Enzyme-Mediated Carbon Dioxide Fixation: Catalytic Mechanisms and Computational Insights

Enzyme-Mediated Carbon Dioxide Fixation: Catalytic Mechanisms and Computational Insights
Kai Wen 1 Jingxuan Zhu 1,* Quanshun Li 1,2,*
Received: 30 October 2025 Revised: 06 November 2025 Accepted: 12 November 2025 Published: 17 November 2025

© 2025 The authors. This is an open access article under the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/).
1. Introduction
Modern industry increasingly relies on traditional fossil fuels, which emit large amounts of CO2 [1,2]. In 2024 alone, global CO2 emissions reached 3.74 billion tons [3]. These emissions could raise Earth’s average temperature by 1.5 °C in the next five years, worsening climate problems and endangering sustainable development [4]. Therefore, researchers must develop advanced carbon conversion technologies that reduce CO2 emissions while sustaining industrial productivity. Recently, researchers have focused on carboxylation reactions to convert CO2 into valuable products [5]. CO2, besides being recognized as a greenhouse gas, also serves as an abundant one-carbon resource [6]. However, the carbon atom in CO2 remains fully oxidized and forms very stable covalent bonds with oxygen atoms. Thermodynamically, CO2 formation via oxidation is more favorable than its reduction. Therefore, converting CO2 requires activating its carbon atom with extra electrons or energy [7].
Traditional organic methods remain the most widely used strategies for CO2 conversion [8,9]. However, these approaches typically rely on harsh conditions, such as electrocatalysis, high temperatures, or strong reducing agents, to activate the carbon atom in CO2 [10,11]. As a result, such methods consume large amounts of energy and impose significant economic and environmental burdens due to their dependence on synthetic reagents. To overcome these limitations, advances in biotechnology and computational tools have significantly improved our understanding of biological CO2 conversion mechanisms [12,13]. Nearly all natural CO2 fixation processes depend on enzymes that facilitate electron transfer through cofactors, metal ions, or catalytic amino acid residues [14]. These components reduce the carbon atom in CO2, rendering it reactive for subsequent biochemical transformations [15]. Inspired by these natural pathways, researchers are now exploring enzyme-catalyzed carboxylation reactions that incorporate carboxyl groups into substrates using CO2 as the carbon source in vitro [15]. Compared to traditional chemical approaches, enzyme-mediated biocatalysis operates under mild conditions, uses low-cost reagents, and generates high-value compounds with fewer side-products. These advantages position enzymatic strategies as promising solutions to both energy and environmental challenges.
Recent advances in enzyme engineering have led to the discovery and isolation of more enzymes that catalyze CO2 conversion [16]. These enzymes fall into four categories based on their reliance on cofactors or metal ions: cofactor-independent enzymes [17], metal-dependent enzymes [18], prFMN-dependent enzymes [19], and NAD(P)H-dependent enzymes [20]. Like chemical catalysis, enzyme-catalyzed CO2 fixation transfers electrons to CO2 to lower the activation energy required for CO2 conversion. However, the structural complexity of enzymes and their specificity for cofactors make their catalytic mechanisms far more intricate than those of traditional chemical reactions [21]. Therefore, developing high-performance enzymes requires a deeper understanding of how they function at the atomic level [22].
To address this complexity, computer-aided techniques such as MD simulations [23] and QM calculations [24] have become essential tools for studying enzyme catalysis. Each technique serves a distinct role: MD simulations capture large-scale processes such as enzyme conformational changes and substrate binding [25], while QM calculations model bond formation and cleavage events during catalysis [26]. In this review, we first introduce the basic principles and common applications of MD and QM methods, including their use in analyzing active site flexibility, identifying substrate pathways, and characterizing transition states in CO2-converting enzymes. We then present detailed examples that show how these techniques elucidate the catalytic mechanisms of CO2-converting enzymes, along with key computational parameters. Overall, this review summarizes the significant contributions of MD and QM methods to understanding enzymatic mechanisms involved in CO2 fixation, providing valuable guidance for discovering and engineering high-performance CO2-converting enzymes.
2. Computer-Aided Techniques in Mechanism Studies
2.1. Molecular Dynamics Simulation
MD simulations use physical laws to model how particles move in a system (Figure 1A) [27]. It predicts particle interactions and solves their equations of motion to generate time-dependent trajectories. For biomacromolecules such as proteins, lipids, and nucleic acids, classical Newtonian mechanics typically describes most systems accurately [28]. MD simulations provide atom-level details and help interpret protein functions at atomic resolution. Enzymes in aqueous environments, in particular, show dynamic behaviors that make them ideal targets for MD studies [29].
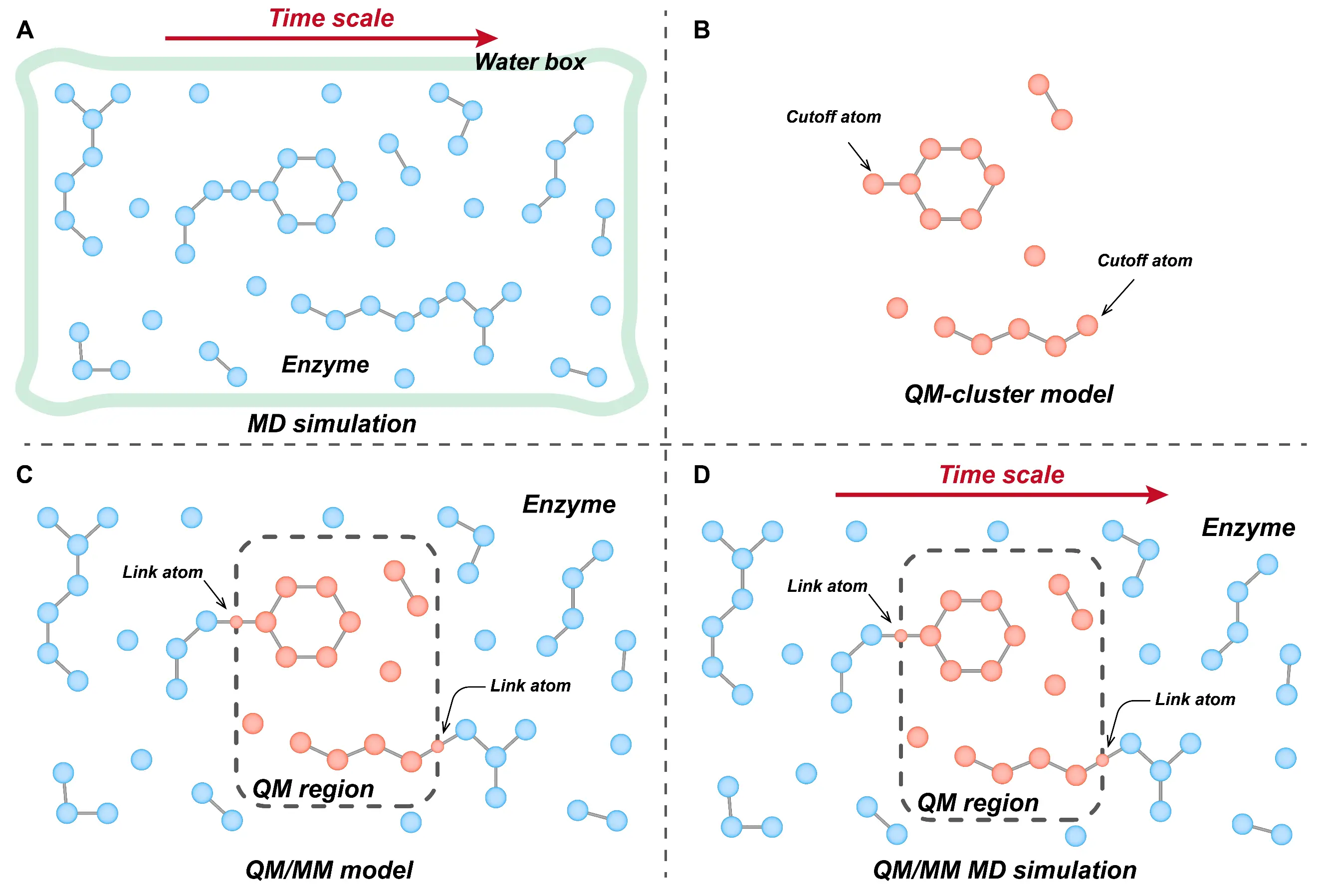
Figure 1. Computer-aided techniques of enzyme catalytic mechanisms, including MD simulations (A), cluster models (B), QM/MM models (C), and QM/MM-MD simulations (D).
Combining MD simulations with experimental data has further strengthened enzyme research [30]. It enables researchers to validate computational predictions against experimental results collected over comparable time scales, thereby deepening our understanding of enzyme dynamics under realistic conditions. Recent advances in algorithms [31], software [32], and hardware [33] have significantly improved the efficiency and scale of MD simulations. These improvements allow researchers to accurately characterize long-timescale events such as substrate binding and release, enzyme conformational shifts, and interactions within multi-enzyme complexes [29]. For example, GPU acceleration now supports full-atom simulations on the microsecond timescale, enabling researchers to capture nearly complete representations of protein motions under physiological conditions [34]. This capability greatly expands the applicability of MD methods in investigating enzymatic mechanisms.
2.2. Quantum Mechanics Calculations
QM methods serve as powerful tools for investigating catalytic reactions within enzymes, especially processes involving bond formation and cleavage [35]. These methods solve the multi-electron Schrödinger equation under the Born-Oppenheimer approximation, providing fundamental information about the energy landscape and atomic structures of reacting species [36]. A key application of QM techniques is to compare energy profiles of competing reaction pathways, which helps elucidate enzyme reactivity and selectivity. To study enzymatic catalytic mechanisms, researchers typically adopt one of two QM-based strategies: the cluster model [37] and the quantum mechanics/molecular mechanics (QM/MM) hybrid model [38]. Each approach offers distinct advantages in balancing computational cost with system complexity.
The cluster model focuses on a small, chemically active region of the enzyme. It uses methods such as density functional theory (DFT) [37], second-order Møller-Plesset perturbation theory (MP2) [39], or density functional tight-binding (DFTB) [40] to treat key atoms—typically including substrates, cofactors, and catalytic residues—at high accuracy (Figure 1B). Due to computational limitations, the cluster model typically includes only a few hundred atoms, requiring truncation of regions not directly involved in catalysis [41]. This simplicity makes the cluster model easy to apply and efficient for modeling localized reactions. However, because it excludes distant residues and the broader protein environment, it cannot account for spatial constraints or polarization effects from the enzyme matrix. To compensate, researchers often constrain atomic positions and adjust the solvent dielectric constant [37].
In contrast, the QM/MM approach preserves the full enzyme structure by partitioning the system into two regions: a QM region encompassing the active site and a MM region describing the surrounding environment (Figure 1C) [42]. Compared with the cluster model, this hybrid method allows a broader atomic scope without significantly increasing computational cost. While more realistic than the cluster model, QM/MM calculations must address interactions across the QM-MM boundary. Currently, techniques such as electrostatic [43], mechanical [44], or polarizable [45] embedding are commonly used to describe QM-MM electrostatic interactions, while link-atom [46], boundary-atom [47], or localized-orbital [48] methods are applied to handle covalent bonds crossing the QM-MM interface. The QM/MM molecular dynamics (QM/MM MD) simulation extends the QM/MM approach by incorporating time-dependent behavior into the static framework (Figure 1D) [49]. In this approach, self-consistent field (SCF) optimization calculates electronic structures of atoms directly involved in chemical transformations, while MM force fields describe the remainder of the system. These forces are then integrated using Newtonian mechanics to generate time-resolved trajectories, providing a dynamic, atomistic view of enzymatic reactions under near-physiological conditions and enabling the evaluation of entropic contributions from enzyme conformational fluctuations [42]. Together, MD and QM-based techniques offer complementary perspectives, such as dynamic and electronic, on enzymatic function. These computational tools have become indispensable for elucidating how enzymes catalyze complex biochemical transformations, including CO2 conversion.
3. Mechanism of Enzymatic CO2 Conversion
By applying the MD and QM techniques described above, researchers have gained detailed insights into enzyme conformational changes, substrate binding events, and catalytic pathways involved in CO2 conversion. These computational approaches help map both the structural evolution and energy landscapes of enzymatic reactions, thereby deepening our mechanistic understanding at an atomic resolution [50]. In the following sections, we review representative studies on the catalytic mechanisms of four major classes of CO2-converting enzymes. For cofactor-independent enzymes, we discuss the mechanism of phenolic acid decarboxylase (PAD). In the case of metal-dependent enzymes, we discuss the catalytic processes of ribulose 1,5-bisphosphate carboxylase/oxygenase (RuBisCO), decarboxylases from the amidohydrolase superfamily (AHS), molybdenum-dependent formate dehydrogenases (Mo-FDH), and carbon monoxide dehydrogenases (CODH). Within the group of NAD(P)H-dependent enzymes, we highlight the catalytic mechanism of crotonyl-CoA carboxylase/reductase (CCR) from the enoyl-CoA carboxylases/reductases (ECR) family. Finally, for prFMN-dependent decarboxylases, we focus on ferulic acid decarboxylase (Fdc1), 3,4-dihydroxybenzoic acid decarboxylases (AroY), and 2,5-furandicarboxylic acid decarboxylase (HmfF).
3.1. Cofactor-Independent Enzymes
PAD, a member of the lyase enzyme class, catalyzes the non-oxidative decarboxylation of toxic phenolic acids in vivo, producing the corresponding para-vinyl derivatives [51]. Structural studies have resolved crystal structures of PADs from several organisms, including Bacillus subtilis (BsPAD) [52], Lactobacillus plantarum (LpPAD) [53], and Bacillus pumilus (BpPAD) [54]. These analyses consistently show that PAD enzymes lack metal ions or cofactors directly involved in catalysis, indicating a cofactor-independent reaction mechanism.
Beyond decarboxylation, recent findings have revealed that PAD also catalyzes the reverse reaction, the carboxylation of hydroxystyrenes, to biosynthesize p-coumaric acid, suggesting its potential role in reversible CO2 conversion [55]. To investigate the structural basis for substrate specificity, Fabián et al. applied molecular docking and MD simulations [56]. They showed that PAD transitions between open and closed conformations during catalysis, but these conformational changes do not hinder substrate binding. To further elucidate the chemical mechanism of PAD, Sheng et al. conducted detailed QM calculations on both decarboxylation and carboxylation pathways [57] (Figure 2 and Figure 3). For the decarboxylation reaction, they constructed a large cluster model based on the PAD crystal structure, including key active site residues and the p-coumaric acid substrate (Figure 2). Their QM analysis identified a plausible stepwise mechanism: after p-coumaric acid binds in the active site, its p-hydroxyl group forms hydrogen bonds with Tyr11 and Tyr13, which assist in its deprotonation. Glu64 then donates a proton to the alkene moiety of p-coumaric acid, facilitating the formation of a quinone methide intermediate. This intermediate subsequently decomposes to release CO2 and generate p-vinylphenol. Notably, before CO2 release, Tyr19 maintains a strong hydrogen bond with the carboxyl group of p-coumaric acid, which helps lower the reaction barrier for decarboxylation. These findings have identified Tyr11, Tyr13, and Glu64 as the key catalytic residues directly involved in bond activation and proton transfer, while Tyr19 contributes to the stabilization of the reactive intermediate and the catalytic microenvironment.
