Photocatalyzed Thiocarbamylation of Alkenyl Radicals via Thiophene Salts

Photocatalyzed Thiocarbamylation of Alkenyl Radicals via Thiophene Salts
Bai-Xi He
1
Xin-Yu Lin
1
Ru-An Chi
2
Qing-Wen Han
2
Jing-Jing Cui
1,2
Zhi-Peng Guan
1,2,3
Zhi-Bing Dong
1,2,3,4,5,6,*

Received: 27 January 2026 Revised: 06 February 2026 Accepted: 25 February 2026 Published: 28 February 2026

© 2026 The authors. This is an open access article under the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/).
1. Introduction
Over the past decade, photochemistry [1,2,3,4] has blossomed into a powerful methodology due to its wide applicability in sustainable free-radical-mediated processes, in which light serves as a cleaner energy source to drive redox cycles with organic molecules, thereby enabling unique chemical transformations [5,6,7,8,9,10]. Numerous examples of highly selective aryl radical formation processes have been achieved in such a green and efficient approach [11,12,13,14,15]. In recent years, the emerging approach for generating vinyl radicals via visible-light drive involves vinyl halides bearing activated groups undergoing single-electron transfer (SET) with the excited state of metal- or organo-based photocatalysts to form vinyl radical precursors, which subsequently undergo oxidation or reduction quenching cycles [16,17,18,19]. However, compared to alkyl and aryl groups, similar applications for alkenyl groups have fallen behind.
This delay primarily stems from the extreme instability of the alkenyl radical intermediates and their strong tendency to abstract hydrogen atoms from suitable sources, such as solvents, leading to the formation of synthetically less valuable reduction products [20]. It was not until the pioneering work of Curran and Stork in the 1980s that the potential of these intermediates was revealed [21,22,23,24]. Building on this foundation, strategies for the direct reduction of vinyl halides rapidly advanced. Alternatively, alkenyl radicals might be formed by the addition of radicals to alkynes, which has been demonstrated in thermal and photochemical atom transfer radical addition (ATRA) processes, the latter mediated by either UV or visible light [25,26,27] (Figure 1a). Research by Oliver Reiser’s group indicates that activating substrates via sensitizer-mediated energy transfer is also feasible as an alternative to photoinduced single-electron transfer (SET) processes [28,29,30]. Such energy transfer processes (EnT) are characterized by a Dexter exchange reaction in which the excited state of the photocatalyst is deactivated to a lower-lying state by transfer of energy to the substrate, which is in turn raised to a higher-energy state [31] (Figure 1b). Subsequently, Manuel Plaza’s team also reported another photocatalytic pathway that achieves alkylation reactions through the participation of vinyl radicals, without requiring a catalyst. This process leverages the photochemical properties of halogen-bonded complexes to generate vinyl-centered radicals, thereby enabling the formation of various C–S bonds [32] (Figure 1c).
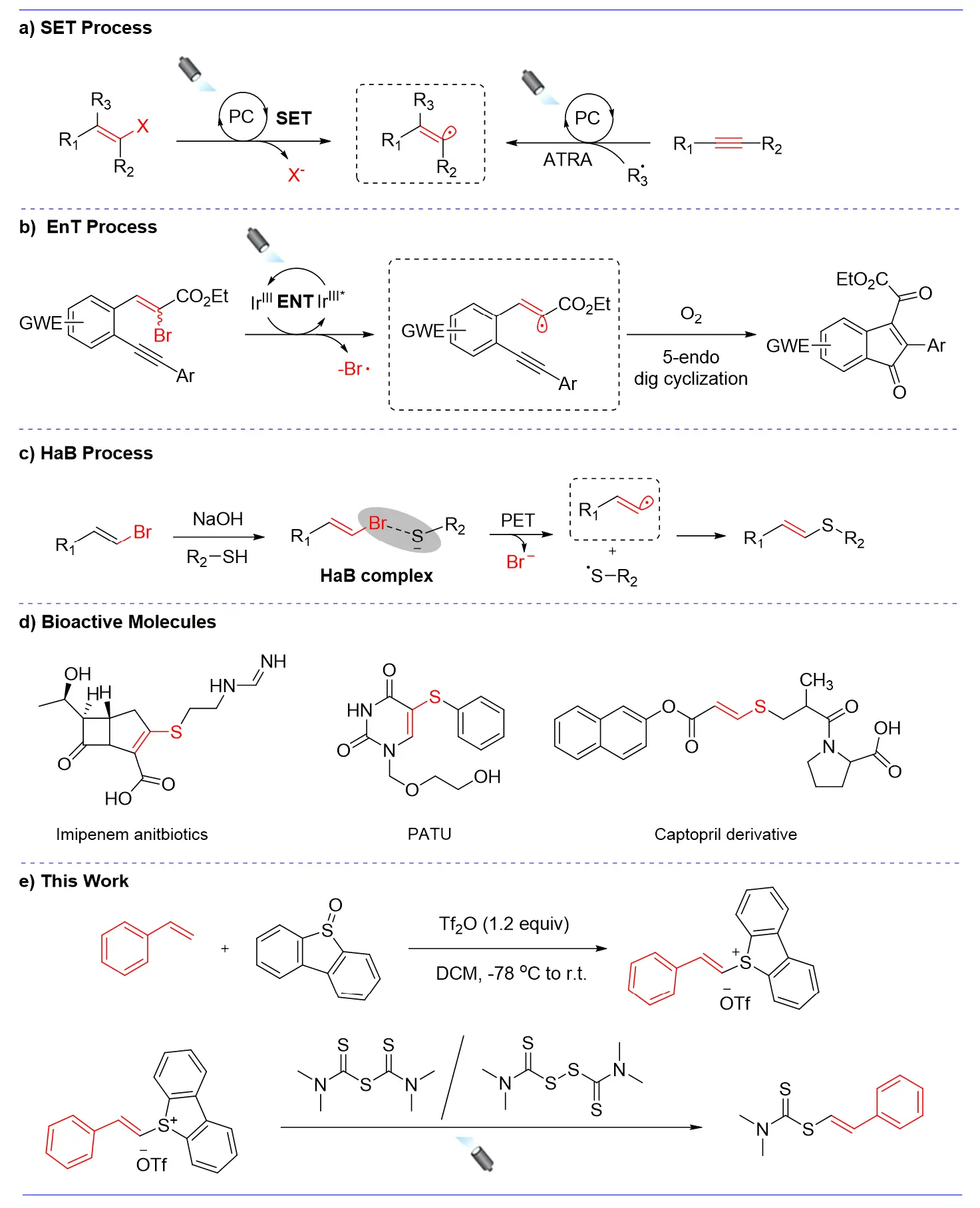
Figure 1. (a) Generation mechanism of alkenyl radicals: single-electron transfer process; (b) Energy transfer process; (c) The process utilizing the HaB complex as an intermediate; (d) Examples of drug molecules and bioactive molecules; (e) Our design.
