1. Introduction
Proteomic analysis is a powerful tool that can provide a systematic understanding of molecular level events of filamentous fungi [
1]. Proteomics includes identification, quantification, localization, modification, interactions, and activities of proteins [
2]. Proteomic analysis of several filamentous fungi genera such as
Aspergilli,
Botrytis,
Neurospora,
Penicillium,
Phanerochaete,
Pleurotus,
Trichoderma,
Lentinus and
Mortierella has already been done [
1]. Concerning functional foods and nutraceuticals studies proteomic analysis has already been conducted about food quality and safety, authentication, microbial toxins and toxins in fermented foods, and food allergens [
2,
3,
4,
5,
6,
7]. The effect of carbon sources on different fungi and the proteomic analysis to estimate at a molecular level what triggers the different production of bioactive compounds, proteins such as enzymes as well as secondary metabolites is very crucial. Comparative proteomic analysis for
Trichoderma reesei grown on different carbon sources offers an understanding of the carbon-dependent induction of extracellular enzymes but also suggests a potential target for metabolic engineering of
T. reesei for cellulase production [
8]. Carbon source-dependent growth effect analysis through proteomics has already been studied in bacteria. The effect of glucose and xylose on butanol production by Clostridium acetobutylicum has revealed a cluster of 22 differential expressed proteins between the two substrates [
9]. Differential expression of proteins related to pyruvate metabolism was observed for Bifidobacterium longum NCC2705 grown on different carbon sources including glucose, fructose, mannose, xylose, ribose, and galactose [
10].
Pleurotus ostreatus or oyster mushroom is a well-studied edible basidiomycota that constitutes a source of protein and polysaccharides for the human diet and is characterized by their high rate of moisture and low calories. Also known as white rot fungi due to the white mycelium that produce and the capability to grow in lignocellulosic substrates [
11]. Submerged cultivation of
P. ostreatus has been used to enhance the production of enzymes, proteins, polysaccharides and lipids in a safer environment in contrast to the traditional cultivation of mushrooms [
12]. In addition, single-cell protein production after optimization of cultivation conditions and using response surface methodology to find the optimum carbon and nitrogen source concentration using a cellulosic hydrolysate obtained after enzymatic hydrolysis of fibre sludge was achieved [
13]. The strain was found to be capable of growing and producing high-value biomass with a rich amino acid profile in a mixture of glucose and xylose and an aspen wood chip hydrolysate composed mostly of glucose and partially of xylose [
14]. Also, the same strain has been used for biomass and laccase production using wine lees as substrate, a wine industry side stream with high phenolic content [
15].
P. ostreatus LGAM 1123 has already been used for polysaccharide production with a high total glucan content using olive mill wastewater [
16].
Lignocellulosic substrates such as lignin, xylan and carboxymethyl cellulose have already been used to characterize the mechanism underlying the response of
P. ostreatus to lignin. Lignin was found to downregulate carbohydrate and energy metabolism whereas xylan and CMC enhanced the carbohydrate metabolism process [
17]. The different expression of bioactive compounds such as amino acid and dipeptides through LC/MS Q-TOF for
P. ostreatus mycelia grown on black poplar wood logs and lignocellulosic byproducts has shown that black poplar wood logs increased the expression of carnitine-based amino acid derivatives and Pro-based dipeptides [
18]. Furthermore, secretome analysis of
P. ostreatus grown on poplar wood, wheat straw and glucose revealed that oxidoreductases were strongly induced on woody and nonwoody substrates, with laccase being the most abundant [
19]. A study on the effect of heat stress on the metabolic response of
P. ostreatus has shown that heat stress enhanced nucleotide and unsaturated fatty acids degradation increased the content of amino acids and vitamins and triggered glycolysis and tricarboxylic acid cycle (TCA) [
20]. Concerning fruiting body production, proteomic analysis contributes to understanding the primordium differentiation in the early development of the fruiting body and the role of Ca
2+ and highlights the differences between the Cap and Stipe development [
21]. Also, the mechanism of CO
2-induced morphological change in
P. ostreatus fruiting body has been proposed [
22]. Finally, a molecular-level explanation of the genomic instability of subcultures of
P. ostreatus and an approach to monitor strain degeneration in industrial cultivation has already been proposed with the aid of proteomic analysis [
23].
The aim of this study is to investigate the effect of glucose and xylose, the main lignocellulosic sugars, on protein dynamics and metabolic adaptation in
P. ostreatus LGAM 1123 submerged cultivation. For this reason, a proteomic analysis of the strain submerged cultivated cells in glucose, xylose, and combinations of them was conducted at the exponential phase. Moreover, intracellular polysaccharides, proteins, lipids and extracellular polysaccharides that are produced from the cells were estimated. Using Gene ontology analysis, the proteins were classified into different metabolic processes and pathways. The proteins were classified into lipid, carbohydrate and amino acid biosynthesis-related proteins using the Volcano plot analysis. The upregulated fungi genes related to amino acid biosynthesis were inserted in the STRING Database to reveal the most upregulated metabolic pathways, through Kyoto Encyclopedia of Genes and Genomes (KEGG) database. The respective enzymes were analyzed to see the expression in each tested conditions.
2. Materials and Methods
2.1. Chemicals and Reagents
Analytical grade chemicals were used in this study. Yeast extract and potato dextrose agar (PDA) were supplied from Neogen Europe Ltd. (Ayr, UK). Zinc sulphate heptahydrate (ZnSO
4·7H
2O), glucose, manganese (II) sulfate heptahydrate (MnSO
4·7H
2O), thiamine hydrochloride (Vitamin B1), ethylenediaminetetraacetic acid disodium salt di-hydrate (EDTA-Na
2·2H
2O) (Sodium EDTA), fructose, xylose, maltose, peptone, bovine serum albumin (BSA), and phenol were supplied from Sigma-Aldrich (St. Louis, MO, USA). Sodium nitrate (NaNO3), di-Potassium hydrogen phosphate anhydrous (dibasic) (K
2HPO
4), potassium chloride (KCl), potassium nitrate (KNO
3), ammonium chloride (NH
4Cl) and ammonium sulfate ((NH
4)
2SO
4) were supplied from AppliChem (Darmstadt, Germany). Magnesium sulfate anhydrous (MgSO
4), calcium chloride dihydrate (CaCl
2·2H
2O), ferrous sulfate heptahydrate (FeSO
4·7H
2O), ammonium molybdate tetrahydrate ((NH
4)Mo7O
2·4H
2O), sucrose, and urea were supplied from Fluka (Buchs, Switzerland). Sodium hydroxide (NaOH) was purchased from Panreac (Barcelona, Spain). Chloroform, acetonitrile and methanol were supplied from Thermo Fisher Scientific (Waltham, MA, USA). Sulfuric acid was supplied from Honeywell Riedel-de Haën, and hydrochloric Acid (HCl) was supplied from Merck (KGaA Darmstadt, Darmstadt, Germany). Iodoacetamide was supplied from Acros Organics (Geel, Belgium). SeraMag carboxylate-modified beads were supplied from GE Life Sciences (Chicago, IL, USA). MS grade Trypsin/LysC was supplied from Promega (Madison, WI, USA).
