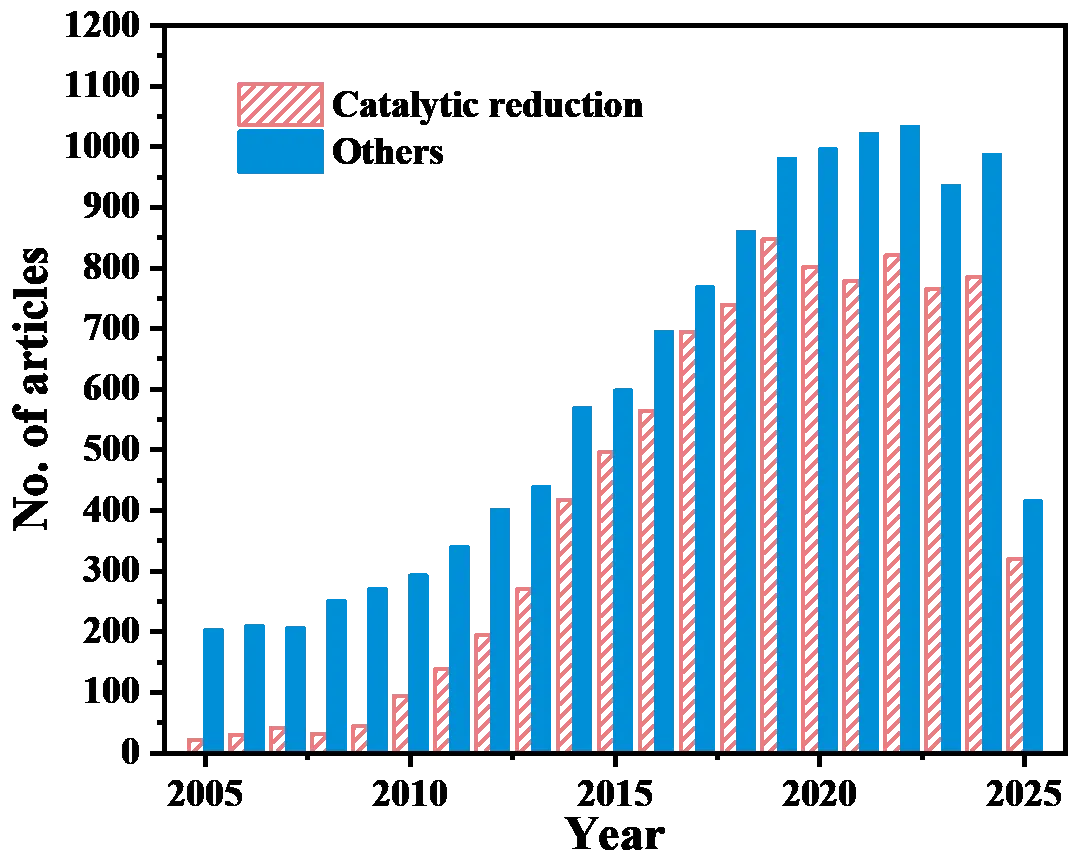
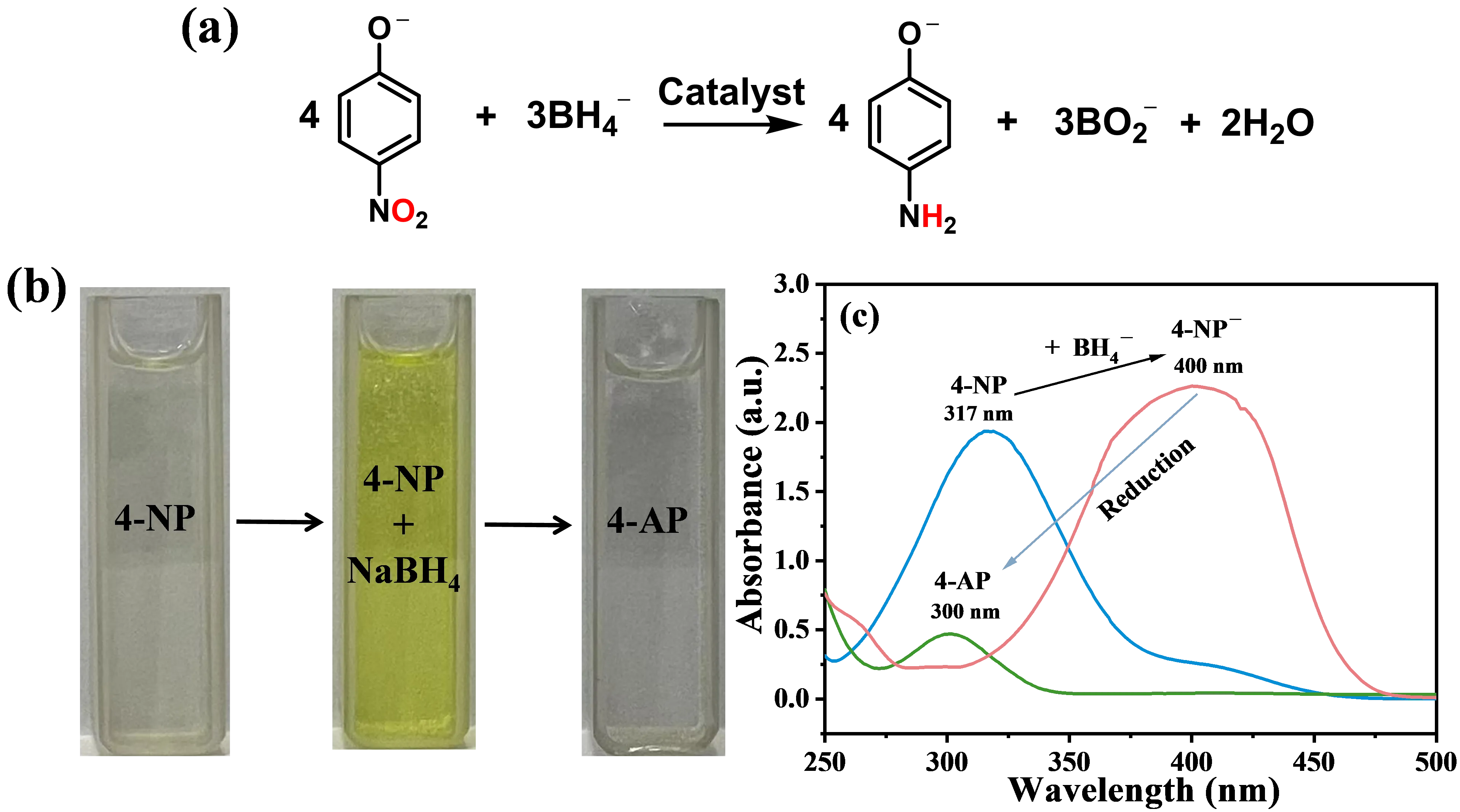
2.1. The Benchmark Reaction: NaBH4 Reduction of 4-NP
4-NP, a representative nitroaromatic pollutant, was first reported by Pel et al. in 2002 to be catalytically reduced to 4-aminophenol (4-AP) in aqueous solution using silver nanoparticles as the catalyst, with sodium borohydride (NaBH
4) as the reductant [
30]. The catalytic reduction of 4-NP to 4-AP using NaBH
4 in aqueous medium has established itself as a benchmark reaction for catalyst evaluation due to its unique experimental and analytical advantages [
18,
19]. As depicted in
a, this reaction exhibits a distinct spectroscopic signature where, under alkaline conditions induced by NaBH
4, 4-NP (pale yellow, λ
max = 317 nm) deprotonates to form the 4-nitrophenolate anion (4-NP
−, bright yellow, λ
max = 400 nm), while the reduction product 4-AP remains colorless (λ
max = 296 nm) (
b). This enables real-time quantitative monitoring of reaction kinetics through straightforward UV-Vis spectroscopy by tracking the decay of the 400 nm absorbance peak without spectral interference (
c). The reaction’s well-defined stoichiometry—evidenced by clear isosbestic points—and operational simplicity in aqueous environments further solidify its status as a robust platform for comparative catalyst assessment. Although initially pioneered with noble metal nanoparticles (e.g., Ag, Au, Pd), the inherent limitations of these systems, including high cost, susceptibility to poisoning, metal leaching, and poor reusability, have intensified the search for sustainable alternatives [
22].
. (<strong>a</strong>) Overall reaction equation for 4-NP reduced to 4-AP by NaBH<sub>4</sub> in aqueous solution. (<strong>b</strong>) The color changes from 4-NP aqueous solution to 4-NP + NaBH<sub>4</sub> aqueous solution to 4-AP aqueous solution. (<strong>c</strong>) UV-Vis. spectrum absorption peak changes from 4-NP aqueous solution to 4-NP + NaBH<sub>4</sub> aqueous solution to 4-AP aqueous solution.
A pivotal advancement occurred in 2011 when Ananthakrishnan et al. demonstrated the first successful metal-free photocatalytic reduction of 4-NP using resin-supported dyes under visible light [
31], marking the debut of C-MFCs for this reaction. Since then, C-MFCs have shown catalytic activity comparable to or exceeding traditional metal-based catalysts. Subsequent years witnessed an explosion in the development and application of diverse C-MFCs architectures, including heteroatom-doped nanocarbons (e.g., N-doped graphene/carbon nanotubes (CNTs) [
32,
33], S-doped graphene (SG) [
34], metal organic framework (MOF)-derived porous carbons, e.g., N-doped [
35], B,N-doped [
36]), biomass-derived carbons from sustainable precursors (e.g., sewage sludge biochar [
37], lignin-derived carbon [
38]), and engineered 3D frameworks (e.g., graphene foams [
39], ionic liquid-derived tri-doped carbons [
40]).
For standardized comparison, catalytic activity is quantified by TOF, representing moles of 4-NP converted per unit mass of catalyst per unit time. The TOF value of the first metal-free photocatalytic reduction of 4-NP using resin-supported organic dyes under visible light was 3.1 × 10
−6 mmol mg
−1 min
−1) [
31]. This seminal work demonstrated the feasibility of C-MFCs for this transformation and ignited significant research interest. Critically, as summarized in
, these C-MFCs achieve TOF spanning 1.3 × 10
−6 to 8.8 × 10
−2 mmol mg
−1 min
−1, rivaling or surpassing benchmark metal catalysts. For instance, N-doped porous reduced graphene oxide (TOF = 8.8 × 10
−2 mmol mg
−1 min
−1) [
41] and N,P-doped multilayer graphene (TOF = 2.4 × 10
−2 mmol mg
−1 min
−1) [
42] outperform Au@C (1.2 × 10
−5 mmol mg
−1 min
−1) and compete effectively with commercial Pd/C (2.8 × 10
−2 mmol mg
−1 min
−1). Beyond their catalytic efficacy, C-MFCs offer compelling advantages of cost-effectiveness through abundant carbon sources (biomass, waste), enhanced sustainability by avoiding critical/precious metals, structural stability and reusability against poisoning and leaching due to the robust sp
2 carbon frameworks, and tunable electronic and surface properties via defect engineering.
.