In fact, the methods currently available for generating alkenyl radicals remain relatively limited, with most focusing on the formation of C–C bonds, while studies on C(sp2)–S bond construction are comparatively scarce. Given that candidate drugs such as imipenem [33], PATU [34], and captopril [35] are progressively entering clinical trials, the synthesis of alkenyl sulfides has garnered significant attention from chemists and pharmacologists (Figure 1d). These compounds can be used in materials science [36,37] and are cherished products of contemporary interest. Furthermore, alkenyl-thio groups serve as crucial intermediates in total syntheses [38] and as universal precursors for constructing diverse functionalized molecules during late-stage functionalization of drug molecules [39,40]. Consequently, developing novel, multifunctional synthetic approaches for alkenyl-thio compounds remains of enduring research significance.
Herein, we designed a mild and practical photoredox system that employs styrenyl thiophene salts as multifunctional modules to generate styrenyl radicals. This system successfully established C(sp2)–S bonds with both tetramethylthiuram disulfide (TMTD) and tetramethylthiuram monosulfide (TMTM) (Figure 1e). Compared to prior studies, our approach uses a novel thiophene salt as a substrate and features straightforward synthesis of the starting materials. Additionally, thiophene is inexpensive, readily available, and recyclable, demonstrating excellent atom economy. Furthermore, this strategy has been confirmed as a metal-free coupling reaction involving styrenyl radicals, opening an alternative way to the research of alkenyl radicals.
2. Materials and Methods
2.1. Materials Collection and Pretreatment
All reagents were obtained from commercial suppliers and used without further purification. Our bulbs and slides are purchased from Taobao. The lamp has a power of 30 W and a peak intensity wavelength of 465 nm. The distance from the light source to the irradiation vessel is about 1 cm. The reaction temperature is maintained at room temperature using a fan connected to the device.
2.2. General Procedure for the Synthesis of DBT Salt
Under an argon atmosphere, a 100 mL glass vial equipped with a magnetic stir bar was charged with dibenzothiophene-S-oxide (DBTO) (1.1 equiv.) and anhydrous DCM (50 mL). After cooling to −78 °C, trifluoromethanesulfonic anhydride (Tf2O) (1.2 equiv.) was added dropwise to the reaction mixture. Then the corresponding styrene (5 mmol, 1.0 equiv.) was added dropwise to the reaction system. The mixture was stirred at −78 °C for 1 h, then at room temperature for 4 h. Then the solvent was evaporated in vacuo and the residue washed with dry Et2O (3 × 30 mL) to obtain crude products as a solid. Further purification was achieved by column chromatography on silica with DCM/MeOH as the eluent.
2.3. Characterization
Reactions were monitored by thin layer chromatography (TLC) on a glass pate coated with silica gel with fluorescent indicator (GF254) using UV light. The 1H and 13C nuclear magnetic resonance (NMR) spectra were recorded on a Bruker ADNANCE III 500 MHz (Bruker company, Fällanden, Switzerland) using CDCl3 as solvent with TMS as internal standard. High-resolution mass spectrometric data (HRMS) were obtained using an Agilent 7200 Q-TOF (Agilent Technologies, Santa Clara, CA, USA) and a Bruker MicroTOF-Qll (Bruker company, Switzerland) (APCI, or Electrospray ionization, ESI). Gas Chromatography-Mass Spectrometry (GC-MS) was performed on an Agilent 7820A (Agilent Technologies, USA)with FID detection.
3. Results and Discussion
3.1. Reaction with TMTD
To efficiently synthesize the target compound 3a, we employed styrene thiophene salt 1a and TMTD 2a as model substrates. Under optimal conditions, using 4CzIPN as photocatalyst (5 mol%), KOH as base (2.0 equiv.), dichloromethane (DCM) as solvent, and 465 nm visible light irradiation, the reaction proceeded for 12 h under nitrogen atmosphere, yielding the best results. Here, we systematically investigated the effects of solvent, photocatalyst, base, and light source wavelength on the reaction.