2.2. Microorganism
The basidiomycota
P. ostreatus LGAM 1123 from the strain database of the Laboratory of General and Agricultural Microbiology (Agricultural Microbiology (Agricultural University of Athens, Athens, Greece) was used in this study. The strain was stored at 4 °C in petri dishes containing potato dextrose agar (PDA).
2.3. Media and Growth Conditions
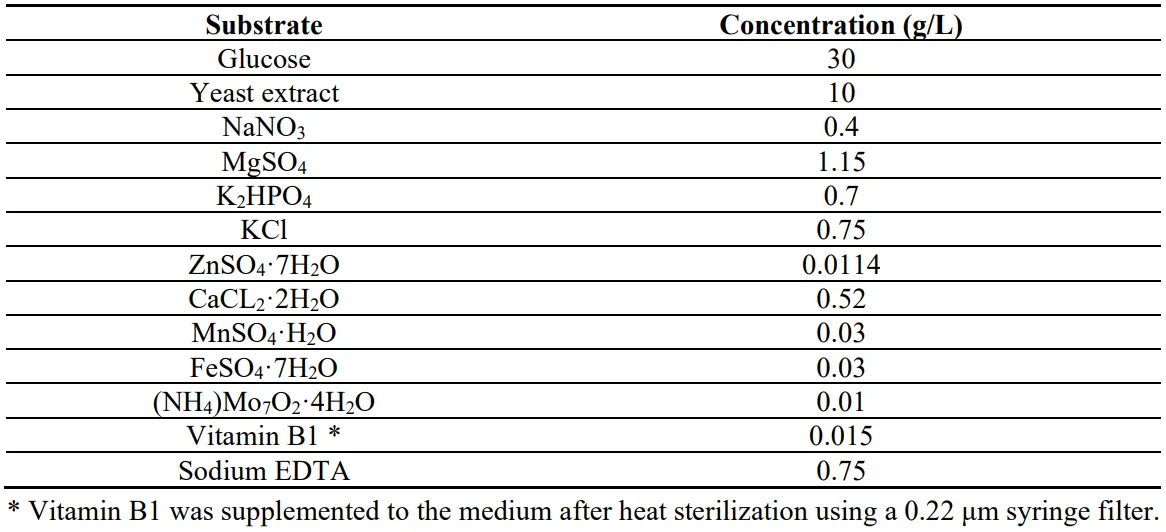
For preculture preparation, a square of 1cm of the mycelium grown in the PDA agar was transferred into an Erlenmeyer flask using an inoculation loop. The flasks contained a 100 mL of the basal medium of the . The pH of the medium was adjusted to 5 and the medium was sterilized in an autoclave at 121 °C for 20 min. The precultures were incubated in a rotary shaker at 150 rpm and 28 °C for twelve days. After the incubation time, five milliliters of the precultures were transferred into a new flask containing the tested conditions. Specifically the conditions tested were 50 g/L glucose and 0 g/L of xylose (100%_Glucose), 35 g/L glucose and 15 g/L xylose (70%Glucose_30%Xylose), 15 g/L glucose and 35 g/L xylose (30%Glucose_70%Xylose) and 0 g/L glucose and 50g/L xylose (100%_Xylose). At the exponential phase, samples were withdrawn for biomass composition analysis and proteomic analysis.
. Basal medium for P. ostreatus LGAM 1123 submerged cultivation.
The biomass of the samples withdrawn from the cultures was estimated after the separation of the supernatant and cell pellets using centrifugation (4000 rpm, 10 min), biomass washed three times and the samples freeze-dried as previously described [
24]. Reducing sugars were determined colorimetrically from the supernatant using DNSA method as previously described using a glucose or xylose standard curve [
24,
25]. Glucose and xylose concentrations were determined separately using an enzymatic method as described previously [
24].
2.5. Protein Estimation
The total protein content was determined from the freeze-dried biomass using the Dumas method, soluble proteins were quantified using the BCA method (Pierce™ BCA protein assay kit) according to the manufacturer’s instructions as previously described [
13].
2.6. Total Lipid Estimation
Total lipids were extracted from the biomass using 2:1 (
v/
v) chloroform: methanol method as described by Folch and modified according to our previous study [
13,
26].
2.7. Polysaccharide Estimation
Ten milligrams of biomass were used for intracellular polysaccharides (IPS) extraction using an ultrasonication (OMNI SONIC RUPTOR 400, Omni International, Kennessaw, GA, USA) at 40% intensity (8 kHz) and 80% pulse until the homogenization of the sample. Cell debris was removed using centrifugation at 4000 rpm for 10 min. The intracellular content was precipitated using ethanol (1:4
v/
v) and was incubated overnight at 4 °C. After the incubation period samples were centrifugated (6000 rpm for 10 min) and the precipitated polysaccharides were kept for analysis. For extracellular polysaccharides (EPS) the supernatant from the culture broth was precipitated as described above. IPS and EPS were determined by the phenol-sulfuric acid method using a glucose standard curve according to Dubois et al. [
27].
2.8. Proteomic Analysis
2.8.1. Cell Lysis and Protein Digestion
One hundred and fifty milligrams of
P. ostreatus frozen cells derived from the culture at the exponential phase were used. The cells were lysed by mixing with 600 μL of a solution containing 4% SDS, 100 mM Tris/HCl pH 7.6, 0.1M DTT (lysis solution) and were incubated at 95 °C for 3 min. Next, the samples were centrifuged at 16K for 20 min and the supernatant was processed through Sp3.
Proteomic sample preparation using the single-pot, solid-phase-enhanced sample preparation Sp3-mediated protein digestion protocol. The supernatant was collected and processed according to the Sp3 protocol [
28] including an alkylation step in the dark for 15 min in 10 mg/mL iodoacetamide (Acros Organics, Geel, Belgium). 20 µg of beads (1:1 mixture of hydrophilic and hydrophobic SeraMag carboxylate-modified beads, GE Life Sciences) were added to each sample in 50% ethanol. The proteins were allowed to bind to the beads for 15 min followed by repeated steps of protein clean-up using a magnetic rack. The beads were washed two times with 80% ethanol and once with 100% acetonitrile (Fisher Chemical, Waltham, MA, USA). The captured on beads proteins were digested overnight at 37 °C under vigorous shaking (1200 rpm, Eppendorf Thermomixer) with 1 ug Trypsin/LysC (MS grade, Promega) prepared in 5 mM ammonium bicarbonate. The next day, the supernatants were collected and the peptides were purified using a modified Sp3 clean-up protocol and finally solubilized in the mobile phase A (0.1% formic acid in water), sonicated and the peptide concentration was determined through absorbance at 280 nm measurement using a nanodrop instrument.
