Hydrogenative Depolymerization of Polyesters Catalyzed by a PN3-Ruthenium Complex Using Both H2 and EtOH as Hydrogen Sources

Hydrogenative Depolymerization of Polyesters Catalyzed by a PN3-Ruthenium Complex Using Both H2 and EtOH as Hydrogen Sources
Yiwei Rao
1,†
Ruiqin Wang
1,†
Li Chen
1
Yanqin Peng
1
Weiran Yang
1
Zhongbao Jian
2
Changguang Yao
1,*

Received: 15 October 2025 Revised: 22 October 2025 Accepted: 11 November 2025 Published: 19 November 2025

© 2025 The authors. This is an open access article under the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/).
1. Introduction
Plastic has become an indispensable type of polymer material in human life due to its advantages, such as easy manufacturing, low cost, good barrier properties, chemical and temperature resistance [1]. The wide use of plastic products has led to their annual production proliferating, as well as the accumulation of a large amount of plastic waste, which poses a deadly threat to life systems [2,3,4]. Over the past 50 years, more than 8 billion tons of plastic waste have accumulated, and it is expected that 12 billion tons by 2050 [1,5]. Plastics degrade very slowly in the natural environment conditions, and the released microplastic fragments and toxic chemicals can cause a threat to public health [6,7,8]. Therefore, it is urgent to develop effective methods to address the negative impact of plastic pollution on human society and the natural environment [9].
Polyester is an important member of the plastic family. Compared with polyolefins, their backbones contain the labile ester linkages, which provide the possibility for degradation and recycling [10,11]. Some polyesters can even degrade in natural environments [3]. However, the production of biodegradable polyester is very low, and the value of the waste polyesters cannot be restored by this method [12,13,14,15]. Therefore, developing green and efficient methods to achieve the controlled recycling of waste polyesters has gradually become a hot topic in both academia and industry [9,16,17].
Traditional methods for polyester degradation, including hydrolysis [18], glycolysis[19,20], alcoholysis [21] and aminolysis [22] generally require the usage of a great deal of solvents, degradation agents, and high reaction pressure. The separation of degradation products from the oligomeric byproducts is often very laborious [23,24]. In recent years, hydrogenative depolymerization has emerged as an excellent alternative method for waste polyesters degradation to produce diols, which cannot be obtained through traditional recycling processes [16,17]. Hydrogenative depolymerization using hydrogen gas as a reducing agent is an atom-economic, green, and sustainable chemical conversion pathway, as H2 can be obtained from renewable resources and does not produce stoichiometric waste during the depolymerization process [16]. The typical catalysts for polyester hydrogenative depolymerization are summarized in Figure 1.
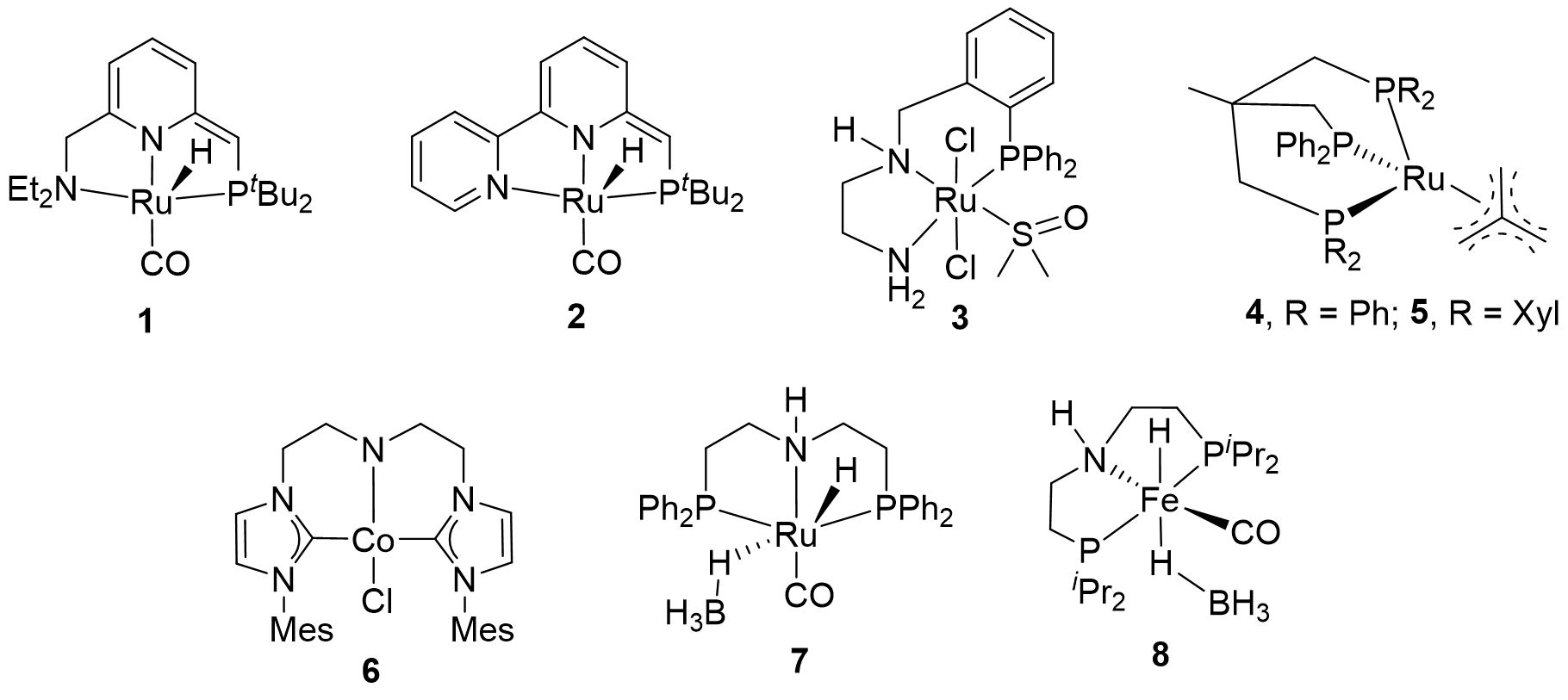
In Robertson and coworkers’ seminal report, they used the versatile Milstein catalysts 1 and 2 to implement the hydrogenative depolymerization of polyesters [25]. Poly(1,10-decanediol) [26] obtained from the dehydrogenative polymerization of 1,10-decanediol was successfully reduced to the corresponding diol in 80% isolated yield under 13.6 atm of hydrogen pressure at 120 °C in the presence of 1. Unfortunately, 1 was unable to depolymerize PLA and fully depolymerize PET even under the elevated temperature (160 °C) and H2 pressure (54.4 atm) conditions. However, PET, ε-CL and polycarbonates could be completely depolymerized into various diols using 2 as a catalyst, probably due to the less sterically bulky dipyridyl backbone [16]. Interestingly, the hydrogenative depolymerization of poly(R-3-hydroxybutyric acid) and poly(3-hydroxypropionic acid) catalyzed by 2 produced only butyric acid and propionic acid. Recently, they used the commercially available Ru-MACHO-BH 7 to catalyze polybutylene succinate depolymerization, and both γ-butyrolactone and 1,4-butanediol were obtained through tuning the hydrogen pressure [27].