Firstly, we evaluated different solvents (Table 1, Entries 1–8). The results showed that the target product 3a was obtained with an optimal yield of 70% in dichloromethane (DCM) (Entry 1). However, in solvents such as acetonitrile (MeCN), 1,2-dichloroethane (DCE), tetrahydrofuran (THF), methanol (MeOH), dimethyl acetate (DMAc), dimethyl sulfoxide (DMSO), and N,N-dimethylformamide (DMF), the target yield was lower than that in DCM. This indicates significant solvent dependence of the reaction, with DCM being the optimal choice. Subsequently, we further screened photocatalysts under DCM solvent conditions (Table 1, Entries 9–14). The catalytic activities of 9-phenylacridine, fac-Ir(ppy)3, Ru(bpy)3Cl2, Ru(bpy)3(PF6)2, and 4DPAIPN exhibited successively lower catalytic activities, while only trace amounts of 3a were detected with Eosin Y. This implies that the reducing power of 4CzIPN best matches the excited-state energy level requirements for this reaction. Additionally, we comprehensively assessed various organic and inorganic bases (Table 1, Entries 15–31). Among these, KOH exhibited the highest yield. And K2CO3, KOtBu, N,N-diisopropylethylamine (DIPEA), 4-dimethylaminopyridine (DMAP), triethylenediamine (DABCO), and N,N,N′,N′-tetramethylethylenediamine (TMEDA) also afforded moderate yields. However, weaker bases such as NaHCO3, NaOAc, Et3N, and pyridine obtained even lower results. To our surprise, 1,8-diazabicyclo[5,4,0]-7-undecene (DBU) and LiCl failed to promote the reaction at all, indicating that maybe both the strength and nucleophilicity of the base are crucial for the reaction. Moreover, we also investigated the effect of light source wavelength on the reaction (Table 1, Entries 32–34). The highest yield was achieved under 465 nm visible light irradiation. Yields decreased significantly at shorter wavelengths, such as 395 nm, 425 nm, and 445 nm. This trend closely matches the absorption spectrum of 4CzIPN, confirming 465 nm as the optimal excitation wavelength for this photocatalyst.
Table 1. Optimization of Conditions for TMTD Reaction.
 |
|||||
|---|---|---|---|---|---|
|
Entry |
Solvent (2 mL) |
Photocatalyst (5 mol%) |
Base (2 equiv.) |
Light (nm) |
Yield a |
|
1 |
DCM |
4CzIPN |
KOH |
465 |
70% |
|
2 |
MeCN |
4CzIPN |
KOH |
465 |
40% |
|
3 |
DCE |
4CzIPN |
KOH |
465 |
66% |
|
4 |
THF |
4CzIPN |
KOH |
465 |
57% |
|
5 |
MeOH |
4CzIPN |
KOH |
465 |
35% |
|
6 |
DMAc |
4CzIPN |
KOH |
465 |
50% |
|
7 |
DMSO |
4CzIPN |
KOH |
465 |
53% |
|
8 |
DMF |
4CzIPN |
KOH |
465 |
57% |
|
9 |
DCM |
9-Phenylacridine |
KOH |
465 |
30% |
|
10 |
DCM |
Eosin Y |
KOH |
465 |
trace |
|
11 |
DCM |
fac-Ir(ppy)3 |
KOH |
465 |
32% |
|
12 |
DCM |
Ru(bpy)3Cl2 |
KOH |
465 |
43% |
|
13 |
DCM |
Ru(bpy)3(PF6)2 |
KOH |
465 |
50% |
|
14 |
DCM |
4DPAIPN |
KOH |
465 |
28% |
|
15 |
DCM |
4CzIPN |
DBU |
465 |
N.R. b |
|
16 |
DCM |
4CzIPN |
K2CO3 |
465 |
55% |
|
17 |
DCM |
4CzIPN |
KOtBu |
465 |
50% |
|
18 |
DCM |
4CzIPN |
DIPEA |
465 |
48% |
|
19 |
DCM |
4CzIPN |
DMAP |
465 |
46% |
|
20 |
DCM |
4CzIPN |
NaHCO3 |
465 |
17% |
|
21 |
DCM |
4CzIPN |
NaOAc |
465 |
23% |
|
22 |
DCM |
4CzIPN |
Et3N |
465 |
19% |
|
23 |
DCM |
4CzIPN |
Cs2CO3 |
465 |
25% |
|
24 |
DCM |
4CzIPN |
Py |
465 |
15% |
|
25 |
DCM |
4CzIPN |
K3PO4 |
465 |
26% |
|
26 |
DCM |
4CzIPN |
Na2CO3 |
465 |
21% |
|
27 |
DCM |
4CzIPN |
Na2S2O8 |
465 |
trace |
|
28 |
DCM |
4CzIPN |
DABCO |
465 |
40% |
|
29 |
DCM |
4CzIPN |
TMEDA |
465 |
41% |
|
30 |
DCM |
4CzIPN |
CsF |
465 |
trace |
|
31 |
DCM |
4CzIPN |
LiCl |
465 |
N.R. |
|
32 |
DCM |
4CzIPN |
KOH |
395 |
30% |
|
33 |
DCM |
4CzIPN |
KOH |
425 |
35% |
|
34 |
DCM |
4CzIPN |
KOH |
445 |
40% |
Reaction conditions: 1a (0.2 mmol, 1 equiv.), 2a (0.6 mmol, 3 equiv.), PC (5 mol%), base (2 equiv.), in solvent (2 mL), room temperature, 12 h, LED (x nm).a Isolated yield of products. (see Supplementary Information Table S1, Table S2, Table S3, Table S4, Table S5, Table S6 and Table S7 for more details). b N.R. = No reaction.