Comparison of catalytic performance for 4-NP reduction using metal-based catalysts and C-MFCs.
| Catalysts Used in 4-NP Reduction |
C4-NP (mmol/L) |
CNaBH₄:C4-NP |
Volume of Reaction (mL) |
mcat. (mg) |
Conversion Rate(%) |
Conversion Time (min) |
TOF (mmol mg−1 min−1) |
Ref. |
| Commercial Pd/C (5 wt%) |
20 |
100 |
3 |
1 |
100 |
2.17 |
2.8 × 10−2 |
[42] |
| Au@C |
0.1 |
100 |
3 |
5 |
100 |
5 |
1.2 × 10−5 |
[43] |
| Ag@C nanofiber |
0.06 |
41.7 |
60 |
1 |
100 |
8 |
4.5 × 10−4 |
[44] |
| Ni@Pd |
0.1 |
833 |
3 |
0.4 |
99.8 |
4 |
1.8 × 10−4 |
[45] |
| Eosin Y immobilized resin |
0.2 |
250 |
30.00 |
30.00 |
93 |
60 |
3.1 × 10−6 |
[31] |
| N-doped graphene |
0.0588 |
100 |
8.50 |
0.14 |
100 |
21 |
6.1 × 10−5 |
[32] |
| N-doped CNTs |
0.2195 |
100 |
2.05 |
0.15 |
100 |
18.67 |
1.6 × 10−4 |
[33] |
| Hydrothermally treated GO |
0.0535 |
200 |
3.30 |
0.16 |
100 |
20 |
5.5 × 10−5 |
[46] |
| L-ascorbic acid reduced GO-4 |
0.1 |
766 |
5 |
2.5 |
98.2 |
25 |
7.9 × 10−6 |
[47] |
| L-ascorbic acid reduced GO-2 |
0.1 |
766 |
5 |
2.5 |
100 |
60 |
3.3 × 10−6 |
[48] |
| S, N-doped CNTs |
0.067 |
298.5 |
3 |
0.0093 |
100 |
10 |
2.2 × 10−3 |
[49] |
| MOF-derived PNC |
1.8 |
95.3 |
100 |
3 |
100 |
10 |
6.0 × 10−3 |
[35] |
| N-doped graphene |
166.67 |
10 |
3 |
2 |
98 |
180 |
1.3 × 10−3 |
[50] |
| Hollow mesoporous carbon spheres |
0.1 |
100 |
2 |
0.09 |
100 |
19 |
5.8 × 10−5 |
[51] |
| Organic capping agents functionalized rGO |
0.0662 |
150 |
3.02 |
0.05 |
100 |
36 |
1.1 × 10−4 |
[52] |
| 3D GQDs/rGO |
0.196 |
53 |
2.55 |
0.048 |
99 |
10 |
1.0 × 10−3 |
[53] |
| Sulfurized Graphene |
0.02 |
100 |
40 |
1 |
100 |
50 |
1.6 × 10−5 |
[34] |
| PNC |
0.08 |
100 |
40 |
10 |
100 |
240 |
1.3 × 10−6 |
[54] |
| 3D N-doped graphene foam |
0.1 |
400 |
2.6 |
0.15 |
100 |
18 |
7.4 × 10−5 |
[39] |
| S, N-doped hollow carbon nanosphere/graphene aerogel |
0.651 |
317.5 |
3.07 |
0.05 |
90 |
8 |
4.5 × 10−3 |
[55] |
| N-doped rGO meshes |
0.0014 |
32,857 |
12.48 |
0.12 |
96 |
8 |
1.7 × 10−5 |
[56] |
| EPOP |
0.36 |
220 |
40 |
3 |
100 |
35 |
1.4 × 10−4 |
[11] |
| N, P-doped multilayer graphene |
20 |
100 |
3 |
1 |
100 |
2.53 |
2.4 × 10−2 |
[42] |
| N-doped multilayer graphene |
20 |
100 |
3 |
1 |
100 |
4.5 |
1.3 × 10−2 |
[42] |
| MOF-derived B, N-doped porous carbon |
0.08 |
625 |
25 |
1 |
94 |
20 |
9.4 × 10−5 |
[36] |
| N-doped GQDs |
0.129 |
100 |
3.1 |
0.02 |
99.5 |
8 |
2.5 × 10−3 |
[57] |
| Porous crimped graphitic carbon nitride |
0.0719 |
183.1 |
50 |
1 |
100 |
8 |
4.5 × 10−4 |
[58] |
| P, N, F-doped rGO |
0.144 |
220.7 |
100 |
1 |
100 |
1.83 |
7.9 × 10−3 |
[40] |
| B, N, F-doped rGO |
0.144 |
220.7 |
100 |
1 |
100 |
2.33 |
6.2 × 10−3 |
[40] |
| 3D N-doped holey graphene |
20 |
100 |
4 |
2.7 |
100 |
0.667 |
3.3 × 10−2 |
[59] |
| Radish derived PNC |
0.1 |
800 |
25 |
1 |
100 |
12 |
1.7 × 10−4 |
[60] |
| Cellulose derived N, P-doped carbon |
0.5 |
100 |
30 |
30 |
80 |
20 |
2.0 × 10−5 |
[61] |
| Lignin derived N-doped carbon |
0.0802 |
400 |
201.6 |
5 |
100 |
0.82 |
3.9 × 10−3 |
[38] |
| B, N-doped hollow mesoporous carbon |
1.8 |
1111 |
100 |
3 |
100 |
3.5 |
8.6 × 10−4 |
[62] |
| Pumpkin-derived PNC |
0.1 |
800 |
25 |
2 |
100 |
8 |
1.3 × 10−4 |
[63] |
| MOF-derived B, N-doped porous carbons |
0.1 |
2500 |
25 |
3 |
95.2 |
20 |
4.0 × 10−5 |
[64] |
| Alga derived PNC |
0.2 |
400 |
100 |
10 |
100 |
15 |
1.3 × 10−4 |
[65] |
| Sewage sludge derived PNC |
0.2 |
200 |
50 |
10 |
100 |
8 |
1.3 × 10−4 |
[37] |
| B, N-doped porous nanocarbon |
0.1 |
400 |
50 |
5 |
99.7 |
9 |
1.1 × 10−4 |
[66] |
| Less edge defect CNFs |
0.025 |
10 |
24 |
40 |
50 |
60 |
5.2 × 10−6 |
[67] |
| N-doped porous rGO |
0.2 |
800 |
3.5 |
0.004 |
100 |
2 |
8.8 × 10−2 |
[41] |
| Eggplant derived PNC |
1 |
200 |
25 |
1 |
100 |
2 |
1.2 × 10−2 |
[68] |
| N-doped fiberboard derived carbon |
0.105 |
396.5 |
190 |
10 |
100 |
10 |
2.0 × 10−4 |
[69] |
| Bamboo pulp derived N, P-doped HPCF |
0.5 |
100 |
30 |
5 |
80 |
5 |
4.8 × 10−4 |
[70] |
| Orange peel derived N-doped carbon |
0.05 |
1000 |
3.5 |
1 |
100 |
5 |
3.0 × 10−5 |
[71] |
2.3. Fundamental Design Principles and Challenges for C-MFCs
2.3.1. Thermodynamic Feasibility and Kinetic Barriers
The reduction of 4-NP to 4-AP in aqueous media is thermodynamically favorable, driven by the significant potential difference between the reducing agent (borohydride) and the substrate. The standard electrode potentials are E
0 = −0.76 V
vs. SHE for the 4-NP/4-AP couple and E
0 = −1.33 V
vs. SHE for the BH
4−/H
3BO
3 couple [
18,
72]. However, the reaction faces substantial kinetic limitations under uncatalyzed conditions. A primary barrier arises from the electrostatic repulsion between the anionic 4-nitrophenolate ion (4-NP
−, formed in alkaline NaBH
4 solution) and the BH
4− anion, creating a high activation energy barrier and rendering the spontaneous reaction extremely slow [
73].
2.3.2. Role of Metal-Free Catalysts
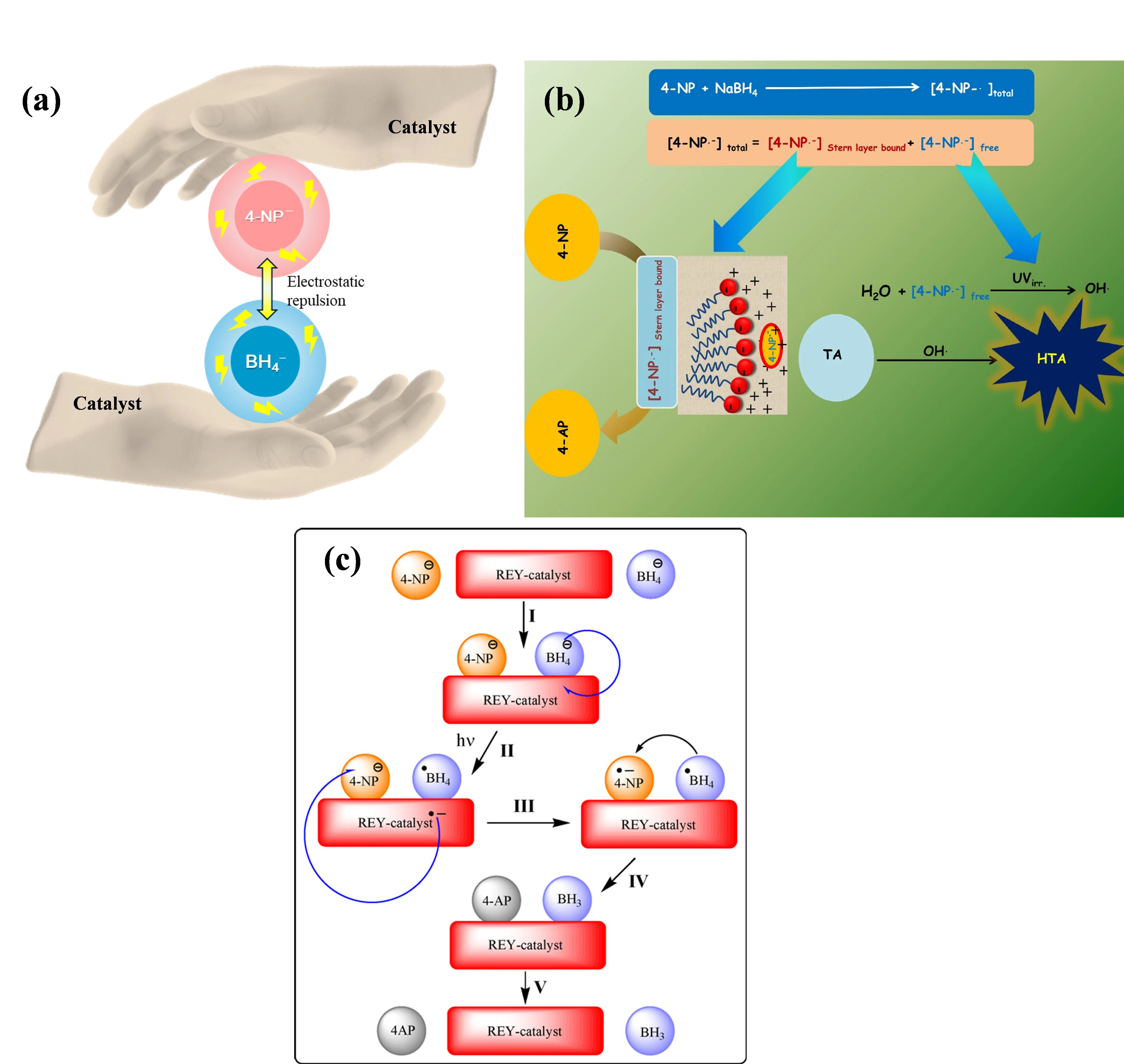
Catalysts overcome these kinetic constraints by providing an alternative reaction pathway with a lower activation energy. They facilitate the close proximity of the reactants and mitigate electrostatic repulsion, effectively lowering the energy barrier for the reduction process (illustrated conceptually in
a). Metal-free catalysts can function in both homogeneous and heterogeneous systems:
Homogeneous Catalysis: Demonstrated in systems like anionic surfactant sodium dodecyl sulfate (SDS) micelles above the critical micelle concentration (cmc). The anionic micelles electrostatically adsorb 4-NP
− within their Stern layer (
b). Here, BH
4− donates electrons, potentially via a Na
+-stabilized anionic radical intermediate (4-NP
·−), enabling multi-step reduction to 4-AP [
74]. While effective, homogeneous catalysts face significant drawbacks: sensitivity to aqueous impurities (e.g., urea, salts) and challenges in separation and recovery post-reaction, limiting their practicality for water treatment applications. Additionally, their well-defined active sites (e.g., dyes) are less relevant to the core focus of this review on carbon defect engineering.