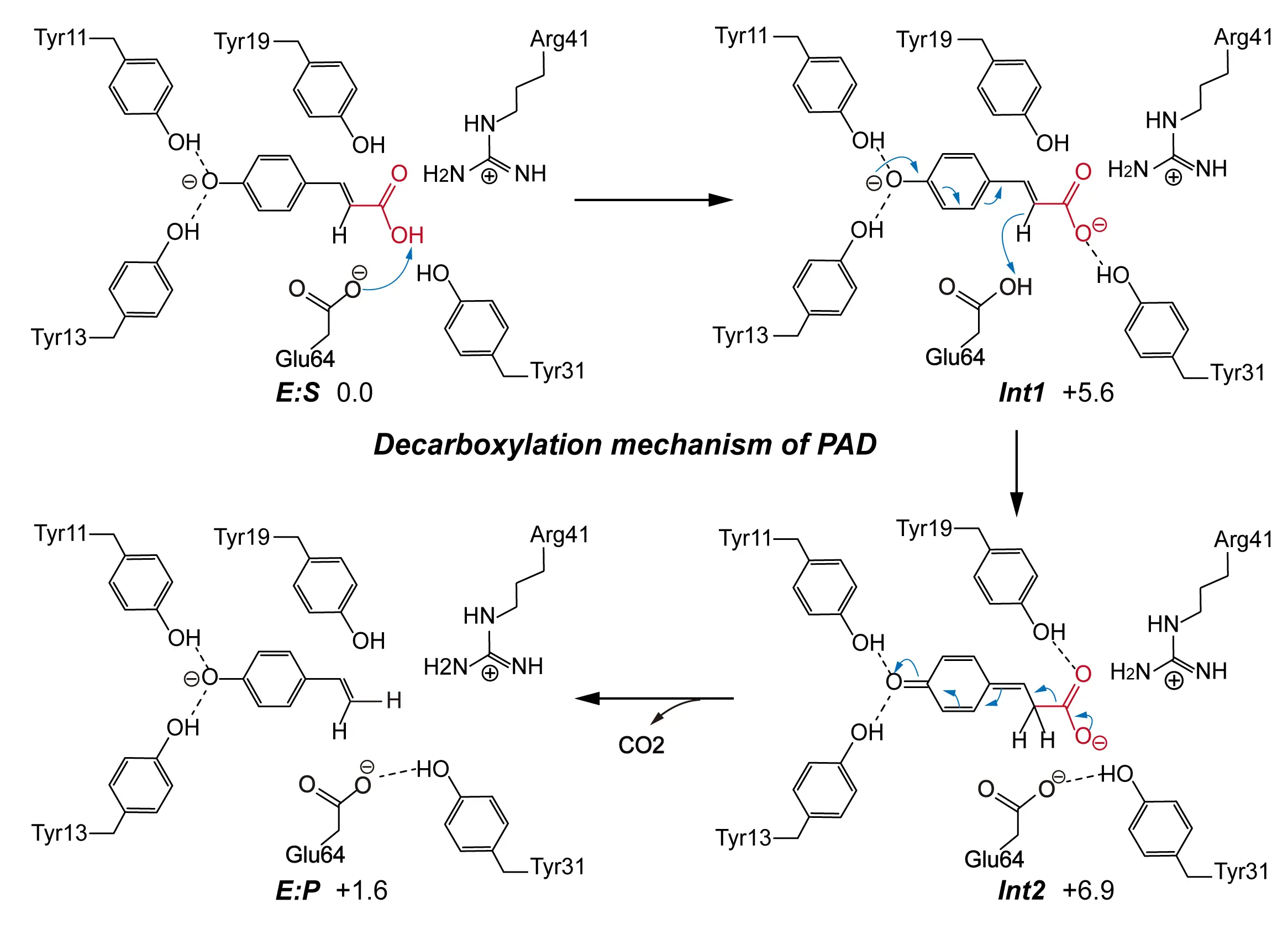
Figure 2. A reaction mechanism proposed on the basis of cluster model for the decarboxylation reaction catalyzed by PAD. The detailed reaction process is shown, including the decarboxylating group (red), the direction of electron transfer (blue arrows), weak interactions (dashed lines), and the free energy of the species involved. Adapted from reference [57], under CC BY-NC 4.0 license.
Although decarboxylation and carboxylation are thermodynamically reversible based on their substrates and products, their underlying mechanisms and the roles of active site residues differ significantly [58]. For the carboxylation reaction, the researchers constructed a cluster model similar to that used for decarboxylation and applied QM calculations to examine the reaction steps in detail (Figure 3A). In the model, p-vinylphenol, the carboxylation substrate, was placed in its deprotonated form, with its phenolic oxygen positioned toward Tyr11 and Tyr13 to mimic the hydrogen-bonding pattern observed during carboxylation. Importantly, the study proposed that under physiological conditions, bicarbonate, rather than molecular CO2, serves as the actual carbon source. In the first step of the reaction, bicarbonate accepts a proton and dissociates into CO2 and water, with Glu64 facilitating this proton transfer by acting as a general acid. In the subsequent step, the deprotonated p-vinylphenol enables its vinyl β-carbon to perform a nucleophilic attack on the newly released CO2, forming a quinone methide intermediate. Glu64 then switches roles and functions as a general base to deprotonate the intermediate, ultimately yielding p-coumaric acid as the final product. Mechanistically, the formation of CO2 from bicarbonate constitutes the rate-limiting step in the carboxylation reaction, with an energy barrier of 19.5 kcal/mol. In contrast, this step occurs spontaneously during the decarboxylation process. This discrepancy explains the markedly lower catalytic efficiency observed in the carboxylation direction (Figure 3B).
Overall, the proposed mechanism provides a new perspective for understanding reversible decarboxylases. It demonstrates that decarboxylation and carboxylation should not be treated as mirror-image processes, even if they involve the same substrates and products. This distinction becomes especially important in the context of CO2-converting enzymes, many of which catalyze reversible reactions but exhibit low efficiency in the carboxylation direction. Therefore, elucidating the mechanistic differences between these two directions is essential for guiding the rational design of enzyme variants with improved CO2 fixation performance.
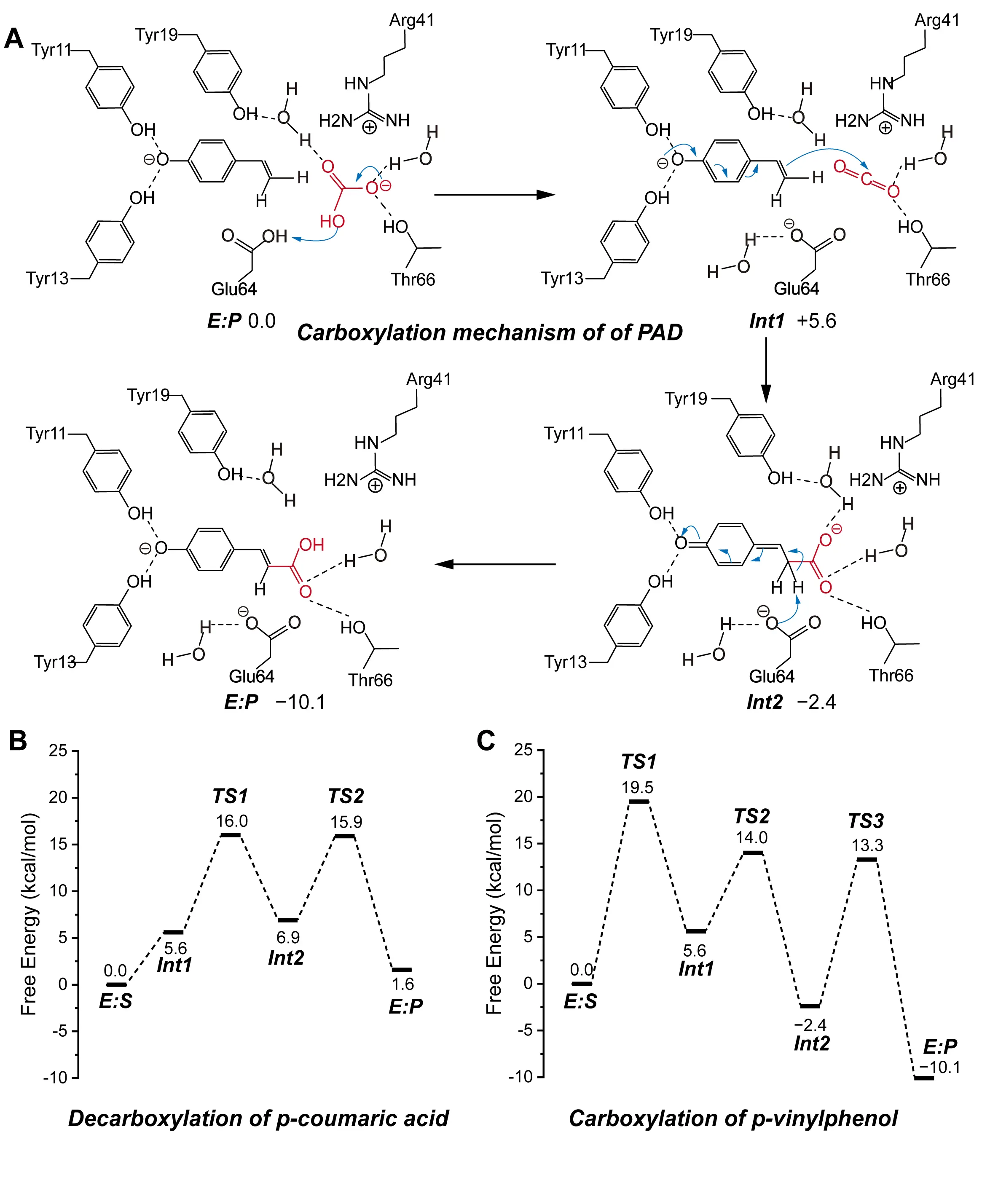
Figure 3. Catalytic mechanism of PAD. (A) A reaction mechanism was proposed on the basis of a cluster model for the carboxylation reaction catalyzed by PAD. The detailed reaction process is shown, including the carboxylating group (red), the direction of electron transfer (blue arrows), weak interactions (dashed lines), and the free energy of the species involved. (B) The calculated energy profile of the decarboxylation reaction energies. (C) The calculated energy profile of the carboxylation reaction energies. These composite figures are adapted from images in references [57,58]. Permissions have been obtained from the corresponding publishers, or the figures are used under applicable Creative Commons licenses.
3.2. Metal-Dependent Enzymes
Metal ions play essential roles in the catalytic functions of many enzymes. Within enzymatic systems, metal ions can act as electron donors or acceptors during redox reactions, or activate coordinating atoms to enhance substrate reactivity [59]. In CO2-converting enzymes, divalent metal ions often form coordination complexes with either the substrate or the CO2 molecule, thereby polarizing specific atoms and facilitating chemical transformations [18]. Among metal-dependent CO2-converting enzymes, two major superfamilies dominate: RuBisCO and AHS. These enzymes typically rely on divalent metals such as magnesium or zinc to stabilize intermediates or promote nucleophilic attack on CO2. In contrast, CO2-reducing enzymes often incorporate transition metals such as molybdenum in Mo-FDH, or nickel and iron in CODH, to enable multi-electron reduction steps. Drawing inspiration from natural systems, researchers have also developed artificial metal-based enzymes using elements like zinc or rubidium to catalyze direct CO2 reduction. While these artificial catalysts offer innovative strategies for carbon transformation, the current review focuses specifically on natural CO2-converting enzymes [60]. Therefore, mechanistic studies of artificial systems are not included in the following sections.
3.2.1. Ribulose 1,5-Bisphosphate Carboxylase/Oxygenase (RuBisCO)
RuBisCO plays a central role in the Calvin cycle by catalyzing the fixation of CO2 onto ribulose-1,5-bisphosphate (RuBP) [61]. As one of the most abundant proteins on Earth, RuBisCO is essential to the global carbon cycle and serves as a cornerstone for developing enzymatic CO2 conversion strategies. Despite its importance, RuBisCO exhibits relatively low catalytic efficiency, prompting extensive mechanistic studies aimed at identifying the rate-limiting steps in its carboxylation reaction [62]. The overall catalytic mechanism of RuBisCO involves several sequential steps: enolization of RuBP, addition of CO2 and water, cleavage of the C2–C3 bond, and final protonation at the C2 position, resulting in the production of two molecules of 3-phosphoglycerate (3-PGA) [63]. Understanding these steps has been the focus of decades of biochemical and computational investigation of RuBisCO’s reaction mechanism.
In 1998, Cleland et al. proposed an early model in which a lysine residue functions as a base to abstract a proton from RuBP during the enolization step [64]. Later, Tcherkez et al. incorporated isotope effect studies and suggested that His294 facilitates proton transfer by interacting with the O3 atom of RuBP [65]. Building on these hypotheses, Cummins et al. in 2018 revised the mechanism using high-resolution crystallographic data [66,67]. They identified that the carbamylated lysine residue, Kcx201, serves as the primary base to deprotonate RuBP, and further demonstrated that the resulting water molecule can act as a nucleophile attacking the C3 position of the intermediate. This leads to the formation of an unstable intermediate, 2-carboxy-3-keto-D-arabinitol-1,5-bisphosphate (CKABP), which undergoes C2–C3 bond cleavage. Phosphate groups then assist in protonation at C2, yielding two 3-PGA molecules. Their findings highlighted Kcx201 and His294 as the key residues mediating proton transfer, and proposed that these steps proceed in a concerted and rate-limiting manner.
More recently, in 2022, Douglas et al. employed QM/MM MD simulations to map the minimum free energy pathway of CO2 fixation [68]. They compared the energy barriers associated with proton transfer from the C3 hydroxyl group to either His294 or Kcx201. Their calculations revealed that direct proton transfer from the C3 hydroxyl group to Kcx201 involves a lower energy barrier than transfer to His294. They systematically examined different protonation states and showed that when Kcx201 is protonated and His294 is deprotonated, the energy barrier for forming the C–C bond between the enediol intermediate and CO2 molecular drops to 4.3 kcal/mol. These results suggest that Kcx201 acts as the principal proton abstractor, while His294 facilitates its re-ionization by accepting a proton. In addition to active-site chemistry, metal ions also influence RuBisCO’s catalytic efficiency. Douglas et al. showed that replacing Mg2+ with Zn2+, which has accessible d orbitals, increases the energy barrier for the CO2 addition step, indicating that metal identity affects the electronic environment of the active site.
