Immunoprofiling of Alcohol-Activated Hepatic Stellate Cells Reveals Mechanisms of Immune Evasion through NK/T Lymphocyte Checkpoint Signaling

Immunoprofiling of Alcohol-Activated Hepatic Stellate Cells Reveals Mechanisms of Immune Evasion through NK/T Lymphocyte Checkpoint Signaling
Received: 31 July 2025 Revised: 17 September 2025 Accepted: 30 September 2025 Published: 10 October 2025

© 2025 The authors. This is an open access article under the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/).
1. Introduction
Alcohol-associated liver disease (AALD) accounts for nearly half of all cirrhosis cases in the United States and represents a major public health problem and economic burden worldwide [1,2,3,4,5,6]. AALD is manifested as a spectrum of clinical disorders ranging from steatosis (fatty liver) to alcohol-associated steatohepatitis (ASH), a combination of steatosis and inflammation. While ASH is reversible, if untreated, it may progress to cirrhosis and hepatocellular carcinoma. ASH is associated with high rates of mortality: up to 40% of severe ASH patients die within six months of clinical diagnosis [7,8]. Currently there are no FDA-approved therapies for AALD, underscoring the urgent need for deeper disease understanding and the approval of new treatments.
Hepatic stellate cells (HSC) are central mediators of AALD onset, progression, and severity. Following long-term heavy alcohol consumption, quiescent HSC (qHSC) activate and transdifferentiate into myofibroblast-like (Myo) cells, which secrete collagen and other extracellular matrix components and promote liver inflammation through the release of inflammatory cytokines, contributing to liver injury [9,10,11,12]. These observations have proposed that selective elimination of activated HSC (aHSC) may offer a therapeutic benefit.
Harnessing the innate immune system, particularly natural killer (NK) cells, may represent a promising strategy to selectively target and eliminate aHSC. The conversion of quiescent HSC to aHSC is accompanied by a transient increase in expression of NK cell activating ligand retinoic acid inducible gene 1 (RAE1), corresponding with vulnerability of early-activated HSC to NK cell killing. In contrast, chronically activated HSCs lose their cytoplasmic stores of retinol and do not produce RA and RAE1, impairing NK cell cytotoxicity against HSC [13,14,15,16]. Chronic alcohol consumption also impairs the immune surveillance and cytotoxicity of NK cells by arresting NK cell development at the CD27+CD11b+ stage [17,18]. Alcohol-induced downregulation of NKG2D, TRAIL, and interferon-(IFN)-𝛾 expression has also been detected on NK cells and is associated with the activation of hepatic stellate cells [19].
Intriguingly, knockdown of the NK cell inhibitory receptor iKIR stimulates NK targeting of activated HSC [20], highlighting the potential for harnessing the immune system to target and eliminate aHSC/Myo. However, the mechanisms underlying immune evasion by HSC remain incompletely understood, limiting therapeutic development.
In this study, we profiled the expression of a panel of immune-modulating ligands expressed by HSC, and show that CD80, a key immune checkpoint ligand, is dynamically expressed during HSC activation. While antibody blockade of the CD80/CTLA-4 checkpoint facilitates aHSC targeting by CD8+ T cells in vitro, combined blockade of CTLA-4, TIGIT, and PD-1 checkpoints does not lead to resolution of alcohol-induced liver fibrosis or amelioration of liver function. Using single-cell RNA-seq profiling, we show that checkpoint blockade reduces the burden on aHSC but expands activation of Th17 T-cells and increases hepatic infiltration of inflammation-promoting immune subsets. Furthermore, the inverse therapeutic approach--immune suppression via combination engagement of the TIGIT/PD-1 checkpoints-- likewise fails to reduce fibrosis or restore liver function. These observations highlight the nuanced and multifaceted role of the immune system in AALD, and suggest that highly selective targeting of aHSC/Myo may be necessary to significantly ameliorate liver function.
2. Materials and Methods
2.1. Flow Cytometry
Mouse HSC were isolated from liver tissue (n = 3 mice) using retrograde pronase-collagenase perfusion of the liver and subsequent density-gradient centrifugation [21], followed by staining with fluorescently labeled antibodies specific for individual checkpoint ligands. The following antibodies were used: CD155-FITC (ThermoFisher #2H7CD155, Waltham, MA, USA), CD274/PD-L1-Alexa 488 (ThermoFisher 53-5983-42), and CD80-PE (ThermoFisher clone 2D10.4, #12-0809-42). FACS was performed using a BD FACSAria III (BD Biosciences, Franklin Lakes, NJ, USA). HSCs were also measured for UV autofluorescence at 328 nm.
2.2. Western Immunoblotting
Plasma membrane proteins and cytoplasmic proteins were fractionated from mouse HSC samples using the Cell Surface Isolation Kit (Thermo Scientific #A44390, Waltham, MA, USA). Proteins were resolved by SDS-PAGE and analyzed by immunoblotting using the following antisera: CD80 (BioXcell InVivoMAb #BE0134, Lebanon, NH, USA); Sodium Potassium ATPase (Abcam ab185065, Cambridge, UK); and actin (ThermoFisher #MA5-11869). Immunoblots were developed using secondary antibodies conjugated to HRP, followed by imaging using the ChemiDoc system (BioRad, Hercules, CA, USA).
2.3. Cytotoxicity Assay
Individual groups of NK cells or CD8+ T cells were cultivated for 6 h with HSC target cells in the absence or presence of anti-CTLA-4 blocking antisera or isotype-matched control IgG (15 µg/mL). Cells were then harvested and dyed with propidium iodide PI (Sigma-Aldrich, St. Louis, MO, USA), a dye that only enters cells with compromised membranes. The percentages of PI+ cells in each group were measured with flow cytometry.
2.4. Mouse Model of Acute-on-Chronic Alcohol Model
Male B6 mice (8 weeks old) were fed ad libitum for 2 weeks with a liquid diet containing two different levels of cholesterol and saturated fat: 2.32 g/L cholesterol and 23.2 g/L lard (DYET#710142) or 46% reduced cholesterol and lard content (#710383). The diet is then switched to those containing ethanol (#710362 or #710384) for ethanol-fed mice. Ethanol content was gradually increased from 1% (v/v) on day 1 to 4.35% (v/v) on day 12 until the end of feeding. A weekly binge of alcohol is given from the second week of ethanol feeding, via a stomach tube at the initial dose of 3.5 g/kg, which is gradually increased to 4.5 g/kg. For control mice, an isocaloric dextrose solution is given as a binge.
2.5. In Vivo Checkpoint Modulation
For ICB treatments, mice were administered a cocktail of blocking antisera for CTLA-4 (Novus Biologicals #1003705, 25 µg, Centennial, CO, USA), TIGIT (Invivogen mab10-010, 40 µg, San Diego, CA, USA), and PD-1 (Invivogen mpd1-mab-15-50, 25 µg), or isotype-matched non-specific control IgG (BioXCell #BE0083), infused biweekly into mice in each of the alcohol or control feeding arms via i.p. injection, commencing at week 3 of each diet regimen. For the reciprocal series of in vivo checkpoint agonist experiments, mice were administered antisera specific for TIGIT (BioXCell InVivoMAb #BE0274, 30 µg) and recombinant PD-L1 extracellular domain (Sino Biological #50010-M08H, 40 mg, Paoli, PA, USA).
2.6. Immunohistochemistry and Immunofluorescence
Formalin-fixed liver tissue sections were stained with H&E for histological analysis or Sirius Red for collagen deposition. Sections were separately stained for CD69 (anti-CD69 clone H1.2F3, Biotin, eBioscience, San Diego, CA, USA), followed by streptavidin-HRP (Invitrogen S911). Slides were imaged using a ZeissAxioScan.z1 microscope and images analyzed using Zeiss Zen 3.9 software. For CD69, >10 fields across two technical replicate slides were scored by scientists blinded to the slide identify. The collagen area/portal vein area was quantified by digital imaging with NIH Scion Image.
2.7. Serum Chemistry Analysis
Serum was analyzed for alanine aminotransferase (ALT) and aspartate aminotransferase (AST) by blood chemistry panel (Antech Diagnostics, Inc., Fountain Valley, CA, USA) to assess liver function.