Heterogeneous Catalysis: Some heterogeneous C-MFCs require external energy inputs (e.g., light), as seen with resin-supported dye photocatalysts [
31]. These systems propose mechanisms involving reactant adsorption, electron transfer from BH
4− to the catalyst, photoexcitation, electron transfer to 4-NP
−, protonation, and product desorption (
c). While exhibiting activity and reusability, the need for external energy limits their scope. As summarized in
, major researches predominantly focus on heterogeneous C-MFCs, utilizing materials such as heteroatom-doped graphene, CNTs, GQDs, MOF-derived heteroatom-doped carbons, 3D porous biomass-derived carbons, and 3D graphene architectures. The catalytic activity for nitrophenol reduction fundamentally relies on efficient electron transfer. In current studies on C-MFCs for the catalytic reduction of 4-NP with NaBH
4 in aqueous solution, it is widely accepted that the sp
2-hybridized carbon framework provides the essential conductive pathway and forms the backbone for active site creation. In contrast, sp
3 amorphous carbon domains typically lack intrinsic catalytic activity but play crucial auxiliary roles, such as forming conductive 3D networks that enhance accessibility to the active sp
2 sites. Therefore, our discussion of active sites will focus exclusively on modifications within the sp
2 carbon structures.
2.3.3. The Need for Defect Engineering
Pristine sp
2 carbon exhibits limited adsorption capacity for the anionic 4-NP
− and BH
4− ions. Defect engineering strategies, encompassing heteroatom doping (single, dual, tri-atom), edge defect creation, and pore defect generation, address this fundamental limitation. These strategies modulate the electronic density distribution, strengthen reactant adsorption, accelerate electron transfer kinetics, and thereby activate the otherwise inert sp
2 carbon matrix. The performance is governed by the synergistic optimization of active site density/accessibility through defect engineering, mass transport efficiency via structural design, and bulk electron conductivity inherent to the sp
2 network. Despite promising advances, persistent ambiguities regarding precise reduction mechanisms (e.g., H
2 vs. H
· vs. H
+ pathways) and incomplete structure-activity correlations remain central challenges for the rational design of next-generation C-MFCs. The following sections systematically review the heterogeneous C-MFCs’ active sites and their proposed reaction pathways, categorized according to the defect engineering approaches derived from these design principles.
. (<strong>a</strong>) Schematic representation of electrostatic repulsion between the anionic 4-NP<sup>−</sup> and BH<sub>4</sub><sup>−</sup> ions and the role of catalyst, and (<strong>b</strong>) formation and stabilization of anion radical of 4-NP where the Stern layer-bound anion radical converts to 4-AP, and the free anion radical is responsible for fluorescent HTA formation from non-fluorescent TA [<a href="#B74" class="html-bibr">74</a>]. Reproduced with permission. Copyright 2017, Elsevier Publications. (<strong>c</strong>) Possible reaction mechanism of the present photocatalytic reduction of 4-NP by resin-supported dyes under visible light irradiation [<a href="#B31" class="html-bibr">31</a>]. Reproduced with permission. Copyright 2011, Elsevier Publications.
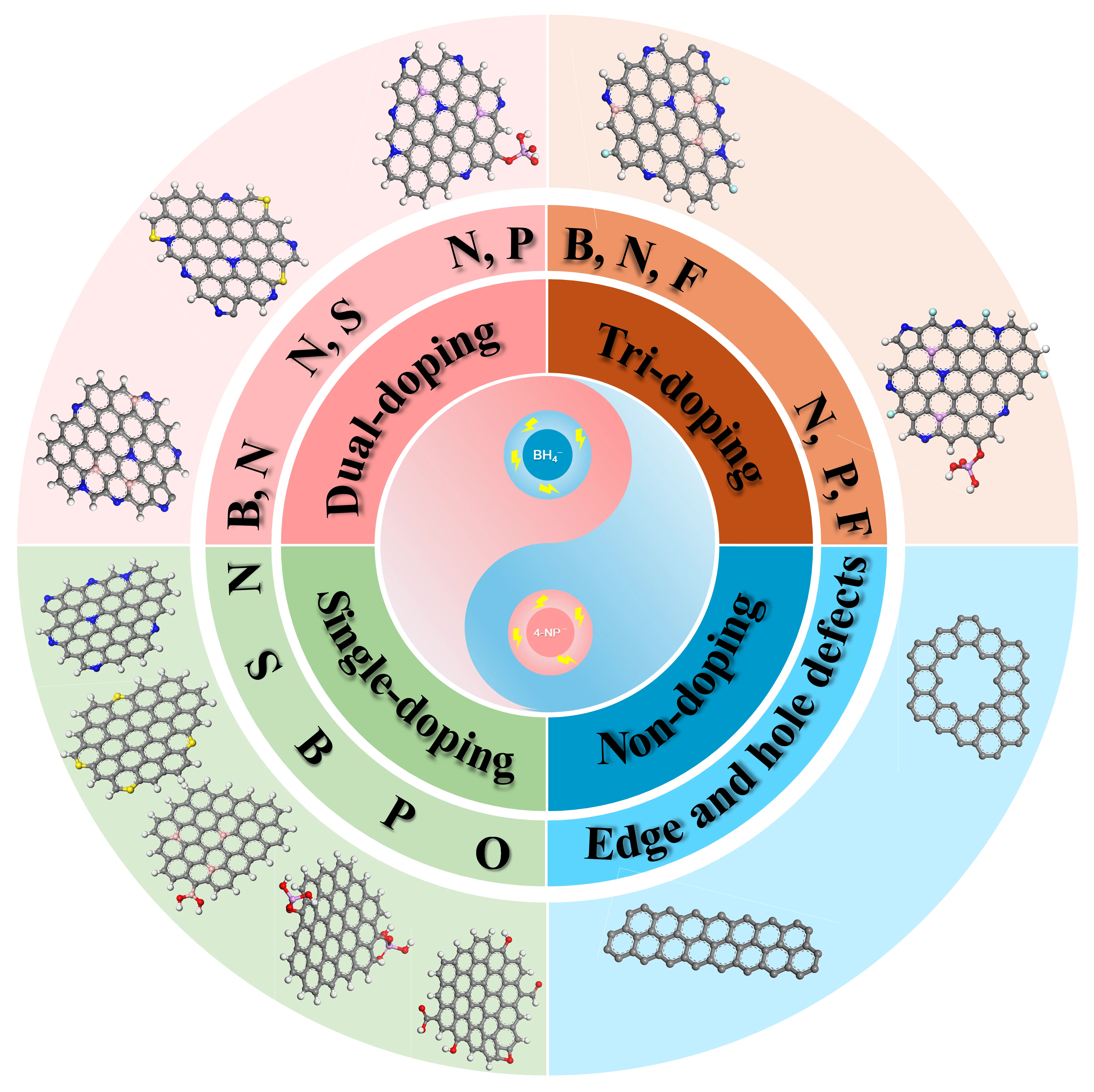
In carbon materials, structural “defects” refer to deviations from an ideal, perfectly periodic atomic arrangement [
75]. While defects can impair intrinsic properties reliant on crystallinity (e.g., the exceptional electrical conductivity of pristine graphene), defect engineering strategically exploits these features to precisely control functionality [
7]. Extensive research on C-MFCs for 4-NP reduction in aqueous NaBH
4 systems unequivocally identifies catalytically active sites localized at defects within sp
2-hybridized carbon frameworks. As systematically classified in
, these defects fall into two primary categories:
- (1)
-
Doped defects: Generated by the intentional introduction of heteroatoms into the sp2-carbon lattice, which modulate electronic properties, charge distribution, and surface reactivity. This review addresses doped defects in the following hierarchical order based on compositional complexity:
Single-atom doping (N, S, B, P, O)
Dual-atom co-doping (e.g., B,N; N,S; N,P)
Tri-atom co-doping (e.g., B,N,F; N,P,F)
- (2)
-
Non-doping defects: Comprise intrinsic structural imperfections without heteroatom incorporation, primarily:
Edge defects (Zigzag/Armchair configurations at sheet peripheries)
Pore defects (Vacancies formed by multi-atom loss, e.g., via etching)
The subsequent sections (3.1–3.4) dissect the formation mechanisms, atomic/electronic structures, and structure-activity relationships governing these defect types, elucidating their distinct roles in activating the sp
2 carbon matrix for efficient nitrophenol reduction.
. Schematic classification framework for defect engineering in carbon-based metal-free catalysts (C-MFCs) applied to nitrophenol reduction. The pink, gray, blue, red, cyan, white, yellow and purpel balls stand for B, C, N, O, F, H, S and P atoms, respectively.
Single-atom doping introduces heteroatoms (N, S, B, P, O) into the sp
2-carbon lattice, creating localized electronic perturbations that activate the inert carbon matrix. The catalytic efficacy is governed by the dopant’s electronegativity, bonding configuration, and resultant charge redistribution, which modulates reactant adsorption and electron transfer kinetics.
3.1.1. Nitrogen Doping
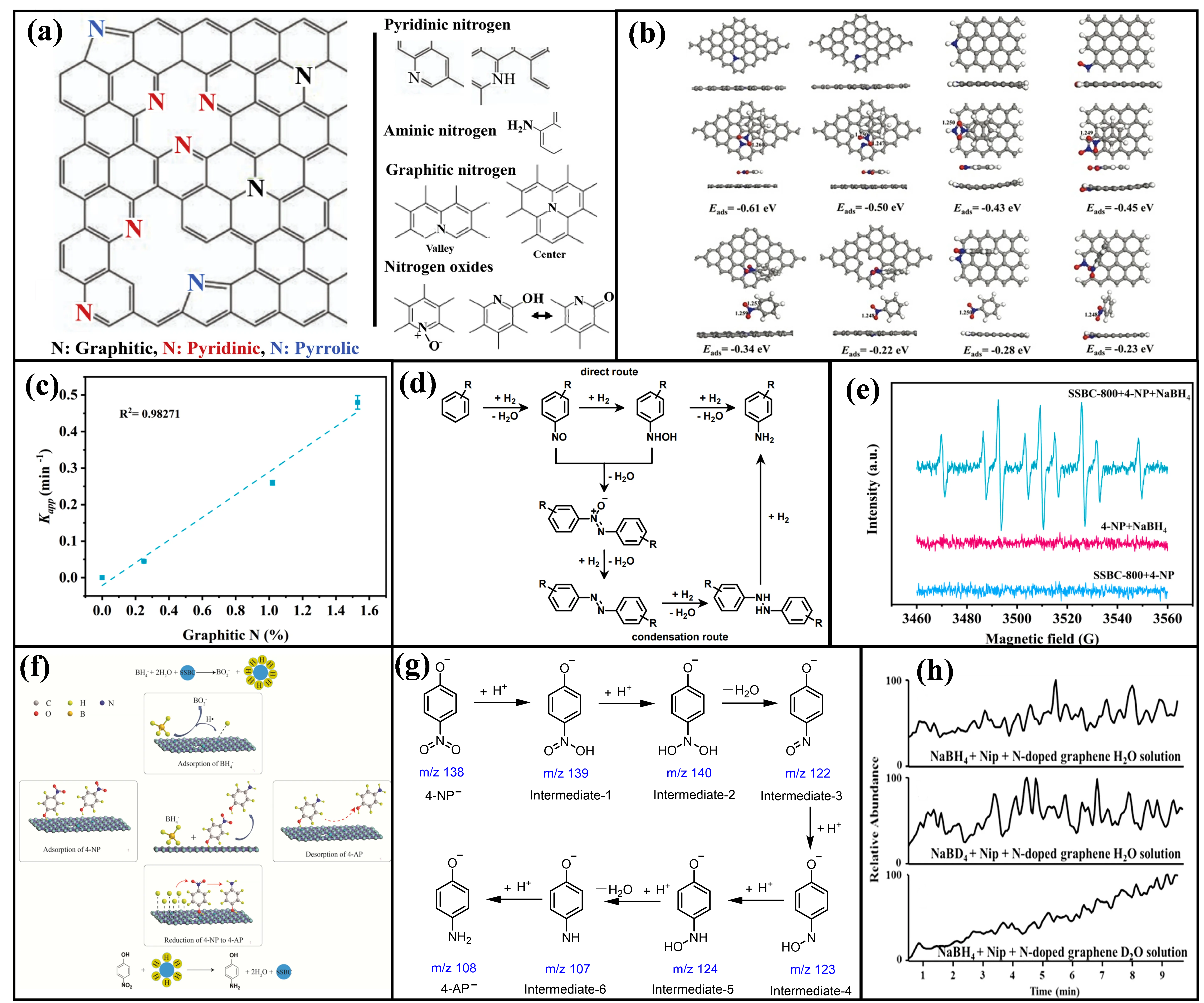
Nitrogen doping, the most extensively studied approach, involves substituting carbon atoms or incorporating nitrogen into defect sites, forming stable chemical bonds. Within the conjugated carbon network, nitrogen doping manifests in three primary configurations depending on the local bonding environment (
a) [
76]: Pyridinic N (N-6): Positioned at the edges of hexagonal rings, this configuration donates one p-electron to the π-conjugated system while retaining one lone electron pair within the ring plane. Pyrrolic N (N-5): Incorporated into five-membered ring structures (e.g., pyrrolic rings), this form contributes two p-electrons to the π-conjugation. Graphitic/ quaternary N (N-Q): This configuration substitutes carbon atoms within the graphene lattice, bonding covalently with three adjacent carbon atoms to form an integral part of the extended sp
2 network [
77,
78].