Moreover, Hendawy and coworkers emphasized the role of CO2 transport as a potential rate-limiting step [69]. Their MD simulations demonstrated that transient hydrogen bonding and electrostatic interactions on the enzyme surface can trap CO2, hindering its access to the active site. They also proposed that van der Waals interactions between CO2 and the Mg2+ ion may induce a “flipping” mechanism, which obstructs CO2 entry into the catalytic pocket. These findings suggest that both catalytic events and substrate delivery processes limit RuBisCO’s overall reaction rate.
In summary, the most widely accepted carboxylation mechanism of RuBisCO involves the following steps. Initially, Kcx201 abstracts a proton from the C3 position of RuBP, forming an enediolate intermediate. Subsequently, a CO2 molecule located within the active site adds to the C2 position of the intermediate, generating the CKABP intermediate, which undergoes hydration of CKABP and C2–C3 bond cleavage. With the help of phosphate groups, this ultimately yields two molecules of 3-PGA (Figure 4). Ongoing efforts to characterize these steps not only clarify the molecular basis of RuBisCO’s limited efficiency but also provide a foundation for rationally engineering RuBisCO variants with improved catalytic performance.
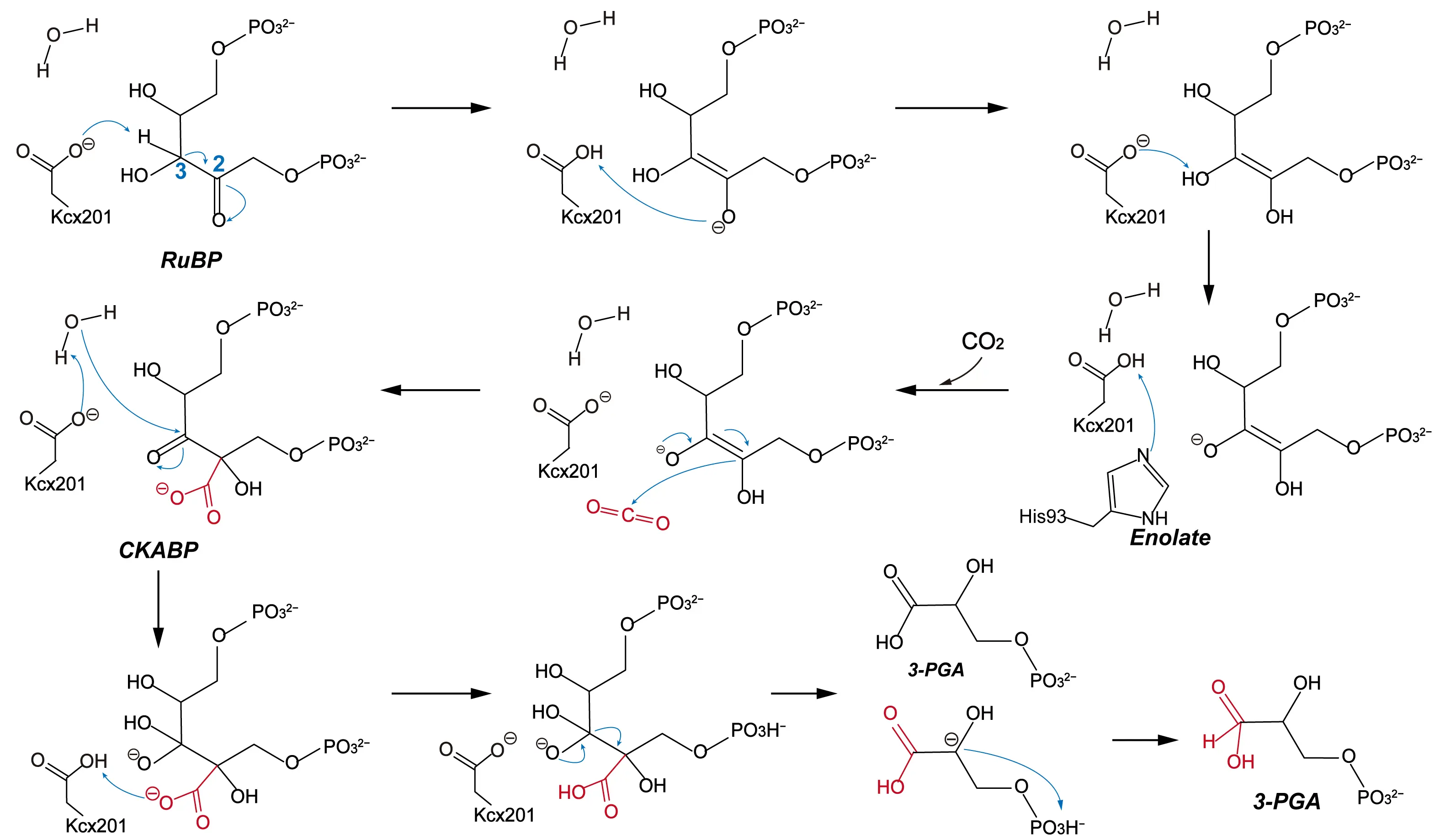
Figure 4. Proposed carboxylation mechanism of RuBisCO based on recent studies. The detailed reaction process is shown, including the carboxylating group (red), the direction of electron transfer (blue arrows), and the free energy of the species involved.
3.2.2. Amidohydrolase Superfamily (AHS)
In recent years, several members of the AHS have been identified to exhibit promiscuous carboxylation activity (Figure 5A) [70]. To investigate the underlying catalytic mechanisms, Sheng et al. conducted detailed QM studies on representative AHS enzymes, including 5-carboxyvanillate decarboxylase (LigW) [71], γ-resorcylate decarboxylase (γ-RSD) [72], 2,3-dihydroxybenzoic acid decarboxylase (2,3-DHBD) [73], salicylic acid decarboxylase (SDC) [74], and iso-orotate decarboxylase (IDCase) (Figure 5B) [75]. They constructed cluster models of each enzyme’s active site and performed QM calculations to examine the decarboxylation and reverse carboxylation pathways. In these studies, the QM region used for cluster model calculations included one CO2 molecule.
Their results revealed a conserved reaction mechanism shared across all five enzymes. First, an aspartate residue protonates the aromatic carboxylic acid substrate, and then CO2 is released. The carboxylation reactions proceed via the exact reverse steps of the decarboxylation process. In both directions, a divalent metal ion in the active site coordinates the substrate and stabilizes the developing negative charge on the intermediate, thereby lowering the activation energy for proton transfer. Importantly, the calculated energy barriers for CO2 release during decarboxylation remained below 11 kcal/mol, indicating that CO2 dissociation is not rate-limiting. In contrast, CO2 addition during carboxylation exhibited significantly higher barriers, making it the rate-limiting step for all enzymes studied. Notably, IDCase displayed the highest energy barrier (17.9 kcal/mol) for CO2 addition, resulting in a near-complete loss of carboxylation activity (Figure 5C). In a complementary study, Hu et al. explored the substrate scope and structural adaptability of SDC [76]. Their simulations showed that SDC possesses an open catalytic cavity that can accommodate a wide range of 2-hydroxybenzoic acid derivatives. Using salicylic acid and ginkgolic acid as model substrates, they demonstrated that forming a tetrahedral coordination geometry, centered on the divalent metal ion, is essential for stable substrate binding (Figure 5D,E).
Overall, members of the AHS exhibit a high degree of catalytic promiscuity, supported by both mechanistic flexibility and structural adaptability. These features make AHS enzymes attractive candidates for the biosynthesis of structurally diverse carboxylated aromatic compounds. Mechanistic insights from QM studies not only enhance our understanding of CO2 fixation chemistry but also provide a rational basis for engineering biocatalysts with tailored specificity and improved efficiency.
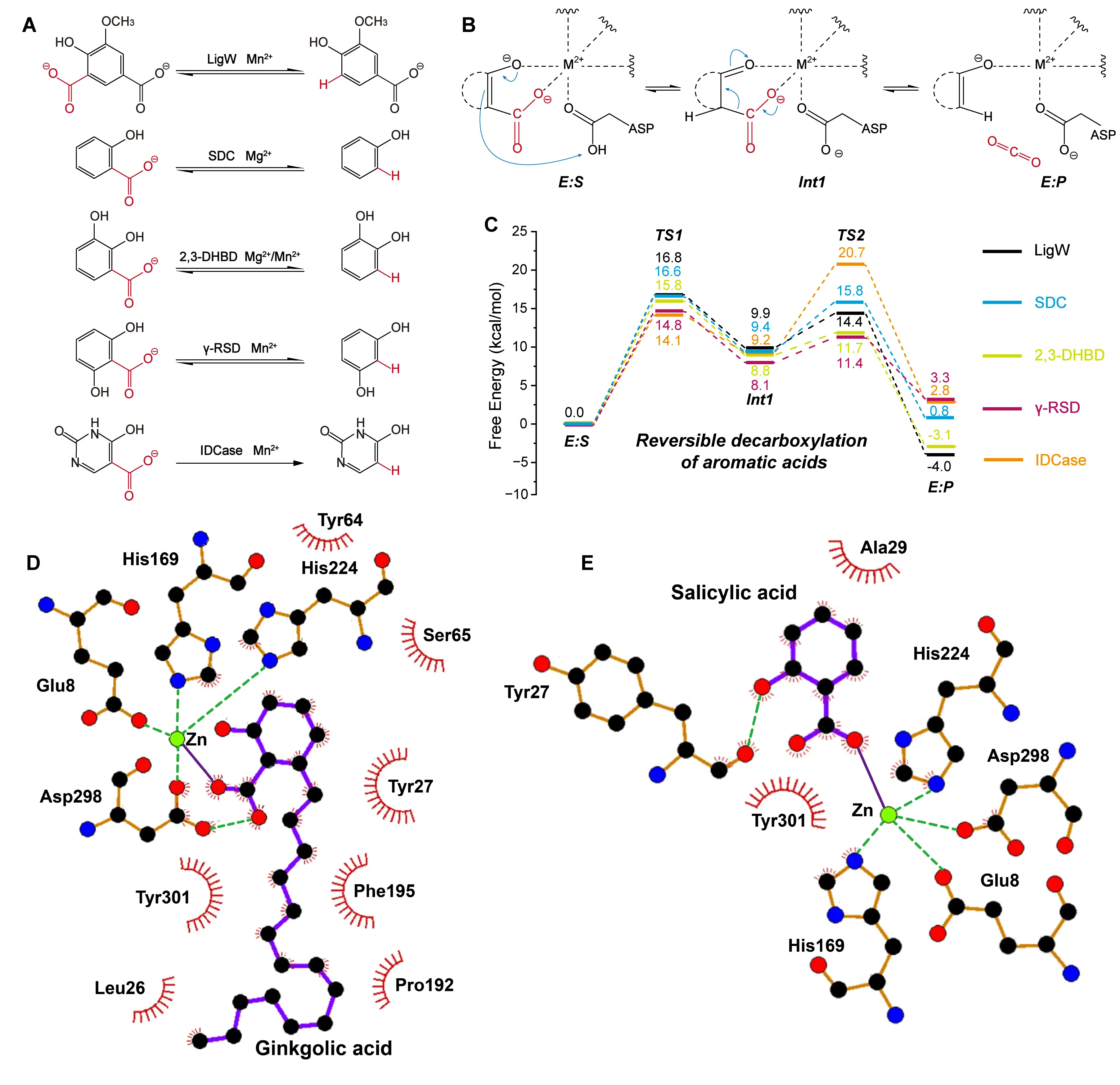
Figure 5. Catalytic and substrate binding mechanisms of AHS members. (A) Decarboxylation and carboxylation reactions catalyzed by AHS family members, including LigW, γ-RSD, 2,3-DHBD, SDC, and IDCase. The group undergoing the carboxylation reaction is highlighted in red. (B) Proposed general reversible decarboxylation mechanism shared by AHS family enzymes. The detailed reaction process is shown, including the decarboxylating group (red), the direction of electron transfer (blue arrows), weak interactions (dashed lines), and the free energy of the species involved. (C) Energy profiles of reversible decarboxylation reactions catalyzed by AHS enzymes, including LigW, γ-RSD, 2,3-DHBD, SDC, and IDCase. Ginkgolic acid (D) and salicylic acid (E) participate in the formation of a tetrahedral coordination geometry within the active site of SDC. This composite figure is adapted from images in references [71,72,73,74,75,76]. Permissions have been obtained from the corresponding publishers, or the figures are used under applicable Creative Commons licenses.
3.2.3. Molybdenum-Dependent Formate Dehydrogenases (Mo-FDH)
Mo-FDH catalyzes the reversible conversion between CO2 and formate. To reduce CO2 to formate, the active site must supply one proton and two electrons. Crystallographic analyses have revealed that the Mo-FDH active site contains a Mo6+ ion in an oxidized state, coordinated by two molybdopterin guanine dinucleotide (MGD) cofactors, a cysteine or selenocysteine residue, and a terminal sulfido ligand [77]. These ligands form a six-coordinate geometry around the Mo center, establishing the structural basis for redox reactivity. Dong et al. investigated the oxidation of formate catalyzed by Mo-FDH using a combination of QM/MM and cluster model approaches [78]. Their results indicated that formate binds via interaction with a cysteine residue in the second coordination sphere and subsequently donates a proton and electron to the Mo center. This study established key roles for outer-sphere residues in facilitating electron transfer during formate oxidation.
To examine the reverse CO2 reduction catalyzed by Mo-FDH, Siegbahn developed a constructed cluster model containing a CO2 molecule for quantum mechanical calculations [79] (Figure 6A). Instead of explicitly modeling electron and proton sources, they initialized their calculations using a pre-activated Mo5+ species, generated by manually introducing one proton and one electron to the Mo6+ coordination complex. In their proposed mechanism, the sulfur atom of Cys196 forms a bond with the carbon of CO2, while the Mo center undergoes a spin-state transition from triplet to singlet. Simultaneously, the surrounding active-site environment donates a second electron to stabilize the intermediate. The reaction proceeds through hydride transfer and ultimately releases formate. Their calculations identified this hydride transfer step as rate-limiting, with a computed energy barrier of 16.0 kcal/mol (Figure 6B).