2.8. scRNA-Seq Library Preparation
Liver non-parenchymal cells (NPC) were isolated from mouse treatment cohorts (n = 3 mice per cohort) through Percoll gradient centrifugation, and NPCs were collected. Single cell suspensions were counted and centrifuged at 200× g for 5 min at 4 °C. Pellets were resuspended in lysis buffer (250 mM sucrose, 25 mM KCl, 5 mM MgCl2, 10mM Tris pH 8.0, 225 1 mM DTT, 0.2 U/mL RNasin (Promega #N2511, Fitchburg, WI, USA), 0.005 U/mL DNase I (ThermoFisher Scientific # EN0521), 0.1% Triton X-100, sterile H2O). Extraction of nuclei was confirmed using the SYTOX Green nuclear stain (ThermoFisher Scientific # S7020), with ~ 100% efficiency across all lines. Nuclei were used as input into the Parse Biosciences Evercode Fixation Kit as per manufacturer instructions. Individual sub-libraries composed of >10,000 nuclei each were sequenced, targeting 20,000 reads per nucleus. Resultant FASTQ files were processed using the Parse Biosciences provided split-pipe 1.2.1 software package to perform alignment to the GRCm38 (mm10) reference genome, and subsequent unique molecular identifier (UMI) counts with sample barcode demultiplexing. Individual sub-libraries were processed separately before being merged. CellBender 0.3.2 30 was also used to test for doublets in the dataset. Mitochondrial expression and the number of features identified for each nucleus were calculated using Seurat 5.1.
2.9. scRNA-Seq Data Analysis
The single-cell RNA-seq dataset was processed, explored, and visualized using Trailmaker (Parse Biosciences, Seatlle, WA, USA). Unfiltered count matrices were uploaded to Trailmaker, and dead or dying cells were removed by filtering droplets with high mitochondrial content. Outliers in the distribution of the number of genes vs. the number of transcripts were removed by fitting a linear regression model. Cells with a high probability of being doublets were filtered out using the scDblFinder method. Data normalization, principal-component analysis (PCA), and data integration using Harmony were performed on data from high-quality cells. Clusters were identified using the Leiden method, and a Uniform Manifold Approximation and Projection (UMAP) embedding was calculated to visualize the results. Cluster-specific marker genes were identified by comparing cells of each cluster to all other cells using the presto package implementation of the Wilcoxon rank-sum test. Trailmaker sums counts for each cell type or cluster across biological replicates to create pseudobulk samples, and then applies the voom function in the limma package to model the mean-variance trend and generate weights for each gene in the pseudobulk count matrix, enabling standard linear modeling to identify differentially expressed genes between conditions for differential abundance analysis. The Benjamini-Hochberg (BH) procedure controls the false discovery rate (FDR) for multiple comparison corrections.
2.10. Statistical Analysis
All experiments were repeated at least three times. Data are reported as mean ± s.d. unless otherwise stated. Analyses for statistical significance were performed in GraphPad Prism 6 (GraphPad Software, Inc., La Jolla, CA, USA) and Microsoft Excel using Student’s t-test, one-way ANOVA with Tukey’s multiple comparisons test, Chi-square analysis and Fisher’s exact test as appropriate. Images represent typical experimental results.
3. Results
To gain additional insight into immune modulation by HSC, we profiled the expression of immune checkpoint ligands by HSC through a range of activation states, from freshly isolated, qHSC to late-activated (day 14) aHSC. Our analysis revealed that CD80, a high-affinity ligand of the CTLA-4 checkpoint receptor, is dynamically expressed on both qHSC and late-activated HSC, while levels transiently decrease in early-activated (day 5) HSC (Figure 1A). In contrast, the checkpoint ligands PD-L1 and CD155 were not expressed above background levels in aHSC (Figure S1). Immunoblotting of a membrane-enriched HSC fraction using anti-CD80 antisera confirmed a dynamic increase in CD80 expression during HSC activation (Figure 1B). CTLA-4 competes with the CD28 activating receptor for binding B7 ligands such as CD80. Increased CTLA-4:B7 binding transmits a net negative signal, limiting IL-2 production, and diminishing T cell proliferation and survival [22,23]. These findings therefore suggest a potential role for the CD80/CTLA-4 checkpoint in evasion of lymphocyte surveillance and targeting.
To test this experimentally, we performed in vitro cytotoxicity assays in which HSC at different stages of activation were co-incubated with CD8+ T effector cells, in the presence of either CTLA-4 blocking antisera or isotype-matched control antisera. For each tested HSC: T cell ratios, the addition of CTLA-4 blockade enhanced CD8+ T destruction of activated (day 14) HSC, but did not affect T cell targeting of HSC at an early-intermediate activation state (day 5) (Figure 1C).
Given this increase in aHSC targeting following exposure to combination checkpoint blockade, we next sought to evaluate the impact of CTLA-4 blockade on liver function and fibrosis in a mouse model of acute-on-chronic AALD, utilizing an alcohol feeding regimen that closely recapitulates key aspects of clinical AALD [24,25]. Given that CD28 is co-regulated by the PD-1 checkpoint [26] and that the mechanistic convergence of the TIGIT and PD-1 inhibitory pathways necessitates co-blockade to optimize lymphocyte responses [27], we pursued the combined antibody blockade of CTLA-4/TIGIT/PD-1 checkpoints to maximize therapeutic potential.
Mice were allowed ad lib consumption of a high-cholesterol, high-fat liquid diet (HCFD) containing either isocalorically substituted maltose-dextrins or 4.0% ethanol for 10 weeks. A binge bolus dose (4 g/kg) of ethanol was given weekly from the second week during the feeding (Figure 2A). This weekly ad lib HCFD+Alc+Binge model robustly induces increased plasma ALT, hepatitis with monocyte infiltration in ~50% of the mice after 8 weeks, and activation of HSC and pericellular and perisinusoidal liver fibrosis [28]. Mice were dosed twice weekly for six weeks with combined CTLA-4/ TIGIT/PD-1 blockade or i = non-targeting control antisera, and liver function was then monitored by measurement of serum ALT/AST, and by Sirius Red fibrosis staining of sectioned liver tissues recovered at the experimental endpoint.
We observed marked elevations of plasma AST and ALT in alcohol-fed mice (Figure 2B), as expected in this acute-on-chronic model of alcohol consumption [28,29]. However, immune checkpoint blockade (ICB) treatment did not reduce serum AST or ALT (Figure 2B). Furthermore, Sirius Red staining revealed modestly increased fibrosis levels in the checkpoint blockade cohort. However, the measured increase did not achieve statistical significance (Figure 2C, D), while promoting infiltration of activated, CD69+ NK and T lymphocytes, as determined by immunohistochemistry (Figure S2). Collectively, these observations indicate that combination immune checkpoint therapy does not confer a therapeutic benefit in the setting of acute-on-chronic alcohol consumption and may deepen the severity of liver injury.
To better understand how immune checkpoint blockade impacts the liver non-parenchymal cell (NPC) landscape in the setting of acute-on-chronic alcohol consumption, we performed droplet-free single-cell RNA sequencing (scRNA-seq) analysis of NPCs isolated from dissociated mouse livers (n = 2 mice per condition). We obtained 43,647 single-cell transcriptomes (225 median transcripts/cell), enabling high-resolution cell subtype profiling and the comparison of NPC following acute-on-chronic alcohol feeding and following treatment with the immune checkpoint blockade (Figure 3A,B). We applied strict filtering on data, including expressed genes and unique molecular identifiers (UMIs), and performed additional signal normalization and scaling using strict statistical cutoffs.