In pristine sp
2 carbon materials, carbon atoms typically exhibit a slightly negative to neutral charge [
79]. Introducing any nitrogen configuration, possessing higher electronegativity (χ
N = 3.04
vs. χ
C = 2.55), disrupts this equilibrium. The nitrogen dopant induces significant charge redistribution within the lattice, generating localized positive charge centers, particularly on adjacent carbon atoms [
80]. This electrostatic modification facilitates the capture of the negatively charged phenolate oxygen (-O
−) of the 4-nitrophenolate ion (4-NP
−) by these positively charged sites (e.g., ortho-carbon atoms relative to the dopant). Beyond this electrostatic attraction of the phenolate group, density functional theory (DFT) calculations further demonstrate that N-doped porous carbon derived from MOFs can adsorb 4-NP via direct interaction with its nitro group. This adsorption significantly elongates the N-O bond from 1.26 Å to 1.34 Å, thereby weakening the bond and activating the nitro group for subsequent reduction [
35].
Notably, the catalytic activity varies significantly among the different N configurations, with graphitic N exhibiting the highest activity. DFT studies consistently report adsorption energies (E
ads) of 4-NP or 4-NP
− on various N configurations ranging from −0.40 eV to −0.85 eV. Among these, N-Q sites demonstrate the strongest adsorption affinity for nitrophenolate species. This theoretical prediction aligns well with experimental observations, which show a positive correlation between the catalytic activity of N-doped carbon materials and their graphitic N content. For instance, Fan Yang et al. conducted DFT calculations comparing the adsorption structures of nitroaromatics on graphitic N, pyridinic N, pyrrolic N, and pyridinic N-oxide sites (
b) [
50]. Their results revealed that a parallel adsorption configuration on graphitic N was the most stable (exhibiting the lowest E
ads) and resulted in the longest N-O bond within the nitro group. This bond elongation indicates facilitated cleavage, thereby lowering the reaction energy barrier and promoting the reduction reaction. Subsequent catalytic performance testing confirmed that N-doped graphene with the highest graphitic N content indeed exhibited the strongest catalytic activity. Similarly, Xiaoya Ren et al. synthesized N-doped biochar derived from sewage sludge via pyrolysis and modulated its graphitic N content by varying the pyrolysis temperature [
37]. Analysis of the active sites revealed a strong linear correlation (
R2 = 0.98) between the graphitic N content (determined by XPS peak deconvolution) and the apparent rate constant (
Kapp) for the catalytic reduction reaction (
c).
While the catalytic reduction of 4-NP by NaBH
4 in aqueous systems using N-doped C-MFCs appears deceptively simple, the dominant reaction pathway remains a subject of persistent ambiguity. Three distinct mechanisms, categorized by their proposed hydride transfer species, are proposed and elaborated as follows:
- (1)
-
H2-Based Reduction Pathway: Based on GC-MS detection of key intermediates such as hydroxylamine, hydrazine, and azoxybenzene, Fan Yang et al. put forward direct and condensation routes (d) [50]. In the direct route, the sequential reduction process is: nitroso compound → hydroxylamine → 4-AP. The condensation route involves the coupling of a nitroso compound with a hydroxylamine molecule, resulting in an azo compound, which can account for the formation of complex by-products like azo compounds. While this mechanism can explain byproduct formation, critical limitations undermine this model: (i) No experimental evidence confirms H2 involvement. (ii) Control experiments by Kong et al. demonstrated that N-doped graphene cannot catalyze H2 reduction of 4-NP [73]. (iii) The mechanism fails to elucidate how N-sites activate/generate H2 or describe surface-specific processes.
- (2)
-
H·-Based (Hydrogen Radical) Reduction Pathway: Xiaoya Ren et al. utilized 5,5-dimethyl-1-pyrroline N-oxide (DMPO) as an H· trapping agent [37]. When NaBH4 was added, they detected a characteristic signal (triplet of triplets, aH = 22.57 G, aN = 16.62 G) ascribed to the DMPO-H adduct (e). Combining with Langmuir-Hinshelwood kinetics, they proposed the pathway in f. BH4− adsorbs onto the graphitic N active site on the N-doped sp2 carbon surface, forming a surface-bound hydride complex. Simultaneously, 4-NP adsorbs, creating a bimolecular adsorption configuration. The graphitic N site facilitates the dissociation of the B-H bond in BH4−, generating active hydrogen species (H· radicals). These active H· attack the nitro group (-NO2) of 4-NP. Concurrently, an electron transfers from BH4− to the C-MFCs surface (supported by the electrochemical current response), and the C-MFCs act as an electron mediator, transferring electrons to the adsorbed 4-NP molecule. Thus, 4-NP is stepwise reduced to 4-AP, desorbs, and the active site regenerates. This mechanism introduces important concepts such as the “bimolecular adsorption configuration” and the “electron mediator” role, elucidating the microscopic mechanism of N-doped site activation for BH4− dissociation and electron transfer. However, the direct detection of surface reduction intermediates is lacking, leaving the reduction sequence partially unresolved.
. (<strong>a</strong>) Various configurations of N atoms doped in a graphene layer [<a href="#B76" class="html-bibr">76</a>]. Reproduced with permission. Copyright 2018, Elsevier Publications. (<strong>b</strong>) The structures of nitroarenes adsorbed on graphitic N, pyridinic N, pyrrolic N, and N-oxides. The gray, blue, red and white balls stand for C, N, O and H atoms, respectively [<a href="#B50" class="html-bibr">50</a>]. (<strong>c</strong>) Relationship between <em>K<sub>app</sub></em> and graphitic N content [<a href="#B37" class="html-bibr">37</a>]. (<strong>d</strong>) Schematic of H<sub>2</sub>-based reduction pathway [<a href="#B50" class="html-bibr">50</a>]. Reproduced with permission. Copyright 2016, Royal Society of Chemistry. (<strong>e</strong>) EPR spectra of DMPO-H adducts formed in the presence of sewage sludge-derived biochar (SSBC-800) + 4-NP, 4-NP + NaBH<sub>4</sub> and SSBC-800 + 4-NP + NaBH<sub>4</sub> [<a href="#B37" class="html-bibr">37</a>]. Reproduced with permission. Copyright 2021, Elsevier Publications. (<strong>f</strong>) Hydrogen radical reduction pathway. (<strong>g</strong>) H⁺-based reduction pathway [<a href="#B73" class="html-bibr">73</a>]. (<strong>h</strong>) Hydrogen isotope labeling experiment [<a href="#B73" class="html-bibr">73</a>]. Reproduced with permission. Copyright 2017, Elsevier Publications.
- (3)
-
H⁺-Based (Proton) Reduction Pathway: Xiangkai Kong et al. employed paper-assisted ultrasonic spray ionization mass spectrometry (PAUSI-MS) combined with DFT calculations to propose the six-step [73], water assisted pathway shown in g: 4-NP−→monohydroxy intermediate (m/z 139)→dihydroxy intermediate (m/z 140)→4-nitrosophenol (m/z 122)→hydroxylamine intermediate (m/z 123)→intermediate-5 (m/z 124)→intermediate-6 (m/z 107)→4-AP. This pathway involves the release of two water molecules, with no condensation route (no azo intermediates detected). Significantly, using isotopic labeling (D2O and NaBD2, h), they demonstrated that the amine hydrogen in the final 4-AP originates from water molecules (via H⁺), not BH4−. This does not undermine the role of BH₄⁻; they suggest that N-doped sites promote the generation of H− from BH4−. This H− then attacks water to produce reactive H+ species. The innovative PAUSI-MS technique enabled the direct capture of several intermediates, strengthening the evidence for these steps. The isotopic experiments definitively resolved the controversy regarding the amine hydrogen source. However, the specific mechanism by which N-doped sites facilitate the generation of H− from BH4− requires further clarification.
3.1.2. Sulfur Doping
Sulfur doping differs from mere physical sulfur adsorption, fundamentally involves the incorporation of sulfur atoms into the lattice structure of sp
2 carbon-based materials such as graphene and graphitic carbon nitride (g-C
3N
4) [
81]. This process occurs via the substitution of carbon atoms or bonding to edge carbon atoms, thereby altering the inherent electronic structure and surface properties of the material [
82]. In the specific context of catalyzing the reduction of 4-NP using C-MFCs, sulfur doping has been achieved through two primary synthesis routes: ball-milling sulfur powder with chemically modified graphite [
34] and thermal polycondensation using trithiocyanuric acid as a precursor to prepare S-doped g-C
3N
4 [
83].
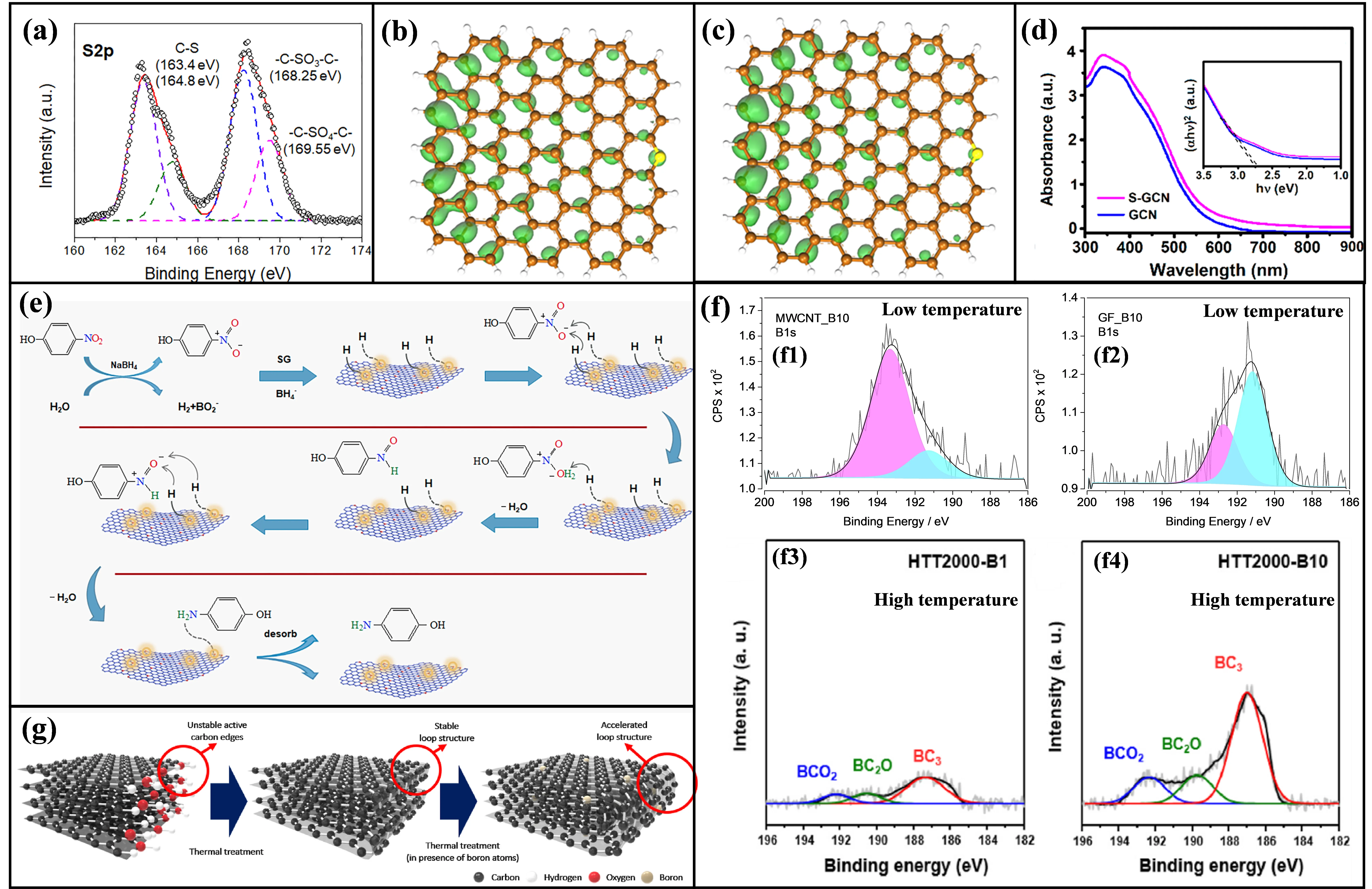
Analysis of chemical states by XPS spectroscopy, as shown in
a, indicates that sulfur within these sp
2 carbon networks primarily exists in two forms: thiophenic sulfur (C-S-C, characterized by bands at 163.4 eV and 164.8 eV) and oxidized sulfur species (e.g., C-SO
3-C or C-SO
4-C, with bands observed between 168–170 eV) [
34,
81,
82]. Critically, the incorporation of sulfur atoms induces significant changes in the electron distribution across the carbon matrix. DFT calculations provide quantitative evidence for this modification, revealing that sulfur doping enhances polarization of the material’s frontier molecular orbitals, specifically the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) (
b,c) [
34]. This polarization results in asymmetric electron density regions centered around the sulfur atoms, an effect originating from the hybridization between sulfur’s 3p orbitals and carbon’s 2p orbitals. Consequently, adjacent carbon atoms acquire increased electron density, making them potential catalytic active sites. For S-doped g-C
3N
4, a key additional benefit is bandgap engineering; where the bandgap narrows from 2.72 eV in the pristine material to approximately 2.66 eV upon sulfur incorporation. This narrowing is attributed to the introduction of mid-gap states (
d), which effectively promote the separation of photogenerated electron-hole pairs, ultimately enhancing the material’s conductivity [
83].
