1. Introduction
Primary biliary cholangitis (PBC) is an immune-mediated disease of unknown etiology and multifactorial pathophysiology characterized by the presence of serum antimitochondrial antibodies (AMAs), and histologically by chronic non-suppurative destructive cholangitis of small bile ducts followed by extensive bile duct loss and biliary cirrhosis [
1,
2]. Incidence and prevalence are variable worldwide with incidence rates per 100,000 population ranging from 0.84 to 2.75 and prevalence per 100,000 ranging from 1.91 to 40.2 [
3,
4]. In the island of Crete, prevalence was found to be 36.5 cases per 100,000 population with a mean incidence of 2.1 per 100,000, one of the highest in Europe [
5].
Emphasis has been given to autoimmunity, and the critical factor responsible for disease pathogenesis is considered to be the presence of autoreactive CD4
+ and CD8
+ lymphocytes infiltrating the liver of patients. However, PBC unlike other autoimmune diseases, does not respond to immunosuppressive drugs but rather to ursodeoxycholic acid (UDCA), a bile salt that induces HCO
3− rich choleresis or to obeticholic acid (OCA) a farnesoid X receptor agonist [
6].
Earlier studies indicated that genetically predisposed patients develop PBC when epigenetic mechanisms reduce the expression of the Cl
−/HCO
3− exchanger AE2 in cholangiocytes and disrupt the protective alkaline umbrella that prevents the penetration of toxic bile salts into cholangiocytes. AE2 reduction causes increased intracellular pH, leading to activation of soluble adenylyl cyclase (sAC), and sensitization of biliary endothelial cells (BECs) to bile salt-induced apoptosis. Moreover, mitophagy was inhibited by cytosolic alkalization and stimulated by acidification. AE2 deficiency therefore, may disturb mitophagy (a specific form of autophagy) in BECs, leading to the accumulation of damaged mitochondria, oxidative stress, endoplasmic reticulum stress (ER stress) and aberrant presentation of immune-reactive mitochondrial antigens to the immune cells [
6,
7]. In addition, the decreased expression of AE2 was correlated with dysregulated autophagy and cellular senescence in bile duct lesions in PBC [
8].
Lysosomes contain proteolytic and lipolytic enzymes operating at acid environment and are implicated in various cellular functions such as lipid metabolism and the final stages of autophagy. More than 60 different acid lysosomal hydrolases such as proteases, nucleases, glycosidases, and lipases have been identified [
9]. The most abundant lysosomal proteases are cathepsins B and D [
6]. They were named after the Greek word “Kathepsini” which literally means “digest” by the Nobel Prize winner Richard Willstätter nearly a century ago [
10]. Lysosomal enzymes are translocated and activated in the cytoplasm after lysosomal membrane permeabilization (LMP) [
11]. Cathepsins B and D retain their activity at cytosolic pH [
12]. Cathepsin D has been implicated in many inflammatory diseases such as inflammatory bowel disease [
13,
14] and autophagy [
15].
Reviews of cysteine cathepsins in the pathobiology of liver diseases have recently been published [
16,
17].
Lipid hydrolases are enzymes involved in lipid degradation. Lipid droplets are taken up by autophagosomes and degraded in lysosomes in a process called lipophagy [
18]. Lysosomal acid lipase (LAL) is a critical enzyme in lipophagy being the only lipase active at acidic pH [
19]. The complex of non-specific esterases may also be involved in lipophagy [
20]. The International Union of Biochemistry recommends that these esterases should be designated as carboxylesterases (EC 3.1.1.1) [
20].
Most importantly, lysosomal enzymes have been implicated in cell signaling. The lysosome is now described as a coordinator of signals regulating important cell functions such as growth, proliferation, and differentiation [
21].
No information exists on the activity of lysosomal enzymes in the liver of PBC patients. On the other hand, autophagy and senescence dysregulation has been proposed as important pathogenetic mechanisms in PBC [
22,
23,
24]. Moreover, proteolytic cathepsins have been used as indicators of autophagy in vascular and brain diseases [
25,
26,
27]. Therefore, we studied the activity of certain lysosomal hydrolases in liver tissue of PBC patients to identify possible abnormalities of the late stage of autophagy that might help to clarify the role of lysosomes in the pathogenesis of PBC. Intervention in lysosomes is feasible as lysosomotropic agents are available [
28]. Therefore, results of the present study may have therapeutic implications.
2. Materials and Methods
2.1. Patients
Twenty-five patients (23 females, age 35–60) with PBC, 7 on early stages (I or II), 11 on late stages (III or IV) were classified according to Ludwig [
29]. Seven patients (3 early stage and 4 late stage) on UDCA treatment for at least one year were also included. PBC diagnosis was established according to the guidelines of the European Association for the Study of the Liver (EASL) for PBC (cholestatic biochemistry, anti-mitochondrial antibodies by immunofluorescence, anti-M2 by Elisa and a compatible liver biopsy) [
30].
As disease controls, thirty-four patients with Alcoholic Liver Disease (20 males, age 25–57 years, 14 with fatty liver, 8 with acute alcoholic hepatitis, 12 with cirrhosis) were studied. Criteria for diagnosis were a daily alcohol consumption ≥40 g and ≥20 g of ethanol for men and women respectively for at least 5 years, and absence of other chronic liver diseases. Twenty-five patients with chronic viral liver disease (17 males, age 28–61, 16 with HBV, 9 with HCV, 13 with chronic moderate or severe hepatitis, 12 with cirrhosis) were also included. Cirrhosis was compensated (Child-Pugh A) in all patients. Five normal liver biopsies were used as controls. Disease controls and normal liver biopsies were from consecutive patients undergone a liver biopsy for diagnostic purposes.
All measurements were done before any treatment was administered. Demographics and laboratory values of the patients are presented in Table 1.
Table 1. Demographics and laboratory data of participants.
Sequential sera samples from six patients with acute alcoholic hepatitis were used to follow the fluctuations of enzymes according to disease progress. These patients were not on steroids, but antibiotics and supportive enteral nutrition were given.
Informed consent in writing was obtained from all patients. The research protocol was approved by the Ethics Committee of the University Hospital (3468/B/2022 and 9113/2022) and the study was performed in accordance with the principles of the Dec-laration of Helsinki.
2.2. Materials and Methods
Dithiothreitol, bovine hemoglobin, trypsin, a-N-benzoyl-DL-arginine-2-naphtylamide (BANA), sodium dodecylsulfate (SDS), Triton X-100,
o-phenanthroline, disopropyl fluorophosphates (DFP), 2-naphtlamide, α-naphtyl acetate, α-naphthol,
p-chloromercuribenzoic acid, cysteine hydrochloride and Tris base were all purchased from Sigma Chemical Co Ltd, London, UK. Ethylenediamine tetraacetic acid, trichloroacetic acid, dioxane and bovine serum albumin were obtained from BDH Chemicals Ltd, Pool, UK, and together with all other chemical and solvents were of analytical grade. 4-methylumbelliferyl oleate and 4-methylumbelliferone were purchased from Koch-Light (Suffolk, UK).
2.2.1. Sample Preparation
The liver biopsy specimens were washed with 0.9% NaCl, blotted and kept at −40 °C until used. They were homogenized in distilled ice-cold water containing 0.1% Triton X-100 using a hand driven Teflon pestle and a glass homogenizer. Cellular debris was removed by centrifugation at 600 g for 5 min and the supernatants were used for enzyme assays. Approximately one-third of the tissue obtained was separated for enzyme determination, and the remainder was fixed in 10% formaldehyde for histological examination.
2.2.2. Cathepsin D
14C-labelled hemoglobin was prepared by the incubation of beef hemoglobin with K14CNO as previously described [
31]. The specific activity of the hemoglobin ranged between 2500 to 3000 dpms/mg and could be kept indefinitely at −20 °C without any increase in the blank values. The incubation mixture contained 100 μL of the substrate (16mg
14C-Hb/mL), 100 μL 1.0 M sodium formate buffer pH 3.1 and 100 μL of liver homogenate. Addition of 100 μL of 50% trichloroacetate after incubation for 60 min at 37 °C stopped the reaction and the incubate was centrifuged in a Beckman microfuge. 0.1 mL of the supernatant was added to 15 mL of liquid scintillation cocktail and counted for radioactivity in a Packard scintillation counter. Enzyme activity was expressed as dpm per mg protein per hour. The blank contained bovine serum albumin (0.5 mg/mL) instead of the homogenate.