3.2. Reaction with TMTM
To further optimize this thiocarbonylation reaction, we systematically investigated the effects of solvent, photocatalyst, and light source wavelength on reaction yield using thiocarbamate 1a and TMTM 2b as model substrates under a nitrogen atmosphere, aiming to obtain optimal reaction parameters. First of all, using 4CzIPN as the photocatalyst under 465 nm, we evaluated various solvents (Table 2, Entries 1–18). Results showed that the 3a was obtained in 80% yield in DCE, representing the highest yield among all solvents. MeCN, MeOH, H2O, PhCF3, PE, N-methylpyrrolidone (NMP), and Et3N yielded only trace amounts of product or no product at all. DCM, THF, DMAc, DMSO, DMF, acetone, and other solvents yielded the product in moderate to good yields, but all were lower than those obtained with DCE. Solvents such as ethyl acetate (EA), 1,4-dioxane, and toluene yielded lower results. These findings present significant solvent dependency in the reaction, with the non-protonic halogenated solvent DCE emerging as the optimal choice. Besides, after establishing DCE as the optimal solvent, photocatalysts were also screened (Table 2, Entries 19–27). Surprisingly, fac-Ir(ppy)3 yielded only 28%, while 4DPAIPN afforded 20%. Photocatalysts including [Ir(dtbbpy)(ppy)2](PF6), Ru(bpy)3Cl2, Ru(bpy)3(PF6)2, 9-Phenylacridine, Acridine, Rosebengalsodium, and Eosin Y detected only trace amounts of product (Table 2, Entries 20–26). These results indicate that the excited-state redox properties of 4CzIPN most closely match the electron transfer mechanism of this reaction. In addition, screening of the light source (Table 2, Entries 28–33) revealed the highest reaction yield under 465 nm visible light irradiation. Yields decreased significantly when the wavelength was shortened to 395 nm, 405 nm, 425 nm, or 445 nm. At 365 nm, only trace amounts of 3a were detected, while no product formation occurred at 550 nm. This trend closely aligns with the absorption spectrum of 4CzIPN, further validating 465 nm as the optimal excitation wavelength for this photocatalyst. In summary, the optimal reaction conditions are: 4CzIPN as photocatalyst (5 mol%), DCE as solvent, irradiation with 465 nm visible light, and reaction under a nitrogen atmosphere.
Table 2. Optimization of Conditions for TMTM Reaction.
 |
||||
|---|---|---|---|---|
|
Entry |
Solvent (2 mL) |
Photocatalyst (5 mol%) |
Light (nm) |
Yield a |
|
1 |
DCE |
4CzIPN |
465 |
80% |
|
2 |
MeCN |
4CzIPN |
465 |
trace |
|
3 |
DCM |
4CzIPN |
465 |
55% |
|
5 |
THF |
4CzIPN |
465 |
57% |
|
6 |
MeOH |
4CzIPN |
465 |
trace |
|
7 |
DMAc |
4CzIPN |
465 |
61% |
|
8 |
DMSO |
4CzIPN |
465 |
64% |
|
9 |
DMF |
4CzIPN |
465 |
75% |
|
10 |
Acetone |
4CzIPN |
465 |
61% |
|
11 |
EA |
4CzIPN |
465 |
16% |
|
12 |
H2O |
4CzIPN |
465 |
trace |
|
13 |
PhCF3 |
4CzIPN |
465 |
trace |
|
14 |
1,4-dioxane |
4CzIPN |
465 |
15% |
|
15 |
Toluene |
4CzIPN |
465 |
12% |
|
16 |
PE |
4CzIPN |
465 |
trace |
|
17 |
NMP |
4CzIPN |
465 |
trace |
|
18 |
Et3N |
4CzIPN |
465 |
trace |
|
19 |
DCE |
fac-Ir(ppy)3 |
465 |
28% |
|
20 |
DCE |
[Ir(dtbbpy)(ppy)2](PF6) |
465 |
trace |
|
21 |
DCE |
Ru(bpy)3Cl2 |
465 |
trace |
|
22 |
DCE |
Ru(bpy)3(PF6)2 |
465 |
trace |
|
23 |
DCE |
9-Phenylacridine |
465 |
trace |
|
24 |
DCE |
Acridine |
465 |
trace |
|
25 |
DCE |
Rosebengalsodium |
465 |
trace |
|
26 |
DCE |
Eosin Y |
465 |
trace |
|
27 |
DCE |
4DPAIPN |
465 |
20% |
|
28 |
DCE |
4CzIPN |
395 |
50% |
|
29 |
DCE |
4CzIPN |
405 |
54% |
|
30 |
DCE |
4CzIPN |
425 |
61 |
|
31 |
DCE |
4CzIPN |
445 |
50 |
|
32 |
DCE |
4CzIPN |
365 |
trace |
|
33 |
DCE |
4CzIPN |
550 |
N.R. b |
Reaction conditions: 1a (0.2 mmol, 1 equiv.), 2b (0.6 mmol, 5 equiv.), PC (5 mol%), in solvent (2 mL), room temperature, 12 h, LED (x nm). a Isolated yield of products. (see Supplementary Information Table S1, Table S2, Table S3, Table S4, Table S5, Table S6 and Table S7 for more details). b N.R. = No reaction.
3.3. Substrate Expansion and Control Experiments
Building upon the optimization of reaction conditions for TMTD and TMTM as radical acceptors, we successfully obtained the target product 3a containing a styrenyl sulfide structure with excellent yields of 70% and 80%. Notably, the reaction between TMTM and thiophene salts proceeds efficiently under photocatalysis without requiring any base. Research on vinyl radicals remains scarce, particularly methodologies for constructing C–S bonds via styrenyl radicals. Inspired by Wu’s group [12], we attempted to generate styrenyl radicals using styrenyl thiophene salts and successfully reacted them with TMTD/TMTM. This establishes a new strategy for organic synthesis methodologies involving styrenyl radical-mediated C–S bond formation and presents fresh possibilities for future developments.
We conducted further reactivity studies on other substrates under standard conditions using TMTD as the substrate, with irradiation by a 465 nm blue light in the presence of 4CzIPN and KOH. And we find that when using tetraethylthioacetamide 2c and tetrabenzylthioacetamide 2d as radical acceptors, the corresponding target alkenyl sulfides 3b and 3c were efficiently obtained, demonstrating the validity of our reaction paradigm (Figure 2a).