2.8.2. LC-MS/MS Analysis
Samples were run on a liquid chromatography-tandem mass spectrometry (LC-MS/MS) setup consisting of a Dionex UltimateRSLC online with a Thermo Q Exactive HF-X Orbitrap mass spectrometer Thermo Scientific, (Waltham, MA, USA). Peptidic samples were directly injected and separated on a 25 cm-long analytical C18 column (PepSep, 1.9 μm
3 beads, 75 µm ID, Bruker) using a one-hour long run, starting with a gradient consisting of 7% Buffer B (0.1% Formic acid in 80% Acetonitrile) to 35% for 40 min and followed by an increase to 45% in 5 min and a second increase to 99% in 0.5 min and then kept constant for equilibration for 14.5 min. The flow rate was set to 400 nL/min in the sample loading phase and lowered to 250 nL/min in the main phase of sample analysis. A full MS was acquired in profile and positive mode using a mass spectrometer, operating in the scan range of 375–1400
m/
z using 120 K resolving power with an AGC of 3 × 10
6 and max IT of 60 ms followed by data independent analysis using 8 Th windows (39 loop counts) with 15 K resolving power with an AGC of 3 × 10
5 and max IT of 22 ms and a normalized collision energy (NCE) of 26.
2.8.3. Data Analysis
Orbitrap raw data was analyzed in DIA-NN 1.8.1 (Data-Independent Acquisition by Neural Networks) [
29] through searching against the
P. ostreatus Uniprot database containing 12173 proteins using the library-free mode of the software and allowing up to two tryptic missed cleavages. A spectral library was created from the DIA runs and used to reanalyze them. DIA-NN default settings have been used with oxidation of methionine residues and acetylation of the protein N-termini set as variable modifications and carbamidomethylation of cysteine residues as fixed modification. N-terminal methionine excision was also enabled. The match between runs (MBR) feature was used for all analyses and the output (precursor) was filtered at 0.01 FDR and finally, the protein inference was performed on the level of genes using only proteotypic peptides. The generated results were processed statistically and visualized in the Perseus software (1.6.15.0).
The LFQ intensities were transformed to logarithmic. Filtering of at least 70% valid values in at least one group was performed. The remaining NaN intensities were imputed—replaced by a normal distribution, assuming that the corresponding protein is low abundant in the sample. The replicas were grouped (two biological and two technical) and ANOVA testing was performed for the comparison of all four conditions (100%_Glucose, 70%Glucose_30%Xylose, 30%Glucose_70%Xylose, 100%_Xylose) using permutation-based FDR of 0.05%. Proteins with statistically significant changes were kept and normalized through Z-scoring, before performing Euclidean hierarchical clustering of rows and columns (average). For the comparisons of glucose versus xylose, a two-sided t-test of the grouped proteins was performed using
p values for truncation. The FDR threshold value was 0.05 and
S0 = 0.1. Volcano plots were constructed by plotting the t-test difference versus the negative Log of the
p values for each protein.
To obtain additional protein information for subsequent functional validation, all of the differentially expressed proteins were subjected to a global protein network analysis using the STRING tool (version 12) (http://string.embl.de/) string [
30]. STRING tool provides the link between overexpressed protein and KEGG pathways. KEGG pathway databases (https://www.kegg.jp/kegg/pathway.html) were used to color overregulated and downregulated proteins in specific metabolic pathways [
31].
2.8.4. Deposition
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE [
32] partner repository with the dataset identifier PXD049213.
2.9. Statistical Analysis
The data were expressed as mean ± standard deviation from experiments that were conducted in triplicate. All statistical analyses were conducted using IBM SPSS statistics (version 28.0.1.0, IBM Corporation, Armonk, NY, USA), using the one-way analysis of variance (ANOVA) statistical test with Tukey’s multiple range test (with
p-values < 0.05 being regarded as significant). Proteomic analysis results were processed statistically and visualized in the Perseus software (1.6.15.0).
3. Results
3.1. Biomass Composition
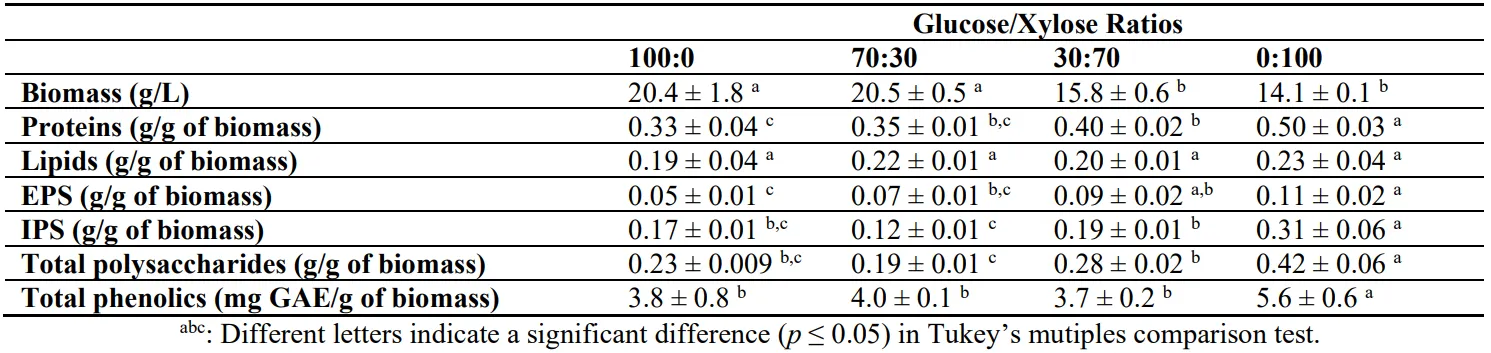
Pleurotus ostreatus was submerged cultivated in sole glucose (100% Glucose), sole xylose (100% xylose) and mixtures of them (70%Glucose_30%Xylose, 30%Glucose_70%Xylose). After eight days of cultivation, biomass was withdrawn from the cultures, the cells were separated from the supernatant and the biomass composition was analyzed in biomass production, protein, lipids, IPS and total phenolics content, while the supernatant was used for extracellular polysaccharide estimation. Glucose promotes the mycelium growth of
P. ostreatus compared to xylose. Biomass production was higher when glucose was used as sole carbon source and as the dominant sugar (70% glucose) reaching a value of up to 20 g/L (). When sole xylose or xylose at a concentration of 70% was used the biomass was lower (
p < 0.05) compared to glucose, reaching a value up to 15 g/L. The protein synthesis was enhanced proportionally with xylose content on the growth medium. The protein content was higher at 100%_Xylose condition (0.50 ± 0.03 g/g of biomass), whereas 30%Glucose_70%Xylose follows with a value of 0.40 ± 0.02 g/g of biomass (
p < 0.05). Lipid content was the same for all the studied conditions (
p > 0.05) with values up to 20%. Also, an analysis of intra and extracellular polysaccharides was conducted, and total polysaccharide content was also estimated. Intracellular polysaccharide content has also been favored by sole xylose condition with a value of 0.31 ± 0.06 g/g of biomass to be observed compared to the value of sole glucose condition, reaching only 0.17 ± 0.01 g/g of biomass EPS has shown a similar xylose dependent production, reaching to lower yields compared to IPS. Overall, the total polysaccharide content presents almost double values in xylose compared to glucose condition, reaching the maximum value of 0.42 ± 0.06 g/g of biomass as shown in . Moreover, the total phenolics estimation revealed that the 100% Xylose condition presents the highest phenolic content in
P. ostreatus cells (
p > 0.05) compared to the other tested conditions that present similar contents.