Complementing these computational insights, Li et al. explored the influence of surrounding amino acid residues using site-directed mutagenesis and MD simulations [80]. Their work revealed that Mo-FDH activity is highly sensitive to perturbations near the metal-binding cofactors. Although mutating Lys44 to Arg44 did not affect cofactor occupancy, MD simulations showed that this change significantly reduced formate binding affinity. These results highlight the importance of precise residue positioning and local charge distribution in maintaining catalytic performance. Together, these studies provide a comprehensive view of the CO2 reduction mechanism catalyzed by Mo-FDH. They demonstrate that metal coordination, electron and proton delivery, spin-state transitions, and the electrostatic landscape of the protein all act in concert to regulate catalytic efficiency.
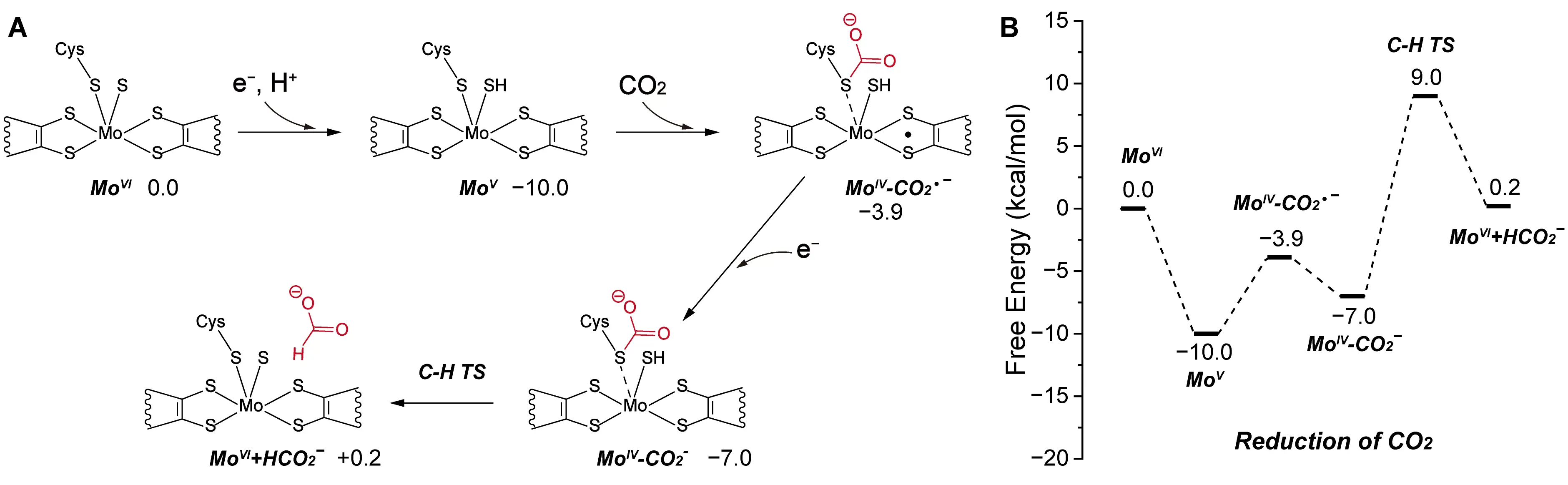
Figure 6. Catalytic mechanism of Mo-FDH. (A) Reaction pathway of CO2 reduction to formate catalyzed by Mo-FDH. The detailed reaction process is shown, including the carboxylating group (red), weak interactions (dashed lines), and the free energy of the species involved. (B) Energy profile of the CO2 reduction process. Adapted from reference [79], Copyright 2022 American Chemical Society and CC-BY 4.0.
3.2.4. Carbon Monoxide Dehydrogenases (CODH)
CODH catalyzes the reversible conversion of CO2 into carbon monoxide (CO) and water, using a NiFe3S4 cluster as its catalytic center. This unique metal-sulfur cluster enables multielectron redox chemistry, which is required for CO2 reduction. To investigate the underlying reaction mechanism, Breglia et al. constructed a detailed cluster model of the active site, incorporating both the first and second coordination spheres surrounding the NiFe3S4 center [81]. They applied QM calculations to probe the electronic structure and proton transfer dynamics involved in catalysis. In CODH active site, a CO2 molecule bridges the Ni and the Fe atoms of the active site. Their simulations suggested that CODH becomes catalytically competent when the His93 residue, located in the second coordination sphere, is doubly protonated. In this protonated state, His93 can donate protons required for the reduction of CO2. The proposed mechanism begins with CO2 binding to the Ni center of the cluster. The molecule then shifts to a bridging coordination position between the Ni and one of the Fe atoms, forming a stable metal-CO2 adduct. Subsequently, two coupled proton and electron transfer steps convert the coordinated CO2 into CO and H2O. The stepwise delivery of protons, mediated by His93, and electrons, supplied by nearby redox centers, facilitates the cleavage of C=O bonds and the formation of the reduced products. This mechanistic model underscores the essential role of the second coordination sphere in modulating proton availability and coordinating substrate activation, both of which are critical for efficient CO2 reduction by CODH (Figure 7A,B).
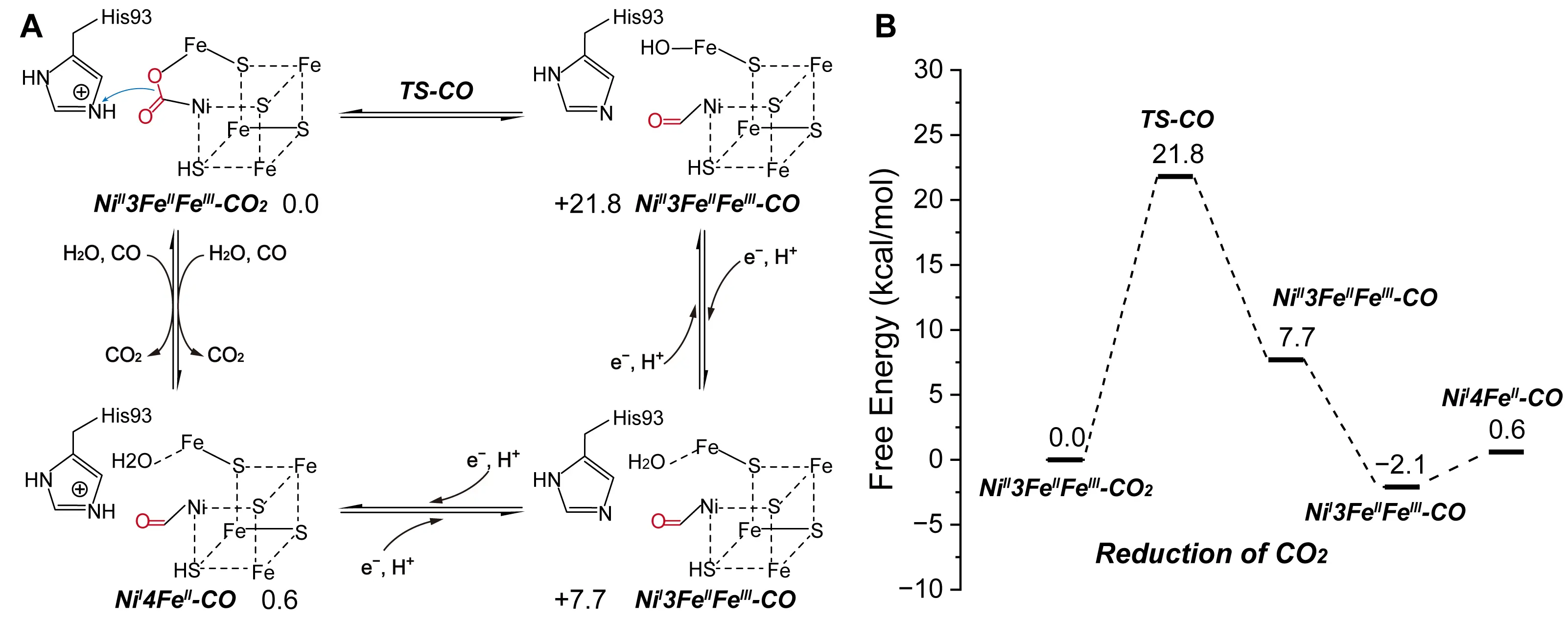
Figure 7. Catalytic mechanism of CODH. Reaction pathway (A) and energy (B) of CO and CO2 interconversion catalyzed by CODH. The detailed reaction process is shown, including the carboxylating group (red), the direction of electron transfer (blue arrows), the iron-sulfur cluster (dashed lines), and the free energy of the species involved. Adapted from reference [81], Copyright 2020 American Chemical Society.
3.3. NAD(P)H-Dependent Enzymes
NAD(P)H-dependent oxidoreductases play critical roles in central metabolism by mediating reversible electron transfer between NAD(P)H and NAD(P)+, often coupling this redox process with the oxidation or reduction of substrates [82]. Among them, a subset has been found capable of reducing CO2, particularly NAD(P)H-dependent formate dehydrogenases (NAD(P)H-FDHs), which catalyze the direct conversion of CO2 to formate [83]. These enzymes show promise in one-carbon metabolism, though their detailed reaction mechanisms remain poorly understood.
More recently, several members of the ECR family, such as CCR [84], hydroxymethylglutaryl-CoA carboxylase (GCC) [85], and medium-chain dehydrogenase/reductase (MDR) [86], have been computationally and experimentally confirmed to catalyze the carboxylation of α,β-unsaturated CoA-thioesters using NAD(P)H as a cofactor. With advancements in cofactor regeneration systems for NAD(P)H, these enzymes hold great promise for large-scale CO2 fixation to produce high-value chemicals. Among these, CCR has been the most extensively characterized and serves as a model for elucidating ECR-catalyzed CO2 fixation mechanisms. Crystallographic studies have revealed that CCR forms a tetramer organized as two dimers, with each dimer containing one open and one closed active site [87]. Upon substrate binding, the open site undergoes a conformational transition to the closed state, thereby enabling catalysis [88]. To explore how these conformational changes influence CO2 binding, Gomez et al. employed MD simulations to evaluate binding free energies [89]. Their results suggest an inter-dimer communication mechanism during the CCR-catalyzed CO2 fixation cycle. Specifically, product release and active-site opening in one dimer trigger active-site closure in the adjacent dimer. Throughout a single catalytic cycle, the four subunits of CCR alternate between open and closed conformations. This alternation ensures that CO2, NADPH, and crotonyl-CoA bind in the open state, while catalysis proceeds in the closed state. Such switching may enhance substrate positioning and CO2 concentration within the open active site, thereby improving CO2 fixation efficiency (Figure 8A).
Following the confirmation of CCR’s carboxylation activity, a series of experimental and theoretical studies have proposed detailed reaction mechanisms. The classic model for CCR involves an initial hydride transfer from NADPH to the β-carbon of crotonyl-CoA, generating an enolate intermediate. This intermediate subsequently attacks CO2, yielding (2S)-ethylmalonyl-CoA [84]. Building on this model, Stoffel et al. used QM/MM calculations to investigate the catalytic roles of key residues [88]. They identified Asn81 as critical for orienting CO2 via direct interaction, while His365 and Glu171 assist indirectly by forming a water-mediated hydrogen-bond network. Additionally, Phe170 stabilizes the reactive enolate intermediate. Interestingly, Rosenthal et al. later discovered a covalent adduct formed between NADPH and crotonyl-CoA during the CCR catalytic cycle [90]. Recabarren subsequently validated this alternative pathway through QM/MM-MD simulations, providing mechanistic evidence for the formation of this novel intermediate [91]. In contrast to the classic hydride transfer mechanism, the adduct formation pathway involves a Michael addition between the C2 atom of NADPH and the Cα atom of the substrate, generating a stable C2-adduct intermediate. In this alternative mechanism, the adduct undergoes carboxylation to form the final product. Free energy profiles indicate that the activation barrier for carboxylating the C2-adduct is comparable to that of the traditional enolate pathway (Figure 8B). In 2024, they further compared the free energy profiles of the direct and C2 mechanisms, providing a detailed summary of the computational methods [92]. They emphasized the crucial role of entropy effects in carboxylation reactions, as CO2 fixation induces an entropy penalty, which must be considered when calculating the free energy profile. To estimate the free energy, they employed a QM/MM MD simulation method that accounts for entropy effects. In the CCR active site, the author docked a gaseous CO2 molecule. First, they used string optimization to search for the minimum free energy path (MFEP) of the two CCR carboxylation mechanisms. They then applied umbrella sampling to explore the conformational space near the MFEP and accurately predict the free energy profile. In this process, they used DFTB3 as the QM method for electronic structure description, CHARMM36 for the MM description of the protein backbone. The free energy profiles revealed that the activation barrier for the C2-addition pathway was nearly identical to that of the direct enol pathway. In the direct mechanism, the proton transfer is the rate-determining step, with the enolate being considered the active intermediate. This intermediate is unstable, requiring only a 6.5 kcal/mol energy barrier to convert into the stable carboxylic acid product. In the C2 mechanism, the transition from the enolate intermediate to the C2 intermediate requires a 1.9 kcal/mol barrier. Furthermore, the C2 intermediate is almost as stable as the carboxylic acid product, which is consistent with experimental results where stable adducts were detected in the presence of CO2. Based on these computational results, the authors proposed that CCR may employ both mechanisms. The enolate pathway enables direct carboxylation, while the C2-adduct route could serve to trap and stabilize the highly reactive enolate intermediate. Since the C2-adduct is more stable, it can protect the enolate from unwanted protonation by ambient acids under conditions of CO2 limitation, thereby indirectly enhancing the efficiency of CO2 fixation.
Together, these studies offer a detailed picture of CCR-catalyzed CO2 fixation, encompassing conformational dynamics, active-site chemistry, and multiple potential reaction pathways. The mechanistic principles derived from CCR provide a valuable foundation for modeling and engineering other ECR family enzymes for improved CO2 utilization.