2.2.3. Cathepsin B1
Hydrolysis of Nα-benzoyl-DL-arginine 2-naphtylamide (BANA), a specific substrate for mammalian liver cathepsin B, was used to measure the enzyme activity as previously described [
32]. A substrate that contained 2.5 mM BANA, 20 mM dithiothreitol in 0.1 M acetate buffer pH 5.1, 20 mM EDTA and 0.1% (
w/
v) triton X was pre-pared. The incubation mixture comprised of 100 μL homogenate or 50 μL serum and 100 μL or 50 μL substrate respectively. The reaction was stopped after 60 min incubation at 37 °C by the addition of 2 mL of glycine-carbonate buffer pH 10.5 to each tube. The fluorescence of the liberated 2-naphthylamide was measured in a Perkin-Elmer spectrofluorometer at 340 nm excitation and 404 nm emission. The blank contained bovine serum albumin (0.5 mg/mL) instead of the homogenate.
Enzyme activity was calculated from a standard curve using pure 2-naphthylamide and was expressed as ng liberated 2-naphthylamide per mg of protein per minute (or per mL serum).
2.2.4. Acid Lipase
The method of Van Berkel et al. was used for the preparation of the substrate and the assay in tissue and serum [
33]. Briefly, 1 mL of 10 mM 4-methylumbelliferyl oleate in hexane and 1 mL of 15 mM lecithin in hexane were mixed and evaporated to dryness under a stream of nitrogen. The residue was resuspended in 25 mL of 2.4 mM sodium taurocholate, sonicated in an ice bath twice for 1 min at 25 kHz and diluted with 0.2 M acetate buffer pH 4.2 to give a 50 mM final substrate concentration. 2 mL of the substrate were mixed with 20 μg of sample protein (or 10 μL of serum) and incubated at 37 °C for 30 min (15 min for serum). The reaction was stopped by adding 200 μL of the incubation mixture to 2 mL of glycine-carbonate buffer pH 10.4. The fluorescence of 4-methylumbelliferone was read at 365 nm excitation and 440 nm emission. Appropriate blanks and 4-methylumbelliferone standard curves were run simultaneously. Enzyme activity was expressed as μg 4-methylumbelliferone per mg protein (or per ml serum) per min.
2.2.5. Neutral Esterase and Acid Esterase
The hydrolysis of α-napthyl acetate was used to determine the activity of these enzymes with a modification of the method of Schaffner et al [
34], using α-naphthyl acetate instead of α-naphthyl palmitate, as previously described in detail [
35,
36]. The fluorescence of liberated α-naphthol was determined at 345 nm excitation and 455 nm emission. Standard curves were created using pure α-naphthol. Enzyme activity was expressed as nmoles of α-naphthol per mg protein per min. Enzyme activity in serum was assessed using the same method and expressed as nmoles of α- naphthol per ml serum per min.
All enzyme assays were linear up to 60 min maximum incubation time and between 10 and 65 μg tissue protein.
2.2.6. Protein Estimations
Total proteins were measured by the method of Lowry et al. using bovine albumin to create the standard curve [
37].
2.3. Statistical Analysis
Results are presented as means ± SD. Box and whiskers plots were used to demonstrate enzyme activities indicating the first quartile, mean, third quartile, the inter-quartile range and outliers. The variance of SDs was tested by the Bartlett test. Comparisons were made by the One-Way Anova with Welch correction in cases of unequal variances. When the Kolmogorov-Smirnov test showed non-canonical distribution of data, the Kruskal Wallis test was used instead.
p < 0.05 was considered statistically significant. All comparisons were with the normal controls. All statistical tests were done with the GraphPad Prism 9.3.0 software.
3. Results
Lysosomes contain more than 60 hydrolases, all implicated in the degradativefinal stage of autophagy [
9]. Cathepsins B and D are the most abundant and better studied proteolytic hydrolases involved in inflammation, fibrosis and autophagy [
6,
38].
They were studied therefore together with three lysosomal enzymes involved in lipid degradation such as acid lipase [
39] and acid and neutral non specific esterases [
36,
40].
3.1. Hepatic Cathepsin D
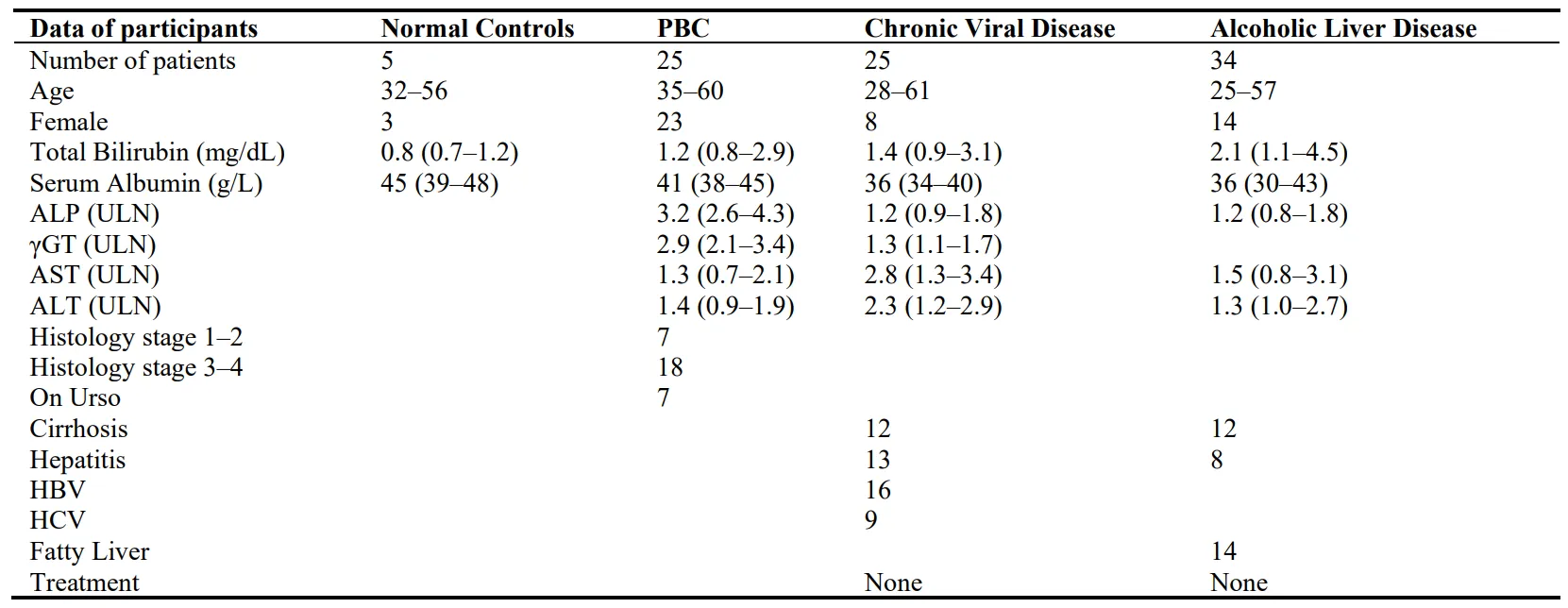
Hepatic cathepsin D activity showed a significant increase in early PBC (39 ± 4.83 dpm × 1000/mg protein/h,
p < 0.0001), late PBC (20.09 ± 3.91,
p = 0.05), viral hepatitis (28.71 ± 4.31,
p < 0.0001) and alcoholic hepatitis (24.55 ± 3.13,
p = 0.0004) compared with normal controls (14.06 ± 2.86). No significant differences were observed in PBC treated with UDCA, viral cirrhosis, fatty liver, and alcoholic cirrhosis (Figure 1).
Figure 1. Measurement of cathepsin D activity in the hepatic tissue of all studied groups, expressed as dpm × 1000/mgPr/h. Box plots indicate the first quartile, mean, third quartile, the inter-quartile range and outliers. Numbers represent patients in each group. (PBC: primary biliary cholangitis; Urso: PBC treated with ursodeoxycholic acid; CVH: Chronic viral hepatitis; Alc Fat: Alcoholic fatty liver; ASH: Alcoholic hepatitis; Alc. Cirrhosis: Alcoholic Cirrhosis).