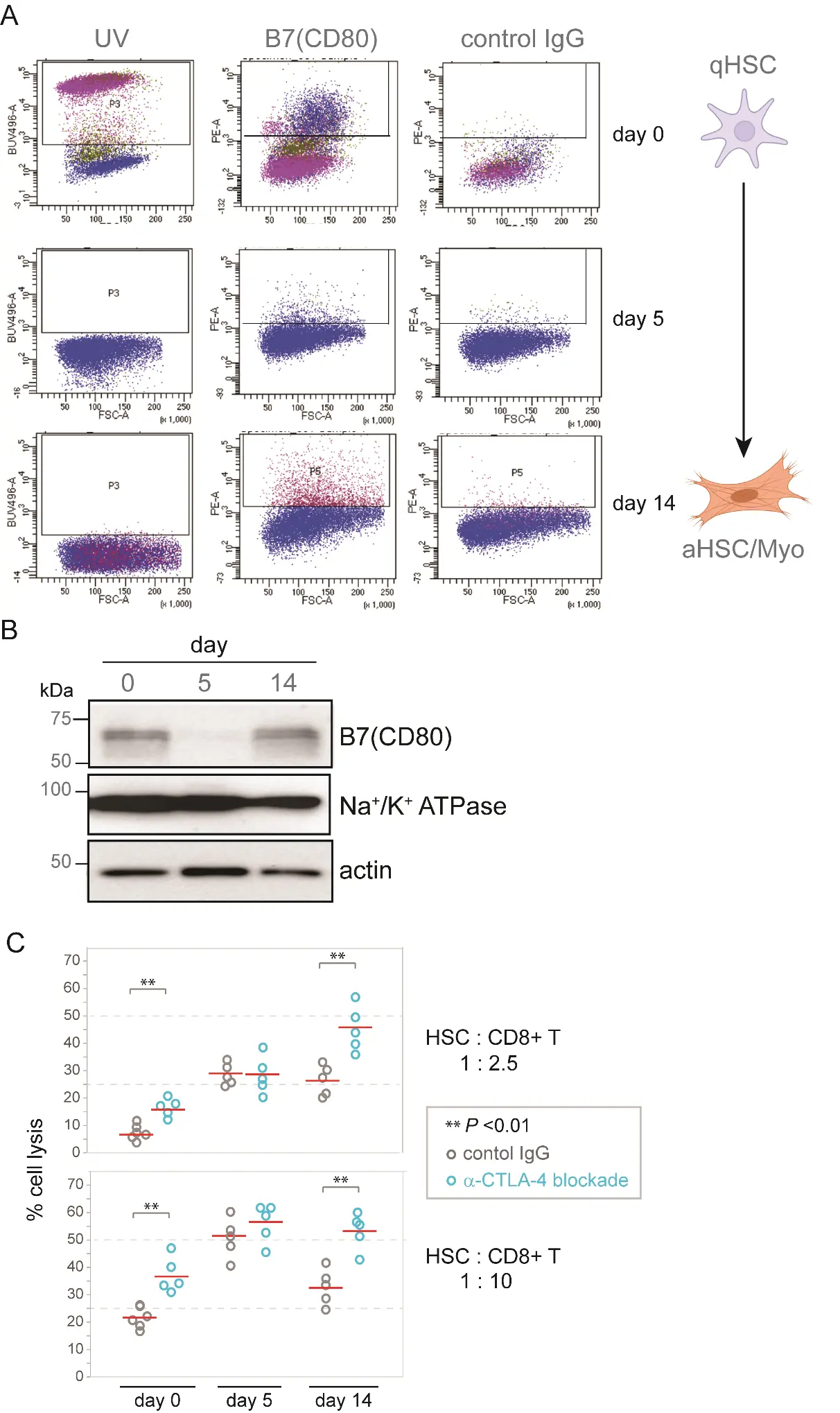
Figure 1. Dynamic expression of B7 (CD80) during HSC activation. (A) FACS profiling of murine HSC showing dynamic expression of lymphocyte checkpoint ligand B7 (CD80) following culture in vitro for the indicated times. (B) Western immunoblot of B7 (CD80) in membrane-enriched subcellular fraction. Na/K+ ATPase and actin, loading controls. (C) In vitro cytotoxicity assay showing lysis of HSC following co-culture with purified CD8+ T cells (2.5:1 T: HSC).
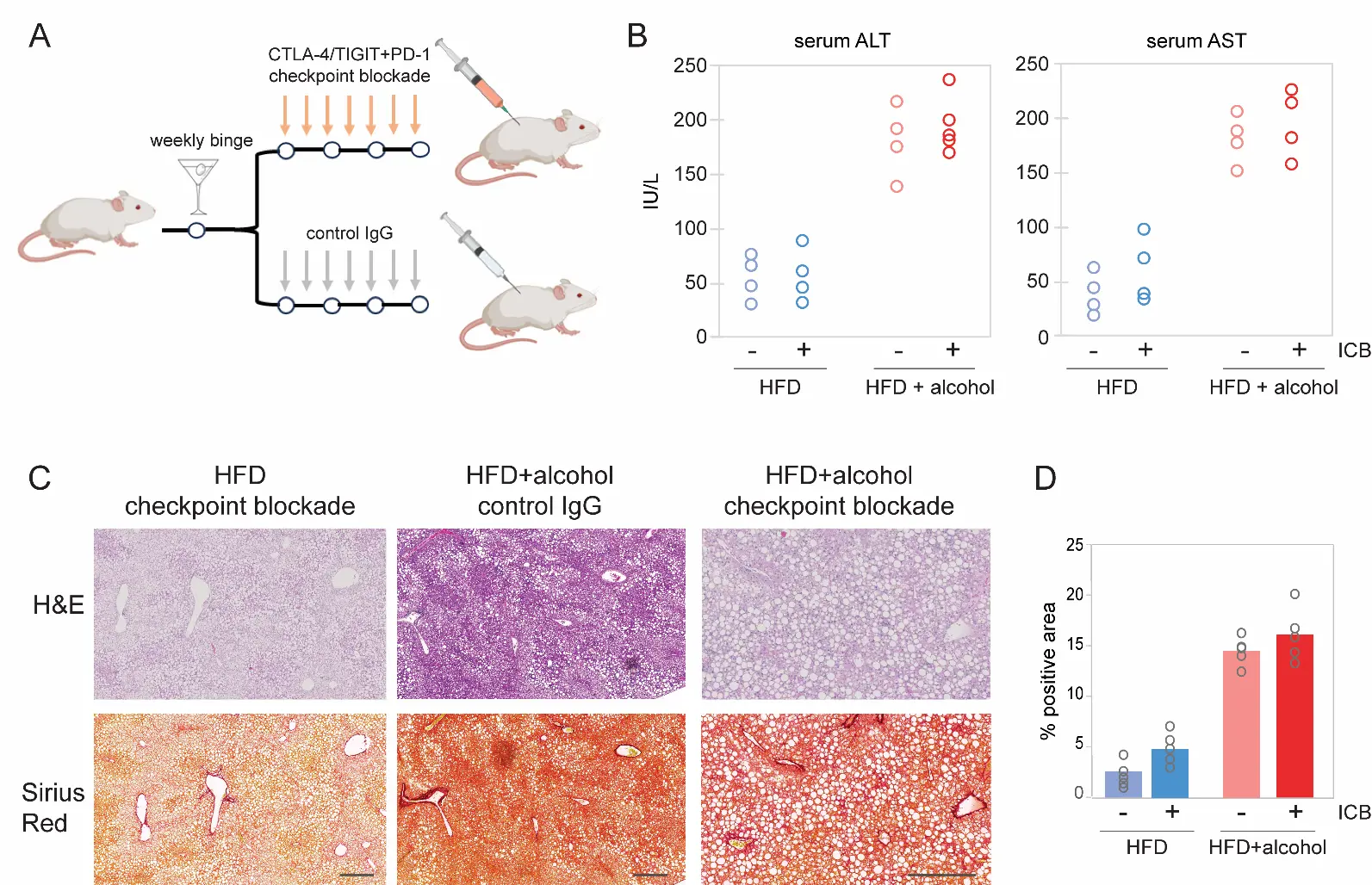
Figure 2. Effect of CTLA-4/TIGIT blockade on alcohol-promoted liver fibrosis. (A) Schematic of the acute-on-chronic mouse model of AALD. Mice were allowed ad lib consumption of a high-cholesterol, high-fat liquid diet (HCFD) containing either isocalorically substituted maltose-dextrins or 4.0% ethanol for 10 weeks. A binge bolus dose (4 g/kg) of ethanol was given weekly from the second week during the feeding. This weekly ad lib HCFD+Alc+Binge model robustly induces increased plasma ALT, hepatitis with monocyte infiltration in ~50% of the mice after 8 weeks, and activation of HSC and pericellular and perisinusoidal liver fibrosis (B) Serum ALT and AST measurements from individual mice at necropsy. Immune checkpoint blockade combination therapy (CTLA-4/TIGIT/PD-1) did not reduce liver enzymes. (C) H&E and Sirius Red staining of mouse livers following treatment with the indicated control or immune checkpoint-blocking antibodies. Scale bars: 200 μm. (D) Quantification of Sirius Red staining in mouse livers following the indicated treatments. HFD, high-fat diet. Data from individual mice are represented as open circles.
We first evaluated how treatment with immune checkpoint blockade impacted the conversion of qHSC into aHSC and subsequently to collagen-secreting Myo. Pseudotime studies characterizing cell developmental programs based on transcriptional similarities have suggested a cell trans-differentiation trajectory from HSC into Myo, with each cell type composed of four distinct subclusters [30,31,32]. Treatment with ICB led to a reduction in the relative abundance of late-activated HSC3 and HSC4 clusters, together with a concomitant depletion of late-stage myofibroblast subpopulations, including Myo III (Egr1, Klf2) and Myo IV (Fbln1, Meg3) (Figure 3C–E).