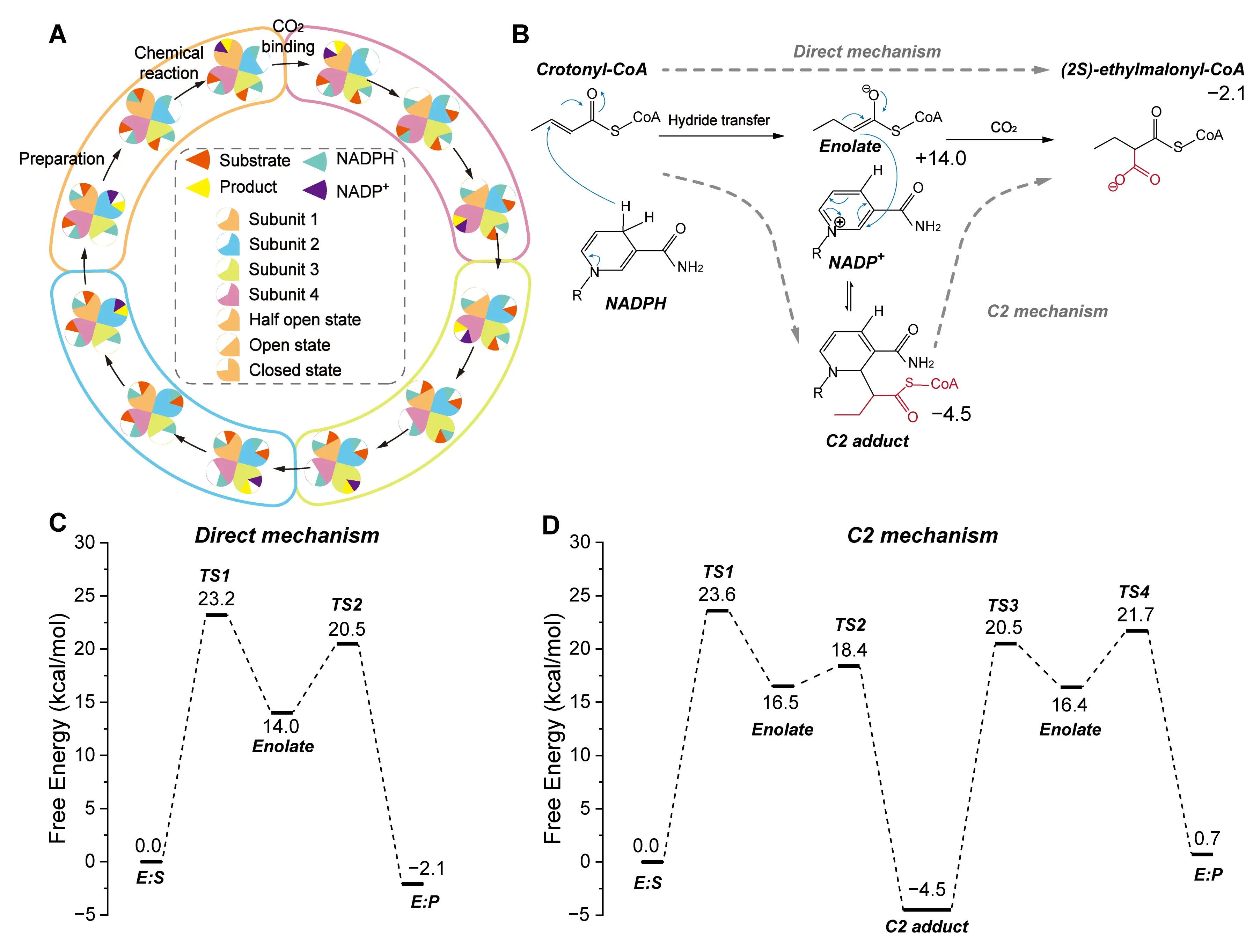
Figure 8. Catalytic and substrate binding mechanisms of CCR. (A) Proposed inter-dimer communication mechanism during the CCR-catalyzed CO2 fixation cycle. (B) Two proposed mechanisms for CCR-catalyzed CO2 conversion: the direct mechanism and the C2 adduct formation mechanism. The detailed reaction process is shown, including the carboxylating group (red), the direction of electron transfer (blue arrows), and the free energy of the species involved. (C) The calculated energy profile of the direct mechanism. (D) The calculated energy profile of the C2 mechanism. This composite figure is adapted from images in references [89,91,92]. Permissions have been obtained from the corresponding publishers.
3.4. PrFMN-Dependent Enzymes
Flavins are among the most abundant cofactors in nature, and flavin-containing enzymes are widely involved in redox-related metabolic pathways across various organisms. Among flavin derivatives, prFMN is uniquely modified at the C4a-N5-C1′ positions and functions as an electron donor that facilitates reversible decarboxylation reactions in members of the UbiD family [19]. UbiD enzymes, which are widely distributed across organisms, catalyze redox-neutral transformations, including the carboxylation of unsaturated compounds such as alkenes, aromatic catechols, and heterocycles [93]. For example, Fdc1 catalyzes the carboxylation of cinnamic acid derivatives [94], while AroY targets aromatic catechol substrates [95]. Additionally, pyrrole-2-carboxylate decarboxylase (HudA) [96], indole-3-carboxylate decarboxylase (InD) [97], and HmfF [98] have been reported to catalyze the carboxylation of heterocyclic substrates such as pyrrole, indole, and furoic acid, respectively. This substrate diversity positions the UbiD family as a promising platform for converting CO2 into valuable chemicals. Although UbiD enzymes act on various substrates, current studies suggest that they share a similar catalytic mechanism [19]. Initially, the substrate forms a covalent adduct intermediate with the prFMN cofactor. A general base then deprotonates the intermediate, enabling nucleophilic attack on CO2 and formation of the carboxylated product. However, the structure of this adduct and the detailed reaction steps can vary among different UbiD enzymes. In the following sections, we discuss the catalytic mechanisms of various UbiD enzymes based on their specific mechanistic features.
Fdc1 was the first UbiD enzyme identified to utilize prFMN cofactor for reversible carboxylation. Payne et al. resolved the crystal structure of Fdc1 and proposed a mechanism involving a 1,3-dipolar cycloaddition between the prFMN cofactor and the unsaturated substrate [94]. This reaction forms a five-membered ring intermediate in which the carboxyl group is unstable. Upon CO2 molecular release, the ring opens, and Glu282 donates a proton to form a second five-membered ring intermediate. A subsequent retro-1,3-dipolar cycloaddition releases the product. The reverse pathway—carboxylation—proceeds by the same steps in reverse order. Subsequent studies employing cluster models and QM/MM methods confirmed that the interaction between the substrate and prFMN in Fdc1 indeed proceeds via a 1,3-dipolar cycloaddition (Figure 9A). Tian et al. further investigated the reaction energetics and identified the deprotonation of the five-membered intermediate, rather than CO2 addition, as the rate-limiting step in the carboxylation direction [99] (Figure 9B).
In addition to chemical steps, structural dynamics play a key role in UbiD catalysis. Beveridge et al. used mass spectrometry and MD simulations to examine how prFMN binding influences Fdc1’s structural behavior [100]. Their findings showed that prFMN binding enhances protein rigidity and strengthens dimeric interactions within the active complex, suggesting that conformational stability is crucial for catalytic performance. Phenazine-1-carboxylic acid decarboxylase (PhdA), another UbiD enzyme, catalyzes the reversible interconversion between phenazine-1-carboxylic acid and phenazine. Datar et al. proposed a catalytic mechanism for PhdA similar to that of Fdc1, which also involves a 1,3-dipolar cycloaddition [101]. Their combined MD and experimental analyses suggested that dynamic transitions between open and closed conformations may represent a general regulatory feature of UbiD enzymes, potentially governing substrate access and product release (Figure 9C,D).
Payer et al. identified AroY as a UbiD enzyme capable of catalyzing the reversible decarboxylation of catechol substrates [95]. Using QM calculations, they proposed a distinct catalytic mechanism in which the Cα atom of the catechol substrate performs a nucleophilic attack on the C1′ position of prFMN, forming a quinone-type adduct linked by a single covalent bond (Figure 10A). CO2 was set as a gas in the molecular form in the calculation. This pathway differs fundamentally from the 1,3-dipolar cycloaddition observed in Fdc1. Their calculations indicated an energy barrier of 23.5 kcal/mol for the nucleophilic attack mechanism, which is significantly lower than that for the cycloaddition route, suggesting that AroY does not proceed via a five-membered ring intermediate (Figure 10B). Mechanistically, the second hydroxyl group on the catechol ring plays a critical role by forming a hydrogen bond with His327, anchoring the substrate within the active site. More importantly, deprotonation of this hydroxyl group enhances substrate nucleophilicity and stabilizes the transition state during the nucleophilic attack. In other words, this deprotonation helps stabilize the positive charge generated during the nucleophilic attack on prFMN, thereby facilitating the reaction.
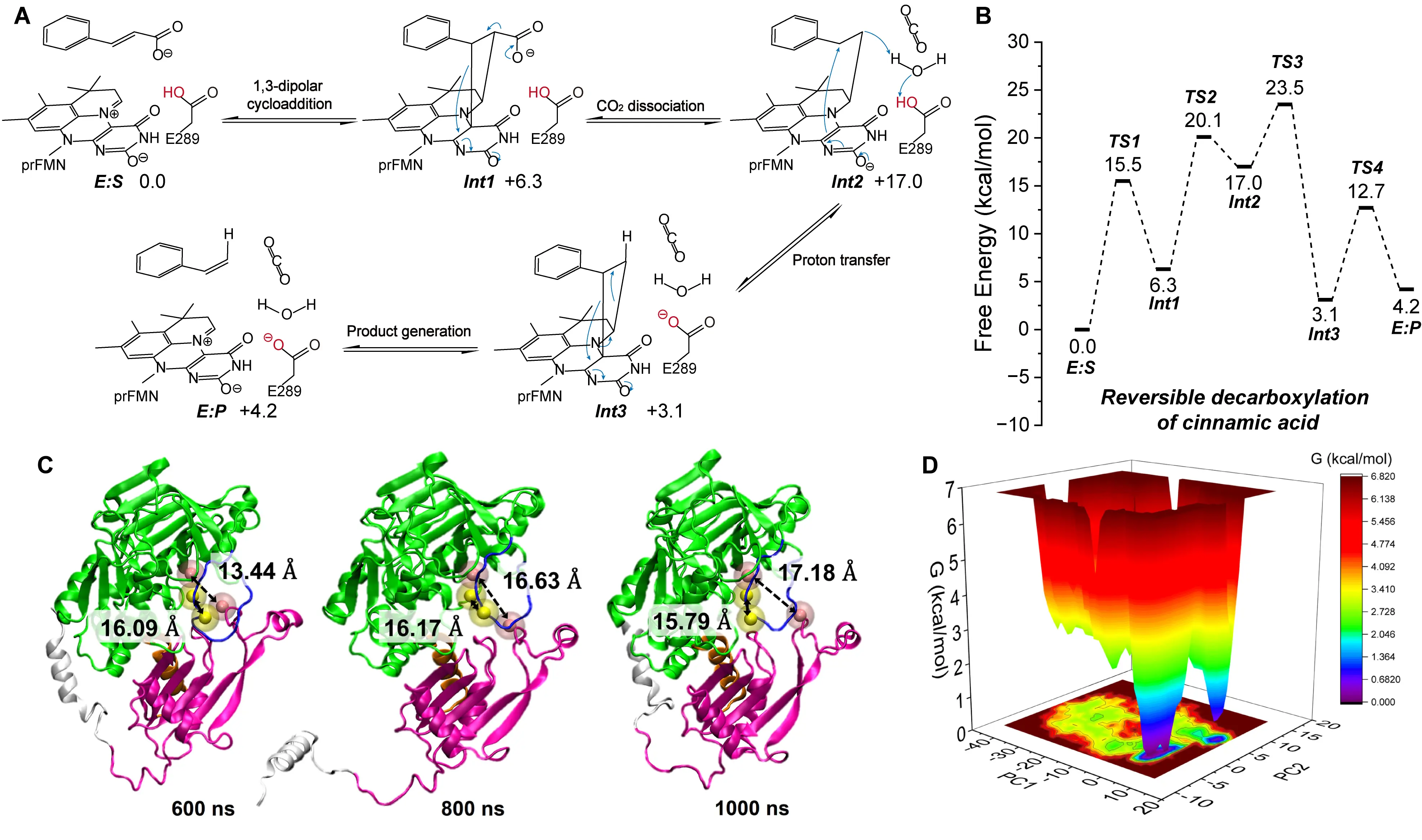
Figure 9. Catalytic mechanisms of Fdc1 and the conformational transition mechanism of PhdA. Reaction pathway (A) and energy profile (B) of carboxylation reaction catalyzed by Fdc1. The detailed reaction process is shown, including the carboxylating group (red), the direction of electron transfer (blue arrows), and the free energy of the species involved. Conformational transition of PhdA from “closed” to “open” observed during MD simulations (C), along with the corresponding free energy landscape (D). This composite figure is adapted from images in references [98,100]. Permissions have been obtained from the corresponding publishers, or the figures are used under applicable Creative Commons licenses.
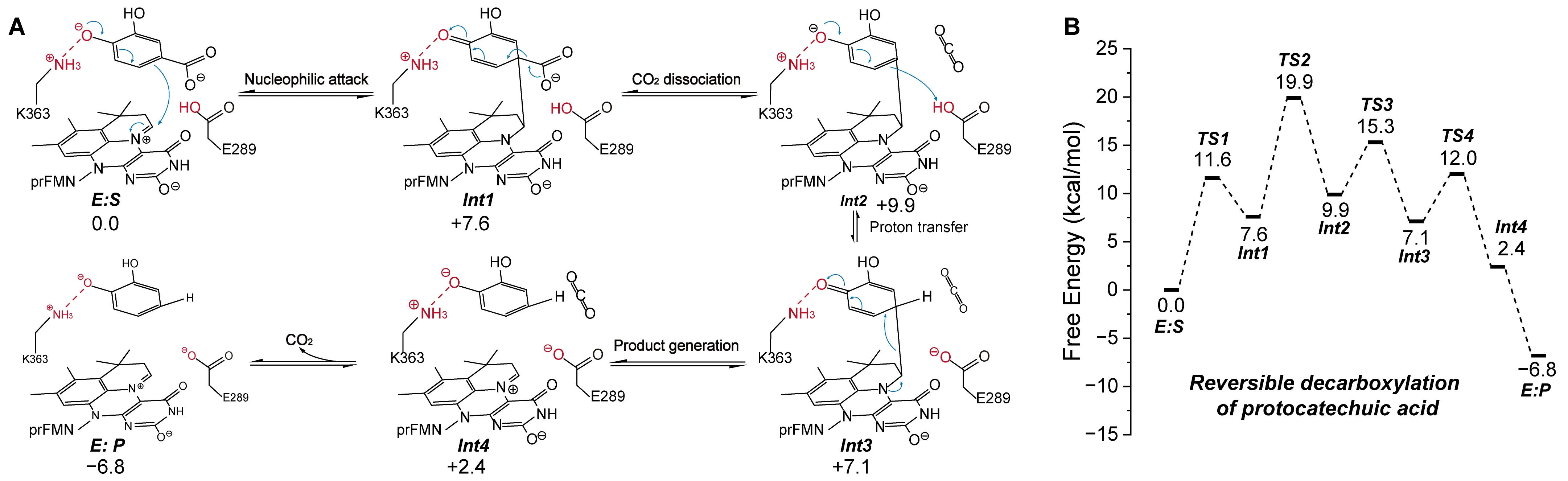
Figure 10. Catalytic mechanisms of AroY. Reaction pathway (A) and energy profile (B) of carboxylation reaction catalyzed by AroY. The detailed reaction process is shown, including the carboxylating group (red), the direction of electron transfer (blue arrows), weak interactions (dashed lines), and the free energy of the species involved. Adapted from reference [95], under CC BY license.