To elucidate the reaction mechanism, we conducted a series of control experiments and radical trapping experiments (Figure 2b, see Supplementary Information for details). When radical inhibitors 2,2,6,6-tetramethyl-1-piperinedinyloxy (TEMPO) and butylated hydroxytoluene (BHT) were added under standard conditions, the target compound 3a was not detected, confirming that the reaction is indeed radical-mediated. Moreover, we successfully captured alkenyl radicals using high-resolution spectroscopy. The reaction failed to proceed in the absence of light and nitrogen, demonstrating the necessity of both light and nitrogen.
As shown in Figure 2c, we scaled up this reaction paradigm. It is gratifying to note that when reacting 10 mmol of 1a with 30 mmol of 2a in a DCE solvent under blue light drive, catalyzed by 4CzIPN and KOH, the target product 3a was obtained in a 73% yield at the gram scale. This scale-up experiment demonstrates the synthetic utility of our designed reaction platform.
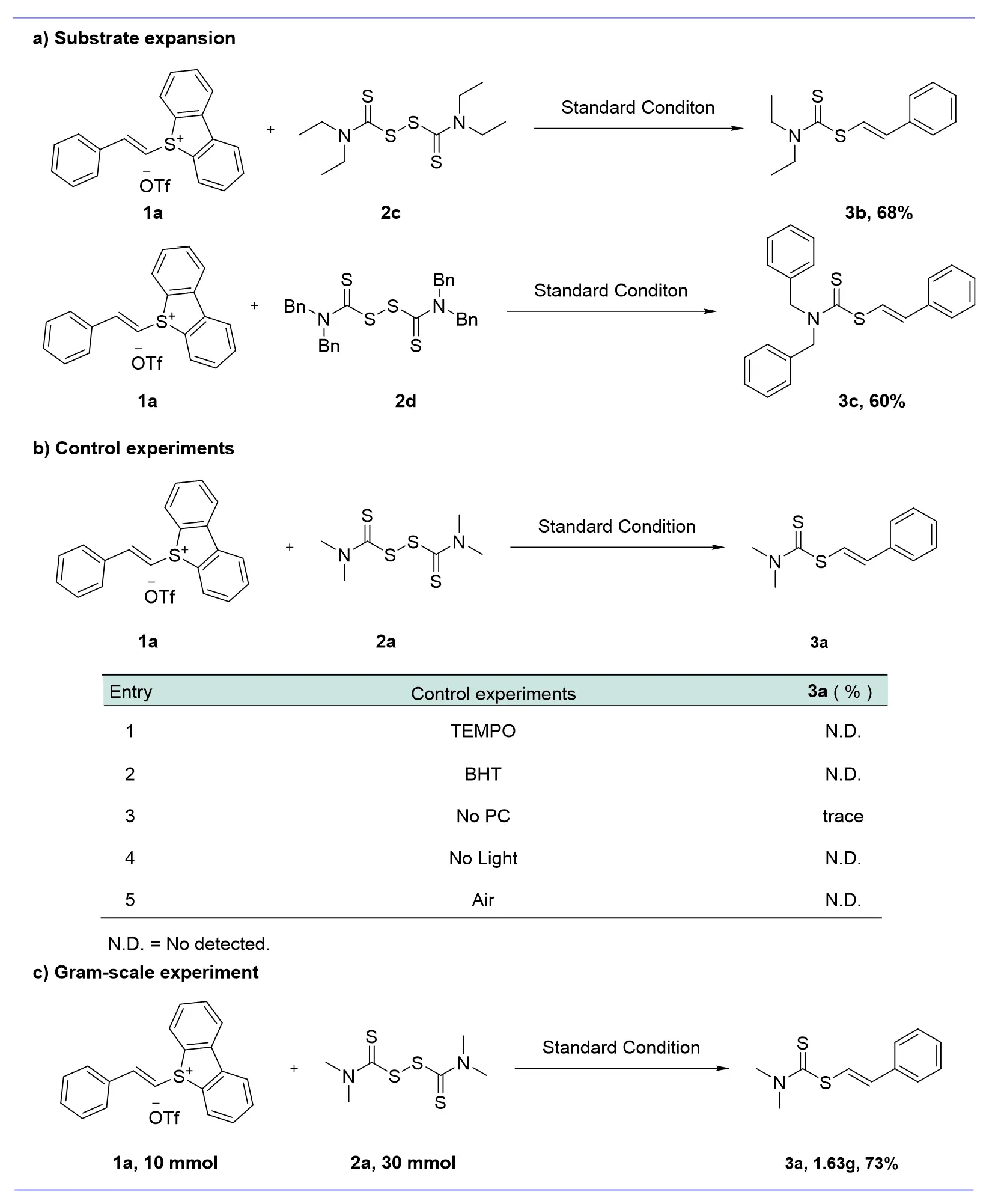
Figure 2. (a) Substrate expansion; (b) Control experiments; (c) Gram-scale experiment: 1a (10 mmol), 2a (30 mmol).