. Biomass per liter of medium and protein, IPS, EPS, total phenolics and lipid content per gr of biomass for P. ostreatus LGAM 1123 cultivation on different glucose/xylose concentrations.
Proteomic analysis of
P. ostreatus LGAM 1123 submerged cultivation using different glucose and xylose concentrations has identified a total of 3545 proteins. Each condition was tested in four replicates and the ANOVA significant proteins are depicted in the heat map (). Extremely different proteomic profiles are observed between glucose and xylose. As we can see cells that have been cultivated in xylose up-regulate a distinct cluster (blue color) of 1296 proteins compared to glucose one that up-regulates a different cluster of 1901 proteins (pink color). The Gene ontology analysis of these two clusters is presented in Table S1. The cluster of proteins that xylose up-regulates has shown that plays an important role in biological processes of organic acid metabolic processes, small molecule metabolic process, cellular ketone metabolic processes and cellular amino acid metabolism (Figure S1). In contrast, the pink cluster that is up-regulated from glucose plays an important role in biological processes like cellular macromolecule biosynthetic process, macromolecule biosynthetic process, and translation (Figure S1). Also, different proteomic profiles are observed in the mixtures, but they seem to be closer to sole glucose condition and to differentiate most with the sole xylose condition.
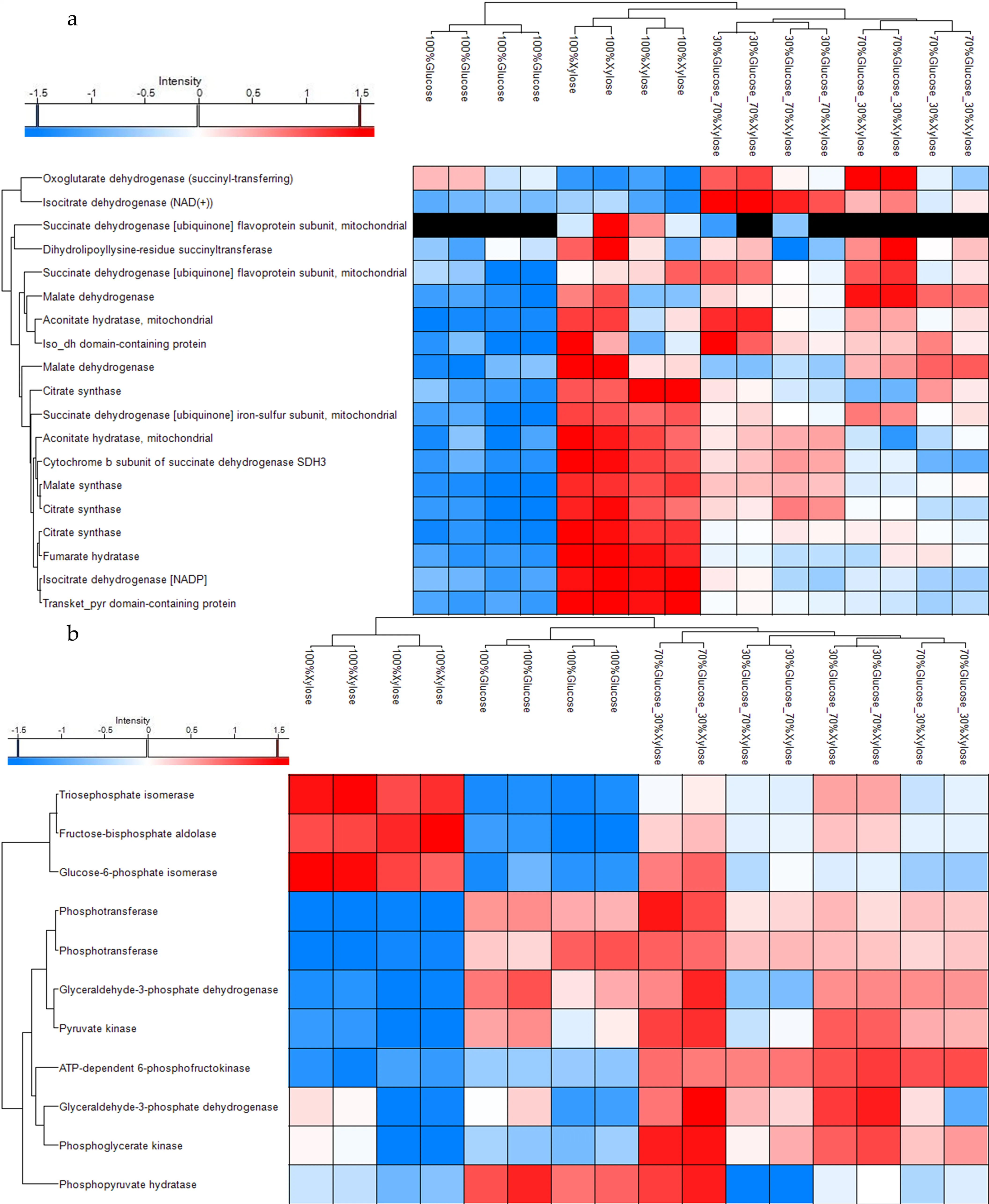
A filter for proteins related to Glycolysis and TCA cycle, the two most important metabolic pathways that are essential for energy production in cells, was conducted for
P. ostreatus LGAM 1123 submerged cultivation as shown in a and 2b. As we can see from the Heat Map (a) 19 proteins were identified from
P. ostreatus biomass that take part in the TCA cycle. 17 of them are upregulated when xylose is used as a carbon source whereas glucose seems to down-regulate almost the total of proteins that participate in the TCA cycle. As concerns the mixtures of glucose and xylose seems that the existence of xylose in the medium up-regulates some of the related proteins. On the other side, glycolysis (b) seems to be upregulated when glucose is contained in the medium, from a total of 11 proteins that related to the glycolysis cycle, 8 of them were upregulated when sole glucose and the mixture of 70%Glucose_30%Xylose were used in the medium. Xylose seems to down-regulate these 8 proteins, and contrarily, up-regulates 3 different proteins in a different cluster. The mixture of 30%Glucose_70%Xylose seems to upregulate almost the total of 11 proteins.
