-
170.
Ahmadvand N, Carraro G, Jones MR, Shalashova I, Noori A, Wilhelm J, et al. Cell-Surface Programmed Death Ligand-1 Expression Identifies a Sub-Population of Distal Epithelial Cells Enriched in Idiopathic Pulmonary Fibrosis.
Cells 2022,
11, 1593.
[Google Scholar]
-
171.
Lv YQ, Cai GF, Zeng PP, Dhlamini Q, Chen LF, Chen JJ, et al. FGF10 Therapeutic Administration Promotes Mobilization of Injury-Activated Alveolar Progenitors in a Mouse Fibrosis Model.
Cells 2022,
11, 2396.
[Google Scholar]
-
172.
Chioccioli M, Liu S, Magruder S, Tata A, Borriello L, McDonough JE, et al. Stem cell migration drives lung repair in living mice.
Dev. Cell 2024,
59, 830–840.e4.
[Google Scholar]
-
173.
El Agha E, Thannickal VJ. The lung mesenchyme in development, regeneration, and fibrosis.
J. Clin. Invest. 2023,
133, e170498.
[Google Scholar]
-
174.
Wu J, Chu X, Chen C, Bellusci S. Role of Fibroblast Growth Factor 10 in Mesenchymal Cell Differentiation During Lung Development and Disease.
Front. Genet. 2018,
9, 545.
[Google Scholar]
-
175.
Konkimalla A, Konishi S, Macadlo L, Kobayashi Y, Farino ZJ, Miyashita N, et al. Transitional cell states sculpt tissue topology during lung regeneration.
Cell Stem Cell 2023,
30, 1486–1502.e9.
[Google Scholar]
-
176.
Higham A, Quinn AM, Cançado JE, Singh D. The pathology of small airways disease in COPD: historical aspects and future directions.
Respir. Res. 2019,
20, 49.
[Google Scholar]
-
177.
Yang J, Antin P, Berx G, Blanpain C, Brabletz T, Bronner M, et al. Guidelines and definitions for research on epithelial-mesenchymal transition.
Nat. Rev. Mol. Cell. Biol. 2020,
21, 341–352.
[Google Scholar]
-
178.
Kheirollahi V, Wasnick RM, Biasin V, Vazquez-Armendariz AI, Chu X, Moiseenko A, et al. Metformin induces lipogenic differentiation in myofibroblasts to reverse lung fibrosis.
Nat. Commun. 2019,
10, 2987.
[Google Scholar]
-
179.
Nasri A, Foisset F, Ahmed E, Lahmar Z, Vachier I, Jorgensen C, et al. Roles of Mesenchymal Cells in the Lung: From Lung Development to Chronic Obstructive Pulmonary Disease.
Cells 2021,
10, 3467.
[Google Scholar]
-
180.
Taghizadeh S, Heiner M, Vazquez-Armendariz AI, Wilhelm J, Herold S, Chen C, et al. Characterization in mice of the resident mesenchymal niche maintaining AT2 stem cell proliferation in homeostasis and disease.
Stem Cells 2021,
39, 1382–1394.
[Google Scholar]
-
181.
Fang Y, Chung SS, Xu L, Xue C, Liu X, Jiang D, et al. RUNX2 promotes fibrosis via an alveolar-to-pathological fibroblast transition.
Nature 2025,
640, 221–230.
[Google Scholar]