Clarke et al. demonstrated the hydrogenation of PET flakes in the presence of 3 and KOtBu under 50 bar of H2 and 110 °C, affording 1,4-benzenedimethanol (BDM) and ethylene glycol (EG) in 53% isolated yield [28]. In 2018, the Klankermayer group investigated the hydrogenative depolymerization of several types of waste polyesters into the corresponding diols catalyzed by 4 and 5 under the activation of bis(trifluoromethanesulfonyl)imide (HNTf2). 4/HNTf2 can catalyze PLA and PCL samples to give 1,2-propanediol and 1,6-hexanediol in 99% yields under 10 MPa of H2 at 140 °C for 16 h. But this system was low efficient for PET degradation, probably arising from the formation of ether byproducts. Replacing the phenyl group in 4 with 3,5-dimethylphenyl, the catalytic counterpart 5/HNTf2 was competent for PET and PBT depolymerization into the relevant diols [29].
In 2019, Liu et al. reported on the hydrogenative depolymerization of PCL. By in-situ mixing of complex 6 and KOtBu, the yield of 1,6-hexanediol was as high as 97% after 16 h of reaction at 30 bar H2 and 100 °C. This is the first example of a base metal non-phosphine complex catalyzing the hydrogenation of polyester to diols, and the authors have conducted in-depth research on the hydrogenation mechanism [30].
Recently, Xie and coworkers developed a novel transesterification/hydrogenation relay strategy and designed a new class of quinaldine-based ruthenium-pincer catalysts with high compatibility with protic solvents, which enables highly efficient hydrogenative polyesters depolymerization in CH3OH. With this protocol, a broad range of commercially available or post-consumer PET sources, and other types of polyester plastics were degraded in excellent yields under very mild conditions [31].
Although the study on dehydrogenative depolymerization of polyesters using H2 as the hydrogen source has made significant progress, it usually involves the use of an expensive high-pressure reactor. To avoid this, de Vries and coworkers reported on the use of the Fe-iPrMACHO-BH complex 8 in the base-free transfer hydrogenation of polyesters into diols with EtOH as a hydrogen source. Under the optimal conditions, 1,6-hexanediol was obtained in 87% isolated yield through depolymerizing Dynacol 7360. This study opened the possibility to recycle polyesters by deconstructing them to their monomers using a completely new, easy, and green strategy [32].
Research in our group is predominantly concerned with waste polyester recycling and preparation of new polymeric materials by utilizing a series of phosphorus-nitrogen pincer complexes [33,34,35]. Herein, we demonstrate an air- and moisture-stable ruthenium PN3-pincer complex, which exhibits excellent activity for the hydrogenation of esters to alcohols [36], catalyzing polyesters upcycling to respective diols under mild conditions by using both H2 and EtOH as hydrogen sources.
2. Materials and Methods
All experiments (if not mentioned otherwise) with metal complexes were carried out under an atmosphere of dry nitrogen in a glovebox or using standard Schlenk techniques. All glassware was rigorously dried. All solvents were distilled from sodium benzophenone ketyl before use. All other chemicals were commercially available and used as received. PN3-Ru complex was prepared according to the literature procedure [36]. NMR spectra were recorded at 400 MHz (1H) and 101 MHz (13C) using a Bruker Avance-400 NMR spectrometer. 1H NMR chemical shifts were referenced to the residual hydrogen signals of the deuterated solvents (7.26 ppm, CDCl3; DMSO-d6, 2.50 ppm), and the 13C NMR chemical shifts were referenced to the 13C signals of the deuterated solvents (CDCl3, 77.16 ppm; DMSO-d6, 39.52 ppm). The data of quantitative ¹³C NMR spectra were acquired with more than 50 mg of depolymerization products solubilized in 0.5 mL of deuterated solvent. All spectra were measured at 25 °C using a 90° pulse (zgig), inverse-gated decoupling, at least 2000 scans, and a delay of 8 s for samples run without relaxation reagent.
2.1. Typical Procedure for Hydrogenative Depolymerization of Polyester with H2
In the glovebox, PN3-Ru complex (24.0 mg, 0.05 mmol), KOtBu (44.9 mg, 0.40 mmol), and solvent (2.0 mL) were added to a glass vial, and the mixture was stirred for 5 min at room temperature to activate the catalyst precursor. Then the mixture was transferred to a high-pressure stainless steel autoclave, equipped with a stir bar, and subsequently polyester (2.0 mmol, referring to its repetition unit) was added. Once the autoclave was sealed and pressurized with hydrogen, the reaction mixture was stirred for several hours at the set temperature. Afterwards, the reaction was cooled in an ice bath, and the pressure was carefully released. The crude reaction mixture was analyzed by 1H and quantitative 13C NMR spectroscopy.
2.2. Hydrogenative Depolymerization of the Mixture of PLA and PET
In the glovebox, PN3-Ru complex (24.0 mg, 0.05 mmol), KOtBu (44.9 mg, 0.40 mmol), and solvent (2.0 mL) were added to a glass vial, and the mixture was stirred for 5 min at room temperature to activate the catalyst precursor. Then the mixture was transferred to a high-pressure stainless steel autoclave, equipped with a stir bar, and subsequently the mixture of PLA and PET (1.0 mmol, 1.0 mmol, respectively, referring to its repetition unit) was added. The autoclave was sealed and pressurized with hydrogen (1 MPa), and the reaction mixture was stirred for 2 h at room temperature. Afterwards, the reaction was cooled in an ice bath, and the pressure was carefully released. The crude reaction mixture was filtered, washed with a little toluene, and the filtrate was analyzed by 1H and quantitative 13C NMR spectroscopy. Then the residual solid, and another dosage of activated catalyst were added to another autoclave again, filled with 4MPa H2 and reacted at 80 °C for 3 h. The crude reaction mixture was also analyzed by 1H and quantitative 13C NMR spectroscopy.
2.3. Typical Procedure for Hydrogenative Depolymerization of Polyester with Ethanol
In the glovebox, PN3-Ru complex (24.0 mg, 0.05 mmol), KOtBu (44.9 mg, 0.40 mmol), polyester (2.0 mmol, referring to its repetition unit), and ethanol (2.0 mL) were added to a Schlenk flask with a stir bar, and subsequently the flask was sealed and stirred for several hours at the set temperature. Afterwards, the reaction was cooled in an ice bath. The crude reaction mixture was analyzed by 1H and quantitative 13C NMR spectroscopy.