3.4. Proposed Mechanism
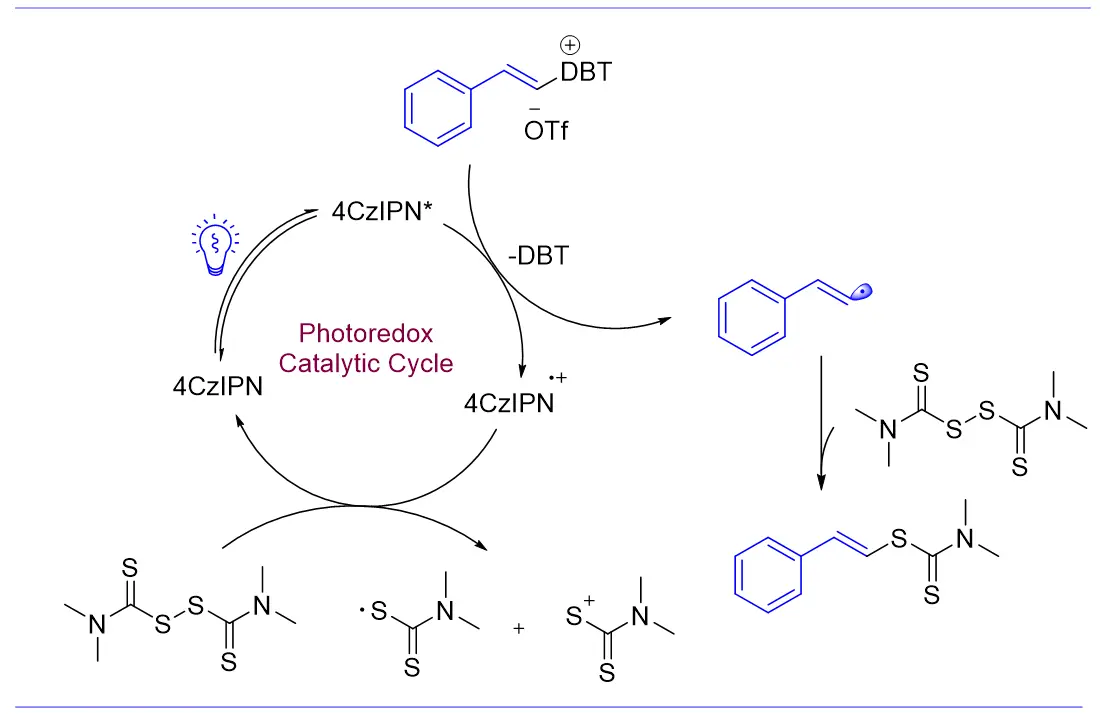
Based on the above studies, we proposed a possible reaction mechanism (Figure 3). Initially, 4CzIPN forms an excited state, 4CzIPN*, under light irradiation. 4CzIPN* undergoes a SET reaction with styrene thiophene salt, producing a radical cation of 4CzIPN and a styrene radical, while releasing recyclable thiophene. Subsequently, the styrene radical reacts with TMTD to produce the target product. The radical cation of 4CzIPN undergoes an SET reaction with TMTD, completing the catalytic cycle.
4. Conclusions
In summary, through a series of extensive and systematic optimization studies, we have achieved the radical coupling reaction between styrene thiophene salts and thioether compounds. Using 4CzIPN as the photocatalyst, styrene thiophene salts as radical donors, and thioamides as acceptors, the desired vinyl sulfide products were obtained in good yields under 465 nm blue light irradiation. Furthermore, our research demonstrates the successful development of a mild, modular photoredox system that directly generates valuable thiocarbamoyl compounds from readily accessible starting materials, specifically styrene. This strategy offers straightforward operation without requiring expensive additives or transition metals, and its mild reaction conditions hold promise as a novel tool in drug discovery and development.
Supplementary Materials
The following supporting information can be found at: https://www.sciepublish.com/article/pii/887, Table S1: Screening of Light (Without PC); Table S2: Screening of Time; Table S3: Screening of equivalence ratio of 1a:2a; Table S4: Screening of equivalence ratio of 1a:2b; Table S5: Screening of time; Table S6: Screening of solvent without PC; Table S7: Control experiments of TMTM.
Acknowledgements
We extend our heartfelt gratitude to the School of Chemistry and Environmental Engineering, Wuhan Institute of Technology, as well as Hubei Three Gorges Laboratory for projects support.
Author Contributions
Methodology, Formal Analysis, Investigation, Writing-Original Draft Preparation, B.-X.H.; Data Curation, Validation, Visualization, X.-Y.L. and J.-J.C.; Writing-Review & Editing, Supervision, Funding Acquisition, R.-A.C., Q.-W.H., Z.-P.G. and Z.-B.D.
Ethics Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All data generated or analyzed during this study are included in this published article.
Funding
This work was supported by the National Natural Science Foundation of China (22401223), the Natural Science Foundation of Hubei Province (2024AFB315), the Science and Technology Research Project of Hubei Provincial Department of Education (Q20231501), the Open and Innovation Fund of Hubei Three Gorges Laboratory (SC240004, SK240010), the Innovation Project of Engineering Research Center of Phosphorus Resources Development and Utilization of Ministry of Education (LCX202403, LCX202504), the Open Research Fund of School of Chemistry and Chemical Engineering, the Henan Normal University (2020ZD02), the Chemical Synthesis and Pollution Control Key Laboratory of Sichuan Province (CSPC202306), the Jiangsu Key Laboratory of Advanced Catalytic Materials and Technology (BM2012110), the Innovative Research Group Project of National Natural Science Foundation of Hubei (2025AFA049), and the Innovative Development Joint Fund of Natural Science Foundation of Hubei Province (2025AFD316), the Open Research Fund of Key Laboratory of Danjiangkou Reservoir Area’s Aquatic Eco-Environment and Health, Hanjiang Normal University (DK12505).