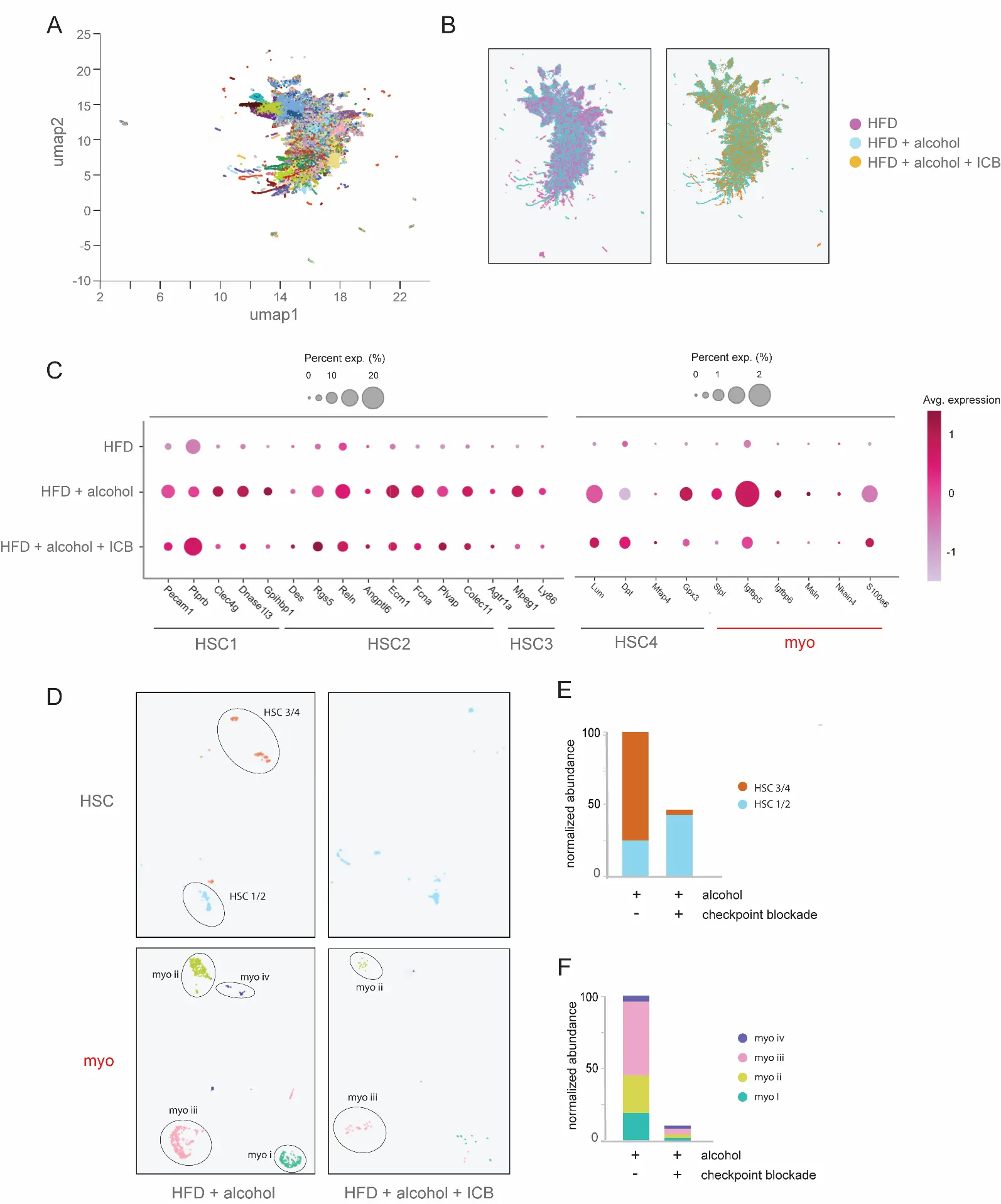
Figure 3. Blockade of the CTLA-4/TIGIT co-inhibitory axis induces elimination of aHSC/Myo. (A) scRNA-seq profiling of mouse liver non-parenchymal cells following treatment with ICB of CTLA-4/TIGIT/PD-1, showing total cell subclusters (n = 3 mice per treatment cohort). (B) Comparative representation of cell subclusters following high-fat diet (HFD) and alcohol-feeding with checkpoint blockade or control IgG treatment regimens. (C) Depletion of aHSC and Myo gene expression signatures following immune checkpoint blockade therapy. The size of dots indicates the percentage of expressed cells, and the color indicates the average scaled expression level. (D) Cluster analysis showing loss of aHSC and Myo subpopulations following immune checkpoint blockade therapy. (E) Relative abundance of HSC subclusters in the absence or presence of immune checkpoint blockade. (F) Depletion of Myo subclusters following ICB treatment.
Given the reduced representation of alcohol-activated HSC and late-stage Myo subpopulations without a corresponding reduction in fibrosis or improved liver function, we next sought to profile changes in the liver immune landscape following ICB treatment. NK and T cells serve as major effectors of ICB treatment, and are each composed of multiple cellular subtypes that can either promote or suppress activating and inflammatory responses [33,34,35]. We therefore compared differences in the relative representation of T cell subclusters, annotated through the expression of canonical marker genes associated with the expression of known cell population-specific transcript signatures. In particular, we found that ICB increased the abundance of activated (IFN-gamma, T-bet, CXCR3, CCR5) CD8+ T cells [36], as well as pro-fibrotic Th1 and Th17 (IL-17R, CCR6+) T cells (Figure 4A). In contrast, cell clustering analysis revealed suppression of quiescent (CD27+, CD45+) CD8+ T cells, along with a near-complete loss of CD25+ FoxP3+ Treg (Figure 4A). This analysis additionally revealed elevated expression of T cell activation markers (Tnf, Gzmb, CD69) and diminished levels of inhibitory genes (CD244, GITR/AITR) in response to ICB treatment (Figure 4B).
Concomitant with these changes, scRNA-seq profiling revealed increased infiltration of monocytes and monocyte-derived macrophages (Mo/MoMF), including M1 polarized and Ly-6C+ macrophages, which favor Th1 pro-inflammatory responses and are associated with perisinusoidal and pericellular liver fibrosis, and severe ASH [37,38] (Figure 4C). Relative expression levels for selected cell lineage marker genes for these subtypes are shown in Figure 4D. In agreement with previous reports demonstrating cross-communication between activated HSC and KC [39,40], ICB treatment also diminished expression of genes associated with activated Kupffer cell subsets, including the KC2 marker Bmp2, and the KC3 markers Nr2f2 and Kdr (Figure 4E). Thus, while ICB diminishes fibrosis-inducing, late-stage aHSC and Myo, this effect may be offset by increased infiltration and activation of pro-inflammatory hepatic NK and T cell and macrophage subpopulations.