Although the catalytic reaction mechanism for S-doped graphene (denoted as SG) remains less comprehensively studied than for nitrogen doping, a plausible pathway for the NaBH
4 reduction of 4-NP is illustrated schematically in
e [
34]. This mechanism involves the following steps: (1) Hydrolysis of reductant: NaBH
4 undergoes hydrolysis in water to generate BH
4−. (2) Electron transfer and H formation: Electrons transfer from BH
4− o the SG surface, generating active hydrogen species (H
∗). (3) Adsorption of reactant: 4-NP molecules adsorb onto SG, likely facilitated by electron-rich carbon sites adjacent to sulfur atoms (as evidenced by DFT-calculated polarization in
b,c. (4) Stepwise reduction: Adsorbed 4-NP is reduced by H
∗ intermediates via a transient
∗4-hydroxylaminophenol
∗ species, rapidly hydrogenated further to form 4-AP. (5) Desorption and regeneration: The 4-AP product desorbs, regenerating active sites for subsequent catalytic cycles. This desorption step is critical for maintaining catalytic continuity, leveraging the asymmetric electron density induced by S-doping (
b,c) to optimize site accessibility.
. (<strong>a</strong>) High resolution S 2p XPS band of SG [<a href="#B34" class="html-bibr">34</a>]. (<strong>b</strong>) HOMO and (<strong>c</strong>) LUMO of SG [<a href="#B34" class="html-bibr">34</a>]. The brown, yellow, and white balls stand for C, S, and H atoms, respectively. Reproduced with permission. Copyright 2017, ACS Publications. (<strong>d</strong>) UV−vis diffuse-reflectance spectroscopy of sulfur-containing graphitic carbon nitride (S-GCN) and GCN (inset: plots of (αhν)<sup>2</sup> vs hν) [<a href="#B83" class="html-bibr">83</a>]. Reproduced with permission. Copyright 2018, ACS Publications. (<strong>e</strong>) Proposed route for SG catalytic reduction of 4-NP to 4-AP by using NaBH<sub>4</sub> as the reducer [<a href="#B34" class="html-bibr">34</a>]. Reproduced with permission. Copyright 2017, ACS Publications. (<strong>f</strong>) High resolution B 1s XPS band of (<strong>f1</strong>) low temperature B-doped MWCNTs, (<strong>f2</strong>) B-doped graphene fibers [<a href="#B84" class="html-bibr">84</a>], Reproduced with permission. Copyright 2018, WILEY Publications. (<strong>f3</strong>) high temperature B-doped carbon nanofibers (low weight boric acid), (<strong>f4</strong>) high temperature B-doped carbon nanofibers (high weight boric acid) [<a href="#B67" class="html-bibr">67</a>]. (<strong>g</strong>) Schematic diagrams of loop formation in the carbon nanofibers in the presence of boron atoms [<a href="#B67" class="html-bibr">67</a>]. Reproduced with permission. Copyright 2023, Elsevier Publications.
3.1.3. Boron Doping
Boron doping has also been reported to enhance the catalytic activity of carbon materials in the reduction of 4-NP to 4-AP, primarily by the incorporation of boron atoms into the carbon skeleton [
85]. Critically, the manner of boron incorporation and its subsequent impact on edge defects in C-MFCs vary significantly with processing temperatures (low
vs. high). A representative approach developed by Bruno Jarrais et al. utilizes H
3BO
3 as the boron source, with doping achieved through a combined ball-milling and pyrolysis process conducted at moderate temperatures (600–800 °C) [
84]. XPS analysis (
(f1,f2)) confirms that boron primarily exists as boronic functional groups (C-BO
2) and B
2O
3, with characteristic binding energies of 191.2 eV and 193.0 eV, respectively. These species are predominantly anchored at carbon edge sites or defects. Importantly, this low-temperature doping strategy generates B-O-C bonding configurations on the sp² carbon surface, inducing a localized positive charge on ortho-carbon atoms. This modification facilitates the preferential adsorption of the electron-rich nitro group in 4-NP, thereby lowering the reaction energy barrier.
In contrast, Choi et al. demonstrated that at substantially higher temperatures (2000 °C), boron atoms achieve ultrahigh diffusion rates, enabling substitutional doping within the carbon lattice to form BC
3 configurations (identified by a distinct B 1s band at 187 eV;
(f3,f4)) [
67]. Crucially, this doping mechanism drives extensive reconstruction of carbon edge structures. Boron acts as a “ring-closing promoter”, facilitating the conversion of unstable edges into multilayer loops (
g). While this passivation stabilizes the structure by reducing dangling bonds, it concurrently diminishes catalytic activity (TOF: 5.2 × 10
−6 mmol mg
−1 min
−1). Notably, the catalytic function of these isolated boron sites remains poorly understood due to limited mechanistic studies. Consequently, the atomic-scale mechanisms governing boron-doped systems represent significant uncharted territory. Future work employing in situ spectroscopy and microkinetic DFT modeling is essential to elucidate the role of isolated boron centers in catalytic cycles.
3.1.4. Phosphorus Doping
Phosphorus doping emerged as an effective approach to improve the properties of carbon materials. As an element with multiple oxidation states, phosphorus is widely utilized for the doping and modification of carbon materials [
86]. Industrially, phosphoric acid serves dual roles as an activating agent and a porogen during activated carbon production, facilitating phosphorus incorporation into the carbon skeleton. However, its application in C-MFCs for the catalytic reduction of nitrophenols remains relatively underexplored, with most studies focusing on phosphorus co-doping with nitrogen to modulate carbon structure. A representative study by Jarrais et al. incorporated phosphorus via ball-milling and pyrolysis (600–800 °C) [
84], revealing through XPS analysis a dominant P 2p band at 133.9 eV corresponding to phosphate-like moieties (e.g., R-O-PO(OH)
2). Notably, the characteristic band for direct P-C bonding at ~132.6 eV was not detected [
87]. Although phosphorus has lower electronegativity than carbon (χ
P = 2.19
vs. χ
C = 2.55), theoretically enabling charge modulation like N/S/B dopants, its strong affinity for oxygen thermodynamically favors P-O bonds over P-C configurations. Consequently, phosphorus cannot directly tune the carbon host’s electronic properties in the manner of other heteroatoms.
Instead, the phosphate groups (R-O-PO(OH)
2) predominantly serve as Brønsted acid sites. These acidic sites enhance the dissociation of NaBH
4, thereby promoting the generation of active hydrogen species (H
∗). Nevertheless, a key limitation arises from the tendency of these phosphate groups to preferentially adsorb reaction byproducts. This surface adsorption can progressively block active sites, leading to a significant decline in catalytic performance. This propensity for deactivation likely contributes to the limited reports on solely P-doped carbon catalysts for nitrophenol reduction compared to other dopants. Investigations into the synergistic effects of phosphorus co-doping with other heteroatoms will be detailed in the following sections. Strategies to stabilize the phosphate groups or utilize P primarily as a co-dopant might mitigate deactivation issues.
3.1.5. Oxygen Functionality
Oxygen functionality also plays an important role in the enhancement of catalytic activities of carbon materials. Oxygen incorporation into carbon materials is often inevitable due to its presence in the ambient atmosphere [
86]. Oxygen rarely incorporates substitutionally into the graphene lattice under standard conditions, unlike nitrogen, boron, sulfur, or phosphorus. Instead, it exists predominantly as surface functional groups bonded to edge or defect carbon atoms [
24]. The catalytic role of oxygen functional groups in the reduction of 4-NP is complex and varies significantly depending on the specific chemical moiety. Computational studies by Xiang-kai Kong et al. demonstrated that hydroxyl groups (-OH) and alkoxy groups (-OR) generally exert positive effects [
46]. For hydroxyl groups, the electron-withdrawing nature of oxygen induces a localized positive charge on the adjacent ortho-carbon atom, creating an effective adsorption site for the 4-NP
− anion. Furthermore, some studies suggested that -OH sites might also facilitate the capture and activation of NaBH
4 to generate active hydrogen species (H
∗) [
88]. Alkoxy groups, conversely, can disrupt the continuous π-bonding network of the carbon backbone, generating unpaired sp
2 electrons that potentially accelerate electron transfer from NaBH
4 to the adsorbed 4-NP.
Carboxyl groups (-COOH), behave differently. Under basic conditions (typical for NaBH
4 reduction), Carboxyl groups deprotonate to form negatively charged carboxylate groups (-COO
−). These groups repel the similarly charged 4-NP
− anion, significantly hindering reactant adsorption. As experimentally observed by Hu et al. [
47], this repulsion can lead to drastic activity loss; for instance, alkaline treatment of reduced graphene oxide (HGO), enriching surface -COO
− groups, resulted in a ~90% decrease in activity. Additionally, -COO
− groups can act as electron scavengers, competitively consuming electrons intended for reduction, thereby lowering the overall reduction efficiency [
46]. The influence of other oxygen groups is also notable. Carbonyl groups (C=O) in carbon quantum dot photocatalysts can serve as electron acceptors, participating in the separation of photogenerated electron-hole pairs and potentially reacting with NaBH
4 to produce active hydrogen species (e.g., H
·). Conversely, epoxide groups (-O-), located within the graphene basal plane, can create steric hindrance, impeding access of 4-NP to active sites and reducing adsorption energy, as supported by DFT calculations.
3.2. Dual-Atom Doping
Co-doping, the simultaneous incorporation of two heteroatoms into the carbon skeleton, can consistently produce synergistic interactions that surpass the catalytic performance achievable with single-element doping [79,89]. Nevertheless, elucidating the underlying mechanisms remains challenging due to the inherent complexity of these synergistic active sites. The mechanistic interpretations of synergistic site functionality presented below thus represent plausible models derived from experimental and theoretical evidence. Notably, oxygen functionality is ubiquitous in carbon materials due to ambient exposure. Its influence on catalytic reduction in C-MFCs depends critically on both concentration and chemical configuration. Given its variable impact, oxygen is deliberately omitted from systematic discussion in this section, though its role will be explicitly addressed where it demonstrably influences catalytic properties.