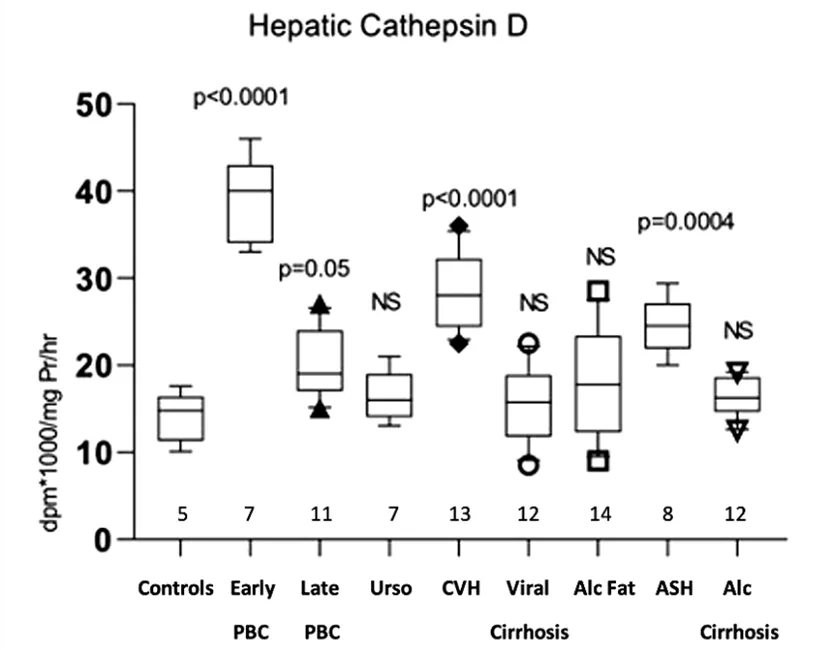
Hepatic cathepsin B1 activity showed a significant increase only in early PBC (225 ± 18.06 ng 2-naphthylamide/mg protein/min,
p < 0.0001) compared with the normal controls (130.4 ± 14.94). Conversely, a significant decrease of hepatic cathepsin B1 was observed in late PBC (66.45 ± 9.73,
p = 0.004), viral cirrhosis (66.67 ± 20.32,
p = 0.004) and alcoholic cirrhosis (74.36 ± 17.84,
p < 0.01). No differences were found in PBC treated with UDCA, viral hepatitis, fatty liver, and alcoholic hepatitis (Figure 2).
Figure 2. Measurement of cathepsin B1 activity in the hepatic tissue of all studied groups, expressed as ng 2-naphthylamide/mgPr/min.
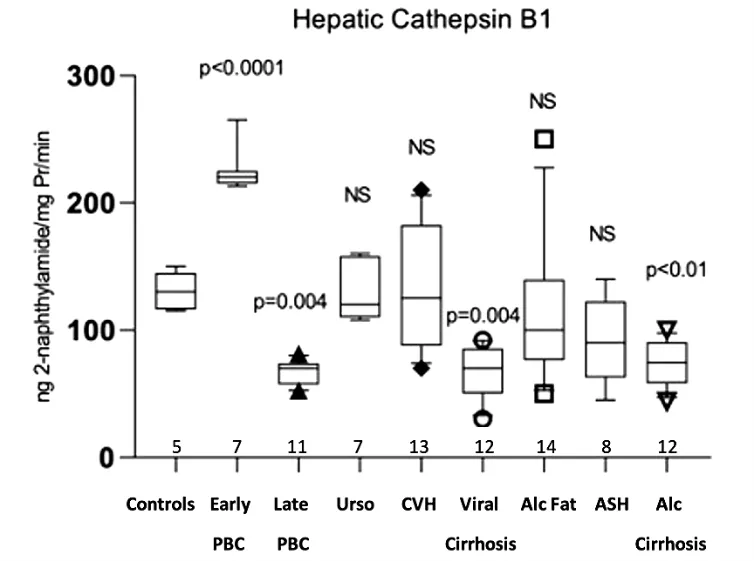
Hepatic acid lipase activity showed a significant increase in early PBC (26.96 ± 3.15 μg 4-methylumbelliferone/mg protein/min,
p = 0.03) compared with the normal controls (21.20 ± 3.92). In contrast, there was a significant decrease in late PBC (15.70 ± 3.67,
p = 0.02), viral cirrhosis (14.03 ± 2.13,
p = 0.001), alcoholic hepatitis (15.69 ± 2.88,
p = 0.03) and alcoholic cirrhosis (14.52 ± 1.62,
p = 0.003). Non- significant differences were observed in PBC treated with UDCA, viral hepatitis, and fatty liver (Figure 3).
Figure 3. Measurement of acid lipase activity in the hepatic tissue of all studied groups, expressed as μg 4-methylumbelliferone/mgPr/min.
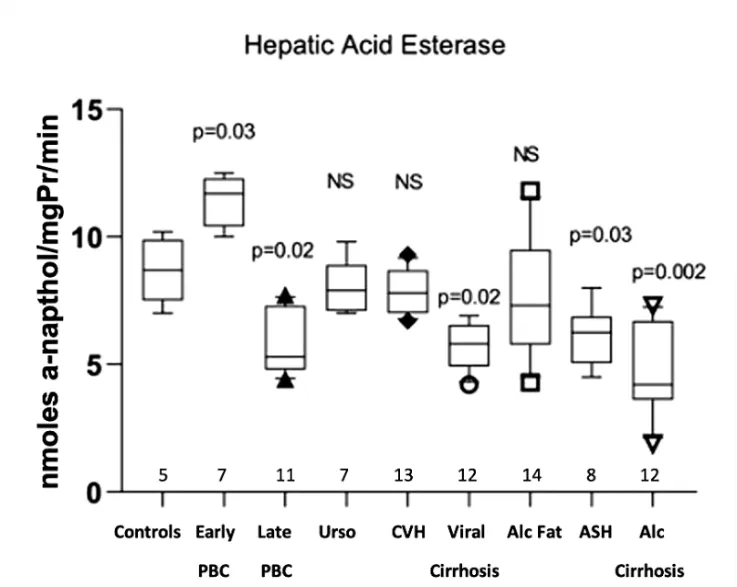
Hepatic acid esterase activity was significantly increased in early PBC (11.39 ± 0.98 μg of α-naphthol/mg protein/min
p = 0.03) compared with the normal controls (8.7 ± 1.26). On the other hand, late PBC (5.9 ± 1.25,
p = 0.02), viral cirrhosis (5.73 ± 0.89,
p = 0.02), alcoholic hepatitis (6.08 ± 1.15,
p = 0.03) and alcoholic cirrhosis (4.69 ± 1.73,
p = 0.002) showed a significantly decreased activity. Non-significant differences were observed in PBC treated with UDCA, in viral hepatitis and in fatty liver (Figure 4).
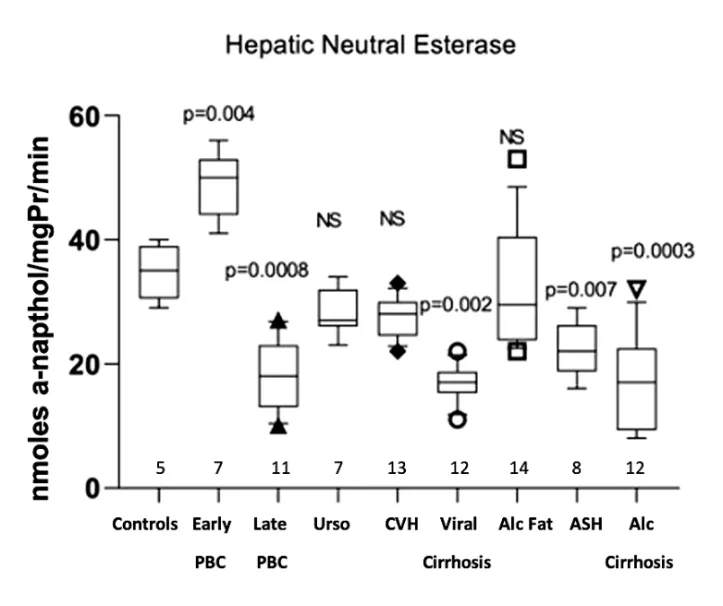
3.5 Hepatic Neutral Esterase
Hepatic neutral esterase activity was significantly increased in early PBC (48.71 ± 5.28 μg of α-naphthol/mg protein/min,
p = 0.004) compared with the normal controls (34.80 ± 4.43). On the contrary, late PBC (18.55 ± 5.75,
p = 0.0008), viral cirrhosis (16.83 ± 2.85,
p = 0.002), alcoholic hepatitis (22.38 ± 4.3,
p = 0.007) and alcoholic cirrhosis (16.75 ± 7.66,
p = 0.0003) showed significantly decreased activity. A non-significant trend towards decreased activity was also observed in PBC treated with UDCA, in chronic viral hepatitis and in fatty liver disease (Figure 5).