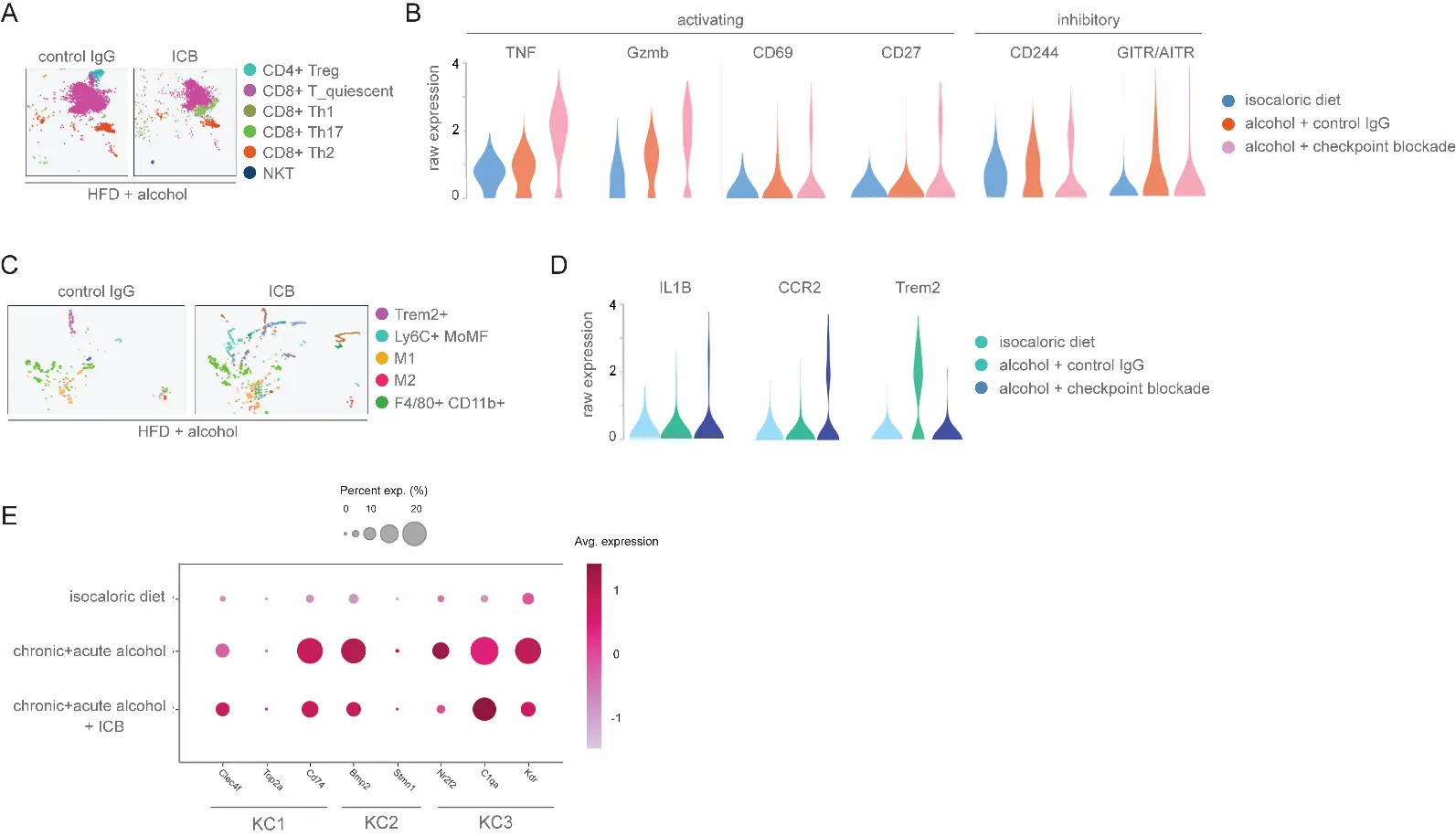
Figure 4. Impact of CTLA-4/TIGIT blockade on hepatic NK and T lymphocyte subsets. (A) scRNA-seq clustering of mouse NK/T/NKT cell populations following treatment with ICB or control antisera. (B) Violin plots showing expression levels of genes implicated in T cell activation or dormancy. Representation of differential cell clustering following the indicated alcohol-feeding and treatment regimens. Showing expression levels in macrophage clusters for genes implicated in pro-inflammatory responses (TNF, Gzmb, CD69, CD27) or suppressive responses (CD244, GITR/AITR). (C) scRNA-seq clustering of mouse macrophage populations following the indicated treatments. Application of ICB therapy leads to increases in M1 and Ly6C+ MoMF subpopulations, and corresponds to increased liver injury. (D) Violin plots showing expression levels in macrophage clusters for genes implicated in pro-inflammatory responses (Il1B, Ccr2) or suppressive responses (Trem2). (E) Dynamic changes in Kupffer cell gene expression signatures following immune checkpoint blockade therapy. The size of dots indicates the percentage of expressed cells, and the color indicates the average scaled expression level.
We therefore sought to determine whether the inverse strategy-immune suppression through checkpoint engagement-could reduce liver fibrosis and restore liver function. Mice were administered a TIGIT/PD-1 checkpoint agonist cocktail designed to broadly suppress NK and T lymphocyte activity (Figure 5A). However, the combination engagement of these checkpoints failed to decrease serum ALT/AST levels (Figure 5B) and did not result in a significant reduction in alcohol-promoted liver fibrosis or steatosis, as determined by histological analysis of mouse liver tissues (Figure 5C,D). We conclude that immune checkpoint engagement does not yield significant restoration of liver function in the setting of alcohol-promoted fibrosis and tissue injury.
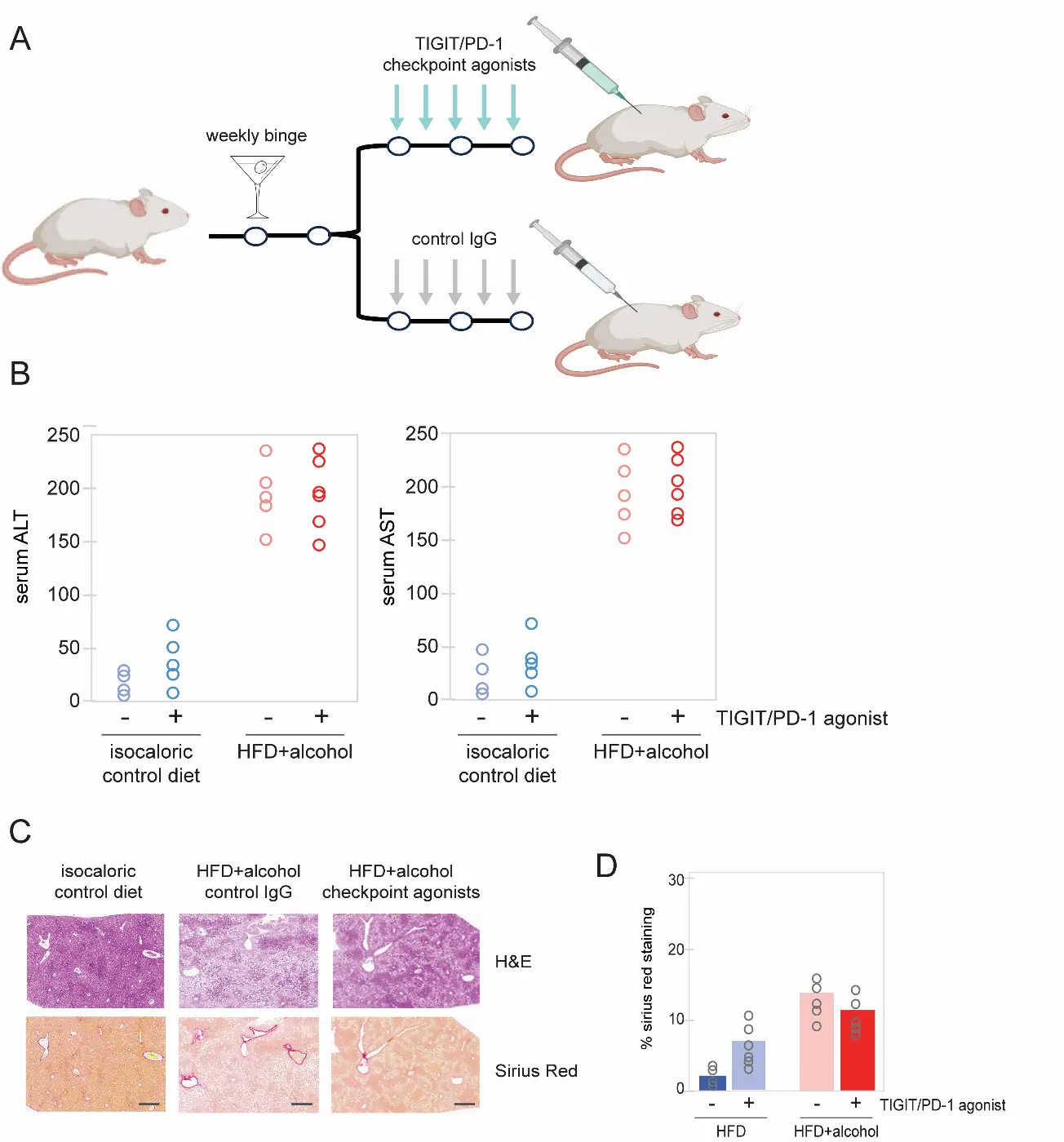
Figure 5. Effect of TIGIT/PD-1 checkpoint engagement on liver function. (A) Schematic of acute-on-chronic mouse model of AALD and checkpoint agonist dosing. (B) Serum ALT and AST levels at necropsy showed that administration of combined TIGIT/PD-1 checkpoint agonists did not lead to a reduction in liver enzymes. (C) H&E (upper panels) and Sirius Red fibrosis staining (lower panels) of mouse livers following treatment with the indicated control or immune checkpoint-agonist antibodies. Scale bars: 200 μm. (D) Quantification of Sirius Red staining in mouse livers following the indicated treatments. HFD, high-fat diet. Data from individual mice are represented as open circles.
4. Discussion
Elimination of aHSC/Myo mitigates liver damage resulting from chronic alcohol consumption, and represents a therapeutic objective for the treatment of AALD [9,10,20,41,42,43]. The recent clinical success of immunotherapy in selectively targeting pre-specified cell populations [44,45,46] has introduced the possible utility of this approach for the treatment of AALD. Here, we show that aHSC can evade immune surveillance through surface expression of the B7/CD80 immunomodulatory ligand, and that combined blockade of the CTLA-4/TIGIT/PD-1 checkpoints stimulates lymphocyte targeting of aHSC/Myo. However, this approach fails to diminish fibrosis or ameliorate liver function, illuminating the multifaceted complexity of the hepatic immune landscape in AALD.