Building on this mechanistic framework, we conducted a systematic study of prFMN-dependent decarboxylases that act on heteroaromatic substrates, including HmfF, InD, and HudA [102]. Experimental and computational results consistently show that these enzymes exhibit markedly lower carboxylation rates compared to their decarboxylation activity, emphasizing the need to identify the rate-limiting steps. To this end, we employed QM/MM MD simulations to examine both directions of the reaction. In the QM region, CO2 is set as the molecular form rather than the bicarbonate form. Our free energy profiles revealed that decarboxylation proceeds via a Wheland-type intermediate, which forms through nucleophilic attack of the heteroaromatic ring on the C1′ atom of prFMN. In contrast, the carboxylation reaction follows a 1,3-dipolar cycloaddition pathway, resulting in a five-membered ring intermediate (Figure 11A–C).
For HmfF, we conducted detailed QM/MM MD simulations to investigate the mechanistic differences between carboxylation and decarboxylation. We propose that the key factor underlying this difference lies in the nucleophilicity of the substrate. Compared to furoic acid, furandicarboxylic acid (FDCA) contains an additional carboxyl group, which reduces the electron density on the aromatic ring and thereby diminishes its nucleophilic reactivity. Upon nucleophilic attack of the substrate on the C1′ atom of prFMN, both reactions proceed through the formation of a Wheland-type intermediate centered on the furan ring. However, the electronic properties of this intermediate differ markedly between substrates. Using cluster model calculations, we compared the electronic population distributions of the prFMN-furoic acid and prFMN-FDCA intermediates within the HmfF active site. Our results showed that the extra carboxyl group in FDCA partially delocalizes and neutralizes the positive charge accumulated on the furan ring, which confers slightly greater stability to the intermediate. By contrast, the intermediate formed from furoic acid exhibits a significantly higher positive charge density on the ring. This destabilization drives the system toward a structural rearrangement, favoring the formation of a five-membered ring species through a 1,3-dipolar cycloaddition. This five-membered ring intermediate imposes critical constraints on the subsequent steps of the carboxylation reaction. Specifically, proton transfer to Glu259 must be coupled with ring cleavage, forming a concerted process that links bond cleavage with proton transfer. Free energy calculations revealed that this coupled event has a high activation barrier of 23.5 kcal/mol, thereby identifying proton transfer as the rate-limiting step in the carboxylation pathway (Figure 11C).
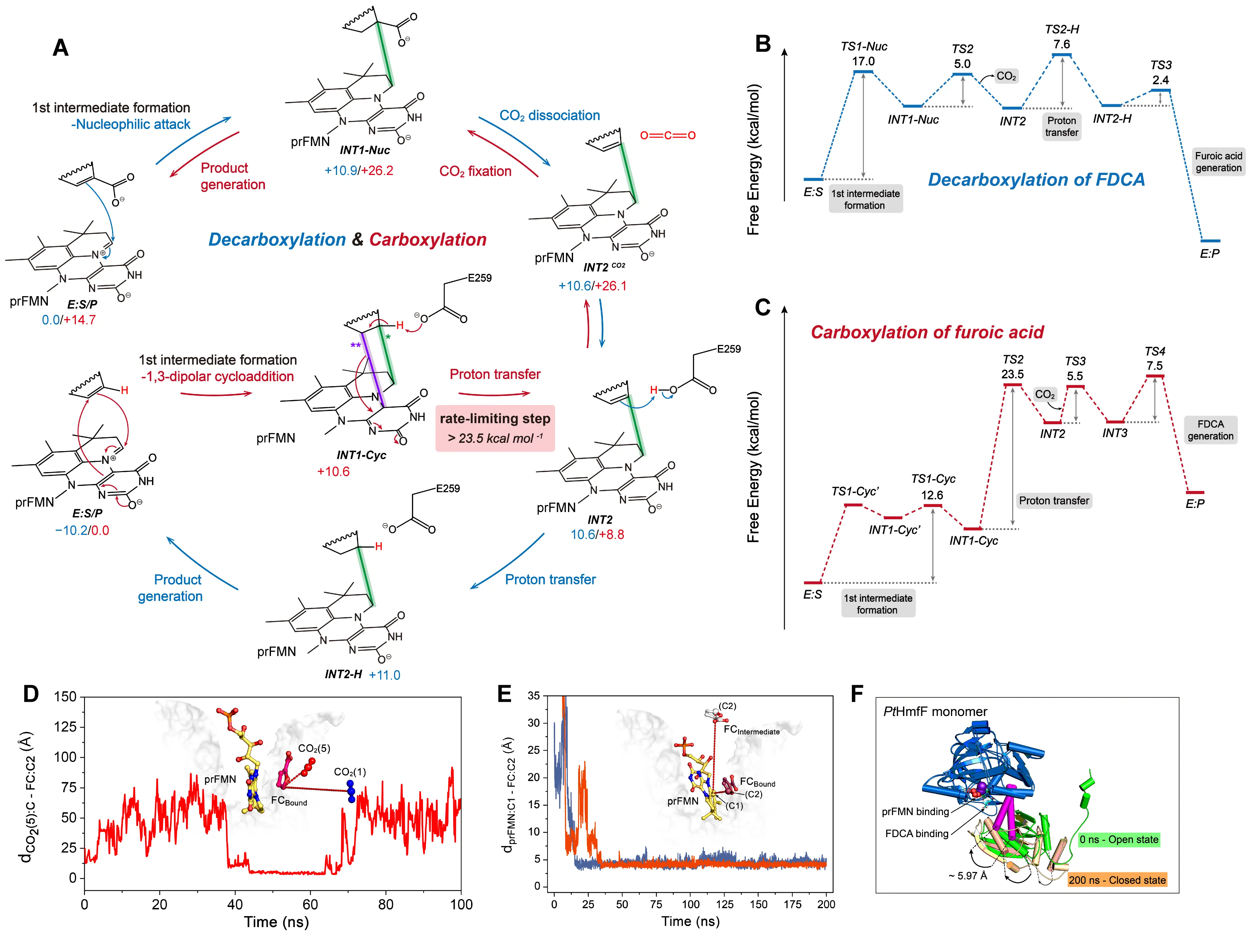
Figure 11. Catalytic and substrate binding mechanisms of HmfF. (A) Decarboxylation and carboxylation pathways catalyzed by prFMN-dependent decarboxylases acting on heteroaromatic substrates. The detailed reaction process is shown, including the carboxylating group (red), weak interactions (dashed lines), and the free energy of the species involved. In carboxylation reaction mechanism, a single asterisk (*) indicates the first bond formed during the formation of the five-membered ring intermediate(INT1-Cyc), while two asterisks (**) indicate the second bond formed. Energy profile of decarboxylation (B), carboxylation (C) reaction catalyzed by HmfF. The complete binding pathways of the two carboxylation substrates of HmfF, including furoic acid (D) and CO2 (E). Conformational transition of HmfF from “closed” to “open” observed during MD simulations (F).
We also employed Gaussian accelerated MD (GaMD) [103] simulations to examine the substrate-binding dynamics of furoic acid and CO2 within HmfF (Figure 11D,E). These simulations indicated that both substrates access the active site without significant steric hindrance from surrounding residues, suggesting that diffusion is not a limiting factor. Instead, the rate-limiting step lies within the chemical transformation itself. Moreover, we observed hinge-region motions between the N-terminal prFMN-binding domain and the C-terminal domain of HmfF (Figure 11F). Given that the substrate binds near this flexible region, the observed inter-domain movement likely governs the transition between “open” and “closed” enzyme states.
These computational findings demonstrate that the catalytic mechanism of UbiD-family enzymes is strongly substrate-dependent. Even small structural modifications, such as the addition of a single carboxyl group, can alter substrate nucleophilicity, affect charge distribution in reaction intermediates, and redirect the overall reaction trajectory. Consequently, different substrates can induce distinct mechanistic outcomes within the same enzyme framework. Furthermore, conformational flexibility around the active site appears to be a conserved feature that enables dynamic regulation of catalysis across the UbiD family.
4. Conclusions and Perspectives
The use of natural enzymes to convert CO2 offers a green, mild, and renewable approach for addressing energy and environmental challenges. In recent years, advances in biotechnology and computational chemistry have led to the discovery and expression of an increasing number of CO2-converting enzymes. These developments have significantly broadened the scope of enzyme-catalyzed synthesis of high-value chemicals.
In this review, we summarized the catalytic mechanisms of four major classes of CO2-converting enzymes: cofactor-independent enzymes, divalent metal-dependent enzymes, NAD(P)H-dependent enzymes, and prFMN-dependent enzymes. We examined both QM studies that elucidate their reaction pathways and MD simulations that reveal how conformational flexibility influences catalytic activity. These studies collectively highlight the mechanistic diversity of CO2 conversion in biology. Rather than relying solely on the formation of a direct covalent bond between CO2 and the substrate, these enzymes achieve CO2 activation and conversion by orchestrating a series of well-timed proton and electron transfers. Specific active-site residues or associated cofactors typically mediate these transfers. In addition, spatial arrangement and weak interactions within the active pocket play essential roles in stabilizing both substrates and reactive intermediates.
Despite their mechanistic sophistication, none of these enzymes has yet been applied at an industrial scale. The main limitation lies in their inherently low carboxylation efficiency. In nature, this level of efficiency suffices for cellular metabolism and survival. However, evolutionary pressures have not favored the optimization of these enzymes for high-throughput carbon fixation, limiting their industrial applicability. To unlock the full potential of biocatalytic carboxylation, it is therefore essential to improve enzyme performance through rational engineering.
A deeper understanding of catalytic mechanisms is crucial for guiding such efforts. Computational studies can identify residues associated with rate-limiting steps by mapping free energy profiles of both substrate binding and chemical transformation. This knowledge enables the rational design of targeted mutations. For instance, site-directed, combinatorial, or iterative saturation mutagenesis can be applied to replace residues that control key reaction barriers, potentially enhancing catalytic turnover. Additionally, MD simulations offer insights into substrate-binding conformations and dynamics, which are valuable for improving binding affinity and broadening substrate scope.
In addition to traditional computational methods, machine learning techniques have increasingly found applications in the field of enzymology. For example, Alphafold [104,105] and RosettaFold [106] have achieved near-experimental accuracy in a variety of protein structure prediction tasks. Pre-trained protein language models such as ESM are capable of transforming enzyme sequences into feature vectors, which can then be used to design predictive models for new functionalities [107]. The developers of DLkcat claim that their model can directly predict changes in functional parameters, such as kcat, due to mutations in enzyme sequences [108]. Unlike traditional computational methods discussed in this review, these machine learning approaches do not require users to have extensive prior knowledge of enzyme structure and function. This is because the models are trained to interpret the relationship between protein sequences, structures, and functions during the training phase. In the field of CO2-fixing enzymes, machine learning methods have also made progress. For instance, Lou et al. utilized Alphafold to predict the structure of phosphoenolpyruvate carboxykinase and conducted MD simulations at 30 °C and 50 °C based on this predicted structure [109]. Using Rosetta, they performed saturation mutagenesis on residues with increased RMSF at high temperatures and obtained a high-activity mutant, C886R. Marchal et al. employed a Gaussian Process (GP) regression model to develop a machine learning model that predicts the catalytic efficiency of acetyl-CoA carboxylase mutants [110]. Based on the predicted mutant rankings, they identified a mutant with a 1000-fold increase in activity. Overall, the application of machine learning in CO2-fixing enzymes remains in its early stages, primarily employed for structure prediction or sequence-function relationship studies. While MD and QM methods have the potential to provide valuable data for training machine learning models by revealing the conformational-function relationship, building an effective mechanism research database requires substantial computational resources, including large-scale MD simulations or QM calculations. For example, Kozinsky recently used machine-learned potentials in biomolecular simulations with 5120 A100 GPUs in parallel [111]. Thus, the greatest challenge in directly using machine learning models to explore the enzyme structure-function relationships involved in traditional mechanistic studies remains the construction of databases.
In summary, we have reviewed how computer-aided techniques contribute to elucidating the catalytic principles of CO2-fixing enzymes. These mechanistic insights not only deepen our understanding of natural biocatalysts but also provide a strong theoretical foundation for engineering more efficient enzymes and discovering novel CO2-converting systems with enhanced performance for future biotechnological applications.
Statement of the Use of Generative AI and AI-Assisted Technologies in the Writing Process
During the preparation of this manuscript, the author used ChatGPT(OpenAI) in order to improve the writing quality. After using this tool, the author reviewed and edited the content as needed and takes full responsibility for the content of the published article.
Author Contributions
Conceptualization, K.W., J.Z. and Q.L.; Writing—Original Draft, K.W.; Writing—Review & Editing, J.Z. and Q.L.; Supervision, J.Z. and Q.L.; Project Administration, J.Z. and Q.L.
Ethics Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data supporting this study are included within the article.
Funding
This research was funded by the National Natural Science Foundation of China (32571468, 32201030, U24A20365 and 32271319), the Science and Technology Department of Jilin Province (20230402041GH), the Development and Reform Commission of Jilin Province (2023C015), the Fundamental Research Funds of the Central Universities, China (2024-JCXK-11), and the Innovation Program for Graduate Students of Jilin University (2025CX127).