3. Results and Discussion
3.1. Hydrogenative Depolymerization of PLA
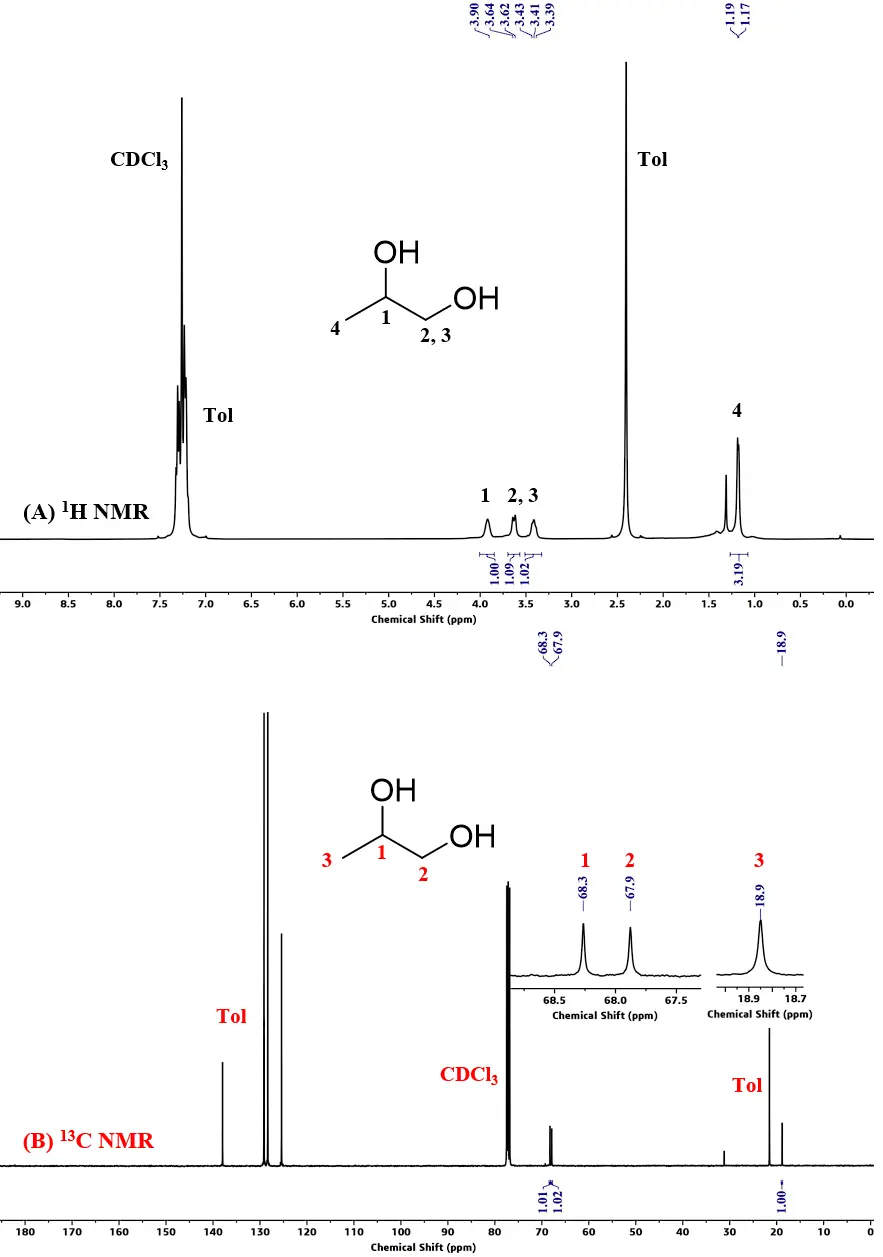
In Robertson’s seminal work, polyesters’ depolymerization needed a high temperature (>120 °C) and high pressure of hydrogen for a long reaction time [25]. Therefore, we started our experiment on PLA degradation in toluene under 4 MPa H2 pressure at 120 °C using 2.5 mol% of PN3-Ru and 20 mol% KOtBu as activator. After 48 h, the polymer was converted to 1,2-propanediol (PG) quantitatively. As shown in Figure 2A, the signals at 3.90, 3.60 and 3.40 ppm in the 1H NMR spectrum of the reaction mixture after the reaction completion are attributed to the methine and methylene protons of PG, and the methyl protons resonate at 1.15 ppm. Correspondingly, the three carbons of PG could be found in the quantitative 13C NMR spectrum (Figure 2B) at 68.3, 67.9 and 18.9 ppm with the integral ratio of 1:1:1. No PLA residual was found in both the 1H and 13C NMR spectra. Reducing the reaction time to 6 h, PLA was still completely converted. Further optimizing the reaction conditions, we found that PG was generated in more than 99% yield under 1 MPa H2 pressure at room temperature for 2 h. Declining the hydrogen pressure to 0.5 MPa or shortening the reaction time to 1 h led to the incomplete conversion (Figure S2 and Figure S3). In the absence of H2, no product was detected. A slight decrease in the yield (96%) was observed when reducing the catalyst loading to 2.0 mol%, and the tendency further increased when reducing the catalyst loading to 1.25 mol%. Interestingly, decreasing the feeding ratio of PN3-Ru: KOtBu to 1:4, the yield of PG dramatically declined to 74%, and nearly no product was observed under the ratio of 1:1. As shown in Table 1, an excess of base is needed, indicating the role of KOtBu include not only activating the pre-catalyst, but also scavenging the trace moisture from the reaction system. This is consistent with the reports in the literature [37,38,39]. When KOtBu was replaced by K2CO3 (Table 1, Entry 21), there was also no product observed under the ratio of PN3-Ru: K2CO3 to 1:8.
Table 1. Hydrogenative depolymerization of PLA with PN3-Ru complex a.
 |
|||||
|---|---|---|---|---|---|
|
Entry |
solv. (mL) |
PH2 (MPa) |
t (h) |
T (°C) |
Yield (%) b |
|
1 |
Toluene |
4 |
48 |
120 |
>99 |
|
2 |
Toluene |
4 |
6 |
120 |
>99 |
|
3 |
Toluene |
3 |
6 |
120 |
>99 |
|
4 |
Toluene |
2 |
6 |
120 |
>99 |
|
5 |
Toluene |
1 |
6 |
120 |
>99 |
|
6 |
Toluene |
1 |
6 |
100 |
>99 |
|
7 |
Toluene |
1 |
6 |
80 |
>99 |
|
8 |
Toluene |
1 |
6 |
60 |
>99 |
|
9 |
Toluene |
1 |
6 |
r.t. |
>99 |
|
10 |
Toluene |
1 |
3 |
r.t. |
>99 |
|
11 |
Toluene |
1 |
2 |
r.t. |
>99 |
|
12 |
Toluene |
1 |
1 |
r.t. |
86 |
|
13 |
Toluene |
0.5 |
2 |
r.t. |
83 |
|
14 c |
Toluene |
- |
2 |
r.t. |
N.R.d |
|
15 e |
Toluene |
1 |
2 |
r.t. |
96 |
|
16 f |
Toluene |
1 |
2 |
r.t. |
46 |
|
17 g |
Toluene |
1 |
2 |
r.t. |
74 |
|
18 h |
Toluene |
1 |
2 |
r.t. |
Trace |
|
19 i |
Toluene |
1 |
2 |
r.t. |
N.R. |
|
20 j |
Toluene |
1 |
2 |
r.t. |
N.R. |
|
21 k |
Toluene |
1 |
2 |
r.t. |
N.R. |
|
22 |
1,4-Dioxane |
1 |
2 |
r.t. |
>99 |
|
23 |
Anisole |
1 |
2 |
r.t. |
>99 |
|
24 |
THF |
1 |
2 |
r.t. |
>99 |
|
25 l |
Toluene |
1 |
2 |
r.t. |
>99 |
a Condition: PN3-Ru (0.05 mmol), KOtBu (0.40 mmol), solvent (2.0 mL), PLA (2.0 mmol, calculated on the repetition unit). PN3-Ru was activated with KOtBu for 5 min before transferring to an autoclave containing PLA. b yield was determined by 1H and quantitative 13C NMR spectra. c N2 (1 MPa). d N.R. stands for No Reaction. e [PLA]:[PN3-Ru]:[KOtBu] = 50:1:8. f [PLA]:[PN3-Ru]:[KOtBu] = 80:1:8. g [PLA]:[PN3-Ru]:[KOtBu] = 40:1:4. h [PLA]:[PN3-Ru]:[KOtBu] = 40:1:1. i [PLA]:[PN3-Ru]:[KOtBu] = 40:1:0. j [PLA]:[PN3-Ru]:[KOtBu] = 40:0:8. k KOtBu was placed by K2CO3. l PN3-Ru (0.25 mmol), KOtBu (2.0 mmol), solvent (8.0 mL), PLA (1.44 g, 20.0 mmol, calculated on the repetition unit), PN3-Ru activated with KOtBu for 5 min before transferring to an autoclave containing PLA.