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
-
Zhu T, Wu C, Zhang X, Yang Z, Mu N, Zhou W, et al. Generation of 2,2-difluoro-4-sulfonylthiochromanes under visible light irradiation and their biological evaluations. Chin. Chem. Lett. 2026. DOI:10.1016/j.cclet.2026.112468 [Google Scholar]
-
Lin X-Y, Xie W-H, Zhou S, Hu S-Y, Yang B-Y, Cui J-J, et al. Photocatalyzed site-selective aryl C–H sulfinamidation via thianthracene salts. Green Synth. Catal. 2026. DOI:10.1016/j.gresc.2025.12.013 [Google Scholar]
-
Miao T-T, Liang R-B, Wang X, Xiao Y, Tong Q-X, Zhong J-J. Defluorinative alkene-carbonyl coupling enabled by photoredox catalysis: An access to allylic alcohols. Org. Lett. 2025, 28, 468–473. DOI:10.1021/acs.orglett.5c04919 [Google Scholar]
-
Liang R-B, Wang Y-J, Miao T-T, Wang Y-J, Wang S-W, Wang Q, et al. Photoredox radical-polar crossover reaction of gem-difluoroalkenes: A facile access to diverse difluoroalkyl sulfides. ChemCatChem 2024, 17, e202401556. DOI:10.1002/cctc.202401556 [Google Scholar]
-
Berger F, Plutschack MB, Riegger J, Yu W, Speicher S, Ho M, et al. Site-selective and versatile aromatic C−H functionalization by thianthrenation. Nature 2019, 567, 223–228. DOI:10.1038/s41586-019-0982-0 [Google Scholar]
-
Kornfilt DJP, MacMillan DWC. Copper-catalyzed trifluoromethylation of alkyl bromides. J. Am. Chem. Soc. 2019, 141, 6853–6858. DOI:10.1021/jacs.9b03024 [Google Scholar]
-
Sang R, Korkis SE, Su W, Ye F, Engl PS, Berger F, et al. Site-selective C−H oxygenation via aryl sulfonium salts. Angew. Chem. Int. Ed. 2019, 58, 16161–16166. DOI:10.1002/anie.201908718 [Google Scholar]
-
He F-S, Bao P, Tang Z, Yu F, Deng W-P, Wu J. Photoredox-catalyzed α-sulfonylation of ketones from sulfur dioxide and thianthrenium salts. Org. Lett. 2022, 24, 2955–2960. DOI:10.1021/acs.orglett.2c01132 [Google Scholar]
-
Jia H, Ritter T. α-Thianthrenium carbonyl species: The equivalent of an α-carbonyl carbocation. Angew. Chem. Int. Ed. 2022, 61, e202208978. DOI:10.1002/anie.202208978 [Google Scholar]
-
Dewanji A, van Dalsen L, Rossi-Ashton JA, Gasson E, Crisenza GEM, Procter DJ. A general arene C–H functionalization strategy via electron donor–acceptor complex photoactivation. Nat. Chem. 2023, 15, 43–52. DOI:10.1038/s41557-022-01092-y [Google Scholar]
-
Cai Y, Chatterjee S, Ritter T. Photoinduced copper-catalyzed late-stage azidoarylation of alkenes via arylthianthrenium salts. J. Am. Chem. Soc. 2023, 145, 13542–13548. DOI:10.1021/jacs.3c04016 [Google Scholar]
-
Zhang W, Liu T, Ang H-T, Luo P, Lei Z, Luo X, et al. Modular and practical 1,2-aryl(alkenyl) heteroatom functionalization of alkenes through iron/photoredox dual catalysis. Angew. Chem. 2023, 135, e202310978. DOI:10.1002/ange.202310978 [Google Scholar]
-
Liu T, Li T, Tea Z-Y, Wang C, Shen T, Lei Z, et al. Modular assembly of arenes, ethylene and heteroarenes for the synthesis of 1,2-arylheteroaryl ethanes. Nat. Chem. 2024, 16, 1705–1714. DOI:10.1038/s41557-024-01560-7 [Google Scholar]
-
Qi W, Gu S, Xie L-G. Reductive radical-polar crossover enabled carboxylative alkylation of aryl thianthrenium salts with CO2 and styrenes. Org. Lett. 2024, 26, 728–733. DOI:10.1021/acs.orglett.3c04183 [Google Scholar]
-
Yan F, Li Q, Fu S, Yang Y, Yang D, Yao S, et al. Single-electron-transfer-generated aryl sulfonyl ammonium salt: Metal-free photoredox-catalyzed modular construction of sulfonamides. ACS Catal. 2024, 14, 5227–5235. DOI:10.1021/acscatal.4c00816 [Google Scholar]
-
Sheng W, Shi Z, Li H, Xu Z, Liu Q, Chen J. Visible-light-induced cascade radical sulfonation/annulation/isomerization reactions of propargyl chalcones with sodium sulfinates: Regioselective synthesis of sulfonyl 2H-chromene and 2H-quinoline derivatives. Green Synth. Catal. 2025. DOI:10.1016/j.gresc.2025.11.007 [Google Scholar]
-
Zhang Y, Xu Z-Y, Fu R, Chen K, Hao W-J, Jiang B. Photocatalytic kharasch-type cyclization cascade for accessing polyhalogenated 1-indanones and indenes. Org. Lett. 2026, 28, 1386–1391. DOI:10.1021/acs.orglett.5c05236 [Google Scholar]
-
Zhang K, Zhang J, He Q, Hu J, Jing S. Synthesis of γ-iodo-allylic diboronic esters via atom transfer radical addition of (diborylmethyl)iodide to alkynes. Org. Lett. 2025, 27, 4152–4157. DOI:10.1021/acs.orglett.5c00730 [Google Scholar]
-
Tang J-B, Bian J-Q, Zhang Z, Cheng Y-F, Qin L, Gu Q-S, et al. Synthesis of axially chiral vinyl halides via Cu(I)-catalyzed enantioselective radical 1,2-halofunctionalization of terminal alkynes. ACS Catal. 2024, 15, 502–513. DOI:10.1021/acscatal.4c06672 [Google Scholar]
-
Piedra HF, Plaza M. Advancements in visible-light-induced reactions via alkenyl radical intermediates. Photochem. Photobiol. Sci. 2024, 23, 1217–1228. DOI:10.1007/s43630-024-00580-z [Google Scholar]