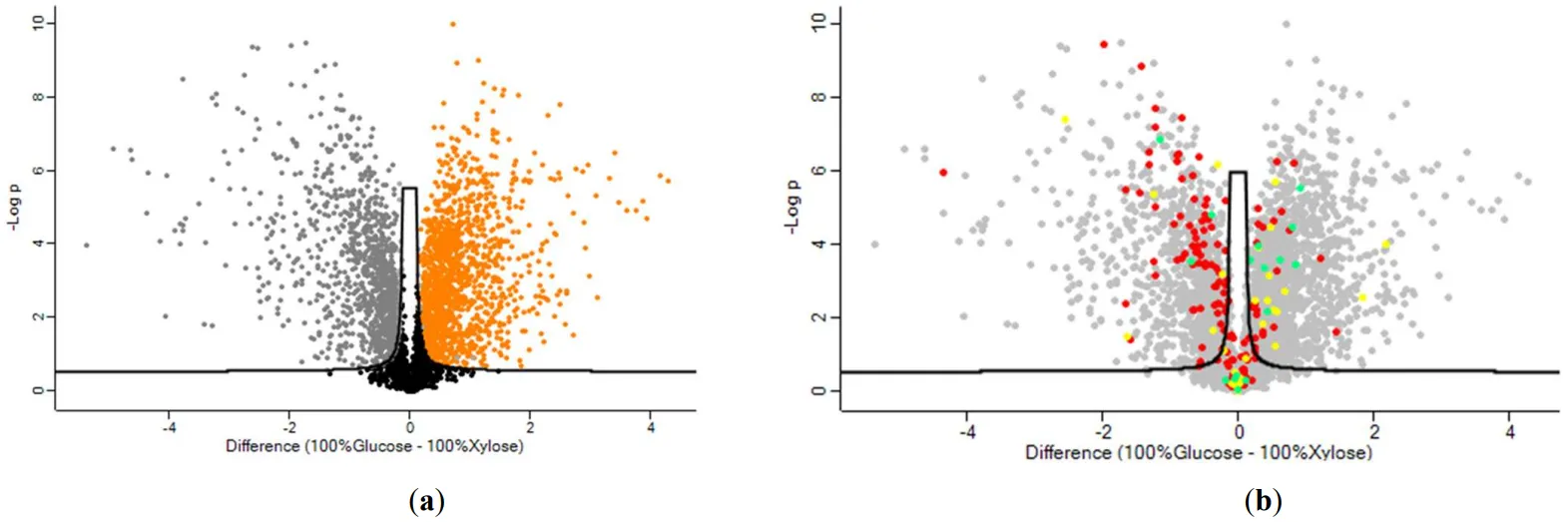
The distribution between differently regulated proteins, when glucose and xylose were used, is shown in the Volcano plot (a) and an annotation analysis of fundamental biological processes is presented in Table S2. In total 2461 proteins were significantly different, 1041 of which upregulated when xylose was used whereas 1420 upregulated proteins were identified for glucose. 190 proteins related to the protein metabolic process, 142 of which were significantly different and 100 of them upregulated when glucose was used in the culture medium and only 42 were upregulated when xylose was used as a carbon source. In addition, 30 proteins related to Nucleotide biosynthesis were detected, 14 of which upregulated when xylose was used and 16 when glucose was used. Twenty-four proteins concerning stress response were identified. Eleven of them upregulated when xylose was used and 13 when glucose was used as a carbon source. Also, an annotation for redox process-related proteins was conducted and resulted in a total of 13 significantly different proteins 5 and 8 of which were upregulated when glucose and xylose were used respectively.
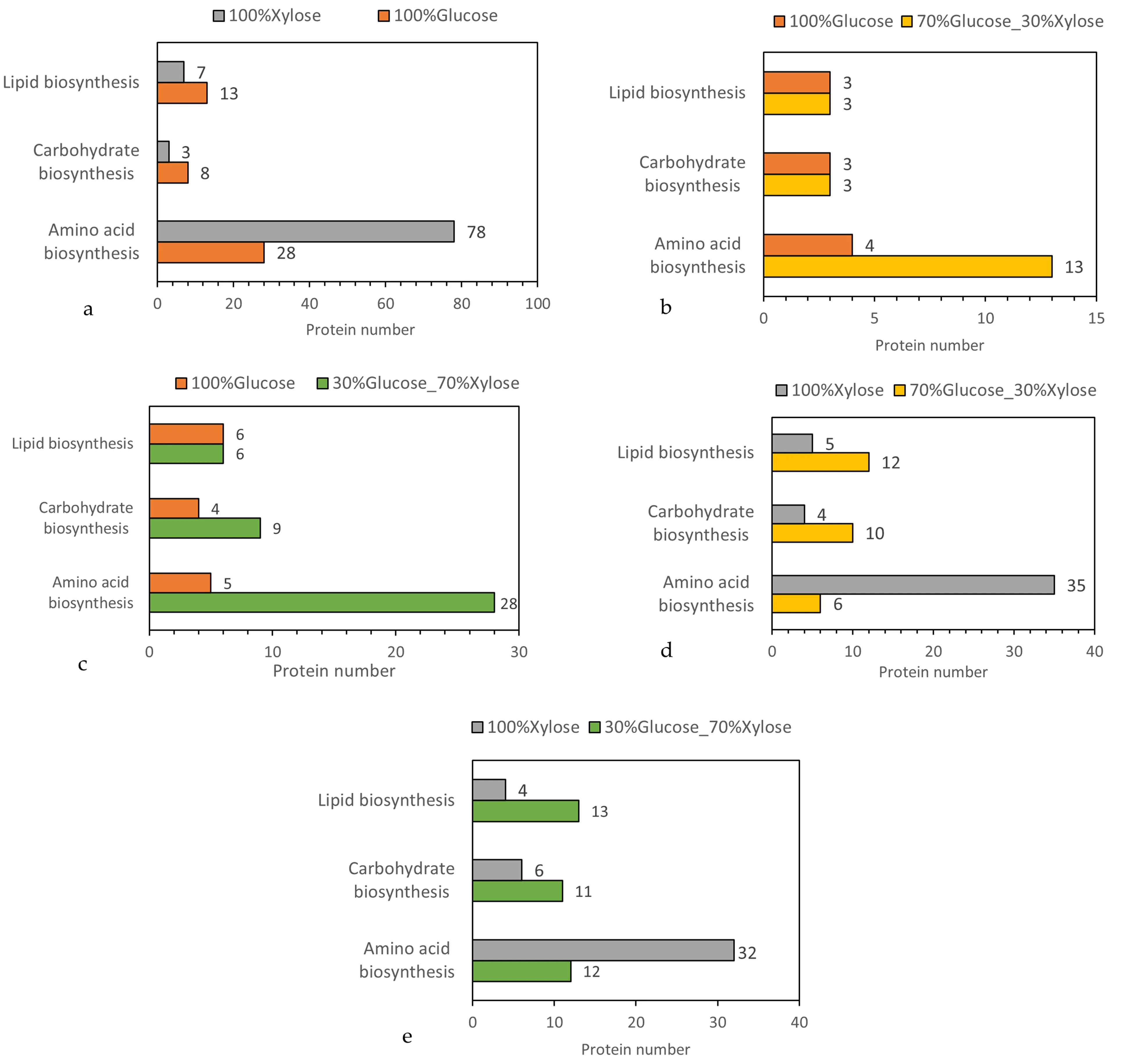
The effect of glucose and xylose on key metabolic pathways of amino acid, lipid, and carbohydrate biosynthesis are depicted in b and . A number of 128 proteins are responsible for amino acid biosynthesis, 104 of which are significantly different. When xylose was used 78 of them were up-regulated and only 28 were up-regulated when glucose was used as a carbon source for
P. ostreatus LGAM 1123 submerged cultivation. For carbohydrate biosynthesis 16 proteins were identified, 11 of which were significantly different, 8 were up-regulated when glucose was used and 3 were up-regulated when xylose was used. For lipid biosynthesis 27 proteins were responsible with 20 of them being significantly different. 13 were upregulated when glucose was used and 7 when xylose was used. The table that presents the up and down-regulated proteins and their categorization from the Gene Ontology biological process for sole glucose condition compared to xylose are given in Table S2.