Moreover, the PN3-Ru complex or KOtBu could not catalyze individually the hydrogenative depolymerization. Switching the solvent to 1,4-dioxane, anisole, and THF, the reaction proceeded smoothly in a quantitative yield. These results disclosed that the PN3-Ru catalytic system is very efficient for PLA depolymerization into PG. For a larger-scale experiment (PLA, 1.44 g), the PG was also successfully obtained in a 99% yield (Table 1, Entry 25), demonstrating that the catalytic system has a promising prospect for industrial application.
3.2. Hydrogenative Depolymerization of Other Polyesters
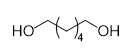
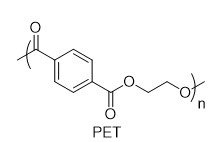
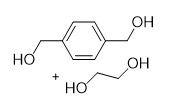
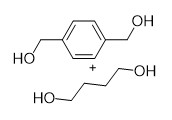
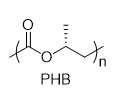
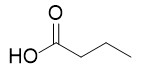
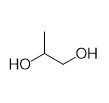
With this active catalytic system in hand, we set out to investigate the hydrogenative depolymerization of other polyesters. As shown in Table 2, under the optimal conditions, the aliphatic polyester PCL could be converted to 1,6-hexanediol in a little longer time (Table 2, Entry 1). However, depolymerization of PBS needed higher H2 pressure (4 MPa), higher temperature (80 °C) and a longer reaction time (3 h) to achieve complete conversion (Table 2, Entry 2). Under the same conditions as PBS, aromatic polyesters PET and PBT could also be degraded to the respective diols in more than 99% yields (Table 2, Entries 4 and 5). Similar to Robertson’s report [25] PHB depolymerized to butyric acid in 50% yield rather than 1,3-butanediol, probably arising from the formation of a transient six-membered ring and which subsequently generates crotonyl chain-end via β-elimination. The crotonyl chain-end is relatively stable because of the conjugation effect of the carbon-carbon double bond and the ester bond, and further transforms into crotonic acid, which is finally hydrogenated into butyric acid by the ruthenium complex (Figure S33). Based on these results, we further investigated the upcycling of the post-consumer PLA and PET products. The PLA sucker and PET bottle were cut into flakes and directly subjected to the autoclaves without any further pretreatment (Figure 3). Under the identical reaction conditions, the corresponding diols were generated nearly quantitatively, indicating the tolerance of the catalyst for additives and impurities in the consumer products.
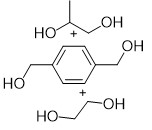
Table 2. Hydrogenative depolymerization of various polyesters with PN3-Ru complex a.
|
Entry |
Polyester |
PH2 (MPa) |
t (h) |
T (°C) |
Product(s) |
Yield (%) b |
|---|---|---|---|---|---|---|
|
1 |
 |
1 |
3 |
r.t. |
 |
>99 |
|
2 |
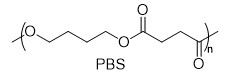 |
4 |
3 |
80 |
 |
>99 |
|
3 |
 |
4 |
3 |
80 |
 |
>99 |
|
4 |
 |
4 |
3 |
80 |
 |
>99 |
|
5 |
 |
4 |
6 |
120 |
 |
50 |
|
6 |
post-consumer PLA |
1 |
2 |
r.t. |
 |
>99 |
|
7 |
post-consumer PET |
4 |
3 |
80 |
|
95 |
|
8 |
PLA, PET |
- |
- |
- |
 |
99 |
|
9 |
PLA, PET, PP, PS, PE |
- |
- |
- |
|
99 |
a Condition: PN3-Ru (0.05 mmol), KOtBu (0.40 mmol), Toluene (2.0 mL), polyesters (2.0 mmol, calculated on the repetition unit). PN3-Ru was activated with KOtBu for 5 min before transferring to an autoclave containing polyester. b yield was determined by 1H and quantitative 13C NMR spectra.

Figure 3. (a) Post-consumer PLA sulker. (b) fragment of (a). (c) Post-consumer PET bottle. (d) fragment of (c).