Alcohol-promoted immune dysfunction is manifested through suppression of NK and T lymphocyte surveillance, and hepatic infiltration of pro-inflammatory monocytes, macrophages, and effector T cells, which drive fibrosis and tissue injury together. Our findings reveal that combined blockade of the CTLA-4/TIGIT/PD-1 axis stimulates targeting of distinct aHSC subtypes (HSC2-4) while diminishing hepatic Treg, contributing to hepatic injury [47,48]. However, Foxp3+ Treg also serve to maintain immune homeostasis by inhibiting pro-fibrotic Th17 and CD8+ T cells [49,50]. Treg cells also suppress activation of Th2 cells and infiltrating Ly‐6ChighCCR2high monocytes/macrophages, reducing chronic inflammation and attenuating hepatic fibrosis [51,52]. Thus, checkpoint blockade may promote an imbalance of Th17/Treg, leading to excess immune activation, offsetting the potential therapeutic benefit from eliminating aHSC/Myo [49,53]. In addition, while we did not observe significant side effects in animal cohorts, off-target effects and immune-mediated adverse events may blunt the efficacy of checkpoint blockade [54,55,56].
Interestingly, our findings reveal that the inverse approach—immune suppression via combined engagement of the TIGIT/PD-1 checkpoints—likewise fails to confer a significant therapeutic benefit. These observations are consistent with recent reports linking activation of multiple types of immune cells—T cells, NKT cells, Kupffer cells, and mucosal-associated invariant T cells—with liver function recovery in alcohol-promoted tissue injury. For instance, Koda and colleagues reported a direct role for CD8+ tissue-resident memory CD8+ T (CD8+ Trm) cells in resolving liver fibrosis, demonstrating that a reduction of liver CD8+ Trm cells significantly delayed recovery from fibrosis, while adoptive transfer of CD8+ Trm protected mice from fibrosis progression [57]. Similarly, disease severity in AALD patients corresponds with impaired innate adaptive responses and increased expression of T cell exhaustion markers [58,59]. Two such markers—LAG-3 and TIM-3—are part of a “second wave” of inhibitory receptors frequently co-expressed on exhausted or dysfunctional T cells alongside CTLA-4, PD-1, and TIGIT, and blocking them simultaneously may represent a potentially synergistic approach to restore T cell function [33,60,61].
Our data suggest that precision targeting of aHSC/Myo, rather than one-dimensional activation or blockade of checkpoints, may offer a greater therapeutic index. Recent clinical advances in targeting pre-defined cell populations using bi-specific T cell engagers [62,63,64,65], as well as the development of engineered NK and T cells expressing highly specific chimeric antigen receptors (CAR) [44,63,66,67,68,69], highlight the potential of these emerging platforms for the selective elimination of the aHSC/Myo disease hub. Recent single-cell transcriptome analysis of HSC/Myo has uncovered surface antigens expressed by aHSC/Myo, which may be targeted through these approaches [30,70]. Future studies will explore the feasibility and therapeutic efficacy of these next-generation precision targeting approaches for treating AALD and diseases of related etiology, such as metabolic dysfunction-associated steatohepatitis (MASH).
5. Conclusions
While combined checkpoint blockade promotes immune targeting of the aHSC/Myo cellular hub, it is insufficient as a standalone approach to reverse alcohol-related liver fibrosis. Therefore, alternative modalities that enable precision targeting of aHSC/Myo may be necessary to achieve therapeutic gains.
Supplementary Materials
The following supporting information can be found at: https://www.sciepublish.com/article/pii/710, Figure S1: FACS profiling of immune checkpoint ligands in HSC; Figure S2: Immunostaining of mouse liver sections.
Acknowledgments
We thank Qihong Yang, Stephanie Pan, and the Southern California Research Center for ALPD and Cirrhosis for excellent technical assistance with animal experiments and cell isolation.
Author Contributions
Conceptualization, A.K. and D.E.F.; Methodology, A.K., P.H. and D.E.F.; Statistical Analysis, Y.C.; Writing—Original Draft Preparation, A.K. and D.E.F; Writing, S.B. and D.E.F.; Visualization, S.B. and D.E.F.
Ethics Statement
The study was conducted according to the guidelines of the Declaration of Helsinki, and approved by the Institutional Animal Care and Use Committee of the University of Southern California (Protocol #21365, approved 28 January 2022).
Informed Consent Statement
Not applicable.
Data Availability Statement
Single-cell RNA-seq data, including unfiltered matrices, and combined and sublibrary reports, have been deposited to Mendeley Data (doi:10.17632/5b9vd5fdtn.1).
Funding
This research was funded by grant 1R21AA029741-01 from the National Institutes of HealthH/National Intitute on Alcohol Abuse and Alcoholism, the Robert E. and May R. Wright Foundation Trust, and the James H. Zumberge Faculty Research and Innovation Fund to D.E.F.
Declaration of Competing Interests
The authors declare no competing interests.
References
- Anouti A, Mellinger JL. The Changing Epidemiology of Alcohol-Associated Liver Disease: Gender, Race, and Risk Factors. Semin Liver Dis. 2023, 43, 50–59. doi:10.1055/a-2000-6680. [Google Scholar]
- Mellinger JL. Epidemiology of Alcohol Use and Alcoholic Liver Disease. Clin. Liver Dis. 2019, 13, 136–139. doi:10.1002/cld.806. [Google Scholar]
- Julien J, Ayer T, Bethea ED, Tapper EB, Chhatwal J. Projected prevalence and mortality associated with alcohol-related liver disease in the USA, 2019-40: a modelling study. Lancet Public Health 2020, 5, e316–e323. doi:10.1016/S2468-2667(20)30062-1. [Google Scholar]
- Tapper EB, Parikh ND. Mortality due to cirrhosis and liver cancer in the United States, 1999-2016: observational study. BMJ 2018, 362, k2817. doi:10.1136/bmj.k2817. [Google Scholar]
- Tampaki M, Tsochatzis E, Lekakis V, Cholongitas E. Prevalence, characteristics and outcomes of patients with metabolic and alcohol related/associated liver disease (MetALD): A systematic review and meta-analysis. Metabolism 2025, 163, 156101. doi:10.1016/j.metabol.2024.156101. [Google Scholar]
- Ciardullo S, Mantovani A, Morieri ML, Muraca E, Invernizzi P, Perseghin G. Impact of MASLD and MetALD on clinical outcomes: A meta-analysis of preliminary evidence. Liver Int. 2024, 44, 1762–1767. doi:10.1111/liv.15939. [Google Scholar]
- Huang DQ, Mathurin P, Cortez-Pinto H, Loomba R. Global epidemiology of alcohol-associated cirrhosis and HCC: trends, projections and risk factors. Nat. Rev. Gastroenterol. Hepatol. 2023, 20, 37–49. doi:10.1038/s41575-022-00688-6. [Google Scholar]
- Vannier AGL, Shay JES, Fomin V, Patel SJ, Schaefer E, Goodman RP, et al. Incidence and Progression of Alcohol-Associated Liver Disease After Medical Therapy for Alcohol Use Disorder. JAMA Netw. Open 2022, 5, e2213014. doi:10.1001/jamanetworkopen.2022.13014. [Google Scholar]
- Kamm DR, McCommis KS. Hepatic stellate cells in physiology and pathology. J. Physiol. 2022, 600, 1825–1837. doi:10.1113/JP281061. [Google Scholar]