Concerning mixture conditions comparison with sole glucose and sole xylose, we can conclude that the mixtures have variations with both glucose and xylose, whereas the differentiation in the presence of sole xylose is greater (). The Volcano plot between 70%Glucose_30%Xylose and 100%Glucose analysis determined 967 significantly different proteins, 361 were upregulated when Glucose was used and 606 when 70%Glucose_30%Xylose mixture was used. For amino acid biosynthesis, 13 proteins were upregulated in 70%Glucose_30%Xylose and 4 were upregulated in glucose. As concerns carbohydrate biosynthesis 6 significantly different proteins were detected, 3 proteins were upregulated in Glucose and 3 in 70%Glucose_30%Xylose. The same distribution was detected for lipid biosynthesis. The comparison between 30%Glucose_70%Xylose and 100%Glucose resulted in an output of 1741 significantly different proteins,580 of them belong to 100%Glucose condition and 1161 to 30%Glucose_70%Xylose. For amino acid biosynthesis, 5 proteins were upregulated in Glucose condition whereas 28 proteins were upregulated in 30%Glucose_70%Xylose. The annotation for carbohydrate biosynthesis resulted in an output of 13 significantly different proteins, 4 were upregulated when glucose was used and 9 when 30%Glucose_70%Xylose was used in the culture medium. Also, the mixtures were compared to the sole xylose condition. The comparison between the 70%Glucose_30%Xylose and 100%Xylose resulted in 2123 significantly different proteins of which 1366 belong to 70%Glucose_30%Xylose and 757 to 100%Xylose condition. As we can see from d more proteins related to lipid and carbohydrate biosynthesis are presented in 70%Glucose_30%Xylose condition whereas 100%Xylose condition presents a strong difference for proteins related to amino acid biosynthesis (35 for 100%Xylose and 6 for 70%Glucose_30%Xylose). The comparison between the 30%Glucose_70%Xylose and 100%Xylose results in an output of 2274 significantly different proteins, 668 belong to 100%Xylose and 1606 to 30%Glucose_70%Xylose condition. The comparison between mixtures and sole xylose condition is depicted in detail in c,d. As we can see from e more proteins related to lipid and carbohydrate biosynthesis are presented in 30%Glucose_70%Xylose condition whereas 100%Xylose condition presents a strong difference for proteins related to amino acid biosynthesis (32 for 100%Xylose and 12 for 30%Glucose_70%Xylose). In total, we observed that proteins related to amino acid biosynthesis were differentiated most for all the tested conditions.
. Clustered heat map of P. ostreatus LGAM 1123 substrate-specific proteomic signatures. Average hierarchical clustering and heatmap visualization of the ANOVA significant proteins. (The LFQ intensity values were Log2 transformed, imputed and Z-scored). Up-regulated proteins (rows) are marked in red and down regulated proteins in blue. Missed proteins are marked in black. Each column represents a sample.
. Clustered heat map of P. ostreatus LGAM 1123 substrate-specific proteomic signatures for proteins related to (a) TCA cycle and (b) Glycolysis cycle. Average hierarchical clustering and heatmap visualization of the ANOVA significant proteins. (The LFQ intensity values were Log2 transformed, imputed and Z-scored). Up-regulated proteins (rows) are marked in red and down regulated proteins in blue. Missed proteins are marked in dark. Each column represents a sample.
. Volcano plots demonstrate the magnitude (x-axis) and significance (y-axis) of the protein comparisons between glucose versus xylose. (a) significantly altered proteins when xylose is used (grey color), significantly altered proteins when glucose is used (orange color) and statistically insignificant proteins (black color) (b) Red color indicates proteins that participate in cellular amino acid biosynthesis, yellow color represents proteins of lipid biosynthesis, and green color proteins responsible for carbohydrate biosynthesis.
. Categorization of protein in amino acid biosynthesis related proteins, carbohydrate biosynthesis related proteins and lipid biosynthesis related proteins from t-test between two tested conditions each time, as observed by the Volcano plot analysis. (a: 100%Glucose vs 100% Xylose, b:100%Glucose vs 70%Glucose_30%Xylose, c:100%Glucose vs 30%Glucose_70%Xylose, d: 100%Xylose vs 30%Xylose), e: 100%Xylose vs 30%Glucose_70%Xylose.