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
-
Ramirez-Corredores MM. Sustainable production of CO2-derived materials. npj Mater. Sustain. 2024, 2, 35. doi:10.1038/s44296-024-00041-9. [Google Scholar]
-
Vega LF, Bahamon D, Alkhatib III. Perspectives on advancing sustainable CO2 conversion processes: trinomial technology, environment, and economy. ACS Sustain. Chem. Eng. 2024, 12, 5357–5382. doi:10.1021/acssuschemeng.3c07133. [Google Scholar]
-
Friedlingstein P, O’Sullivan M, Jones MW, Andrew RM, Hauck J, Landschützer P, et al. Global carbon budget 2024. Earth Syst. Sci. Data 2025, 17, 965–1039. doi:10.5194/essd-17-965-2025. [Google Scholar]
-
Deng Z, Zhu B, Davis SJ, Ciais P, Guan D, Gong P, et al. Global carbon emissions and decarbonization in 2024. Nat. Rev. Earth Environ. 2025, 6, 231–233. doi:10.1038/s43017-025-00658-x. [Google Scholar]
-
Faba L, Ordóñez S. Carboxylation reactions for the sustainable manufacture of chemicals and monomers. RSC Sustain. 2024, 2, 3167–3182. doi:10.1039/D4SU00482E. [Google Scholar]
-
O’Keeffe S, Garcia L, Chen Y, Law RC, Liu C, Park JO. Bringing carbon to life via one-carbon metabolism. Trends Biotechnol. 2025, 43, 572–585. doi:10.1016/j.tibtech.2024.08.014. [Google Scholar]
-
Yang D, Li S, He S, Zheng Y. Can conversion of CO2 into fuels via electrochemical or thermochemical reduction be energy efficient and reduce emissions? Energy Conv. Manag. 2022, 273, 116425. doi:10.1016/j.enconman.2022.116425. [Google Scholar]
-
Zhao L, Hu H, Wu A, Terent’ev AO, He L, Li H. CO2 capture and in-situ conversion to organic molecules. J. CO2 Util. 2024, 82, 102753. doi:10.1016/j.jcou.2024.102753. [Google Scholar]
-
Song Q, Ma R, Liu P, Zhang K, He L. Recent progress in CO2 conversion into organic chemicals by molecular catalysis. Green Chem. 2023, 25, 6538–6560. doi:10.1039/D3GC01892J. [Google Scholar]
-
Yan Z, Liu W, Liu X, Shen Z, Li X, Cao D. Recent progress in electrocatalytic conversion of CO2 to valuable C2 products. Adv. Mater. Interfaces 2023, 10, 2300186. doi:10.1002/admi.202300186. [Google Scholar]
-
Lei G, Wang Z, Xia S, Fan Y, Zhao K, Zhao Z, et al. Recent progress in thermal catalytic conversion of CO2: Insights into synergies with alkane or biomass transformations. Fuel 2025, 381, 133366. doi:10.1016/j.fuel.2024.133366. [Google Scholar]
-
Bachleitner S, Ata Ö, Mattanovich D. The potential of CO2-based production cycles in biotechnology to fight the climate crisis. Nat. Commun. 2023, 14, 6978. doi:10.1038/s41467-023-42790-6. [Google Scholar]
-
Liu B, Lin B, Su H, Sheng X. Quantum chemical studies of the reaction mechanisms of enzymatic CO2 conversion. Phys. Chem. Chem. Phys. 2024, 26, 26677–26692. doi:10.1039/D4CP03049D. [Google Scholar]
-
Liu X, Li L, Zhao G, Xiong P. Optimization strategies for CO2 biological fixation. Biotechnol. Adv. 2024, 73, 108364. doi:10.1016/j.biotechadv.2024.108364. [Google Scholar]
-
Bierbaumer S, Nattermann M, Schulz L, Zschoche R, Erb TJ, Winkler CK, et al. Enzymatic conversion of CO2: From natural to artificial utilization. Chem. Rev. 2023, 123, 5702–5754. doi:10.1021/acs.chemrev.2c00581. [Google Scholar]
-
Yuan L, Bonku EM, Yang Z-H. Biocatalysis-driven CO2 valorization: Innovations and sustainable strategies in conversion and utilization. Carbon Capture Sci. Technol. 2025, 15, 100437. doi:10.1016/j.ccst.2025.100437. [Google Scholar]
-
Terholsen H, Huerta-Zerón HD, Möller C, Junge H, Beller M, Bornscheuer UT. Photocatalytic CO2 reduction using CO2-binding enzymes. Angew. Chem. Int. Edit. 2024, 63, e202319313. doi:10.1002/anie.202319313. [Google Scholar]
-
Deng Y, Wang JX, Ghosh B, Lu Y. Enzymatic CO2 reduction catalyzed by natural and artificial metalloenzymes. J. Inorg. Biochem. 2024, 259, 112669. doi:10.1016/j.jinorgbio.2024.112669. [Google Scholar]
-
Bloor S, Michurin I, Titchiner GR, Leys D. Prenylated flavins: structures and mechanisms. FEBS J. 2023, 290, 2232–2245. doi:10.1111/febs.16371. [Google Scholar]
-
Gupta N, Sarkar A, Biswas SK. In situ NADH regeneration coupled to enzymatic CO2 reduction: from fundamental concepts to recent advances. J. Environ. Chem. Eng. 2025, 13, 116657. doi:10.1016/j.jece.2025.116657. [Google Scholar]
-
Studer A, Curran DP. The electron is a catalyst. Nat. Chem. 2014, 6, 765–773. doi:10.1038/nchem.2031. [Google Scholar]
-
Callender R, Dyer RB. The dynamical nature of enzymatic catalysis. Acc. Chem. Res. 2015, 48, 407–413. doi:10.1021/ar5002928. [Google Scholar]
-
Hollingsworth SA, Dror RO. Molecular dynamics simulation for all. Neuron 2018, 99, 1129–1143. doi:10.1016/j.neuron.2018.08.011. [Google Scholar]
-
Świderek K, Bertran J, Zinovjev K, Tuñón I, Moliner V. Advances in the simulations of enzyme reactivity in the dawn of the artificial intelligence age. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2025, 15, e70003. doi:10.1002/wcms.70003. [Google Scholar]
-
Moradi S, Nowroozi A, Aryaei Nezhad M, Jalali P, Khosravi R, Shahlaei M. A review on description dynamics and conformational changes of proteins using combination of principal component analysis and molecular dynamics simulation. Comput. Biol. Med. 2024, 183, 109245. doi:10.1016/j.compbiomed.2024.109245. [Google Scholar]
-
Acevedo O, Jorgensen WL. Advances in quantum and molecular mechanical (QM/MM) simulations for organic and enzymatic reactions. Acc. Chem. Res. 2010, 43, 142–151. doi:10.1021/ar900171c. [Google Scholar]
-
Berendsen HJC, Postma JPM, van Gunsteren WF, DiNola A, Haak JR. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. doi:10.1063/1.448118. [Google Scholar]
-
Karplus M, McCammon JA. Molecular dynamics simulations of biomolecules. Nat. Struct. Biol. 2002, 9, 646–652. doi:10.1038/nsb0902-646. [Google Scholar]
-
Ensign DL, Kasson PM, Pande VS. Heterogeneity even at the speed limit of folding: large-scale molecular dynamics study of a fast-folding variant of the villin headpiece. J. Mol. Biol. 2007, 374, 806–816. doi:10.1016/j.jmb.2007.09.069. [Google Scholar]
-
Bernetti M, Bussi G. Integrating experimental data with molecular simulations to investigate RNA structural dynamics. Curr. Opin. Struct. Biol. 2023, 78, 102503. doi:10.1016/j.sbi.2022.102503. [Google Scholar]
-
Larsson P, Hess B, Lindahl E. Algorithm improvements for molecular dynamics simulations. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2011, 1, 93–108. doi:10.1002/wcms.3. [Google Scholar]
-
Hernández-Rodríguez M, Rosales-Hernández MC, Mendieta-Wejebe JE, Martínez-Archundia M, Basurto CJ. Current tools and methods in molecular dynamics (MD) simulations for drug design. Curr. Med. Chem. 2016, 23, 3909–3924. doi:10.2174/0929867323666160530144742. [Google Scholar]
-
Rapaport DC. GPU molecular dynamics: algorithms and performance. J. Phys. Conf. Ser. 2022, 2241, 12007. doi:10.1088/1742-6596/2241/1/012007. [Google Scholar]
-
Lee TS, Cerutti DS, Mermelstein D, Lin C, LeGrand S, Giese TJ, et al. GPU-accelerated molecular dynamics and free energy methods in Amber18: Performance enhancements and new features. J. Chem Inf. Model. 2018, 58, 2043–20502. doi:10.1021/acs.jcim.8b00462. [Google Scholar]
-
Siegbahn PEM, Blomberg MRA. Transition-metal systems in biochemistry studied by high-accuracy quantum chemical methods. Chem. Rev. 2000, 100, 421–438. doi:10.1021/cr980390w. [Google Scholar]
-
Pople JA. Nobel lecture: Quantum chemical models. Rev. Mod. Phys. 1999, 71, 1267–1274. doi:10.1103/RevModPhys.71.1267. [Google Scholar]
-
Sheng X, Himo F. The quantum chemical cluster approach in biocatalysis. Acc. Chem. Res. 2023, 56, 938–947. doi:10.1021/acs.accounts.2c00795. [Google Scholar]
-
Sousa SF, Ribeiro AJM, Neves RPP, Brás NF, Cerqueira NMFSA, Fernandes PA, et al. Application of quantum mechanics/molecular mechanics methods in the study of enzymatic reaction mechanisms. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2017, 7, e1281. doi:10.1002/wcms.1281. [Google Scholar]
-
Suardíaz R, Lythell E, Hinchliffe P, van der Kamp M, Spencer J, Fey N, et al. Catalytic mechanism of the colistin resistance protein MCR-1. Org. Biomol. Chem. 2021, 19, 3813–3819. doi:10.1039/D0OB02566F. [Google Scholar]
-
Batebi H, Imhof P. Phosphodiester hydrolysis computed for cluster models of enzymatic active sites. Theor. Chem. Acc. 2016, 135, 262. doi:10.1007/s00214-016-2020-8. [Google Scholar]
-
Visser SPD, Wong HPH, Zhang Y, Yadav R, Sastri CV. Tutorial review on the set-up and running of quantum mechanical cluster models for enzymatic reaction mechanisms. Chem. Eur. J. 2024, 30, e202402468. doi:10.1002/chem.202402468. [Google Scholar]
-
Clemente CM, Capece L, Martí MA. Best practices on QM/MM simulations of biological systems. J. Chem Inf. Model. 2023, 63, 2609–2627. doi:10.1021/acs.jcim.2c01522. [Google Scholar]
-
Dohn AO. Multiscale electrostatic embedding simulations for modeling structure and dynamics of molecules in solution: A tutorial review. Int. J. Quantum Chem. 2020, 120, e26343. doi:10.1002/qua.26343. [Google Scholar]
-
Bakowies D, Thiel W. Hybrid models for combined quantum mechanical and molecular mechanical approaches. J. Phys. Chem. 1996, 100, 10580–10594. doi:10.1021/jp9536514. [Google Scholar]
-
Bondanza M, Nottoli M, Cupellini L, Lipparini F, Mennucci B. Polarizable embedding QM/MM: The future gold standard for complex (bio)systems? Phys. Chem. Chem. Phys. 2020, 22, 14433–14448. doi:10.1039/D0CP02119A. [Google Scholar]
-
Nakamura Y, Takahashi N, Okamoto M, Uda T, Ohno T. Link molecule method for quantum mechanical/molecular mechanical hybrid simulations. J. Chem. Phys. 2007, 225, 1985–1993. doi:10.1016/j.jcp.2007.03.001. [Google Scholar]
-
Xiao C, Zhang Y. Design-atom approach for the quantum mechanical/molecular mechanical covalent boundary: A design-carbon atom with five valence electrons. J. Chem. Phys. 2007, 127, 124102. doi:10.1063/1.2774980. [Google Scholar]
-
Kairys V, Jensen JH. QM/MM boundaries across covalent bonds: A frozen localized molecular orbital-based approach for the effective fragment potential method. J. Phys. Chem. A 2000, 104, 6656–6665. doi:10.1021/jp000887l. [Google Scholar]
-
Ahmadi S, Barrios Herrera L, Chehelamirani M, Hostaš J, Jalife S, Salahub DR. Multiscale modeling of enzymes: QM-cluster, QM/MM, and QM/MM/MD: A tutorial review. Int. J. Quantum Chem. 2018, 118, e25558. doi:10.1002/qua.25558. [Google Scholar]
-
Lonsdale R, Harvey JN, Mulholland AJ. A practical guide to modelling enzyme-catalysed reactions. Chem. Soc. Rev. 2012, 41, 3025–3038. doi:10.1039/c2cs15297e. [Google Scholar]
-
Cavin JF, Dartois V, Diviès C. Gene cloning, transcriptional analysis, purification, and characterization of phenolic acid decarboxylase from bacillus subtilis. Appl. Environ. Microbiol. 1998, 64, 1466–1471. doi:10.1128/AEM.64.4.1466-1471.1998. [Google Scholar]
-
Frank A, Eborall W, Hyde R, Hart S, Turkenburg JP, Grogan G. Mutational analysis of phenolic acid decarboxylase from Bacillus subtilis (BsPAD), which converts bio-derived phenolic acids to styrene derivatives. Catal. Sci. Technol. 2012, 2, 1568–1574. doi:10.1039/c2cy20015e. [Google Scholar]
-
Rodríguez H, Angulo I, Rivas BDL, Campillo N, Páez JA, Muñoz R, et al. p-Coumaric acid decarboxylase from Lactobacillus plantarum: Structural insights into the active site and decarboxylation catalytic mechanism. Proteins Struct. Funct. Bioinform. 2010, 78, 1662–1676. doi:10.1002/prot.22684. [Google Scholar]
-
Matte A, Grosse S, Bergeron H, Abokitse K, Lau PCK. Structural analysis of Bacillus pumilus phenolic acid decarboxylase, a lipocalin-fold enzyme. Acta Cryst. F 2010, 66, 1407–1414. doi:10.1107/S174430911003246X. [Google Scholar]
-
Wuensch C, Glueck SM, Gross J, Koszelewski D, Schober M, Faber K. Regioselective enzymatic carboxylation of phenols and hydroxystyrene derivatives. Org. Lett. 2012, 14, 1974–1977. doi:10.1021/ol300385k. [Google Scholar]
-
Parada-Fabián JC, Hernández-Sánchez H, Méndez-Tenorio A. Substrate specificity of the phenolic acid decarboxylase from Lactobacillus plantarum and related bacteria analyzed by molecular dynamics and docking. J. Plant Biochem. Biotechnol. 2019, 28, 91–104. doi:10.1007/s13562-018-0466-6. [Google Scholar]
-
Sheng X, Lind MES, Himo F. Theoretical study of the reaction mechanism of phenolic acid decarboxylase. FEBS J. 2015, 282, 4703–4713. doi:10.1111/febs.13525. [Google Scholar]
-
Sheng X, Himo F. Theoretical study of enzyme promiscuity: Mechanisms of hydration and carboxylation activities of phenolic acid decarboxylase. ACS Catal. 2017, 7, 1733–1741. doi:10.1021/acscatal.6b03249. [Google Scholar]
-