- Zhang Y, Wu Y, Shen W, Wang B, Yuan X. Crosstalk between NK cells and hepatic stellate cells in liver fibrosis (Review). Mol. Med. Rep. 2022, 25, 208. doi:10.3892/mmr.2022.12724. [Google Scholar]
- Xiong X, Kuang H, Ansari S, Liu T, Gong J, Wang S, et al. Landscape of Intercellular Crosstalk in Healthy and NASH Liver Revealed by Single-Cell Secretome Gene Analysis. Mol. Cell 2019, 75, 644–660.e5. doi:10.1016/j.molcel.2019.07.028. [Google Scholar]
- Higashi T, Friedman SL, Hoshida Y. Hepatic stellate cells as key target in liver fibrosis. Adv. Drug Deliv. Rev. 2017, 121, 27–42. doi:10.1016/j.addr.2017.05.007. [Google Scholar]
- Gao B, Radaeva S. Natural killer and natural killer T cells in liver fibrosis. Biochim. Biophys. Acta 2013, 1832, 1061–1069. doi:10.1016/j.bbadis.2012.09.008. [Google Scholar]
- Gao B, Radaeva S, Park O. Liver natural killer and natural killer T cells: immunobiology and emerging roles in liver diseases. J. Leukoc. Biol. 2009, 86, 513–528. doi:10.1189/JLB.0309135. [Google Scholar]
- Radaeva S, Wang L, Radaev S, Jeong WI, Park O, Gao B. Retinoic acid signaling sensitizes hepatic stellate cells to NK cell killing via upregulation of NK cell activating ligand RAE1. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 293, G809–G816. doi:10.1152/ajpgi.00212.2007. [Google Scholar]
- Jeong WI, Park O, Gao B. Abrogation of the antifibrotic effects of natural killer cells/interferon-gamma contributes to alcohol acceleration of liver fibrosis. Gastroenterology 2008, 134, 248–258. doi:10.1053/j.gastro.2007.09.034. [Google Scholar]
- Little A, Li Y, Zhang F, Zhang H. Chronic alcohol consumption exacerbates murine cytomegalovirus infection via impairing nonspecific and specific NK activation in mice. FASEB Bioadv. 2018, 1, 18–31. doi:10.1096/fba.1019. [Google Scholar]
- Zhang F, Little A, Zhang H. Chronic alcohol consumption inhibits peripheral NK cell development and maturation by decreasing the availability of IL-15. J. Leukoc. Biol. 2017, 101, 1015–1027. doi:10.1189/jlb.1A0716-298RR. [Google Scholar]
- Byun JS, Yi HS. Hepatic Immune Microenvironment in Alcoholic and Nonalcoholic Liver Disease. Biomed. Res. Int. 2017, 2017, 6862439. doi:10.1155/2017/6862439. [Google Scholar]
- Li T, Yang Y, Song H, Li H, Cui A, Liu Y, et al. Activated NK cells kill hepatic stellate cells via p38/PI3K signaling in a TRAIL-involved degranulation manner. J. Leukoc. Biol. 2019, 105, 695–704. doi:10.1002/JLB.2A0118-031RR. [Google Scholar]
- Mederacke I, Dapito DH, Affò S, Uchinami H, Schwabe RF. High-yield and high-purity isolation of hepatic stellate cells from normal and fibrotic mouse livers. Nat. Protoc. 2015, 10, 305–315. doi:10.1038/nprot.2015.017. [Google Scholar]
- Jin S, Shang Z, Wang W, Gu C, Wei Y, Zhu Y, et al. Immune Co-inhibitory Receptors CTLA-4, PD-1, TIGIT, LAG-3, and TIM-3 in Upper Tract Urothelial Carcinomas: A Large Cohort Study. J. Immunother. 2023, 46, 154–159. doi:10.1097/CJI.0000000000000466. [Google Scholar]
- Zhao J, Li L, Yin H, Feng X, Lu Q. TIGIT: An emerging immune checkpoint target for immunotherapy in autoimmune disease and cancer. Int. Immunopharmacol. 2023, 120, 110358. doi:10.1016/j.intimp.2023.110358. [Google Scholar]
- Mathews S, Xu M, Wang H, Bertola A, Gao B. Animals models of gastrointestinal and liver diseases. Animal models of alcohol-induced liver disease: pathophysiology, translational relevance, and challenges. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 306, G819–G823. doi:10.1152/ajpgi.00041.2014. [Google Scholar]
- Bertola A, Mathews S, Ki SH, Wang H, Gao B. Mouse model of chronic and binge ethanol feeding (the NIAAA model). Nat. Protoc. 2013, 8, 627–637. doi:10.1038/nprot.2013.032. [Google Scholar]
- Hui E, Cheung J, Zhu J, Su X, Taylor MJ, Wallweber HA, et al. T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science 2017, 355, 1428–1433. doi:10.1126/science.aaf1292. [Google Scholar]
- Banta KL, Xu X, Chitre AS, Au-Yeung A, Takahashi C, O'Gorman WE, et al. Mechanistic convergence of the TIGIT and PD-1 inhibitory pathways necessitates co-blockade to optimize anti-tumor CD8+ T cell responses. Immunity 2022, 55, 512–526.e9. doi:10.1016/j.immuni.2022.02.005. [Google Scholar]
- Lazaro R, Wu R, Lee S, Zhu NL, Chen CL, French SW, Xu J, Machida K, Tsukamoto H. Osteopontin deficiency does not prevent but promotes alcoholic neutrophilic hepatitis in mice. Hepatology 2015, 61, 129–140. doi:10.1002/hep.27383. [Google Scholar]
- Xu J, Chi F, Guo T, Punj V, Lee WN, French SW, Tsukamoto H. NOTCH reprograms mitochondrial metabolism for proinflammatory macrophage activation. J. Clin. Investig. 2015, 125, 1579–1590. doi:10.1172/JCI76468. [Google Scholar]
- Krenkel O, Hundertmark J, Ritz TP, Weiskirchen R, Tacke F. Single Cell RNA Sequencing Identifies Subsets of Hepatic Stellate Cells and Myofibroblasts in Liver Fibrosis. Cells 2019, 8, 503. doi:10.3390/cells8050503. [Google Scholar]
- Krenkel O, Puengel T, Govaere O, Abdallah AT, Mossanen JC, Kohlhepp M, et al. Therapeutic inhibition of inflammatory monocyte recruitment reduces steatohepatitis and liver fibrosis. Hepatology 2018, 67, 1270–1283. doi:10.1002/hep.29544. [Google Scholar]
- Su Q, Kim SY, Adewale F, Zhou Y, Aldler C, Ni M, et al. Single-cell RNA transcriptome landscape of hepatocytes and non-parenchymal cells in healthy and NAFLD mouse liver. iScience 2021, 24, 103233. doi:10.1016/j.isci.2021.103233. [Google Scholar]
- Yang AY, Wistuba-Hamprecht K, Greten TF, Ruf B. Innate-like T cells in liver disease. Trends Immunol. 2024, 45, 535–548. doi:10.1016/j.it.2024.05.008. [Google Scholar]
- Li M, Wang L, Cong L, Wong CC, Zhang X, Chen H, et al. Spatial proteomics of immune microenvironment in nonalcoholic steatohepatitis-associated hepatocellular carcinoma. Hepatology 2024, 79, 560–574. doi:10.1097/HEP.0000000000000591. [Google Scholar]
- Peng Y, Wong CC, Yu J. The paradox of immunotherapy in NASH-HCC. Signal Transduct. Target Ther. 2021, 6, 228. doi:10.1038/s41392-021-00654-9. [Google Scholar]
- Ham SD, Abraham MN, Deutschman CS, Taylor MD. Single-cell RNA sequencing reveals Immune Education promotes T cell survival in mice subjected to the cecal ligation and puncture sepsis model. Front. Immunol. 2024, 15, 1366955. doi:10.3389/fimmu.2024.1366955. [Google Scholar]
- Song P, Zhang J, Zhang Y, Shu Z, Xu P, He L, et al. Hepatic recruitment of CD11b+Ly6C+ inflammatory monocytes promotes hepatic ischemia/reperfusion injury. Int. J. Mol. Med. 2018, 41, 935–945. doi:10.3892/ijmm.2017.3315.. [Google Scholar]
- Ramachandran P, Pellicoro A, Vernon MA, Boulter L, Aucott RL, Ali A, et al. Differential Ly-6C expression identifies the recruited macrophage phenotype, which orchestrates the regression of murine liver fibrosis. Proc. Natl. Acad. Sci. USA 2012, 109, E3186–E3195. doi:10.1073/pnas.1119964109. [Google Scholar]
- Yu Z, Xie X, Su X, Lv H, Song S, Liu C, et al. ATRA-mediated-crosstalk between stellate cells and Kupffer cells inhibits autophagy and promotes NLRP3 activation in acute liver injury. Cell Signal 2022, 93, 110304. doi:10.1016/j.cellsig.2022.110304. [Google Scholar]
- Matsuda M, Seki E. Hepatic Stellate Cell-Macrophage Crosstalk in Liver Fibrosis and Carcinogenesis. Semin Liver Dis. 2020, 40, 307–320. doi:10.1055/s-0040-1708876. [Google Scholar]