-
Curran DP, Shen W. Radical translocation reactions of vinyl radicals substituent effects on 1,5-hydrogen transfer reactions. J. Am. Chem. Soc. 2002, 115, 6051–6059. DOI:10.1021/ja00067a021 [Google Scholar]
-
Curran DP, Liu H. 4 + 1 Radical annulations with isonitriles a simple route to cyclopenta fused quinolines. J. Am. Chem. Soc. 1991, 113, 2127–2132. DOI:10.1021/ja00006a033 [Google Scholar]
-
Curran DP, Kim D, Liu HT, Shen W. Translocation of radical sites by intramolecular 1,5 hydrogen atom transfer. J. Am. Chem. Soc. 1988, 110, 5900–5902. DOI:10.1021/ja00225a052 [Google Scholar]
-
Baine GS. Cyclization of vinyl radicals a versatile method for the construction of five- and six-membered rings. J. Am. Chem. Soc. 1982, 104, 2321–2323. DOI:10.1021/ja00372a042 [Google Scholar]
-
Hossain A, Engl S, Lutsker E, Reiser O. Visible-light-mediated regioselective chlorosulfonylation of alkenes and alkynes: Introducing the Cu(II) complex [Cu(dap)Cl2] to photochemical atra reactions. ACS Catal. 2018, 9, 1103–1109. DOI:10.1021/acscatal.8b04188 [Google Scholar]
-
Kippo T, Hamaoka K, Ueda M, Fukuyama T, Ryu I. Radical bromoallylation of alkynes leading to 1-bromo-1,4-dienes. Tetrahedron 2016, 72, 7866–7874. DOI:10.1016/j.tet.2016.05.084 [Google Scholar]
-
Wille U. Radical cascades initiated by intermolecular radical addition to alkynes and related triple bond systems. Chem. Rev. 2012, 113, 813–853. DOI:10.1021/cr100359d [Google Scholar]
-
Pagire SK, Föll T, Reiser O. Shining visible light on vinyl halides: Expanding the horizons of photocatalysis. Acc. Chem. Res. 2020, 53, 782–791. DOI:10.1021/acs.accounts.9b00615 [Google Scholar]
-
Föll T, Rehbein J, Reiser O. Ir(ppy)3-catalyzed, visible-light-mediated reaction of α-chloro cinnamates with enol acetates: An apparent halogen paradox. Org. Lett. 2018, 20, 5794–5798. DOI:10.1021/acs.orglett.8b02484 [Google Scholar]
-
Paria S, Kais V, Reiser O. Visible light-mediated coupling of α-bromochalcones with alkenes. Adv. Synth. Catal. 2014, 356, 2853–2858. DOI:10.1002/adsc.201400638 [Google Scholar]
-
Pagire SK, Reiser O. Tandem cyclisation of vinyl radicals: A sustainable approach to indolines utilizing visible-light photoredox catalysis. Green Chem. 2017, 19, 1721–1725. DOI:10.1039/c7gc00445a [Google Scholar]
-
Piedra HF, Gebler V, Valdés C, Plaza M. Photochemical halogen-bonding assisted carbothiophosphorylation reactions of alkenyl and 1,3-dienyl bromides. Chem. Sci. 2023, 14, 12767–12773. DOI:10.1039/d3sc05263j [Google Scholar]
-
Gomis-Font MA, Cabot G, Sánchez-Diener I, Fraile-Ribot PA, Juan C, Moya B, et al. In vitro dynamics and mechanisms of resistance development to imipenem and imipenem/relebactam in pseudomonas aeruginosa. J. Antimicrob. Chemother. 2020, 75, 2508–2515. DOI:10.1093/jac/dkaa206 [Google Scholar]
-
Al Safarjalani ON, Zhou X-J, Rais RH, Shi J, Schinazi RF, Naguib FNM, et al. 5-(Phenylthio)acyclouridine: A powerful enhancer of oral uridine bioavailability: Relevance to chemotherapy with 5-fluorouracil and other uridine rescue regimens. Cancer Chemother. Pharmacol. 2005, 55, 541–551. DOI:10.1007/s00280-004-0967-y [Google Scholar]
-
Rastkari N, Khoobi M, Shafiee A, Khoshayand MR, Ahmadkhaniha R. Development and validation of a simple and sensitive HPLC-UV method for the determination of captopril in human plasma using a new derivatizing reagent 2-naphthyl propiolate. J. Chromatogr. B 2013, 932, 144–151. DOI:10.1016/j.jchromb.2013.06.019 [Google Scholar]
-
Zhou H, Zhang F, Wang R, Lai W-M, Xie S, Ren W-M, et al. Facile access to functionalized poly(thioether)s via anionic ring-opening decarboxylative polymerization of COS-sourced α-alkylidene cyclic thiocarbonates. Macromolecules 2021, 54, 10395–10404. DOI:10.1021/acs.macromol.1c01475 [Google Scholar]
-
Hu H, Niu G, Wang X, Liu X, Jiang J, Che L, et al. Thermodynamic control of multichannel emission in two-dimensional ruddlesden-popper perovskites. Chin. Chem. Lett. 2026, 112473. DOI:10.1016/j.cclet.2026.112473 [Google Scholar]
-
Li Q, Dong T, Liu X, Lei X. A bioorthogonal ligation enabled by click cycloaddition of o-quinolinone quinone methide and vinyl thioether. J. Am. Chem. Soc. 2013, 135, 4996–4999. DOI:10.1021/ja401989p [Google Scholar]
-
Zhai S, Zhang X, Cheng B, Li H, Li Y, He Y, et al. Synthesis of tetrasubstituted thiophenes via a [3+2] cascade cyclization reaction of pyridinium 1,4-zwitterionic thiolates and activated allenes. Chem. Commun. 2020, 56, 3085–3088. DOI:10.1039/d0cc00262c [Google Scholar]
-
Jeanne-Julien L, Astier E, Lai-Kuen R, Genta-Jouve G, Roulland E. Palladium nanoparticle-catalyzed stereoretentive cross-coupling of alkenyl sulfides with grignard reagents. Org. Lett. 2018, 20, 1430–1434. DOI:10.1021/acs.orglett.8b00208 [Google Scholar]