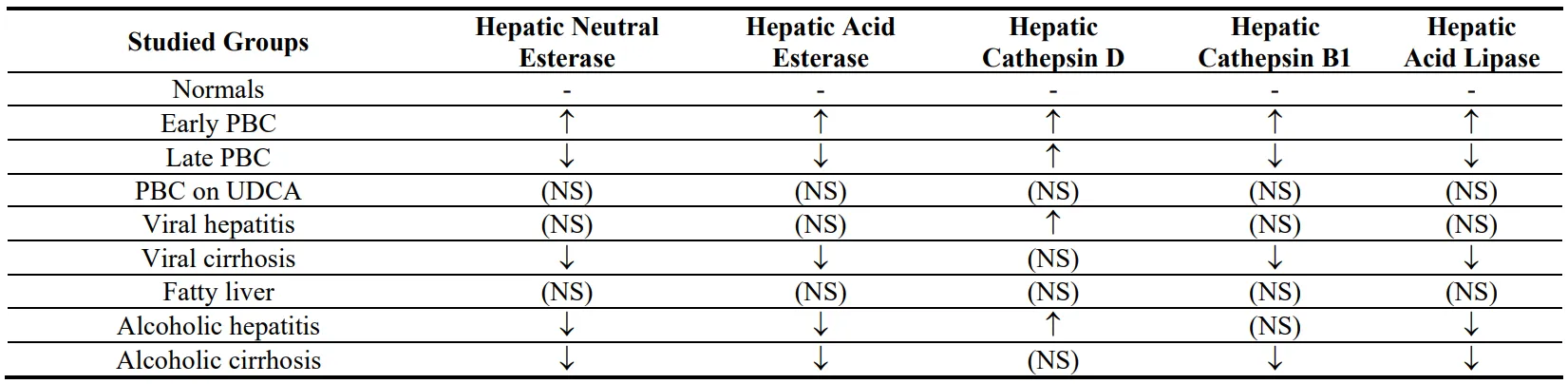
A summary of specific enzyme activity in all studied groups is presented in Table 2.
Figure 4. Measurement of acid esterases activity in the hepatic tissue of all studied groups, expressed as nmoles a-naphthol/mgPr/min.
Figure 5. Measurement of neutral esterase activity in the hepatic tissue of all studied groups, expressed as nmoles α-naphthol/mgPr/min.
Table 2. A summary of the results demonstrating the increase (↑) or decrease (↓) of specific enzymatic activity in all studied groups compared to normal controls.
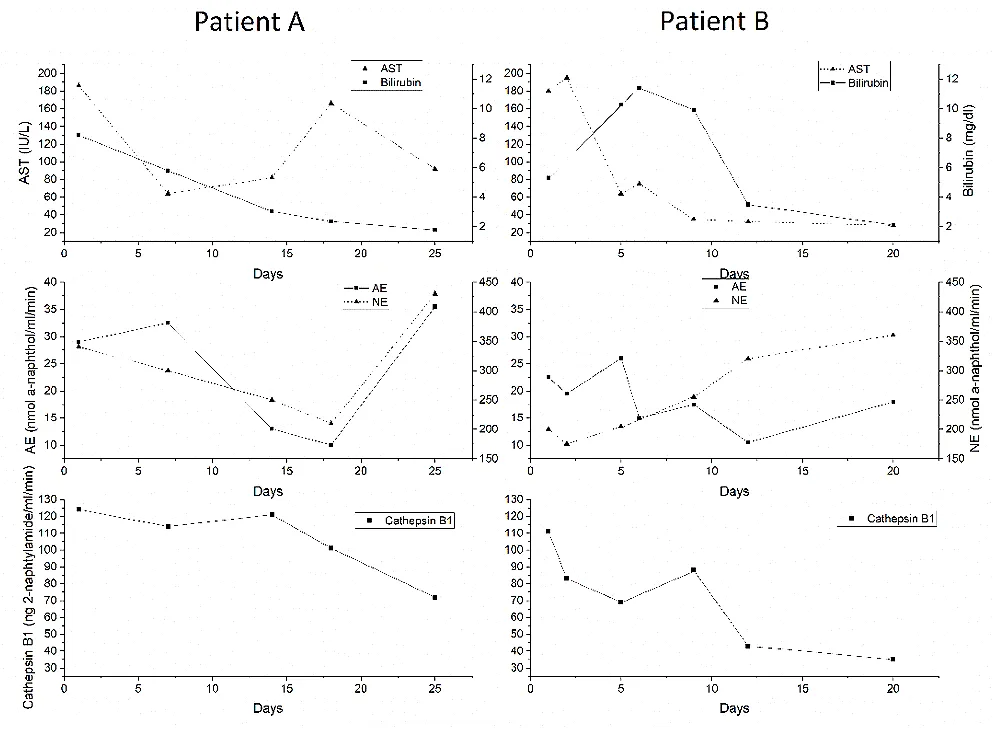
Serum enzymes (Cathepsin D cannot be measured in serum with our method) were sequentially estimated in the only group of patients with an acute progress to see if these enzymes could be used as potential markers of progress monitoring in acute liver disease. Figure 6 demonstrates the results of two patients with severe acute alcoholic hepatitis. Similar changes were observed in the other four patients studied. The upper part of the figure shows the fluctuations in serum bilirubin and AST, while the lower part shows changes in enzyme activity. When the AST is elevated, serum cathepsin B1 activity is high and the esterases are low. As patient A improves, cathepsin B1 decreases, and the esterases particularly the neutral enzyme, increase. Patient B shows that, despite a temporary increase in bilirubin, the constant improvement in AST is accompanied by a constant fall in cathepsin B1 activity and a constant increase in neutral esterase activity. Similar patterns were observed for acid lipase (not shown for clarity) in the other four patients studied.
. Demonstration of the results of two patients with severe acute alcoholic hepatitis. Similar changes were observed in the other four patients studied. The upper part of the figure shows the fluctuations in serum bilirubin and AST, while the lower parts show changes in enzyme activity.
4. Discussion
In the present study we assessed the activity of certain hepatic lysosomal enzymes in PBC and other chronic liver diseases.
All lysosomal enzymes were increased in early PBC including the highest levels of cathepsin D among all the other chronic liver diseases we studied. Increased levels were retained even at the advanced stages of PBC. There are three possible explanations for the increased lysosomal enzymes we found in PBC. They may be explained by either increased senescence or increased final stages of autophagy during early PBC, which gradually decreases with progression of the disease as recently proposed [
41]. These two pathways are interconnected as cellular senescence in biliary endothelial cells is associated with deregulated autophagy [
42,
43]. It is only logical therefore, that an interesting hypothesis implicating deregulated cholangiocyte autophagy connected to cholangiocyte senescence has been proposed to explain the pathogenesis of PBC [
44]. Our findings support this hypothesis.
Senescent cells present a remarkable increase in lysosomal mass and elevated autophagic activity. Proteins previously identified as plasma biomarkers of aging are highly enriched in the lysosomes of senescent cells, including cathepsin D [
45]. The im-portance of senescence in cholangiopathies was recently reviewed [
46].
A third explanation is related to the fact that cathepsin B has been reported to induce apoptosis and ferroptosis [
47,
48,
49], the recently identified form of cell death [
50]. The increased levels of cathepsin B1 we observed in PBC may therefore be associated with the increased apoptosis reported in this disease [
8].
However, there is also an additional simpler explanation for the increased levels of cathepsin D in PBC. Transcription of cathepsin D is activated by estrogens and patients with PBC are almost 90% women [
51].
Another finding in our study was the effect of UDCA on lysosomal enzymes. There were no significant differences of lysosomal enzymes in PBC treated with UDCA compared to normal controls. This implies that UDCA may restore dysregulations in au-tophagic flux reported in cholestasis [
52,
53]. UDCA was reported to reduce ER stress and partially corrected the dysregulated autophagy and senescence [
54,
55,
56], findings compatible with our results. The mechanism by which UDCA may restore lysosomal enzymes is currently unknown. An earlier paper described reduction of the initially increased lysosomal enzymes in PBC after treatment with D-penicillamine but the mechanism was not identified [
57]. Late stage autophagy has been reported to be impaired in human cholestasis by bile acids through the farnesoid X receptor (FXR) induction. Autophagy was induced by UDCA in this study [
55]. It is conceivable therefore, that the modification of the bile acid microenvironment caused by UDCA may be the mechanism of the restoration of the enzymes we found in our study.