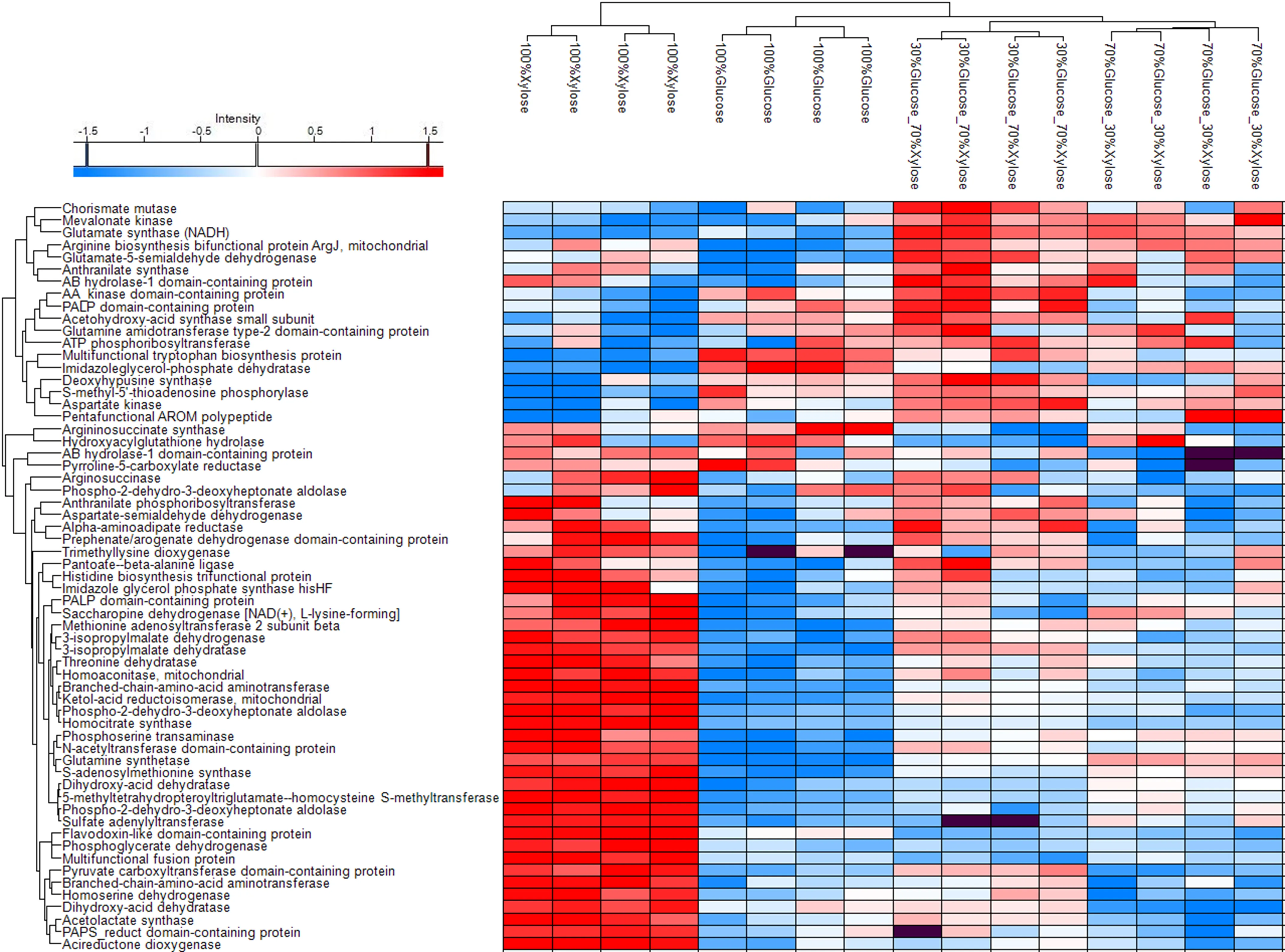
Consequently, an analysis for proteins related to amino acid biosynthesis was conducted to show the best medium condition for the accumulation of amino acids on
P. ostreatus LGAM 1123 biomass as shown in . A total of 61 proteins related to amino acid biosynthesis were identified from which a cluster of 43 proteins was upregulated when sole xylose was used in the medium. Some of the upregulated proteins of xylose are Homocitrate synthase, Sulfate adenylyltransferase, Phospho-2-dehydro-3-deoxyheptonate aldolase, Phospho-2-dehydro-3-deoxyheptonate aldolase, Phosphoglycerate dehydrogenase, Acireductone dioxygenase, Multifunctional fusion protein, Pyruvate carboxyltransferase domain-containing protein, Branched-chain-amino-acid aminotransferase, Homoserine dehydrogenase and Acetolactate synthase. In contrast for the conditions of 70%Glucose_30%Xylose, a different cluster of 18 proteins was upregulated, while the 30%Glucose_70%Xylose condition upregulated some proteins of both the two clusters. The mixture conditions are clustered together with some of the proteins that are upregulated to be AB hydrolase-1 domain-containing protein, Anthranilate synthase, Chorismate mutase, Arginine biosynthesis bifunctional protein ArgJ, Glutamate-5-semialdehyde dehydrogenase, Mevalonate kinase, Glutamate synthase (NADH), Pentafunctional AROM polypeptide, ATP phosphoribosyltransferase and Aspartate kinase. The 100%Glucose condition upregulated the fewest proteins among all the tested conditions, with some of them being Hydroxyacylglutathione hydrolase, AB hydrolase-1 domain-containing protein, Imidazoleglycerol-phosphate dehydratase, Pyrroline-5-carboxylate reductase, Argininosuccinate synthase, and Multifunctional tryptophan biosynthesis protein. From these results, we can conclude that xylose seems to favor the upregulation of a wide range of proteins related to amino acid biosynthesis.
. Clustered heat map of P. ostreatus LGAM 1123 substrate-specific proteomic signatures for proteins related to amino acid biosynthesis. Average hierarchical clustering and heatmap visualization of the ANOVA significant proteins. (The LFQ intensity values were Log2 transformed, imputed and Z-scored). Up-regulated proteins (rows) are marked in red and down regulated proteins in blue. Missed proteins are marked in dark. Each column represents a sample.