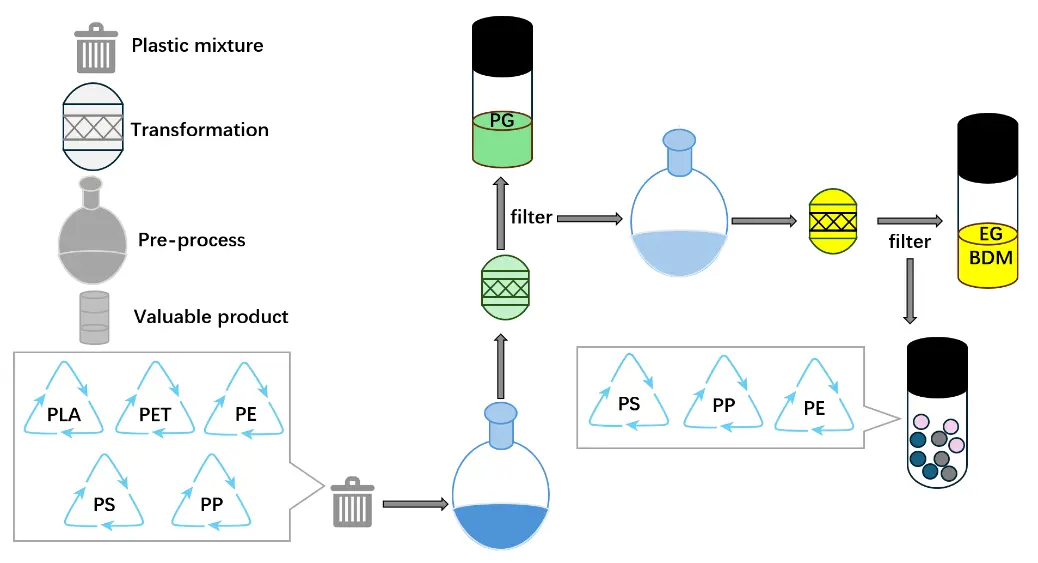
In view of the different degradation temperatures and H2 pressure of PLA and PET, we then started to separate their hydrogenation products. Firstly, 1.0 mmol PET and 1.0 mmol PLA were mixed and reacted at room temperature under 1 MPa H2 for 2 h (Table, Entry 8), and the autoclave was subsequently transferred to a glovebox. The NMR-monitoring experiment disclosed the full conversion of PLA, with marginal PET decomposition being detected under such temperature and pressure. After filtering and washing the residual solid with toluene (to remove the produced PG and the catalyst), another dosage of catalyst and toluene combined with the filter residue were added to another autoclave, again filled with 4 MPa H2 and reacted at 80 °C for 3 h. Finally, full conversion of PET to 1,4-benzenedimethanol and ethylene glycol was achieved. To imitate the realistic post-consumer plastics which usually contain complicated compositions, we further mixed equimolar amounts of PE, PP, PS, PLA and PET and carried out the reaction using the same procedure as that of PLA and PET. High yields of PG, BDM and EG were obtained, leaving polyolefins as untouched residues, as shown in Figure 4. Consequently, our catalytic system shows great promise toward the end-of-life polyesters degradation and recycling.
3.3. Hydrogenative Depolymerization of Polyesters with Alcohol
As described above, the PN3-Ru complex is an excellent catalyst for a variety of aliphatic and aromatic polyesters deconstruction using H2 as a hydrogen source. Nevertheless, high pressures are usually required for catalytic hydrogenation, which results in the use of expensive autoclaves as well. Therefore, exploring safe and cost-effective alternatives to replace H2 as a hydrogen source is crucial in polyester depolymerization. Transfer hydrogenation (TH) is an attractive alternative to classical catalytic hydrogenation, in which isopropanol is the most widely used reagent. For this reason, we first chose isopropanol as a hydrogen source to conduct the TH of PLA. As shown in the 1H NMR spectrum of Figure S21 (Table 3, entry 1), PLA absolutely converted into isopropyl lactate catalyzed by PN3-Ru/KOtBu in the presence of 20 equivalent of isopropanol at room temperature for 2 h, the signals at 4.99, 4.16, 1.33 and 1.20 ppm are attributable to the methine and methyl protons respectively, no any diol signals was observed, suggesting the catalyst is also an effective catalyst for alcoholysis of polyesters. Maintaining otherwise conditions unchanged, elevating the reaction temperature to 80 °C, a set of signals attributed to PG in the 1H NMR spectrum of the reaction mixture could be found, with an NMR yield of 5%. In the meantime, a signal at 2.17 ppm corresponding to acetone could also be observed, meaning that the catalytic system is also an efficient catalyst for TH. Further elevating the temperature to 100 °C, the yield of 1,2-propanediol reached 43%. Switching isopropanol to ethanol and maintaining the other conditions unchanged, PG and ethyl lactate were produced in 7% and 93% yields (Table 3, Entry 4, Fiugre 5a), respectively, at ambient temperature, which rose to 23% and 77% at 80 °C (Table 3, Entyr 5, Figure 5b). When elevating the temperature to 100 °C, all PLA was converted to PG, accompanied by ethanol oxidation to form ethyl acetate production (Table 3, Entry 6, Figure 5c). As shown in the quantitative 13C NMR spectrum, the peaks at 68.0, 67.6 and 18.5 ppm are attributed to the carbons of PG, and the characteristic signal of carbonyl carbon in ethyl acetate resonates at 171.6 ppm; no other byproduct was observed. These results indicated that ethanol is an ideal hydrogen source for TH of PLA in our catalytic system. For the catalytic transfer hydrogenation reaction, isopropanol usually shows better performance than ethanol. However, our catalytic system involves both transesterification and transfer hydrogenation processes. This could be confirmed by the products of isopropyl lactate, acetone, ethyl lactate and ethyl acetate. The transesterification efficiency of isopropanol is much slower than that of ethanol, thus leading to poor reaction [32,40]. When changing the solvent to methanol, the alcoholysis of PLA to afford methyl lactate in 99% yield was achieved at room temperature within 2 h, and no PG was produced under these conditions. Moreover, the yield of PG was negligible as the reaction temperature elevated to 80 °C and even 100 °C, maybe originating from the catalyst poisoning through forming a less reactive carbonyl complex [32].
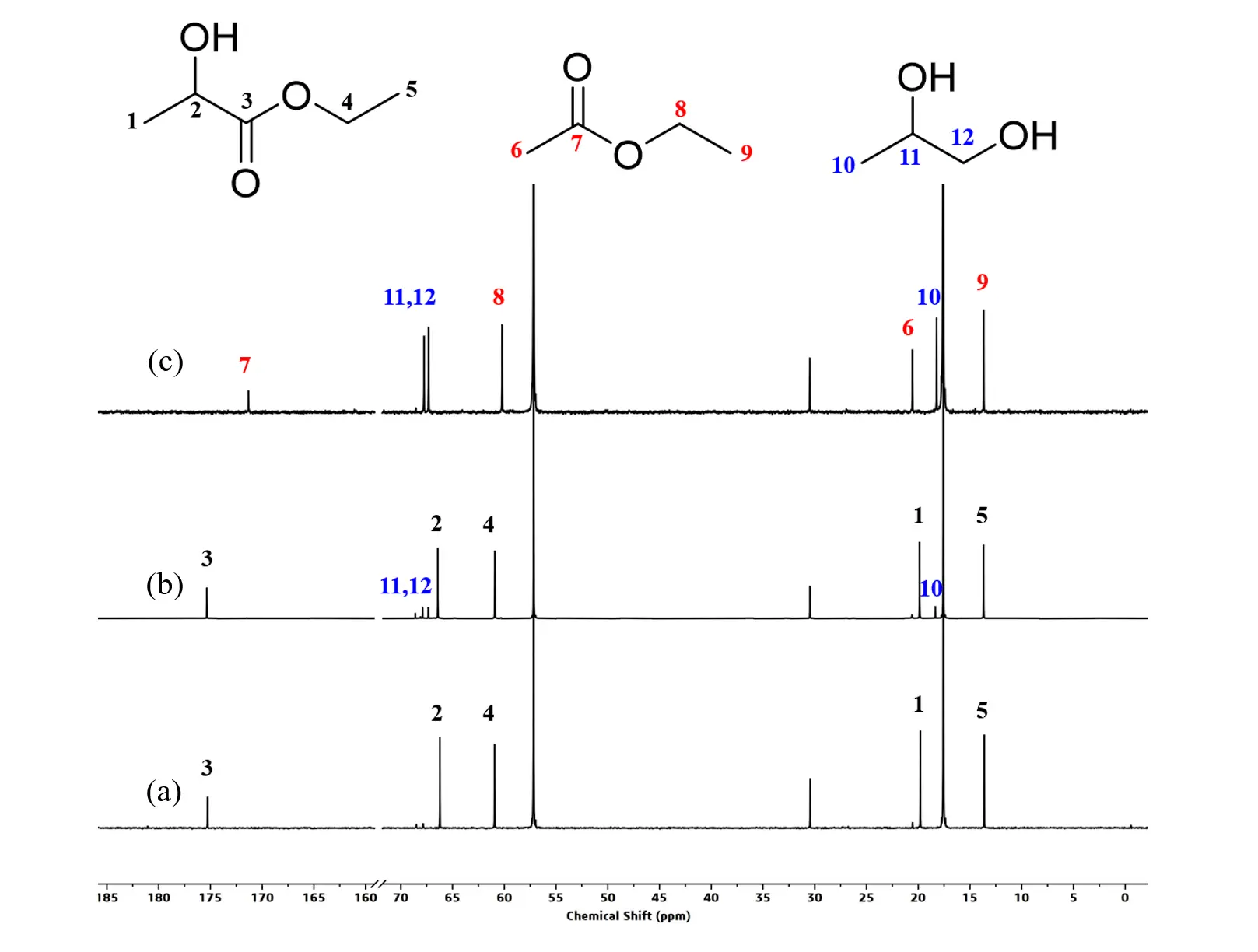
Table 3. Hydrogenative depolymerization of polyesters with alcohol a.