In the other chronic liver diseases, we found a reduction of most hepatic enzymes in patients with advanced fibrosis including the fibrotic stages of PBC. Our findings indicate that a relative cathepsin B deficiency is associated with fibrosis. The role of cathepsin B in liver fibrosis is controversial. Both pro-fibrotic and anti-fibrotic effects have been demonstrated. Cathepsin B deficiency in experimental NASH reduced liver TGF-β1 and therefore fibrosis [
58], while levels of cathepsin B and active TGF-β increased in parallel with the METAVIR stage of hepatic fibrosis in chronic HCV [
59]. Rats with carbon tetrachloride-induced liver fibrosis, increased the hepatic expression of Cathepsin B during fibrogenesis [
60].
The pro-fibrotic action of cathepsin B could be explained by the reported association of cathepsin B with inflammation. A significant association of increased cathepsin B and the severity of hepatic inflammation was observed in the serum of chronic HBV patients [
59]. Cathepsin B has been involved in NLRP3 inflammasome activation [
61]. Our findings are not compatible with this explanation as liver cathepsin B1 was not increased in chronic inflammatory liver diseases such as viral and alcoholic hepatitis.
Our findings showed that reduced cathepsin B may be related to increased matrix deposition in the liver. This could be due to the fact that we studied patients with established fibrosis while most reports on the pro-fibrotic association of cathepsin B are experimental and were observed during the early stages of fibrogenesis.
Our findings on cathepsin B in the other liver diseases we studied, may also be explained by impairment of autophagy. Autophagy is indeed associated with liver fibrosis, and the end result depends on the cell population involved. Autophagy restricts fi-brogenesis acting in hepatocytes, macrophages and sinusoidal endothelial cells (LSECs). However, in hepatic stellate cells (HSC), autophagy favors fibrogenesis [
62,
63,
64,
65]. Cathepsins B and D have been previously used as indicators of autophagy in various diseases such as atherosclerosis and myocardial infarction [
25,
27] and the brain where reduced cathepsin B levels suggested reduced autophagy in parallel with other established markers of autophagy [
26].
In contrast to cathepsin B, we found increased levels of hepatic cathepsin D in the livers of viral and alcoholic hepatitis patients, confirming a relation of this acid protease with inflammation [
66]. Direct evidence connects cathepsin D to inflammation [
67,
68]. Inhibition of cathepsin D attenuated inflammation in experimental non-alcoholic steatohepatitis (NASH) [
69,
70], while increased levels of plasma cathepsin D were found in patients with NASH [
71,
72]. Experimental upregulation of cathepsin D increased lysosomal fragility and conceivably increased lysosomal permeabilization in the liver [
73] inducing necroptosis, apoptosis and ferroptosis. Lysosomal membrane permeabilization may also amplify or initiate cell death signaling in the context of apoptosis or autophagy-dependent cell death, and ferroptosis [
74]. Interestingly many viruses were shown to induce lysosome permeabilization [
75] and this may be connected to the increased cathepsin D levels we found in chronic viral hepatitis.
Slightly different results from our study were recently reported. Liver biopsy sections from patients with chronic liver diseases (HCV, HBV, PBC and NAFLD) were analyzed by immunohistochemistry for expression of cathepsin B, D and L. Suppression of cathepsins B, D and L was not observed in HBV, HCV and PBC. Hepatic inflammation was associated with dysfunction of autophagy as detected by the expression of p62 in hepatocytes [
76]. This slight discrepancy in PBC is likely associated with the fact that, this was a semi-quantitative immunohistochemical study and no separation of fibrotic cases was reported. Moreover, in analogy with our study, there was a non-significant increase in cathepsin D in both chronic viral hepatitis and PBC patients.
Another finding of our study was that in chronic liver disease patients the levels of cathepsin B and D are opposite with the notable exception of PBC. Cathepsin B is usually reduced when cathepsin D is increased. The same pattern was reported in cell cultures and mice [
77].
Lipolytic enzyme abnormalities were also found in our patients. All three lipolytic enzymes were significantly reduced in alcoholic hepatitis and cirrhosis irrespective of etiology. We used methylumbelliferyl oleate as a substrate to measure acid lipase (LAL) activity as this substrate identifies acid lipase deficiency in Wolman’s disease [
40] where cholestasis and cirrhosis have been described [
39].
Our findings agree with recent reports where serum LAL activity was significantly reduced in patients with cryptogenic cirrhosis but also in cirrhosis associated with HCV, HBV and alcohol [
78]. A progressive reduction of LAL activity from simple steatosis to non-alcoholic steatohepatitis and cirrhosis was recently reported [
79]. The same was observed in our study with a progressive reduction of hepatic acid esterase and acid lipase in alcoholic liver disease.
Non-specific esterases are multiple mostly cytoplasmic isoenzymes. However, a close correlation with acid phosphatase was observed in human gallbladder mucosa suggesting a lysosomal localization for both acid and neutral esterase [
36]. Moreover, the similarity of isoenzyme patterns between neutral and acid esterases was suggestive of a single enzyme molecule while the different in vitro response to inhibitors may indicate two different active sites on the same molecule [
36]. This is possibly verified in the present study as abnormalities of the two enzymes parallel each other. A similar situation has been previously suggested for acid lipase, where acid lipase and acid cholesteryl esterase were one protein with two different active sites, although this has been recently questioned [
19,
80]. To the best of our knowledge, there are no reports on non-specific esterases in liver diseases.
We found an interesting result when we tested the possibility that sequential serum measurements of these lysosomal enzymes could be used to monitor disease progression in acute liver disease such as acute alcoholic hepatitis (Figure 6). The initial relative high serum cathepsin B1 levels are constantly reduced as patients improve irrespective of temporary increases of transaminases or bilirubin. Cathepsin B1 in serum is associated with its main inhibitor a2 macroglobulin and the complex is cleared by macrophages [
81]. Therefore, increased serum levels may be compatible with Kupffer cell inability to clear the complex and progressive reduction may be related to macrophage recovery. Non-specific esterases, on the other hand, show a parallel fluctuation with transaminase values and increase as patients improve. It is possible therefore, that their measurement may be useful for assessing macrophage function in liver disease progression or regression. Whether this is indeed clinically useful and true for viral hepatitis or other liver diseases as well, requires extensive additional studies.
A drawback of our study is the fact that no direct comparisons with autophagy markers were done. Therefore, the association of our findings with autophagy deregulation is conjectural and based only in literature reports. Moreover, this is a cross sectional study with a relatively small number of patients in each group. Larger studies are obviously required but it should be noted that liver biopsies are now infrequently performed in PBC and other chronic liver diseases.
Another inherent problem in our study similar to other studies where liver homogenates or serum from patients are used, is the inability to discriminate between hepatocyte and sinusoidal cells changes. This is particularly important in our study. Liver macrophages are a major site of production of the enzymes we studied. Non-specific esterases are genetically determined markers of macrophages [
35,
82]. Cathepsins are increased up to 100 nM in the macrophage secretome [
83]. Therefore, an additional explanation that may interfere in the interpretation of our results is that they may reflect activation or suppression of Kupffer cells. We have recently proposed that Kupffer cells are crucial for the pathogenesis of PBC [
84]. Recent experimental evidence supports the important role of Kupffer cells in PBC pathogenesis. In PBC mice, Kupffer cells are activated and Kupffer cell-derived IL-12 and TNF-a synergistically with NK cell-derived IFN-γ induce biliary epithelial cell damage [
85]. Furthermore, the Clostridium metabolite p-Cresol sulfate (PCS), produced by Clostridium-metabolized tyrosine, reduced PBC inflammation by modulating the polarization of Kupffer cells. Thus, the production of pro-inflammatory cytokines such as IL-1β, TNFα and IL-6 were reduced and the anti-inflammatory factors were increased [
86].
However, our results agree more with the hypothesis that Kupffer cell hypersecretion of cathepsin D may be linked to inflammation, while reduced Kupffer cell secretion of cathepsin B may lead to advanced fibrosis [
87,
88]. Importantly, since lipolytic hydrolases also mark macrophages, their reduced activities we found may also represent Kupffer cell suppression in cirrhosis.