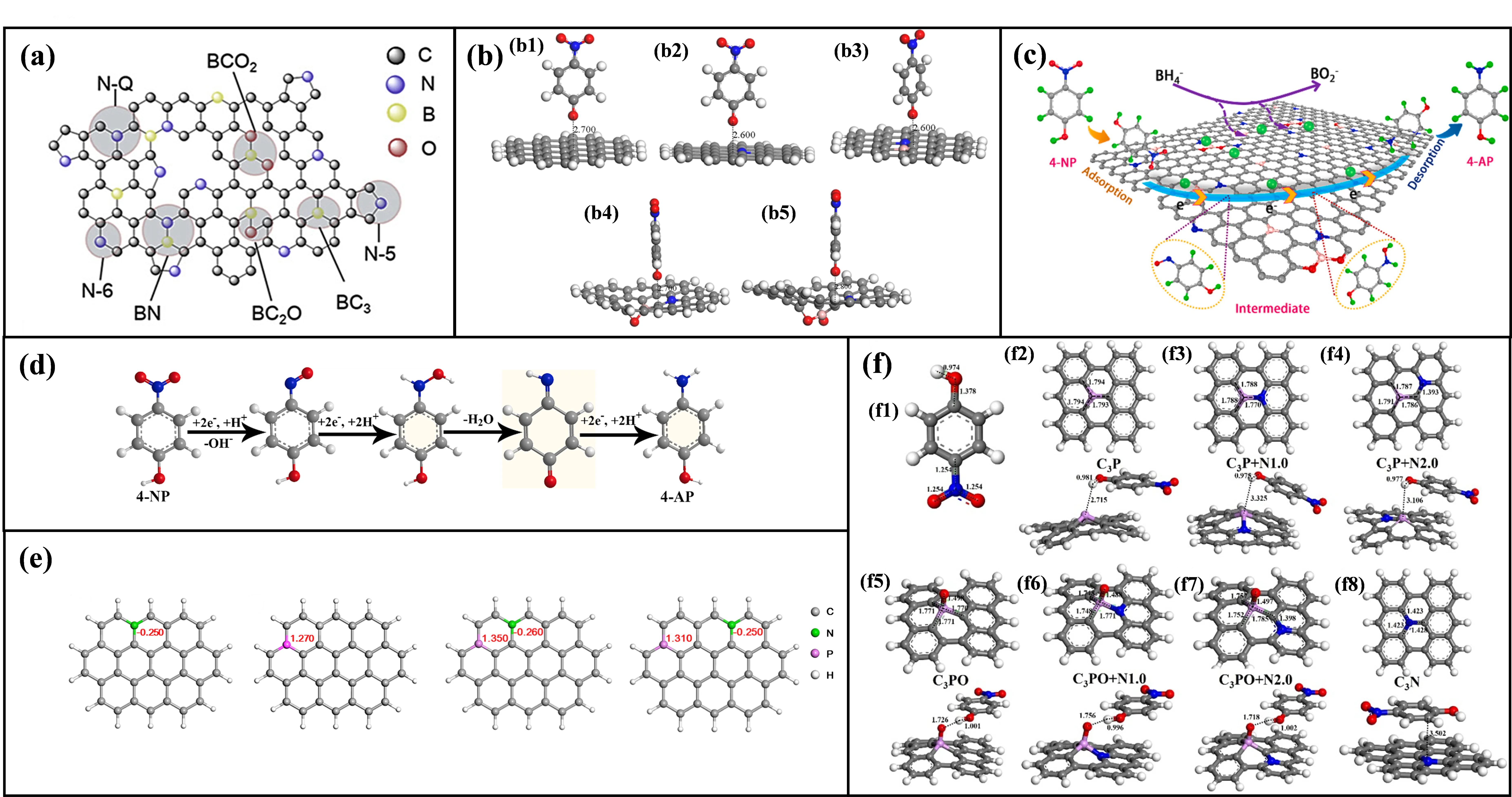
3.2.1. B, N Co-Doping
B, N co-doping is a highly favored strategy for constructing high-performance catalysts due to the comparable atomic sizes of boron and nitrogen to carbon and their potential synergistic effects [
90]. The concurrent incorporation of B (electronegativity χ
B = 2.04) and N (χ
N = 3.04) within the carbon matrix induces significant electronic structure reorganization, substantially enhancing catalytic activity. Specifically, the boron atom, with its lower electronegativity, induces electron deficiency in adjacent carbon atoms, forming Lewis acid sites. Conversely, nitrogen atoms provide localized electron-rich environments, acting as Lewis base sites [
91]. This complementary electronic effect optimizes charge distribution across the catalyst surface. Configurations such as direct B-N bonds (B-N-C) and B-C combined with pyridinic/graphitic nitrogen (
a) synergistically enhance the electron transfer capability of the active sites [
36]. DFT calculations provide compelling support, revealing an exceptionally low adsorption energy (E
ads ≈ −3.95 eV) for 4-NP on specific B-C-N sites (
b), significantly outperforming any single-element-doped counterparts. Furthermore, boron doping concomitantly reduces the material’s electrical resistance, accelerating electron transfer during the catalytic process [
36].
The overall reduction mechanism is illustrated collectively in
c,d [
64,
66]: 4-NP preferentially adsorbs onto the B/N-active sites via the oxygen atom of its -NO
2. Concurrently, NaBH
4 hydrolysis generates active hydrogen species [H] and BO
2− ions [
64]. Electron transfer mediated by the catalyst surface activates the adsorbed 4-NP, reducing its -NO
2 stepwise to a -NO intermediate [
66]. Subsequent hydrogenation by [H] yields hydroxylamine (-NHOH), which is ultimately reduced to the amine (-NH
2) group [
64]. This reaction pathway adheres to the Langmuir-Hinshelwood kinetic model, where the reaction rate exhibits a positive correlation with the surface concentrations of both 4-NP and BH
4− [
36]. Upon completion, the 4-AP product desorbs from the active sites, enabling site regeneration and sustained catalytic cycling.
. (<strong>a</strong>) Schematic illustration of various types of B and N atoms configurations [<a href="#B36" class="html-bibr">36</a>]. Reproduced with permission. Copyright 2019, Elsevier Publications. (<strong>b</strong>) The optimized structures of 4-NP absorbed on various graphene models, (<strong>b1</strong>) pristine graphene, (<strong>b2</strong>) N-Q site of N-co-doped graphene, (<strong>b3</strong>) N-C-B site, (<strong>b4</strong>) N-C-BO sites, (<strong>b5</strong>) N-C-BO<sub>2</sub> site of B and N co-doped graphene [<a href="#B36" class="html-bibr">36</a>]. The gray, blue, pink, red and white balls stand for C, N, B, O and H atoms, respectively. Reproduced with permission. Copyright 2019, Elsevier Publications. (<strong>c</strong>) Proposed reaction mechanism of 4-NP reduction on the B, N-doped C-MFCs [<a href="#B66" class="html-bibr">66</a>]. Reproduced with permission. Copyright 2022, Elsevier Publications. (<strong>d</strong>) The possible catalytic hydrogenation route of 4-NP over B, N-doped C-MFCs [<a href="#B36" class="html-bibr">36</a>]. Reproduced with permission. Copyright 2019, Elsevier Publications. (<strong>e</strong>) Analog structure of N, P-doped graphene along with charge densities [<a href="#B42" class="html-bibr">42</a>]. Reproduced with permission. Copyright 2018, Elsevier Publications. (<strong>f1</strong>) The 4-NP molecular model. The calculated structures of (<strong>f2</strong>) P-doped graphite carbon skeleton, (<strong>f3</strong>) N, P co-doped (in adjacent positions) graphite carbon skeleton, (<strong>f4</strong>) N and P co-doped (separated by one carbon atom) graphite carbon skeleton, (<strong>f5</strong>) O=P-doped graphite carbon skeleton, (<strong>f6</strong>) N and O=P co-doped (in the ortho-position) graphite carbon skeleton, (<strong>f7</strong>) N and O=P co-doped(in the meta-position) graphite carbon skeleton, (<strong>f8</strong>) N-doped graphite carbon skeleton [<a href="#B61" class="html-bibr">61</a>]. The gray, blue, pink, red and white balls stand for C, N, B, O and H atoms, respectively. Reproduced with permission. Copyright 2020, Elsevier Publications.
3.2.2. N, S Co-Doping
Unlike B, N co-doping, N, S co-doping leverages distinct synergistic mechanisms. Firstly, the larger atomic radius of sulfur induces greater distortion within the carbon layers, disrupting electron density equilibrium and generating a higher density of defect sites [
92]. Secondly, the electron-deficient nature of nitrogen combines with the electron-rich character of sulfur, triggering more pronounced charge redistribution within the carbon skeleton and optimizing electron transfer pathways [
49]. Simultaneously, as discussed previously for thiophenic sulfur, its lone-pair electrons stabilize transition states. The sulfur 3p orbitals form weak bonding interactions with BH
4−, facilitating the release of H
−. Furthermore, the p-orbitals of thiophenic sulfur extend the π-conjugated system, enhancing electron delocalization capability. Critically, the electron-withdrawing nature of nitrogen and the electron-donating character of sulfur establish a charge gradient across the material, significantly promoting electron transfer. The synergistic interplay manifests kinetically: The 4-NP molecule adsorbed on the pyridinic N site receives the activated H
− species originating from the sulfur site. This cooperative hydrogen transfer mechanism substantially lowers the reaction energy barrier by 37% (E
a = 24.53 kJ/mol
vs. 39.2 kJ/mol for single-doped counterparts) [
49]. As summarized in
, N, S co-doping C-MFCs exhibits good catalytic activation with a high TOF (2.2 × 10
−3 and 4.5 × 10
−3 mmol mg
−1 min
−1).
3.2.3. N, P Co-Doping
In N, P co-doped C-MFCs, graphitic nitrogen exhibits synergistic interactions with phosphorus incorporated within the carbon skeleton [
70]. Computational analyses reveal that N and P atoms residing within the same hexatomic ring generate a significantly steeper electron density distribution gradient compared to isolated N or P sites (
e) [
42]. Specifically, the charge density on the P atom within this synergistic configuration is calculated at +1.350, contrasting with values of +1.270 or +1.310 for isolated P sites. Correspondingly, the charge density on the N atom in the synergistic structure is −0.260, compared to −0.250 in isolated configurations. This pronounced charge polarization, particularly the strong positive charge center on P, confers exceptionally strong adsorption affinity for the 4-NP
− anion. This N, P co-doping C-MFCs show a super catalytic performance (TOF = 2.4 × 10
−2 mmol mg
−1 min
−1).
Furthermore, a study suggested potential synergy between graphitic N and phosphorus-oxygen functionalities like C
3PO motifs. As depicted in
f, a co-doping site combining a phosphorous oxide group (C
3PO) with an adjacent nitrogen atom (denoted C
3PO + N2.0) exhibits the strongest adsorption energy (E
ads = −0.925 eV) for 4-NP, significantly outperforming single-doped sites [
61]. However, a critical limitation of this particular DFT study warrants attention: the calculations modeled neutral 4-NP molecules, neglecting the anionic form (4-NP
−) prevalent under typical catalytic conditions (basic NaBH
4 solution). Moreover, the computed adsorption energy primarily reflected interaction with the phenolic hydrogen atom of 4-NP, which may not accurately reflect the dominant adsorption mode involving the nitro group observed experimentally. Consequently, the general applicability and validity of this specific conclusion regarding C
3PO + N synergy require further validation through calculations employing more realistic models (
i.e., 4-NP
− adsorbing via its nitro group).
3.3. Tri-Atom Doping
Building on the synergistic effects of dual-atom doping, tri-atom doping further optimizes the electronic structure and surface reactivity of C-MFCs by introducing three distinct heteroatoms. This strategy leverages the complementary electronegativities and bonding characteristics of multiple dopants to induce more intricate charge redistribution within the sp
2 carbon framework, thereby creating a richer landscape of active sites for nitrophenol reduction. Compared to single- or dual-atom doping, tri-atom doping not only strengthens the adsorption of reactants (e.g., 4-nitrophenolate anions and borohydride ions) but also accelerates electron transfer kinetics through cascaded electronic interactions between dopants. Additionally, the introduction of three heteroatoms often facilitates the formation of hierarchical porous structures, which enhance mass transport and active site accessibility—critical factors for boosting catalytic efficiency in aqueous reduction reactions. These combined effects make tri-atom-doped C-MFCs promising candidates for achieving superior catalytic performance in nitrophenol remediation.