|
Entry |
Polyester |
Alcohol |
t (h) |
T(°C) |
Conv. (%) |
Ester(%) |
Diol (%) |
|---|---|---|---|---|---|---|---|
|
1 |
PLA |
iPrOH |
2 |
r.t. |
>99 |
>99 |
0 |
|
2 |
iPrOH |
2 |
80 |
>99 |
95 |
5 |
|
|
3 |
iPrOH |
2 |
100 |
>99 |
57 |
43 |
|
|
4 |
EtOH |
2 |
r.t. |
>99 |
93 |
7 |
|
|
5 |
EtOH |
2 |
80 |
>99 |
77 |
23 |
|
|
6 |
EtOH |
2 |
100 |
>99 |
0 |
>99 |
|
|
7 |
MeOH |
2 |
r.t. |
>99 |
>99 |
0 |
|
|
8 |
MeOH |
2 |
80 |
>99 |
97 |
3 |
|
|
9 |
MeOH |
2 |
100 |
>99 |
88 |
12 |
|
|
10 |
PCL |
EtOH |
8 |
100 |
>99 |
4 |
96 |
|
11 |
PET |
EtOH |
8 |
100 |
>99 |
60 |
40 |
|
12 b |
EtOH |
8 |
100 |
>99 |
0 |
>99 |
a Conditions: PN3-Ru (0.05 mmol), KOtBu (0.40 mmol), Toluene (2.0 mL), polyester (0.5 mmol, calculated on the repetition unit), alcohol (20 equiv.), conversion and yield determined by NMR spectroscopy. b 96 equiv of EtOH with respect to PET.
Therefore, we chose ethanol as a hydrogen source to catalyze other commercial polyesters via TH in the presence of PN3-Ru catalyst. The aliphatic polyester PCL could completely convert into 1,6-hexanediol in 96% yield. For the widely used aromatic polyester PET, BDM and EG were obtained in 40% yields when using 20 eq EtOH as a hydrogen source. Increasing EtOH to 96 eq, the yield of BDM was improved to 96%.
4. Conclusions
The bipyridine-based ruthenium pincer complex exhibited high efficiency for hydrogenative depolymerization of various polyesters into diols under mild conditions. Under the optical conditions, the waste PLA sucker and PET bottle were successfully converted into 1,2-propanediol and 1,4-benzenedimethanol/EG. Through tuning the reaction temperature and H2 pressure, highly pure products of PLA and PET mixture were obtained, which is extended to separate more complicated PLA, PET, PE, PP, and PS mixture, thus indicating the potential practical application value. More strikingly, this catalyst is also capable of catalytic transfer hydrogenation of polyesters into diols in excellent yields by using EtOH as a hydrogen source. This avoids the use of expensive and dangerous hydrogen gas, as well as the harsh reaction conditions. Further investigation of more effective catalytic systems for polyesters depolymerization and direct use of the degradation products for new material preparation is ongoing in our laboratories.
Supplementary Materials
The following supporting information can be found at: https://www.sciepublish.com/article/pii/762, Table S1. The Manufacturer and Purity of Required Chemical Reagent; Figure S1. 1H NMR spectrum (CDCl3) of PLA depolymerization (Table 1, Entry11); Figure S2. 1H NMR spectrum (CDCl3) of PLA depolymerization (Table 1, Entry12); Figure S3. 1H NMR spectrum (CDCl3) of PLA depolymerization (Table 1, Entry13); Figure S4. Quantitative 13C NMR spectrum (CDCl3) of PLA depolymerization (Table 1, Entry 15); Figure S5. Quantitative 13C NMR spectrum (CDCl3) of PLA depolymerization (Table 1, Entry 16); Figure S6. 1H NMR spectrum (CDCl3) of PLA depolymerization (Table 1, Entry 17); Figure S7. 1H NMR spectrum (CDCl3) of PLA depolymerization (Table 1, Entry 18); Figure S8. Quantitative 13C NMR spectrum (CDCl3, 25 °C) of PLA depolymerization. (Table 1, Entry 19); Figure S9. Quantitative 13C NMR spectrum (CDCl3) of PLA depolymerization (Table 1, Entry 20); Figure S10. 1H NMR spectrum (CDCl3) of PLA depolymerization (Table 1, Entry 21); Figure S11. 1H NMR spectrum (CDCl3) of PLA depolymerization (Table 1, Entry 22); Figure S12. 1H NMR spectrum (CDCl3) of PLA depolymerization (Table 1, Entry 23); Figure S13. 1H NMR spectrum (CDCl3) of PLA depolymerization (Table 1, Entry 24); Figure S14. 1H NMR spectrum (CDCl3) of PLA depolymerization (Table 1, Entry 25); Figure S15. 1H NMR spectrum (CDCl3) of PCL depolymerization (Table 2, Entry 1); Figure S16. 1H NMR spectrum (CDCl3) of PBS depolymerization (Table 2, Entry 2); Figure S17. 1H NMR spectrum (CDCl3) of PET depolymerization (Table 2, Entry 3); Figure S18. 1H NMR spectrum (CDCl3) of PBT depolymerization (Table 2, Entry 4); Figure S19. 1H NMR spectrum (CDCl3) of PHB depolymerization (Table 2, Entry 5); Figure S20. 1H NMR spectrum (CDCl3) of PLA, PET depolymerization (Table 2, Entry 8); Figure S21. 1H NMR spectrum (CDCl3) (Table 3, Entry 1); Figure S22. 1H NMR spectrum (CDCl3) (Table 3, Entry 2); Figure S23. Quantitative 13C NMR spectrum (CDCl3) (Table 3, Entry 2); Figure S24. 1H NMR spectrum (CDCl3) (Table 3, Entry 3); Figure S25. Quantitative 13C NMR spectrum (CDCl3, 25 °C). (Table 3, Entry 4); Figure S26. Quantitative 13C NMR spectrum (CDCl3, 25 °C). (Table 3, Entry 5); Figure S27. Quantitative 13C NMR spectrum (CDCl3) (Table 3, Entry 6); Figure S28. 1H NMR spectrum (CDCl3) (Table 3, Entry 7); Figure S29. 1H NMR spectrum (CDCl3) (Table 3, Entry 8); Figure S30. Quantitative 13C NMR spectrum (CDCl3) (Table 3, Entry 9); Figure S31. Quantitative 13C NMR spectrum (CDCl3) (Table 3, Entry 10); Figure S32. 1H NMR spectrum (CDCl3) (Table 3, Entry 12); Figure S33. The possible pathway for converting PHB to butyric acid through Ru catalyst under H2.