As mentioned before, our findings may be also consistent with a dysregulation of the late stages of autophagy in chronic viral and alcoholic liver disease. Future studies should further clarify the matter. Dysregulated autophagy may be the critical factor that possibly dictates the direction of the interrelated pathways of senescence apoptosis and autophagy [
64]. If so, it can be envisaged that inhibitors of the late stages of autophagy like chloroquine or hydroxychloroquine or autophagy activators such as Pevonedistat may be useful as additional treatments in chronic liver diseases [
89].
5. Conclusions
In conclusion, our findings in PBC probably indicate increased senescence in this disease and may explain the combination of increased senescence and increased apoptosis reported in PBC pathogenesis. In the disease controls we used, cathepsin B1 re-duction seems to be associated with advanced fibrosis, while increased cathepsin D seems to be associated and probably involved in the development of necro-inflammation. Treatment with UDCA restores the abnormal values of lysosomal enzymes. Measurement of serum cathepsin B and esterases may be a useful marker of acute liver disease progression, but this requires further documentation and validation.
Author Contributions
This paper has not been published in part or as a whole before nor has been submitted for publication in another journal. All authors significantly contributed to the paper and have read and approved of the submitted manuscript. I.T. interpreted the data and drafted the article. G.N. performed the laboratory measurements and analyzed the data. A.V. acquired the data and critically revised the article. D.S. and M.K. acquired the data and critically revised the article. E.K. was responsible for the conception and design of the study and contributed to the laboratory measurements, analysis and interpretation of data.
Ethics Statement
The study was conducted according to the guidelines of the Declaration of Helsinki, and approved by the Ethics Committee of the University Hospital (protocol codes 3468B and 436/9113; date[s] of approval: [3 September 2022] and [5 May 2022]).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors on request.
Funding
This research received no external funding.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
-
1.
Lleo A, Leung PSC, Hirschfield GM, Gershwin EM. The pathogenesis of primary biliary cholangitis: a comprehensive review.
Semin. Liver Dis. 2020,
40, 34–48.
[Google Scholar]
-
2.
Prieto J, Banales JM, Medina JF. Primary biliary cholangitis: pathogenic mechanisms.
Curr. Opin. Gastroenterol. 2021,
37, 91–98.
[Google Scholar]
-
3.
Trivedi PJ, Hirschfield GM. Recent advances in clinical practice: epidemiology of autoimmune liver diseases.
Gut 2021,
70, 1989–2003.
[Google Scholar]
-
4.
Trivella J, John BV, Levy C. Primary biliary cholangitis: Epidemiology, prognosis, and treatment.
Hepatol. Commun. 2023,
7, e0179.
[Google Scholar]
-
5.
Koulentaki M, Mantaka A, Sifaki-Pistolla D, Thalassinos E, Tzanakis N, Kouroumalis E. Geoepidemiology and space-time analysis of Primary biliary cirrhosis in Crete, Greece.
Liver Int. 2014,
34, e200–e207.
[Google Scholar]
-
6.
Turk B, Turk D, Turk V. Lysosomal cysteine proteases: more than scavengers.
Biochim. Biophys. Acta 2000,
1477, 98–111.
[Google Scholar]
-
7.
Park JW, Kim JH, Kim SE, Jung JH, Jang MK, Park SH, et al. Primary Biliary Cholangitis and Primary Sclerosing Cholangitis: Current Knowledge of Pathogenesis and Therapeutics.
Biomedicines 2022,
10, 1288.
[Google Scholar]
-
8.
Sasaki M, Sato Y, Nakanuma Y. An impaired biliary bicarbonate umbrella may be involved in dysregulated autophagy in primary biliary cholangitis.
Lab. Invest. 2018,
98, 745–754.
[Google Scholar]
-
9.
Trivedi PC, Bartlett JJ, Pulinilkunnil T. Lysosomal Biology and Function: Modern View of Cellular Debris Bin.
Cells 2020,
9, 1131.
[Google Scholar]
-
10.
Willstätter R, Bamann E. Über die Proteasen der Magenschleimhaut. Erste Abhandlung Über die Enzyme der Leukocyten.
Hoppe-Seyler’s Zeitschrift Fur Physiologische Chemie 1929,
180, 127–143.
[Google Scholar]
-
11.
Karch J, Schips TG, Maliken BD, Brody MJ, Sargent MA, Kanisicak O, et al. Autophagic cell death is dependent on lysosomal membrane permeability through Bax and Bak.
Elife 2017,
6, e30543.
[Google Scholar]
-
12.
Wang Y, Wu Q, Anand BG, Karthivashan G, Phukan G, Yang J, et al. Significance of cytosolic cathepsin D in Alzheimer's disease pathology: Protective cellular effects of PLGA nanoparticles against β-amyloid-toxicity.
Neuropathol. Appl. Neurobiol. 2020,
46, 686–706.
[Google Scholar]
-
13.
Hausmann M, Obermeier F, Schreiter K, Spottl T, Falk W, Schölmerich J, et al. Cathepsin D is up-regulated in inflammatory bowel disease macrophages.
Clin. Exp. Immunol. 2004,
136, 157–167.
[Google Scholar]
-
14.
Mijanovic O, Petushkova AI, Brankovic A, Turk B, Solovieva AB, Nikitkina AI, et al. Cathepsin D-Managing the Delicate Balance.
Pharmaceutics 2021,
13, 837.
[Google Scholar]
-
15.
Iwama H, Mehanna S, Imasaka M, Hashidume S, Nishiura H, Yamamura KI, et al. Cathepsin B and D deficiency in the mouse pancreas induces impaired autophagy and chronic pancreatitis.
Sci. Rep. 2021,
11, 6596.
[Google Scholar]
-
16.
Senjor E, Kos J, Nanut MP. Cysteine Cathepsins as therapeutic targets in immune regulation and immune disorders.
Biomedicines 2023,
11, 476.
[Google Scholar]
-
17.
Vidak E, Javoršek U, Vizovišek M, Turk B. Cysteine Cathepsins and their Extracellular Roles: Shaping the Microenvironment.
Cells 2019,
8, 264.
[Google Scholar]
-
18.
Singh R, Cuervo AM. Lipophagy: connecting autophagy and lipid metabolism.
Int. J. Cell. Biol. 2012,
2012, 282041.
[Google Scholar]
-
19.
Pajed L, Wagner C, Taschler U, Schreiber R, Kolleritsch S, Fawzy N, et al. Hepatocyte-specific deletion of lysosomal acid lipase leads to cholesteryl ester but not triglyceride or retinyl ester accumulation.
J. Biol. Chem. 2019,
294, 9118–9133.
[Google Scholar]
-
20.
Uphoff CC, Drexler HG. Biology of monocyte-specific esterase.
Leuk Lymphoma 2000,
39, 257–270.
[Google Scholar]
-
21.
Nowosad A, Besson A. Lysosomes at the Crossroads of Cell Metabolism, Cell Cycle, and Stemness.
Int. J. Mol. Sci. 2022,
23, 2290.
[Google Scholar]
-
22.
Sasaki M, Miyakoshi M, Sato Y, Nakanuma Y. Autophagy may precede cellular senescence of bile ductular cells in ductular reaction in primary biliary cirrhosis.
Dig. Dis. Sci. 2012,
57, 660–666.
[Google Scholar]
-
23.
Sasaki M, Miyakoshi M, Sato Y, Nakanuma Y. Increased expression of mitochondrial proteins associated with autophagy in biliary epithelial lesions in primary biliary cirrhosis.
Liver Int. 2013,
33, 312–320.
[Google Scholar]
-
24.
van de Graaf S, Beuers U. Autophagy—another piece of the puzzle towards understanding primary biliary cirrhosis?
Liver Int. 2014,
34, 481–483.
[Google Scholar]
-
25.
Osonoi Y, Mita T, Azuma K, Nakajima K, Masuyama A, Goto H, et al. Defective autophagy in vascular smooth muscle cells enhances cell death and atherosclerosis.
Autophagy. 2018,
14, 1991–2006.
[Google Scholar]
-
26.
Pla A, Pascual M, Renau-Piqueras J, Guerri C. TLR4 mediates the impairment of ubiquitin-proteasome and autophagy-lysosome pathways induced by ethanol treatment in brain.
Cell Death Dis. 2014,
5, e1066.
[Google Scholar]
-
27.
Wu P, Yuan X, Li F, Zhang J, Zhu W, Wei M, et al. Myocardial Upregulation of Cathepsin D by Ischemic Heart Disease Promotes Autophagic Flux and Protects Against Cardiac Remodeling and Heart Failure.