The outstanding catalytic performance of three-dimensional tri-doped B, N, F-rGO and P, N, F-rGO materials, synthesized via pyrolysis of 1-butyl-3-methylimidazolium ionic liquids (BMIM-BF
4/BMIM-PF
6) and exhibiting high turnover frequencies (TOF > 1.0 × 10
−3 mmol mg
−1 min
−1), is attributed to a combination of synergistic electronic effects and structural advantages [
40]. Primarily, the tri-doping strategy induces pronounced charge redistribution within the carbon framework. Specifically, the incorporation of highly electronegative nitrogen and fluorine atoms generates partial positive charges on adjacent sp
2-hybridized carbon atoms. Conversely, the co-doping of boron or phosphorus, elements possessing lower electronegativity than carbon, results in the dopant atoms carrying a significant positive charge density; this electronic configuration markedly enhances the activation of neighbouring carbon sites. Collectively, this complementary charge modulation synergistically activates the sp
2 carbon skeleton, thereby facilitating the efficient transfer of active hydrogen species critical for the reduction reaction.
Additionally, the exceptional performance stems notably from the unique three-dimensional porous architecture inherently derived from the ionic liquid precursor synthesis route. This interconnected 3D network provides substantial benefits: it offers a high surface area exposing abundant catalytically active sites and significantly enhances mass transport of reactants and products throughout the catalyst matrix. Ultimately, the confluence of optimized electronic structure through multi-heteroatom synergy and the favorable mass-transport properties of the tailored 3D morphology collectively contribute to the observed high catalytic efficiency.
3.4. Non-Doping Defects
Non-doping defects, encompassing edge defects and pore defects, represent intrinsic structural imperfections in sp
2 carbon frameworks that do not involve heteroatom incorporation. These defects arise from deviations from the ideal hexagonal lattice, such as unsaturated carbon atoms at sheet peripheries or vacancies formed by multi-atom loss, and play a pivotal role in activating carbon materials for nitrophenol reduction.
3.4.1. Edge Defects
Edge defects in carbon materials generate abundant unpaired π electrons at their peripheries, significantly accelerating electron transfer kinetics and lowering the formation energy of key reaction intermediates. Concurrently, edge carbon atoms exhibit higher charge density and dangling bonds, serving as active catalytic sites [
93]. Edges within hexagonal carbon networks can be classified into zigzag and armchair configurations [
75]. Within the field of nitrophenol reduction catalysis, numerous studies have demonstrated the catalytic activity inherent to edge defects in sp
2 carbon networks. For instance, professor Xinhe Bao et al. reported that rGO rich in edge defects effectively catalyzed the reduction of nitrobenzene using hydrazine hydrate, achieving performance comparable to some metal catalysts [
94]. Similarly, Huawen Hu et al. found that rGO films enriched with edge defects catalyzed the NaBH
4 reduction of 4-NP [
47,
48]. Jiali Zhang et al. further utilized carbon quantum dots rich in edge defects as active sites, assembling them with rGO into a three-dimensional network exhibiting high catalytic activity for 4-NP reduction [
53]. Notably, Go Bong Choi et al. observed that passivation of graphene edges leads to a marked decline in catalytic reduction performance [
67].
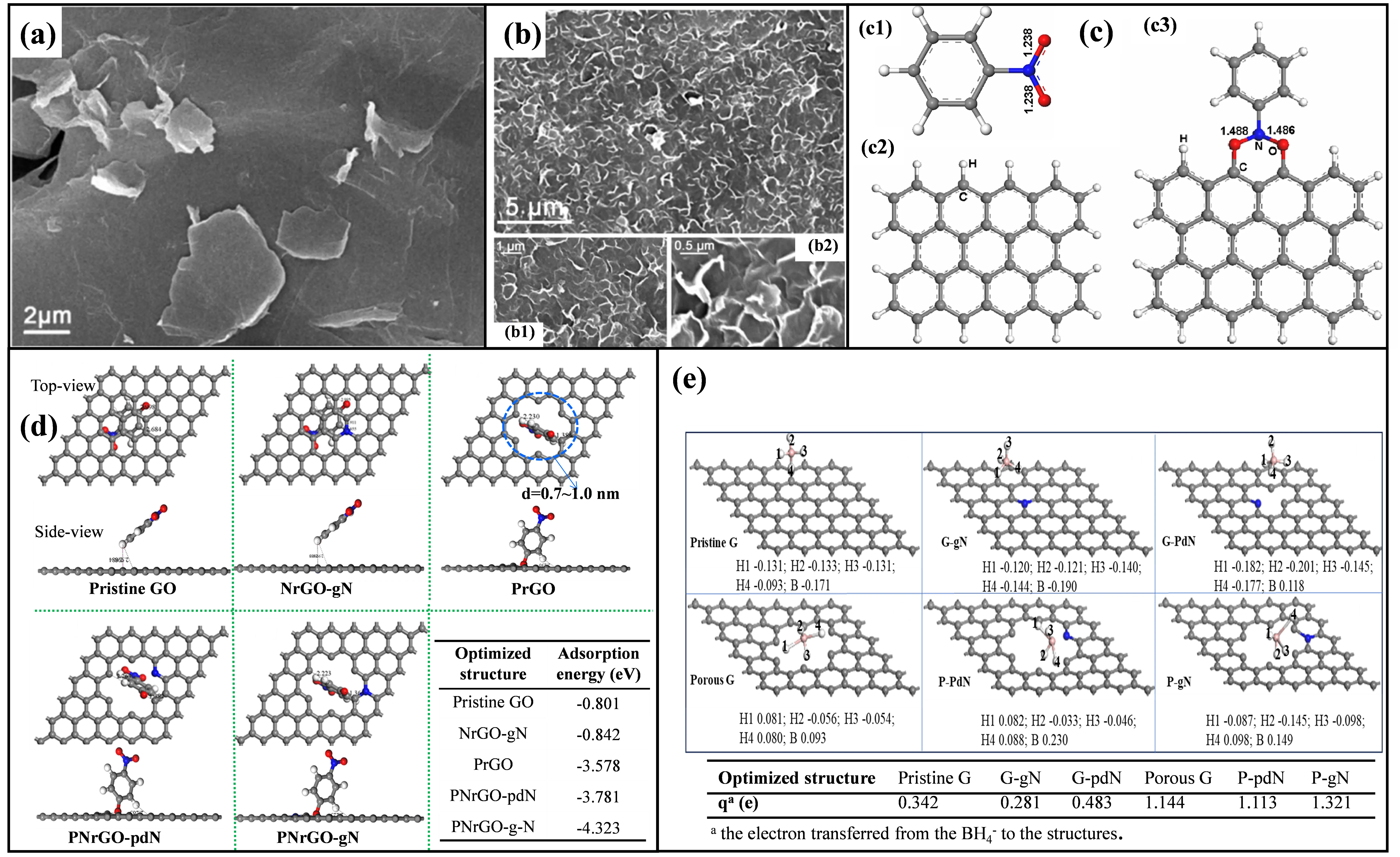
The formation of these edge defects is illustrated in
a,b, showing SEM images of rGO films with and without edge defects [
48]. This defect generation likely originates from the protonation of abundant oxygen-containing functional groups located at GO edges during the reduction process. The resulting electrostatic repulsion induces severe bending of the sp
2 planes. Subsequent reduction and removal of these oxygen groups leaves behind structurally unstable edge defects. Crucially, these defects often lack protective terminal hydrogen atoms, exposing highly reactive dangling bonds. These dangling bonds exhibit strong adsorption affinity for nitrophenols and facilitate activation of the N=O bonds. DFT simulations substantiate this activation mechanism (
c) [
94]. The N=O bond length, measuring 1.238 Å prior to adsorption (
c1), elongates significantly to 1.488 Å upon interaction with a monolayer graphene edge defect. Even adsorption onto multi-layer graphene defects results in substantial bond elongation exceeding 1.47 Å. Analogously, the nitro group of the 4-NP
− anion can be effectively adsorbed onto edge defects, priming it for subsequent reduction.
. SEM images of rGO films (<strong>a</strong>) without and (<strong>b</strong>) with edge defects; (<strong>b1</strong>,<strong>b2</strong>) high resolution image of Figure (<strong>b</strong>) [<a href="#B48" class="html-bibr">48</a>]. Reproduced with permission. Copyright 2015, Royal Society of Chemistry. (<strong>c</strong>) DFT simulations for the adsorption of NP on the edge defects [<a href="#B94" class="html-bibr">94</a>]. Reproduced with permission. Copyright 2011, Royal Society of Chemistry. (<strong>c1</strong>) The optimized structure of the nitrobenzene molecule. (<strong>c2</strong>) Structure of defect free one-layer graphene. (<strong>c3</strong>) The optimized structure of the nitrobenzene molecule reacting with one-layer graphene through two C-O bonds. (<strong>d</strong>) The top view (upper) side view (under) of the adsorption configuration of 4-NP adsorption on different catalyst models: Pristine GO, NrGO-gN, PrGO, PNrGO-pdN, PNrGO-gN. gN: graphitic-N; pdN: pyridinic-N. Table: the adsorption energies of different catalyst-4-NP systems. The gray, blue, red, and white balls stand for C, N, O and H atoms, respectively [<a href="#B41" class="html-bibr">41</a>]. Reproduced with permission. Copyright 2023, Elsevier Publications. (<strong>e</strong>) Charge distributions of pristine graphene, graphitic-N graphene, pyridinic-N graphene, porous graphene, porous pyridinic-N graphene, and porous graphitic-N graphene after BH<sub>4</sub><sup>−</sup> adsorbed on the surface [<a href="#B68" class="html-bibr">68</a>]. Reproduced with permission. Copyright 2024, Elsevier Publications.
3.4.2. Pore Defects
Pore defects form through the loss of multiple carbon atoms within the sp
2 network, typically achieved via chemical etching [
7]. Functionally similar to edge defects, pore defects leverage the catalytic activity of exposed dangling bonds. For example, Wang et al. synthesized porous rGO (PrGO) rich in pore defects through KOH-assisted thermal etching of GO, yielding a material with high catalytic activity for 4-NP reduction (TOF = 8.8 × 10
−2 mmol mg
−1 min
−1) [
41]. As demonstrated by DFT calculations in
d, 4-NP
− adsorbs onto the pore defect with a high adsorption energy (E
ads = −3.578 eV). Further investigations revealed a proposed synergistic effect between pore defects and pyridinic/graphitic nitrogen dopants, enhancing adsorption of both 4-NP
− and BH
4− species while promoting electron transfer efficiency (
e) [
68]. It should be noted that single-atom vacancies (point defects or stone-Wales defects) typically exhibit limited direct catalytic activity due to significant steric hindrance impeding efficient interaction with reactants. Creating larger, well-defined pore structures (meso/macropores) appears more effective than point defects for generating catalytically active sites.
While the intrinsic activity of defect-engineered sites governs the fundamental catalytic potential of C-MFCs, the practical efficacy of these materials is critically dependent on their macroscopic architecture. Two-dimensional (2D) sp
2 carbon materials, such as graphene or carbon nanotube sheets, inherently suffer from strong interlayer van der Waals forces, leading to detrimental restacking. This aggregation drastically reduces accessible active sites, impedes mass transport of reactants (4-NP
− and BH
4−) and products (4-AP), complicates catalyst recovery, and diminishes reusability [
95,
96,
97]. To overcome these limitations, transforming 2D active sites into three-dimensional (3D) architectures has emerged as an essential engineering strategy [
98]. These 3D frameworks mitigate stacking through physical separation of carbon domains, create hierarchical pore networks for efficient diffusion, and provide structural robustness for repeated catalytic cycles. Two complementary synthetic paradigms—bottom-up assembly and top-down reconstruction—enable the rational design of architectures [
29], each offering distinct pathways to integrate defect engineering with morphological control.
4.1. Bottom-up Assembly
The bottom-up strategy utilizes nanoscale carbon-based units (e.g., graphene quantum dots, carbon nanotubes, nanosheets) as building blocks, achieving controllable assembly through intermolecular forces (e.g., π-π stacking, hydrogen bonding) or chemical bonding to form 3D networks with well-defined porous structures.