Acknowledgments
We express our thanks for funding support from the National Natural Science Foundation of China (nos. 22261034, 22061027), the Natural Science Foundation of Jiangxi Province (nos. 20224BAB203023, 20232ACB213008), and the Open Research Fund of State Key Laboratory of Polymer Science and Technology, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences (no. PPCL2024-07).
Author Contributions
Experimental designing, data collection and analysis, writing manuscript, R.W. and Y.R.; supervision, W.Y., Z.J. and C.Y.; formal analysis, R.W. and L.C.; investigation, Y.P.; editing, Y.R. and C.Y.; conceptualization, C.Y. All authors have read and agreed to the published version of the manuscript.
Ethics Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data will be made available on request.
Funding
This work was supported by the National Natural Science Foundation of China (nos. 22261034, 22061027), the Natural Science Foundation of Jiangxi Province (nos. 20232ACB213008, 20224BAB203023), and the Open Research Fund of State Key Laboratory of Polymer Science and Technology, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences (no. PPCL2024-07).
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
-
Geyer R, Jambeck JR, Law KL. Production, use, and fate of all plastics ever made. Sci. Adv. 2017, 3, e1700782. doi:10.1126/sciadv.1700782. [Google Scholar]
-
Shi C, Reilly LT, Phani Kumar VS, Coile MW, Nicholson SR, Broadbelt LJ, et al. Design principles for intrinsically circular polymers with tunable properties. Chem 2021, 7, 2896–2912. doi:10.1016/j.chempr.2021.10.004. [Google Scholar]
-
Coates GW, Getzler YDYL. Chemical recycling to monomer for an ideal, circular polymer economy. Nat. Rev. Mater. 2020, 5, 501–516. doi:10.1038/s41578-020-0190-4. [Google Scholar]
-
Yang S, Du S, Zhu J, Ma S. Closed-loop recyclable polymers: From monomer and polymer design to the polymerization-depolymerization cycle. Chem. Soc. Rev. 2024, 53, 9609–9651. doi:10.1039/d4cs00663a. [Google Scholar]
-
Law KL, Narayan R. Reducing environmental plastic pollution by designing polymer materials for managed end-of-life. Nat. Rev. Mater. 2022, 7, 104–116. doi:10.1038/s41578-021-00382-0. [Google Scholar]
-
Chamas A, Moon H, Zheng J, Qiu Y, Tabassum T, Jang JH, et al. Degradation Rates of Plastics in the Environment. ACS Sustain. Chem. Eng. 2020, 8, 3494–3511. doi:10.1021/acssuschemeng.9b06635. [Google Scholar]
-
Stubbins A, Law KL, Muñoz SE, Bianchi TS, Zhu L. Plastics in the Earth system. Science 2021, 373, 51–55. doi:10.1126/science.abb0354. [Google Scholar]
-
Korley LTJ, Epps TH, Helms BA, Ryan AJ. Toward polymer upcycling-adding value and tackling circularity. Science 2021, 373, 66–69. doi:10.1126/science.abg4503. [Google Scholar]
-
Wang S, Li J, Li X, Tu Y. Recent advances in the chemical recycling of polyesters. Fundam. Res. 2025, 5, 935–950. doi:10.1016/j.fmre.2024.05.014. [Google Scholar]
-
Häußler M, Eck M, Rothauer D, Mecking S. Closed-loop recycling of polyethylene-like materials. Nature 2021, 590, 423–427. doi:10.1038/s41586-020-03149-9. [Google Scholar]
-
Raheem AB, Noor ZZ, Hassan A, Abd Hamid MK, Samsudin SA, Sabeen AH. Current developments in chemical recycling of post-consumer polyethylene terephthalate wastes for new materials production: A review. J. Clean. Prod. 2019, 225, 1052–1064. doi:10.1016/j.jclepro.2019.04.019. [Google Scholar]
-
Ahmed T, Shahid M, Azeem F, Rasul I, Shah AA, Noman M, et al. Biodegradation of plastics: Current scenario and future prospects for environmental safety. Environ. Sci. Pollut. Res. 2018, 25, 7287–7298. doi:10.1007/s11356-018-1234-9. [Google Scholar]
-
Tsuji Y, Yamamoto K, Yamauchi K, Sakai K. Near-Infrared Light-Driven Hydrogen Evolution from Water Using a Polypyridyl Triruthenium Photosensitizer. Angew. Chem. Int. Ed. 2018, 57, 208–212. doi:10.1002/anie.201708996. [Google Scholar]
-
Tang X, Chen EYX. Toward Infinitely Recyclable Plastics Derived from Renewable Cyclic Esters. Chem 2019, 5, 284–312. doi:10.1016/j.chempr.2018.10.011. [Google Scholar]
-
Weng C, Tang X. Circular Polymers from Ring-Opening Polymerization of Five-Membered (Thio)lactones and Derivatives. Chin. J. Chem. 2023, 41, 1603–1607. doi:10.1002/cjoc.202200841. [Google Scholar]
-
Fernandes AC. Reductive depolymerization as an efficient methodology for the conversion of plastic waste into value-added compounds. Green Chem. 2021, 23, 7330–7360. doi:10.1039/D1GC01634B. [Google Scholar]
-
Weng Y, Hong C-B, Zhang Y, Liu H. Catalytic depolymerization of polyester plastics toward closed-loop recycling and upcycling. Green Chem. 2024, 26, 571–592. doi:10.1039/D3GC04174C. [Google Scholar]
-
Ügdüler S, Van Geem KM, Denolf R, Roosen M, Mys N, Ragaert K, et al. Towards closed-loop recycling of multilayer and coloured PET plastic waste by alkaline hydrolysis. Green Chem. 2020, 22, 5376–5394. doi:10.1039/D0GC00894J. [Google Scholar]
-
Wang L, Nelson GA, Toland J, Holbrey JD. Glycolysis of PET Using 1,3-Dimethylimidazolium-2-Carboxylate as an Organocatalyst. ACS Sustain. Chem. Eng. 2020, 8, 13362–13368. doi:10.1021/acssuschemeng.0c04108. [Google Scholar]