Circ. Heart Fail. 2017,
10, e004044.
[Google Scholar]
-
28.
Zeng J, Acin-Perez R, Assali EA, Martin A, Brownstein AJ, Petcherski A, et al. Restoration of lysosomal acidification rescues autophagy and metabolic dysfunction in non-alcoholic fatty liver disease.
Nat. Commun. 2023,
14, 2573.
[Google Scholar]
-
29.
Ludwig J, Dickson ER, McDonald GS. Staging of chronic nonsuppurative destructive cholangitis (syndrome of primary biliary cirrhosis).
Virchows Arch. A Pathol. Anat. Histol. 1978,
379, 103–112.
[Google Scholar]
-
30.
European Association for the Study of the Liver. European Association for the Study of the Liver. EASL Clinical Practice Guidelines: The diagnosis and management of patients with primary biliary cholangitis.
J. Hepatol. 2017,
67, 145–172.
[Google Scholar]
-
31.
Roth JS, Losty T, Wierbicki E. Assay of proteolytic enzyme activity using a
14C-labeled hemoglobin.
Anal. Biochem. 1971,
42, 214–221.
[Google Scholar]
-
32.
Barrett AJ. A new assay for cathepsin B1 and other thiol proteinases.
Anal Biochem. 1972,
47, 280–293.
[Google Scholar]
-
33.
Van Berkel TJ, Vaandrager H, Kruijt JK, Koster JF. Characteristics of acid lipase and acid cholesteryl esterase activity in parenchymal and non-parenchymal rat liver cells.
Biochim. Biophys. Acta 1980,
617, 446–457.
[Google Scholar]
-
34.
Schaffner T, Elner VM, Bauer M, Wissler RW. Acid lipase: a histochemical and biochemical study using triton X100-naphtyl palmitate micelles.
J. Histochem. Cytochem. 1978,
26, 696–712.
[Google Scholar]
-
35.
Kolios G, Valatas V, Psilopoulos D, Petraki K, Kouroumalis E. Depletion of non specific esterase activity in the colonic mucosa of patients with ulcerative colitis.
Eur. J. Clin. Invest. 2002,
32, 265–273.
[Google Scholar]
-
36.
Kouroumalis E, Hopwood D, Ross PE, Bouchier IA. Human gallbladder epithelium: non-specific esterases in cholecystitis.
J. Pathol. 1984,
142, 151–159.
[Google Scholar]
-
37.
Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent.
J. Biol. Chem 1951,
193, 265–275.
[Google Scholar]
-
38.
Wang J, Zheng M, Yang X, Zhou X, Zhang S. The Role of Cathepsin B in Pathophysiologies of Non-tumor and Tumor tissues: A Systematic Review.
J. Cancer 2023,
14, 2344–2358.
[Google Scholar]
-
39.
Menon J, Shanmugam N, Srinivas S, Vij M, Jalan A, Srinivas Reddy M, et al. Wolman's Disease: A Rare Cause of Infantile Cholestasis and Cirrhosis.
J. Pediatr. Genet. 2020,
11, 132–134.
[Google Scholar]
-
40.
Cortner JA, Coates PM, Swoboda E, Schnatz JD. Genetic variation of lysosomal acid lipase.
Pediatr. Res. 1976,
10, 927–932.
[Google Scholar]
-
41.
Panzitt K, Fickert P, Wagner M. Regulation of autophagy by bile acids and in cholestasis—CholestoPHAGY or CholeSTOPagy.
Biochim. Biophys. Acta Mol. Basis Dis. 2021,
1867, 166017.
[Google Scholar]
-
42.
Sasaki M, Ikeda H, Yamaguchi J, Nakada S, Nakanuma Y. Telomere shortening in the damaged small bile ducts in primary biliary cirrhosis reflects ongoing cellular senescence.
Hepatology 2008,
48, 186–195.
[Google Scholar]
-
43.
Sasaki M, Yoshimura-Miyakoshi M, Sato Y, Nakanuma Y. A possible involvement of endoplasmic reticulum stress in biliary epithelial autophagy and senescence in primary biliary cirrhosis.
J. Gastroenterol. 2015,
50, 984–995.
[Google Scholar]
-
44.
Nakanuma Y, Sasaki M, Harada K. Autophagy and senescence in fibrosing cholangiopathies.
J. Hepatol. 2015,
62, 934–945.
[Google Scholar]
-
45.
Rovira M, Sereda R, Pladevall-Morera D, Ramponi V, Marin I, Maus M, et al. The lysosomal proteome of senescent cells contributes to the senescence secretome.
Aging Cell 2022,
21, e13707.
[Google Scholar]
-
46.
Bogert PS, O’Hara SP, LaRusso NF. Cellular senescence in the cholangiopathies.
Curr. Opin. Gastroenterol. 2022,
38, 121–127.
[Google Scholar]
-
47.
Baskin-Bey ES, Canbay A, Bronk SF, Werneburg N, Guicciardi ME, Nyberg SL, et al. Cathepsin B inactivation attenuates hepatocyte apoptosis and liver damage in steatotic livers after cold ischemia-warm reperfusion injury.
Am. J. Physiol. Gastrointest. Liver Physiol. 2005,
288, G396–G402.
[Google Scholar]
-
48.
Colletti GA, Miedel MT, Quinn J, Andharia N, Weisz OA, Kiselyov K. Loss of lysosomal ion channel transient receptor potential channel mucolipin-1 (TRPML1) leads to cathepsin B-dependent apoptosis.
J. Biol. Chem. 2012,
287, 8082–8091.
[Google Scholar]
-
49.
de Castro MA, Bunt G, Wouters FS. Cathepsin B launches an apoptotic exit effort upon cell death-associated disruption of lysosomes.
Cell Death Discov. 2016,
2, 16012.
[Google Scholar]
-
50.
Nagakannan P, Islam MI, Conrad M, Eftekharpour E. Cathepsin B is an executioner of ferroptosis.
Biochim. Biophys. Acta Mol. Cell. Res. 2021,
1868, 118928.
[Google Scholar]
-
51.
Cavaillès V, Augereau P, Rochefort H. Cathepsin D gene is controlled by a mixed promoter, and estrogens stimulate only TATA-dependent transcription in breast cancer cells.
Proc. Natl. Acad. Sci. USA 1993,
90, 203–207.
[Google Scholar]
-
52.
Paumgartner G, Beuers U. Mechanisms of action and therapeutic efficacy of ursodeoxycholic acid in cholestatic liver disease.
Clin. Liver Dis. 2004,
8, 67–81.
[Google Scholar]
-
53.
Ye HL, Zhang JW, Chen XZ, Wu PB, Chen L, Zhang G. Ursodeoxycholic acid alleviates experimental liver fibrosis involving inhibition of autophagy.
Life Sci. 2020,
242, 117175.
[Google Scholar]
-
54.
Amaral JD, Viana RJ, Ramalho RM, Steer CJ, Rodrigues CM. Bile acids: regulation of apoptosis by ursodeoxycholic acid.
J. Lipid Res. 2009,
50, 1721–1734.
[Google Scholar]
-
55.
Panzitt K, Jungwirth E, Krones E, Lee JM, Pollheimer M, Thallinger GG, et al. FXR-dependent Rubicon induction impairs autophagy in models of human cholestasis.
J. Hepatol. 2020,
72, 1122–1131.
[Google Scholar]
-
56.
Sasaki M, Nakanuma Y. Bile Acids and Deregulated Cholangiocyte Autophagy in Primary Biliary Cholangitis.
Dig. Dis. 2017,
35, 210–216.
[Google Scholar]
-
57.
Scott J, Jenkins W, Smith GP, Peters TJ. Hepatic organelle pathology in primary biliary cirrhosis and the response to low-dose D-penicillamine therapy.
Clin. Sci. 1981,
60, 207–212.
[Google Scholar]
-
58.
Fang W, Deng Z, Benadjaoud F, Yang C, Shi GP. Cathepsin B deficiency ameliorates liver lipid deposition, inflammatory cell infiltration, and fibrosis after diet-induced nonalcoholic steatohepatitis.
Transl. Res. 2020,
222, 28–40.
[Google Scholar]
-
59.
Zanelatto ACO, Lacerda GS, Accardo CM, Rosário NFD, Silva AAD, Motta G, et al. Cathepsin B and Plasma Kallikrein Are Reliable Biomarkers to Discriminate Clinically Significant Hepatic Fibrosis in Patients with Chronic Hepatitis-C Infection.