4.1.1. Quantum Dot/Nanosheet Synergistic Assembly
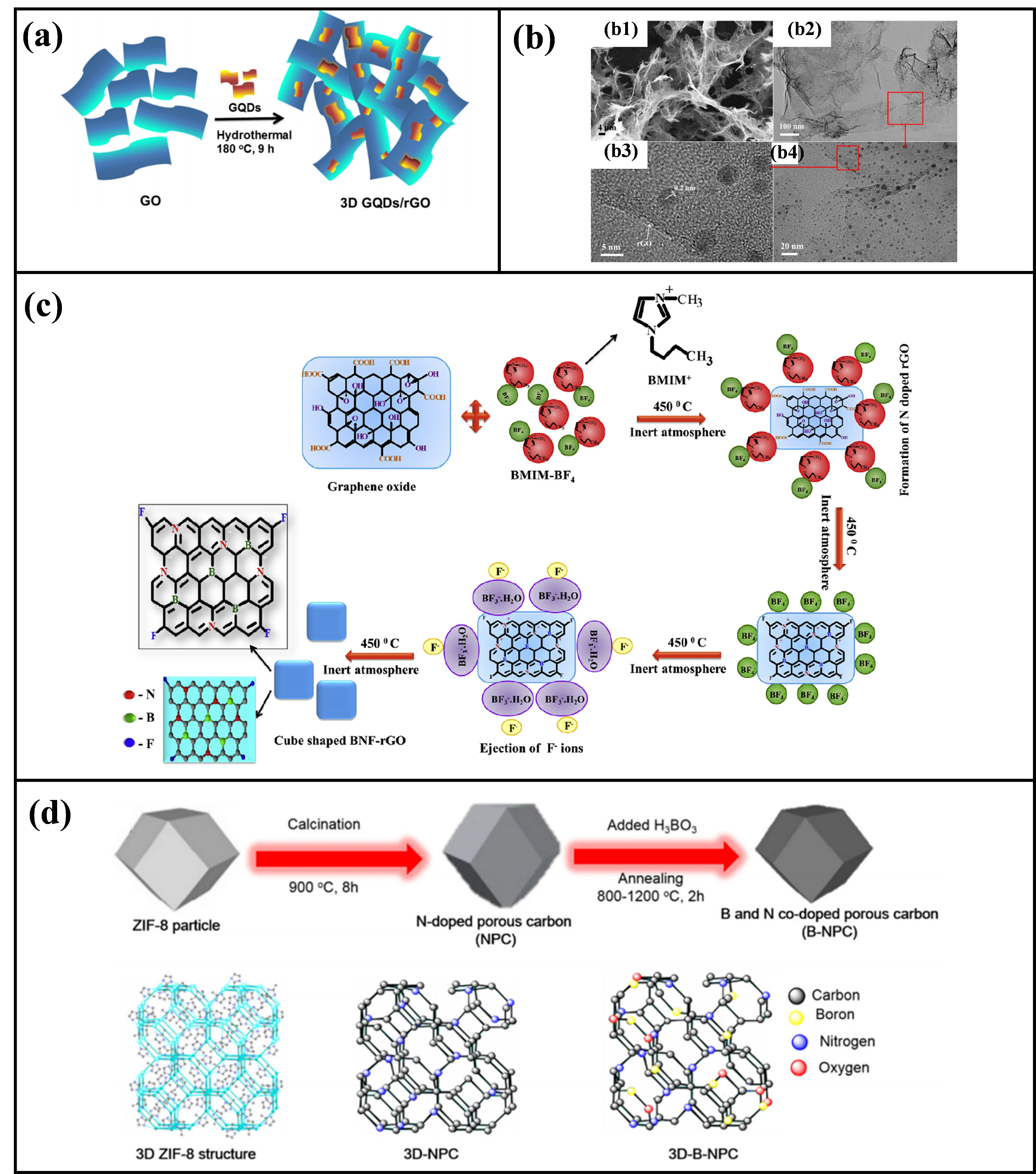
Taking the assembly of graphene quantum dots (GQDs) and reduced graphene oxide (rGO) as an example, GQDs are rich in edge defects and can be uniformly dispersed between rGO layers via π-π interactions. This not only prevents rGO stacking but also acts as “molecular bridges” to facilitate electron transfer. For instance, Zhang et al. assembled GQDs and rGO into a 3D network via hydrothermal method, yielding a robust 3D GQDs/rGO composite (
a,b) [
53], resulting in a TOF of 1.0 × 10
−3 mmol mg
−1 min
−1 for 4-NP reduction, which is 1.7 times that of undispersed GQDs.
. (<strong>a</strong>) Illustration of the formation process of the 3D GQDs/rGO composites. (<strong>b</strong>) SEM (<strong>b1</strong>), TEM (<strong>b2</strong>,<strong>b3</strong>), and HRTEM (<strong>b4</strong>) images of the 3D GQDs/rGO composites [<a href="#B53" class="html-bibr">53</a>]. CC license permits reuse (<strong>c</strong>) Schematic representation of a mechanism for the formation of 3D nanocube shaped BNF-rGO by BMIM-BF<sub>4</sub> ionic liquid [<a href="#B40" class="html-bibr">40</a>]. Reproduced with permission. Copyright 2019, Elsevier Publications. (<strong>d</strong>) Synthesis illustration of ZIF-8-derived 3D B and N co-doped porous carbons (3D-BNPCs) [<a href="#B36" class="html-bibr">36</a>]. Reproduced with permission. Copyright 2019, Elsevier Publications.
4.1.2. Ionic Liquid-Assisted Templating
Ionic liquids (ILs) serve dual roles as solvents and structure-directing agents: their cations can adsorb onto the surface of carbon precursors (e.g., graphene oxide) via electrostatic interactions, while anions regulate the assembly process through hydrogen bonding. Murugesan et al. used 1-butyl-3-methylimidazolium tetrafluoroborate (BMIM-BF
4) as a template to assemble graphene oxide into 3D nanocube structures, which were carbonized to form B, N, F co-doped porous carbons [
40]. Here, the BMIM⁺ cations adsorbed onto the -COO
− groups located at the GO sheet edges, while BF
4− anions formed hydrogen bonds with epoxy/hydroxyl groups. Freezing this mixture at −10 °C created an ice template, and removing the water content resulted in a porous precursor with a locked-in 3D framework. During subsequent heating, hydrolysis products of BF
4− (e.g., [BF
2(OH)
2]
−) coordinated with GO epoxide groups, guiding the carbon atoms towards tetrahedral growth and ultimately forming 60–80 nm cubes (
c). This material demonstrated outstanding catalytic activity for 4-NP reduction, achieving a TOF of 6.17 × 10
−3 mmol mg
−1 min
−1.
4.1.3. Sol-Gel Method Combined with Ice Templating
Gel formation via sol-gel process using carbon precursors (e.g., phenolic resins, biomass derivatives) and cross-linking agents (e.g., formaldehyde), combined with ice templating, can construct 3D structures with interconnected macropores and mesopores. During freezing, ice crystals act as templates, leaving micrometer-scale macroporous channels after sublimation, while the gel network itself generates mesopores during pyrolysis—ultimately forming a hierarchical system of “macroporous transport-mesoporous mass transfer-microporous reaction”. For example, Liu et al. used sodium alginate as a precursor to three-dimensional nitrogen-doped graphene foam (3D-NGF) via freeze-drying and ammonia activation [
39]. The GO foam was heated at 750 °C for 2 h in a mixture of 50% Ar and 50% NH
3 gas to dope nitrogen into sp
2 carbon lattices. The macropores (5–50 μm) accelerate solution convection, and mesopores (2–50 nm) shorten diffusion paths, enabling 98% conversion of 4-NP even at high concentrations (1.0 mmol/L) with no significant activity loss after 10 cycles. This 3D-NGF showed a good catalytic performance with an activation energy 44.3 kJ mol
−1.
4.2. Top-down Reconstruction
The top-down strategy utilizes natural biomass (e.g., wood, straw) or artificial macroscopic materials (e.g., MOFs) as skeletons, retaining their inherent 3D structures through chemical etching and high-temperature carbonization while introducing defective active sites, offering advantages of raw material sustainability and structural stability [
99].
4.2.1. Biomass-Derived 3D Carbons
Biomass (e.g., wood, pumpkin, radish) naturally has a hierarchical porous structure, which can be converted into 3D carbon skeletons after carbonization. Its abundant O and N elements can in-situ dope to form active sites. For instance, Wang et al. used pumpkin as a precursor to prepare N-doped 3D carbon via K
2CO
3 activation and carbonization at 800 °C [
63]. This structure retains the macroporous vascular bundles of pumpkin, and micropores (0.8–2 nm) generated during activation provide abundant adsorption sites. The reduction rate constant of 4-NP reaches 0.568 min
−1. Yang et al. employed eggplant as a carbon source to fabricate 3D N-doped porous carbon using KOH/urea as the pore generator and nitrogen source, exhibiting exceptional 4-NP reduction performance (TOF = 1.23 × 10
−2 mmol mg
−1 min
−1) [
68].
4.2.2. MOF-Derived 3D Porous Carbons
MOFs can be converted into 3D porous carbons via high-temperature carbonization and activation, with their morphology and porous structures adjustable by MOF types (e.g., ZIF-8, HKUST-1) and activators. For example, Van Nguyen et al. used ZIF-8 as a precursor to obtain B, N co-doped 3D porous carbons via boric acid impregnation and high-temperature carbonization and activation (
d) [
36]. This structure inherits the rhombic dodecahedral morphology of ZIF-8, while forming a hierarchical structure of mesopores (5.6 nm) and micropores (0.9 nm). The synergistic doping of B and N induces local positive charge distribution in the carbon framework, enhancing adsorption capacity for 4-NP (adsorption energy: -3.95 eV), with catalytic activity (TOF = 9.4 × 10
−5 mmol mg
−1 min
−1) superior to single N-doped carbons.
4.3. Mechanisms of Catalytic Performance Enhancement by 3D Structures
3D structures enhance the catalytic performance of C-MFCs through multi-dimensional synergistic effects: (a) Active site exposure: Inhibiting layer stacking increases the exposure rate of defect sites, directly increasing the number of reactive centers; (b) Mass transfer optimization: Hierarchical pores (macropores-mesopores-micropores) shorten reactant diffusion distances, enabling 4-NP to maintain rapid mass transfer even at high concentrations (>1 mmol L
−1) and avoiding concentration polarization; (c) Enhanced electron conduction: 3D network structures reduce electron transport resistance; especially with synergistic heteroatom doping, accelerating electron transfer from BH
4− to nitro groups; (d) Stability improvement: The macroscopic framework reduces catalyst loss due to abrasion in stirring or flow systems, extending the cyclic service life to over 10 cycles—far exceeding that of 2D powder materials.
In summary, the construction of 3D architectures is a critical bridge connecting defect engineering and practical applications. Through precise bottom-up assembly and efficient top-down reconstruction, synergistic optimization of active site exposure, mass transfer efficiency, and mechanical stability can be achieved, providing core material support for the large-scale application of C-MFCs in nitrophenol pollution remediation. Future research should further combine in-situ characterization techniques (e.g., in-situ X-ray diffraction, Raman spectroscopy) to reveal the dynamic evolution of active sites in 3D structures, guiding the rational design of higher-performance catalysts.
The work was financially supported by the Fund of National Natural Science Foundation of China (No. 52063016, No. 52372048, No. 52202336), the Fund of Key Laboratory of Advanced Materials of Ministry of Education (No. Advmat-2421).
Investigation, Software, Writing—Original Draft Preparation, X.Y.; Resources, Q.L.; Validation, Conceptualization, Funding, L.W.; Investigation, Funding, Formal analysis, B.Z.; Funding, Formal analysis, Z.W. Supervision, Project administration, Funding, Z.H.; Writing—review & editing, Project administration, Conceptualization, M.W.
Not applicable.
Not applicable.
Relevant information and dates can be made available upon request.
We express our thanks for funding support from the National Natural Science Foundation of China (No. 52063016, No. 52372048, No. 52202336), the Key Laboratory of Advanced Materials of Ministry of Education (No. Advmat-2421).
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.