Riordan JF. The role of metals in enzyme activity. Ann. Clin. Lab. Sci. 1977, 7, 119–129. [Google Scholar]
-
Huber M, Hess CR. Transferring enzyme features to molecular CO2 reduction catalysts. Curr. Opin. Chem. Biol. 2024, 83, 102540. doi:10.1016/j.cbpa.2024.102540. [Google Scholar]
-
Prywes N, Phillips NR, Tuck OT, Valentin-Alvarado LE, Savage DF. Rubisco function, evolution, and engineering. Annu. Rev. Biochem. 2023, 92, 385–410. doi:10.1146/annurev-biochem-040320-101244. [Google Scholar]
-
Bathellier C, Tcherkez G, Lorimer GH, Farquhar GD. Rubisco is not really so bad. Plant Cell Environ. 2018, 41, 705–716. doi:10.1111/pce.13149. [Google Scholar]
-
Sharkey TD. The discovery of rubisco. J. Exp. Bot. 2023, 74, 510–519. doi:10.1093/jxb/erac254. [Google Scholar]
-
Cleland WW, Andrews TJ, Gutteridge S, Hartman FC, Lorimer GH. Mechanism of rubisco: the carbamate as general base. Chem. Rev. 1998, 98, 549–562. doi:10.1021/cr970010r. [Google Scholar]
-
Tcherkez GGB, Bathellier C, Stuart-Williams H, Whitney S, Gout E, Bligny R, et al. D2O solvent isotope effects suggest uniform energy barriers in ribulose-1,5-bisphosphate carboxylase/oxygenase catalysis. Biochemistry 2013, 52, 869–877. doi:10.1021/bi300933u. [Google Scholar]
-
Cummins PL, Kannappan B, Gready JE. Revised mechanism of carboxylation of ribulose-1,5-biphosphate by rubisco from large scale quantum chemical calculations. J. Comput. Chem. 2018, 39, 1656–1665. doi:10.1002/jcc.25343. [Google Scholar]
-
Andersson I. Large structures at high resolution: The 1.6 Å crystal structure of spinach ribulose-1,5-bisphosphate carboxylase/oxygenase complexed with 2-carboxyarabinitol bisphosphate. J. Mol. Biol. 1996, 259, 160–174. doi:10.1006/jmbi.1996.0310. [Google Scholar]
-
Douglas-Gallardo OA, Murillo-López JA, Oller J, Mulholland AJ, Vöhringer-Martinez E. Carbon dioxide fixation in RuBisCO is protonation-state-dependent and irreversible. ACS Catal. 2022, 12, 9418–9429. doi:10.1021/acscatal.2c01677. [Google Scholar]
-
El-Hendawy MM, Garate JA, English NJ, O’Reilly S, Mooney DA. Diffusion and interactions of carbon dioxide and oxygen in the vicinity of the active site of Rubisco: Molecular dynamics and quantum chemical studies. J. Chem. Phys. 2012, 137, 145103. doi:10.1063/1.4757021. [Google Scholar]
-
Payer SE, Faber K, Glueck SM. Non-Oxidative Enzymatic (de)carboxylation of (hetero)aromatics and acrylic acid derivatives. Adv. Synth. Catal. 2019, 361, 2402–2420. doi:10.1002/adsc.201900275. [Google Scholar]
-
Sheng X, Zhu W, Huddleston J, Xiang DF, Raushel FM, Richards NGJ, et al. A combined experimental-theoretical study of the LigW-catalyzed decarboxylation of 5-carboxyvanillate in the metabolic pathway for lignin degradation. ACS Catal. 2017, 7, 4968–4974. doi:10.1021/acscatal.7b01166. [Google Scholar]
-
Sheng X, Patskovsky Y, Vladimirova A, Bonanno JB, Almo SC, Himo F, et al. Mechanism and structure of γ-resorcylate decarboxylase. Biochemistry 2018, 57, 3167–3175. doi:10.1021/acs.biochem.7b01213. [Google Scholar]
-
Hofer G, Sheng X, Braeuer S, Payer SE, Plasch K, Goessler W, et al. Metal ion promiscuity and structure of 2,3-dihydroxybenzoic acid decarboxylase of Aspergillus oryzae. ChemBioChem 2021, 22, 652–656. doi:10.1002/cbic.202000600. [Google Scholar]
-
Chen F, Zhao Y, Zhang C, Wang W, Gao J, Li Q, et al. A combined computational-experimental study on the substrate binding and reaction mechanism of salicylic acid decarboxylase. Catalysts 2022, 14, 807. doi:10.3390/catal12121577. [Google Scholar]
-
Sheng X, Plasch K, Payer SE, Ertl C, Hofer G, Keller W, et al. Reaction mechanism and substrate specificity of iso-orotate decarboxylase: A combined theoretical and experimental study. Front. Chem. 2018, 6, 608. doi:10.3389/fchem.2018.00608. [Google Scholar]
-
Hu Y, Hua Q, Sun G, Shi K, Zhang H, Zhao K, et al. The catalytic activity for ginkgolic acid biodegradation, homology modeling and molecular dynamic simulation of salicylic acid decarboxylase. Comput. Biol. Chem. 2018, 75, 82–90. doi:10.1016/j.compbiolchem.2018.05.003. [Google Scholar]
-
Boyington JC, Gladyshev VN, Khangulov SV, Stadtman TC, Sun PD. Crystal structure of formate dehydrogenase H: catalysis involving Mo, molybdopterin, selenocysteine, and an Fe4S4 cluster. Science 1997, 275, 1305–1308. doi:10.1126/science.275.5304.1305. [Google Scholar]
-
Dong G, Ryde U. Reaction mechanism of formate dehydrogenase studied by computational methods. J. Biol. Inorg. Chem. 2018, 23, 1243–1254. doi:10.1007/s00775-018-1608-y. [Google Scholar]
-
Siegbahn PEM. Energetics for CO2 reduction by molybdenum-containing formate dehydrogenase. J. Phys. Chem. B 2022, 126, 1728–1733. doi:10.1021/acs.jpcb.2c00151. [Google Scholar]
-
Li F, Lienemann M. Stabilization of the catalytically active structure of a molybdenum-dependent formate dehydrogenase depends on a highly conserved lysine residue. FEBS J. 2025, 292, 3165–3179. doi:10.1111/febs.70048. [Google Scholar]
-
Breglia R, Arrigoni F, Sensi M, Greco C, Fantucci P, De Gioia L, et al. First-principles calculations on Ni,Fe-containing carbon monoxide dehydrogenases reveal key stereoelectronic features for binding and release of CO2 to/from the C-cluster. Inorg. Chem. 2021, 60, 387–402. doi:10.1021/acs.inorgchem.0c03034. [Google Scholar]
-
Sellés Vidal L, Kelly CL, Mordaka PM, Heap JT. Review of NAD(P)H-dependent oxidoreductases: Properties, engineering and application. BBA Proteins Proteom. 2018, 1866, 327–347. doi:10.1016/j.bbapap.2017.11.005. [Google Scholar]
-
Sato R, Amao Y. Studies on the catalytic mechanism of formate dehydrogenase from Candida boidinii using isotope-labelled substrate and co-enzyme. Catal. Today 2023, 411, 113796. doi:10.1016/j.cattod.2022.06.011. [Google Scholar]
-
Erb TJ, Brecht V, Fuchs G, Müller M, Alber BE. Carboxylation mechanism and stereochemistry of crotonyl-CoA carboxylase/reductase, a carboxylating enoyl-thioester reductase. Proc. Natl. Acad. Sci. USA 2009, 106, 8871–8876. doi:10.1073/pnas.0903939106. [Google Scholar]
-
Scheffen M, Marchal DG, Beneyton T, Schuller SK, Klose M, Diehl C, et al. A new-to-nature carboxylation module to improve natural and synthetic CO2 fixation. Nat. Catal. 2021, 4, 105–115. doi:10.1038/s41929-020-00557-y. [Google Scholar]
-
Bernhardsgrütter I, Schell K, Peter DM, Borjian F, Saez DA, Vöhringer-Martinez E, et al. Awakening the sleeping carboxylase function of enzymes: Engineering the natural CO2-binding potential of reductases. J. Am. Chem. Soc. 2019, 141, 9778–9782. doi:10.1021/jacs.9b03431. [Google Scholar]
-
Zhang L, Mori T, Zheng Q, Awakawa T, Yan Y, Liu W, et al. Rational control of polyketide extender units by structure-based engineering of a crotonyl-CoA carboxylase/reductase in antimycin biosynthesis. Angew. Chem. Int. Ed. 2015, 54, 13462–13465. doi:10.1002/anie.201506899. [Google Scholar]
-
Stoffel GMM, Saez DA, DeMirci H, Vögeli B, Rao Y, Zarzycki J, et al. Four amino acids define the CO2 binding pocket of enoyl-CoA carboxylases/reductases. Proc. Natl. Acad. Sci. USA 2019, 116, 13964–13969. doi:10.1073/pnas.1901471116. [Google Scholar]
-
Gomez A, Erb TJ, Grubmüller H, Vöhringer-Martinez E. Conformational dynamics of the most efficient carboxylase contributes to efficient CO2 fixation. J. Chem Inf. Model. 2023, 63, 7807–7815. doi:10.1021/acs.jcim.3c01447. [Google Scholar]
-
Rosenthal RG, Ebert MO, Kiefer P, Peter DM, Vorholt JA, Erb TJ. Direct evidence for a covalent ene adduct intermediate in NAD(P)H-dependent enzymes. Nat. Chem. Biol. 2014, 10, 50–55. doi:10.1038/nchembio.1385. [Google Scholar]
-
Recabarren R, Tinzl M, Saez DA, Gomez A, Erb TJ, Vöhringer-Martinez E. Covalent adduct formation as a strategy for efficient CO2 fixation in crotonyl-CoA carboxylases/reductases. ACS Catal. 2023, 13, 6230–6241. doi:10.1021/acscatal.2c05250. [Google Scholar]
-
Recabarren R, Llanos AG, Vöhringer ME. Computational methods for the study of carboxylases: The case of crotonyl-CoA carboxylase/reductase. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 2024; Volume 708, pp. 353–387. [Google Scholar]
-
Marshall SA, Payne KAP, Leys D. The UbiX-UbiD system: The biosynthesis and use of prenylated flavin (prFMN). Arch. Biochem. Biophys. 2017, 632, 209–221. doi:10.1016/j.abb.2017.07.014. [Google Scholar]
-
Payne KAP, White MD, Fisher K, Khara B, Bailey SS, Parker D, et al. New cofactor supports α,β-unsaturated acid decarboxylation via 1,3-dipolar cycloaddition. Nature 2015, 522, 497–501. doi:10.1038/nature14560. [Google Scholar]
-
Payer SE, Marshall SA, Bärland N, Sheng X, Reiter T, Dordic A, et al. Regioselective para-carboxylation of catechols with a prenylated flavin dependent decarboxylase. Angew. Chem. Int. Ed. 2017, 56, 13893–13897. doi:10.1002/anie.201708091. [Google Scholar]
-
Payne KAP, Marshall SA, Fisher K, Rigby SEJ, Cliff MJ, Spiess R, et al. Structure and mechanism of Pseudomonas aeruginosa PA0254/HudA, a prFMN-dependent pyrrole-2-carboxylic acid decarboxylase linked to virulence. ACS Catal. 2021, 11, 2865–2878. doi:10.1021/acscatal.0c05042. [Google Scholar]
-
Gahloth D, Fisher K, Payne KAP, Cliff M, Levy C, Leys D. Structural and biochemical characterization of the prenylated flavin mononucleotide-dependent indole-3-carboxylic acid decarboxylase. J. Biol. Chem. 2022, 298, 101771. doi:10.1016/j.jbc.2022.101771. [Google Scholar]
-
Payne KAP, Marshall SA, Fisher K, Cliff MJ, Cannas DM, Yan C, et al. Enzymatic carboxylation of 2-furoic acid yields 2,5-furandicarboxylic acid (FDCA). ACS Catal. 2019, 9, 2854–2865. doi:10.1021/acscatal.8b04862. [Google Scholar]
-
Tian G, Liu Y. Mechanistic insights into the catalytic reaction of ferulic acid decarboxylase from Aspergillus niger: A QM/MM study. Phys. Chem. Chem. Phys. 2017, 19, 7733–7742. doi:10.1039/C6CP08811B. [Google Scholar]
-
Beveridge R, Migas LG, Payne KAP, Scrutton NS, Leys D, Barran PE. Mass spectrometry locates local and allosteric conformational changes that occur on cofactor binding. Nat. Commun. 2016, 7, 12163. doi:10.1038/ncomms12163. [Google Scholar]
-
Datar PM, Joshi SY, Deshmukh SA, Marsh ENG. Probing the role of protein conformational changes in the mechanism of prenylated-FMN-dependent phenazine-1-carboxylic acid decarboxylase. J. Biol. Chem. 2024, 300, 105621. doi:10.1016/j.jbc.2023.105621. [Google Scholar]
-
Wen K, Tao Y, Jiang W, Jiang L, Zhu J, Li Q. (De)carboxylation mechanisms of heteroaromatic substrates catalyzed by prenylated FMN-dependent UbiD decarboxylases: An in-silico study. Int. J. Biol. Macromol. 2024, 260, 129294. doi:10.1016/j.ijbiomac.2024.129294. [Google Scholar]
-
Miao Y, Feher VA, McCammon JA. Gaussian accelerated molecular dynamics: Unconstrained enhanced sampling and free energy calculation. J. Chem. Theory Comput. 2015, 11, 3584–3595. doi:10.1021/acs.jctc.5b00436. [Google Scholar]
-
Abramson J, Adler J, Dunger J, Evans R, Green T, Pritzel A, et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 2024, 630, 493–500. doi:10.1038/s41586-024-07487-w. [Google Scholar]
-
Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. doi:10.1038/s41586-021-03819-2. [Google Scholar]
-
Baek M, DiMaio F, Anishchenko I, Dauparas J, Ovchinnikov S, Lee GR, et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 2021, 373, 871–876. doi:10.1126/science.abj8754. [Google Scholar]
-
Lin Z, Akin H, Rao R, Hie B, Zhu Z, Lu W, et al. Evolutionary-scale prediction of atomic-level protein structure with a language model. Science 2023, 379, 1123–1130. doi:10.1126/science.ade2574. [Google Scholar]
-
Li F, Yuan L, Lu H, Li G, Chen Y, Engqvist MK, et al. Deep learning-based kcat prediction enables improved enzyme-constrained model reconstruction. Nat. Catal. 2022, 5, 662–672. doi:10.1038/s41929-022-00798-z. [Google Scholar]
-
Lou L, Zhang J, Zhang L, Li S, Li Z, Li Z. Engineering Phosphoenolpyruvate Carboxylase with Improved Activity and Stability for High-Efficiency Carbon Dioxide Fixation. Adv. Synth. Catal. 2025, 367, e70052. doi:10.1002/adsc.70052. [Google Scholar]
-
Marchal DG, Schulz L, Schuster I, Ivanovska J, Paczia N, Prinz S, et al. Machine Learning-Supported Enzyme Engineering toward Improved CO2-Fixation of Glycolyl-CoA Carboxylase. ACS Synth. Biol. 2023, 12, 3521–3530. doi:10.1021/acssynbio.3c00403. [Google Scholar]
-
Kozinsky B, Musaelian A, Johansson A, Batzner S. Scaling the Leading Accuracy of Deep Equivariant Models to Biomolecular Simulations of Realistic Size. In Proceedings of the International Conference for High Performance Computing, Networking, Storage and Analysis, Denver, CO, USA, 12–17 November 2023; pp. 1–12. [Google Scholar]