- Arab JP, Cabrera D, Sehrawat TS, Jalan-Sakrikar N, Verma VK, Simonetto D, et al. Hepatic stellate cell activation promotes alcohol-induced steatohepatitis through Igfbp3 and SerpinA12. J. Hepatol. 2020, 73, 149–160. doi:10.1016/j.jhep.2020.02.005. [Google Scholar]
- Liu X, Rosenthal SB, Meshgin N, Baglieri J, Musallam SG, Diggle K, et al. Primary Alcohol-Activated Human and Mouse Hepatic Stellate Cells Share Similarities in Gene-Expression Profiles. Hepatol. Commun. 2020, 4, 606–626. doi:10.1002/hep4.1483. [Google Scholar]
- Torres S, Abdullah Z, Brol MJ, Hellerbrand C, Fernandez M, Fiorotto R, et al. Recent Advances in Practical Methods for Liver Cell Biology: A Short Overview. Int. J. Mol. Sci. 2020, 21, 2027. doi:10.3390/ijms21062027. [Google Scholar]
- Fraietta JA, Lacey SF, Orlando EJ, Pruteanu-Malinici I, Gohil M, Lundh S, et al. Author Correction: Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat. Med. 2021, 27, 561. doi:10.1038/s41591-021-01248-2. [Google Scholar]
- Lim WA, June CH. The Principles of Engineering Immune Cells to Treat Cancer. Cell 2017, 168, 724–740. doi:10.1016/j.cell.2017.01.016. [Google Scholar]
- Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. doi:10.1056/NEJMoa1709866. [Google Scholar]
- Kasztelan-Szczerbinska B, Zygo B, Rycyk-Bojarzynska A, Surdacka A, Rolinski J, Cichoz-Lach H. Blood concentrations of mediators released from activated neutrophils are related to the severity of alcohol-induced liver damage. PLoS ONE 2023, 18, e0280068. doi:10.1371/journal.pone.0280068.. [Google Scholar]
- Kasztelan-Szczerbinska B, Adamczyk K, Surdacka A, Rolinski J, Michalak A, Bojarska-Junak A, et al. Gender-related disparities in the frequencies of PD-1 and PD-L1 positive peripheral blood T and B lymphocytes in patients with alcohol-related liver disease: a single center pilot study. PeerJ 2021, 9, e10518. doi:10.7717/peerj.10518. [Google Scholar]
- Ma R, Su H, Jiao K, Liu J. Role of Th17 cells, Treg cells, and Th17/Treg imbalance in immune homeostasis disorders in patients with chronic obstructive pulmonary disease. Immun. Inflamm. Dis. 2023, 11, e784. doi:10.1002/iid3.784. [Google Scholar]
- Kasztelan-Szczerbińska B, Surdacka A, Celiński K, Roliński J, Zwolak A, Miącz S, et al. Prognostic Significance of the Systemic Inflammatory and Immune Balance in Alcoholic Liver Disease with a Focus on Gender-Related Differences. PLoS ONE 2015, 10, e0128347. doi:10.1371/journal.pone.0128347. [Google Scholar]
- Wu KJ, Qian QF, Zhou JR, Sun DL, Duan YF, Zhu X, Sartorius K, Lu YJ. Regulatory T cells (Tregs) in liver fibrosis. Cell Death Discov. 2023, 9, 53. doi:10.1038/s41420-023-01347-8. [Google Scholar]
- Ikeno Y, Ohara D, Takeuchi Y, Watanabe H, Kondoh G, Taura K, et al. Foxp3+ Regulatory T Cells Inhibit CCl4-Induced Liver Inflammation and Fibrosis by Regulating Tissue Cellular Immunity. Front. Immunol. 2020, 11, 584048. doi:10.3389/fimmu.2020.584048. [Google Scholar]
- Wang H, Wu T, Wang Y, Wan X, Qi J, Li L, Wang X, Luo X, Ning Q. Regulatory T cells suppress excessive lipid accumulation in alcoholic liver disease. J. Lipid Res. 2019, 60, 922–936. doi:10.1194/jlr.M083568. [Google Scholar]
- Keam S, Turner N, Kugeratski FG, Rico R, Colunga-Minutti J, Poojary R, et al. Toxicity in the era of immune checkpoint inhibitor therapy. Front. Immunol. 2024, 15, 1447021. doi:10.3389/fimmu.2024.1447021. [Google Scholar]
- Yin Q, Wu L, Han L, Zheng X, Tong R, Li L, Bai L, Bian Y. Immune-related adverse events of immune checkpoint inhibitors: a review. Front. Immunol. 2023, 14, 1167975. doi:10.3389/fimmu.2023.1167975. [Google Scholar]
- Zheng L, Lin F, Cai D, Zhang L, Yin C, Qi Y, et al. Single-cell transcriptome sequencing reveals the immune microenvironment in bronchoalveolar lavage fluid of checkpoint inhibitor-related pneumonitis. Cancer Immunol. Immunother. 2025, 74, 128. doi:10.1007/s00262-025-03983-8. [Google Scholar]
- Koda Y, Nakamoto N, Chu PS, Teratani T, Ueno A, Amiya T, et al. CCR9 axis inhibition enhances hepatic migration of plasmacytoid DCs and protects against liver injury. JCI Insight 2022, 7, e159910. doi:10.1172/jci.insight.159910. [Google Scholar]
- Highton AJ, Schuster IS, Degli-Esposti MA, Altfeld M. The role of natural killer cells in liver inflammation. Semin Immunopathol. 2021, 43, 519–533. doi:10.1007/s00281-021-00877-6. [Google Scholar]
- Markwick LJ, Riva A, Ryan JM, Cooksley H, Palma E, Tranah TH, et al. Blockade of PD1 and TIM3 restores innate and adaptive immunity in patients with acute alcoholic hepatitis. Gastroenterology 2015, 148, 590–602.e10. doi:10.1053/j.gastro.2014.11.041. [Google Scholar]
- Aggarwal V, Workman CJ, Vignali DAA. LAG-3 as the third checkpoint inhibitor. Nat. Immunol. 2023, 24, 1415–1422. doi:10.1038/s41590-023-01569-z. [Google Scholar]
- Kong X, Zhang J, Chen S, Wang X, Xi Q, Shen H, Zhang R. Immune checkpoint inhibitors: breakthroughs in cancer treatment. Cancer Biol. Med. 2024, 21, 451–72. doi:10.20892/j.issn.2095-3941.2024.0055. [Google Scholar]
- Lin CT, Wu SJ, Liao CH, Weng RR, Lin CM. The novel CD16A/anti-CD3 bifunctional protein, eCD16A/anti-CD3-BFP, redirects T cell cytotoxicity toward antibody-bound target cells. Hum. Vaccin. Immunother. 2025, 21, 2447141. doi:10.1080/21645515.2024.2447141. [Google Scholar]
- Rujirachaivej P, Siriboonpiputtana T, Choomee K, Supimon K, Sangsuwannukul T, Songprakhon P, et al. Engineered T cells secreting αB7-H3-αCD3 bispecific engagers for enhanced anti-tumor activity against B7-H3 positive multiple myeloma: a novel therapeutic approach. J. Transl. Med. 2025, 23, 54. doi:10.1186/s12967-024-05923-z. [Google Scholar]
- Bisio M, Legato L, Fasano F, Benevolo Savelli C, Boccomini C, Nicolosi M, et al. Bispecific Antibodies for Lymphoid Malignancy Treatment. Cancers 2024, 17, 94. doi:10.3390/cancers17010094. [Google Scholar]
- Efimov GA, Kruglov AA, Khlopchatnikova ZV, Rozov FN, Mokhonov VV, Rose-John S, et al. Cell-type-restricted anti-cytokine therapy: TNF inhibition from one pathogenic source. Proc. Natl. Acad. Sci. USA 2016, 113, 3006–3011. doi:10.1073/pnas.1520175113. [Google Scholar]
- Goulding J, Yeh WI, Hancock B, Blum R, Xu T, Yang BH, et al. A chimeric antigen receptor uniquely recognizing MICA/B stress proteins provides an effective approach to target solid tumors. Med 2023, 4, 457–477.e8. doi:10.1016/j.medj.2023.04.004. [Google Scholar]
- Daher M, Basar R, Gokdemir E, Baran N, Uprety N, Nunez Cortes AK, et al. Targeting a cytokine checkpoint enhances the fitness of armored cord blood CAR-NK cells. Blood 2021, 137, 624–636. doi:10.1182/blood.2020007748. [Google Scholar]
- Le Saux O, Ray-Coquard I, Labidi-Galy SI. Challenges for immunotherapy for the treatment of platinum resistant ovarian cancer. Semin Cancer Biol. 2021, 77, 127–143. doi:10.1016/j.semcancer.2020.08.017. [Google Scholar]
- Liu E, Marin D, Banerjee P, Macapinlac HA, Thompson P, Basar R, et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N. Engl. J. Med. 2020, 382, 545–553. doi:10.1056/NEJMoa1910607. [Google Scholar]
- Wen Y, Lambrecht J, Ju C, Tacke F. Hepatic macrophages in liver homeostasis and diseases-diversity, plasticity and therapeutic opportunities. Cell. Mol. Immunol. 2021, 18, 45–56. doi:10.1038/s41423-020-00558-8. [Google Scholar]