-
Fuentes CA, Gallegos MV, García JR, Sambeth J, Peluso MA. Catalytic Glycolysis of Poly(ethylene terephthalate) Using Zinc and Cobalt Oxides Recycled from Spent Batteries. Waste Biomass Valorization 2020, 11, 4991–5001. doi:10.1007/s12649-019-00807-6. [Google Scholar]
-
Pham DD, Cho J. Low-energy catalytic methanolysis of poly(ethyleneterephthalate). Green Chem. 2021, 23, 511–525. doi:10.1039/D0GC03536J. [Google Scholar]
-
Demarteau J, Olazabal I, Jehanno C, Sardon H. Aminolytic upcycling of poly(ethylene terephthalate) wastes using a thermally-stable organocatalyst. Polym. Chem. 2020, 11, 4875–4882. doi:10.1039/D0PY00067A. [Google Scholar]
-
Thiounn T, Smith RC. Advances and approaches for chemical recycling of plastic waste. J. Polym. Sci. 2020, 58, 1347–1364. doi:10.1002/pol.20190261. [Google Scholar]
-
Vollmer I, Jenks MJF, Roelands MCP, White RJ, van Harmelen T, de Wild P, et al. Beyond Mechanical Recycling: Giving New Life to Plastic Waste. Angew. Chem. Int. Ed. 2020, 59, 15402–15423. doi:10.1002/anie.201915651. [Google Scholar]
-
Krall EM, Klein TW, Andersen RJ, Nett AJ, Glasgow RW, Reader DS, et al. Controlled hydrogenative depolymerization of polyesters and polycarbonates catalyzed by ruthenium(II) PNN pincer complexes. Chem. Commun. 2014, 50, 4884–4887. doi:10.1039/C4CC00541D. [Google Scholar]
-
Hunsicker DM, Dauphinais BC, Mc Ilrath SP, Robertson NJ. Synthesis of High Molecular Weight Polyesters via in Vacuo Dehydrogenation Polymerization of Diols. Macromol. Rapid Commun. 2012, 33, 232–236. doi:10.1002/marc.201100653. [Google Scholar]
-
Johnson ML, Fine RL, Stankowski DS, Koch CA, Limoges KA, Robertson NJ. Highly selective pressure-dependent (transfer) hydrogenative depolymerization of polybutylene succinate. Chem. Commun. 2024, 60, 702–705. doi:10.1039/D3CC05239G. [Google Scholar]
-
Fuentes García JA, Smith S, Scharbert MT, Carpenter I, Cordes DB, Slawin AMZ, et al. On the functional group tolerance of ester hydrogenation and polyester depolymerisation catalysed by ruthenium complexes of tridentate aminophosphine ligands. Chem.-Eur. J. 2015, 21, 10851–10860. doi:10.1002/chem.201500907. [Google Scholar]
-
Westhues S, Idel J, Klankermayer J. Molecular catalyst systems as key enablers for tailored polyesters and polycarbonate recycling concepts. Sci. Adv. 2018, 4, eaat9669. doi:10.1126/sciadv.aat9669. [Google Scholar]
-
Shao Z, Zhong R, Ferraccioli R, Li Y, Liu Q. General and Phosphine-Free Cobalt-Catalyzed Hydrogenation of Esters to Alcohols. Chin. J. Chem. 2019, 37, 1125–1130. doi:10.1002/cjoc.201900292. [Google Scholar]
-
Hu Y, Zhang SY, Xu JF, Liu Y, Yu AAI, Qian J, et al. Highly Efficient Depolymerization of Waste Polyesters Enabled by Transesterification/Hydrogenation Relay Under Mild Conditions. Angew. Chem. Int. Ed. 2023, 62, e202312564. doi:10.1002/anie.202312564. [Google Scholar]
-
Farrar-Tobar RA, Wozniak B, Savini A, Hinze S, Tin S, de Vries JG. Base-Free Iron Catalyzed Transfer Hydrogenation of Esters Using EtOH as Hydrogen Source. Angew. Chem. Int. Ed. 2019, 58, 1129–1133. doi:10.1002/anie.201810605. [Google Scholar]
-
Xu H-D, Deng H-J, Zeng H-M, Yang W, Yao C. Direct Coupling of Biobased Bifunctional Hydroxy-Aldehyde Monomers to Chemically Recyclable Alipharomatic Polyesters via Dehydrogenative Polycondensation. Macromolecules 2024, 57, 1546–1555. doi:10.1021/acs.macromol.3c02504. [Google Scholar]
-
Xu W-M, Yu Y-D, Ma M-X, Xu H-D, Wang R-Q, Pan Y-P, et al. Green Synthesis of Chemically Recyclable Polyesters via Dehydrogenative Copolymerization of Diols. Chin. J. Polym. Sci. 2023, 41, 1206–1214. doi:10.1007/s10118-023-2903-9. [Google Scholar]
-
Chen L, Li L, Rao YW, Yang WR, Jian ZB, Yao CG. Progress in the Preparation of Polar Polymer Materials via Dehydrogenative Polymerization. Acta Polym. Sin. 2025, 56, 551–563. doi:10.11777/j.issn1000-3304.2024.24183. [Google Scholar]
-
Chen T, Li HF, Qu SL, Zheng B, He LP, Lai ZP, et al. Hydrogenation of Esters Catalyzed by Ruthenium PN3-Pincer Complexes Containing an Aminophosphine Arm. Organometallics 2014, 33, 4152–4155. doi:10.1021/om5005491. [Google Scholar]
-
Bruneau-Voisine A, Wang D, Dorcet V, Roisnel T, Darcel C, Sortais J-B. Mono-N-methylation of anilines with methanol catalyzed by a manganese pincer-complex. J. Catal. 2017, 347, 57–62. doi:10.1016/j.jcat.2017.01.004. [Google Scholar]
-
Hamilton RJ, Bergens SH. Direct Observations of the Metal−Ligand Bifunctional Addition Step in an Enantioselective Ketone Hydrogenation. J. Am. Chem. Soc. 2008, 130, 11979–11987. doi:10.1021/ja8034812. [Google Scholar]
-
van Putten R, Uslamin EA, Garbe M, Liu C, Gonzalez-de-Castro A, Lutz M, et al. Non-Pincer-Type Manganese Complexes as Efficient Catalysts for the Hydrogenation of Esters. Angew. Chem. Int. Ed. 2017, 56, 7531–7534. doi:10.1002/anie.201701365. [Google Scholar]
-
Dubey A, Khaskin E. Catalytic Ester Metathesis Reaction and Its Application to Transfer Hydrogenation of Esters. ACS Catal. 2016, 6, 3998–4002. doi:10.1021/acscatal.6b00827. [Google Scholar]