Microorganisms 2022,
10, 1769.
[Google Scholar]
-
60.
Manchanda M, Das P, Gahlot GPS, Singh R, Roeb E, Roderfeld M, et al. Cathepsin L and B as Potential Markers for Liver Fibrosis: Insights From Patients and Experimental Models.
Clin. Transl. Gastroenterol. 2017,
8, e99.
[Google Scholar]
-
61.
Campden RI, Zhang Y. The role of lysosomal cysteine cathepsins in NLRP3 inflammasome activation.
Arch. Biochem. Biophys. 2019,
670, 32–42.
[Google Scholar]
-
62.
Allaire M, Rautou PE, Codogno P, Lotersztajn S. Autophagy in liver diseases: Time for translation?
J. Hepatol. 2019,
70, 985–998.
[Google Scholar]
-
63.
Gual P, Gilgenkrantz H, Lotersztajn S. Autophagy in chronic liver diseases: the two faces of Janus.
Am. J. Physiol. Cell. Physiol. 2017,
312, C263–C273.
[Google Scholar]
-
64.
Hung TM, Hsiao CC, Lin CW, Lee PH. Complex Cell Type-Specific Roles of Autophagy in Liver Fibrosis and Cirrhosis.
Pathogens 2020,
9, 225.
[Google Scholar]
-
65.
Kouroumalis E, Voumvouraki A, Augoustaki A, Samonakis DN. Autophagy in liver diseases.
World J. Hepatol. 2021,
13, 6–65.
[Google Scholar]
-
66.
Mahapatra KK, Mishra SR, Behera BP, Patil S, Gewirtz DA, Bhutia SK. The lysosome as an imperative regulator of autophagy and cell death.
Cell. Mol. Life Sci. 2021,
78, 7435–7449.
[Google Scholar]
-
67.
Khurana P, Yadati T, Goyal S, Dolas A, Houben T, Oligschlaeger Y, et al. Inhibiting Extracellular Cathepsin D Reduces Hepatic Steatosis in Sprague⁻Dawley Rats.
Biomolecules 2019,
9, 171.
[Google Scholar]
-
68.
Orlowski GM, Colbert JD, Sharma S, Bogyo M, Robertson SA, Rock KL. Multiple Cathepsins Promote Pro-IL-1β Synthesis and NLRP3-Mediated IL-1β Activation.
J. Immunol. 2015,
195, 1685–1697.
[Google Scholar]
-
69.
Houben T, Oligschlaeger Y, Hendrikx T, Bitorina AV, Walenbergh SMA, van Gorp PJ, et al. Cathepsin D regulates lipid metabolism in murine steatohepatitis.
Sci. Rep. 2017,
7, 3494.
[Google Scholar]
-
70.
Yadati T, Houben T, Bitorina A, Oligschlaeger Y, Gijbels MJ, Mohren R, et al. Inhibition of Extracellular Cathepsin D Reduces Hepatic Lipid Accumulation and Leads to Mild Changes in Inflammationin NASH Mice.
Front. Immunol. 2021,
12, 675535.
[Google Scholar]
-
71.
Ding L, De Munck TJI, Oligschlaeger Y, Dos Reis IM, Verbeek J, Koek GH, et al. Myosteatosis in NAFLD patients correlates with plasma Cathepsin D.
Biomol. Concepts 2021,
12, 27–35.
[Google Scholar]
-
72.
Walenbergh SM, Houben T, Rensen SS, Bieghs V, Hendrikx T, van Gorp PJ, et al. Plasma cathepsin D correlates with histological classifications of fatty liver disease in adults and responds to intervention.
Sci. Rep. 2016,
6, 38278.
[Google Scholar]
-
73.
Ke PY. Diverse Functions of Autophagy in Liver Physiology and Liver Diseases.
Int. J. Mol. Sci. 2019,
20, 300.
[Google Scholar]
-
74.
Gao H, Bai Y, Jia Y, Zhao Y, Kang R, Tang D, et al. Ferroptosis is a lysosomal cell death process.
Biochem. Biophys. Res. Commun. 2018,
503, 1550–1556.
[Google Scholar]
-
75.
Aits S, Jäättelä M. Lysosomal cell death at a glance.
J. Cell Sci. 2013,
126, 1905–1912.
[Google Scholar]
-
76.
Fukuo Y, Yamashina S, Sonoue H, Arakawa A, Nakadera E, Aoyama T, et al. Abnormality of autophagic function and cathepsin expression in the liver from patients with non-alcoholic fatty liver disease.
Hepatol. Res. 2014,
44, 1026–1036.
[Google Scholar]
-
77.
Felbor U, Kessler B, Mothes W, Goebel HH, Ploegh HL, Bronson RT, et al. Neuronal loss and brain atrophy in mice lacking cathepsins B and L.
Proc. Natl. Acad. Sci. USA 2002,
99, 7883–7888.
[Google Scholar]
-
78.
Vespasiani-Gentilucci U, Gallo P, Piemonte F, Riva E, Porcari A, Vorini F, et al. Lysosomal Acid Lipase Activity Is Reduced Both in Cryptogenic Cirrhosis and in Cirrhosis of Known Etiology.
PLoS ONE 2016,
11, e0156113.
[Google Scholar]
-
79.
Baratta F, Pastori D, Ferro D, Carluccio G, Tozzi G, Angelico F, et al. Reduced lysosomal acid lipase activity: A new marker of liver disease severity across the clinical continuum of non-alcoholic fatty liver disease?
World J. Gastroenterol. 2019,
25, 4172–4180.
[Google Scholar]
-
80.
Zhang H. Lysosomal acid lipase and lipid metabolism: new mechanisms, new questions, and new therapies.
Curr. Opin. Lipidol. 2018,
29, 218–223.
[Google Scholar]
-
81.
Starkey PM, Barrett AJ. Inhibition by alpha-macroglobulin and other serum proteins.
Biochem. J. 1973,
131, 823–831.
[Google Scholar]
-
82.
Amaral EP, Riteau N, Moayeri M, Maier N, Mayer-Barber KD, Pereira RM, et al. Lysosomal Cathepsin Release Is Required for NLRP3-Inflammasome Activation by Mycobacterium tuberculosis in Infected Macrophages.
Front. Immunol. 2018,
9, 1427.
[Google Scholar]
-
83.
Sobotič B, Vizovišek M, Vidmar R, Van Damme P, Gocheva V, Joyce JA, et al. Proteomic Identification of Cysteine Cathepsin Substrates Shed from the Surface of Cancer Cells.
Mol. Cell. Proteomics 2015,
14, 2213–2228.
[Google Scholar]
-
84.
Kouroumalis E, Notas G. Primary biliary cirrhosis: From bench to bedside.
World J. Gastrointest. Pharmacol. Ther. 2015,
6, 32–58.
[Google Scholar]
-
85.
Fu HY, Bao WM, Yang CX, Lai WJ, Xu JM, Yu HY, et al. Kupffer Cells Regulate Natural Killer Cells Via the NK group 2, Member D (NKG2D)/Retinoic Acid Early Inducible-1 (RAE-1) Interaction and Cytokines in a Primary Biliary Cholangitis Mouse Model.
Med. Sci. Monit. 2020,
26, e923726.
[Google Scholar]
-
86.
Fu HY, Xu JM, Ai X, Dang FT, Tan X, Yu HY, et al. The Clostridium Metabolite P-Cresol Sulfate Relieves Inflammation of Primary Biliary Cholangitis by Regulating Kupffer Cells.
Cells 2022,
11, 3782.
[Google Scholar]
-
87.
Gracia-Sancho J, Guixé-Muntet S. The many-faced role of autophagy in liver diseases.
J. Hepatol. 2018,
68, 593–594.
[Google Scholar]
-
88.
Lodder J, Denaës T, Chobert MN, Wan J, El-Benna J, Pawlotsky JM, et al. Macrophage autophagy protects against liver fibrosis in mice.
Autophagy 2015,
11, 1280–1292.
[Google Scholar]
-
89.
Mohsen S, Sobash PT, Algwaiz GF, Nasef N, Al-Zeidaneen SA, Karim NA. Autophagy Agents in Clinical Trials for Cancer Therapy: A Brief Review.
Curr. Oncol. 2022,
29, 1695–1708.
[Google Scholar]
 George Notas
3
George Notas
3