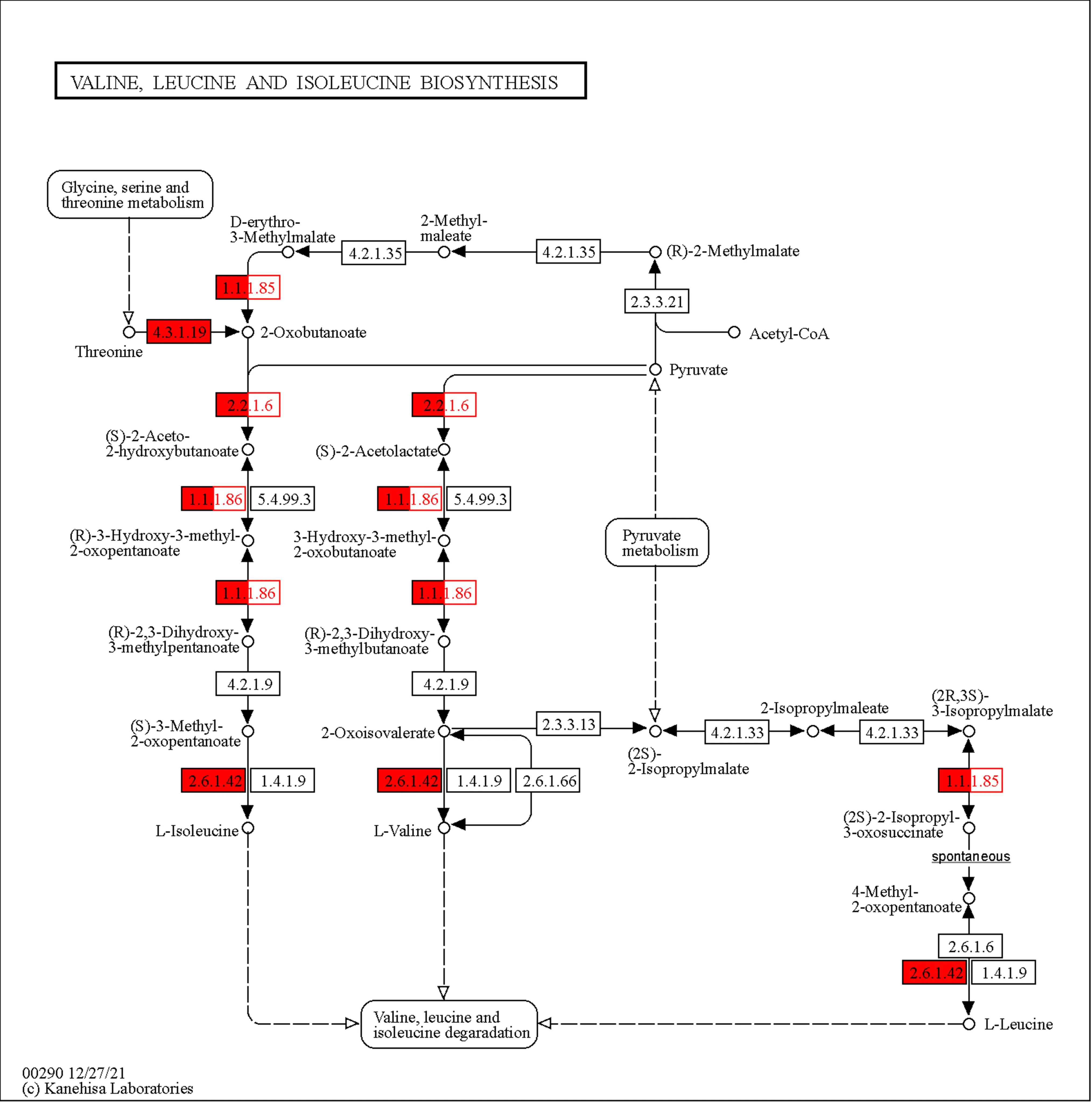
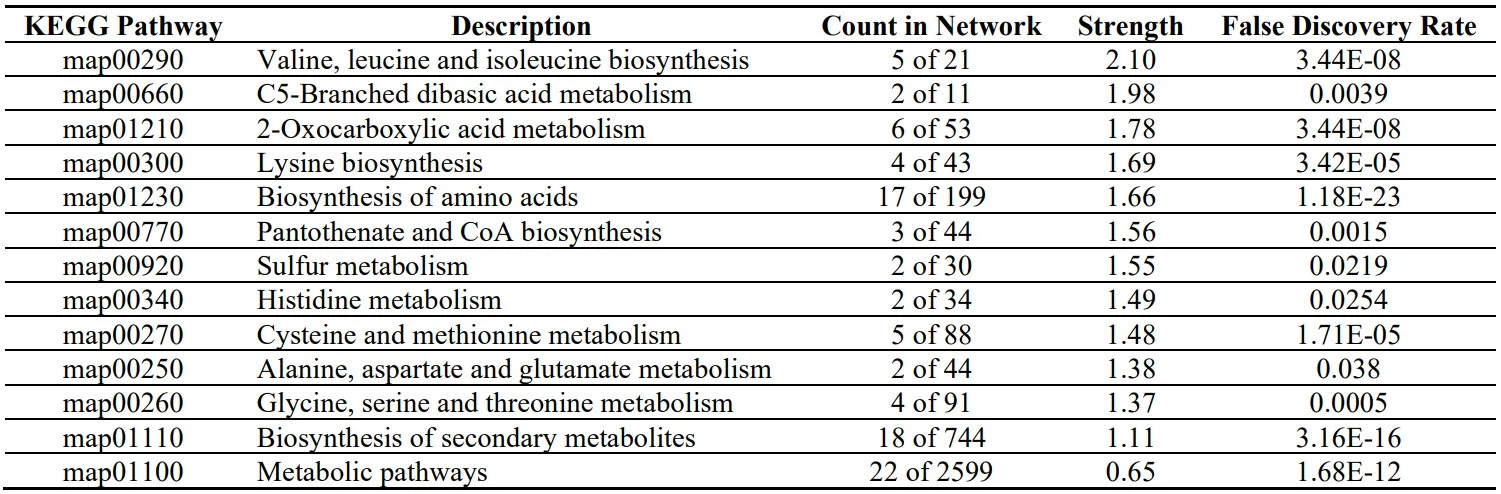
To have a more systematic analysis of the pathways that are related to the up-regulated proteins, the genes that overexpressed were assessed in the STRING Database (https://string-db.org/) to connect them with the pathways from KEGG pathways databases. Since, xylose up-regulates the TCA cycle and amino acid biosynthesis process, the proteins that upregulated from xylose and were responsible for the amino acid biosynthesis process were entered in the STRING Database and the pathways that are stimulated are shown the Table 3. The analysis has shown that the pathway with the highest strength to be upregulated is valine, leucine and isoleucine biosynthesis. This pathway produces essential branched-chain amino acids in protein synthesis, which humans should receive through diet. In addition, pathways with high strength include C5-branched dibasic acid metabolism, 2-Oxocarboxylic acid metabolism, Lysine biosynthesis, and Biosynthesis of amino acids. All of them are strongly related to the amino acid biosynthesis process. The KEGG pathway database and the tool Mapper Color were used to highlight the up-regulated proteins in the pathway. As we can see from Figure 6, the fundamental enzymes that lead to Valine, leucine and Isoleucine biosynthesis and are triggered when xylose is used in the medium are 3-isopropylmalate dehydrogenase (1.1.1.85) and Threonine dehydratase (4.3.1.19), acetolactate synthase (2.2.1.6), Ketol-acid reductoisomerase (1.1.1.86) and Branched-chain-amino-acid aminotransferase (2.6.1.42). The upregulation of the specific enzymes and taking into account Figure 6, we can suggest that for
P. ostreatus LGAM 1123 isoleucine is synthesized mainly from threonine and not from pyruvate, valine from pyruvate, while leucine is synthesized from 3-isopropylmalate.
. KEGG database pathway of Valine Leucine Isoleucine biosynthesis map. The enzymes that are upregulated when xylose is used in the cultivation medium are marked with red.
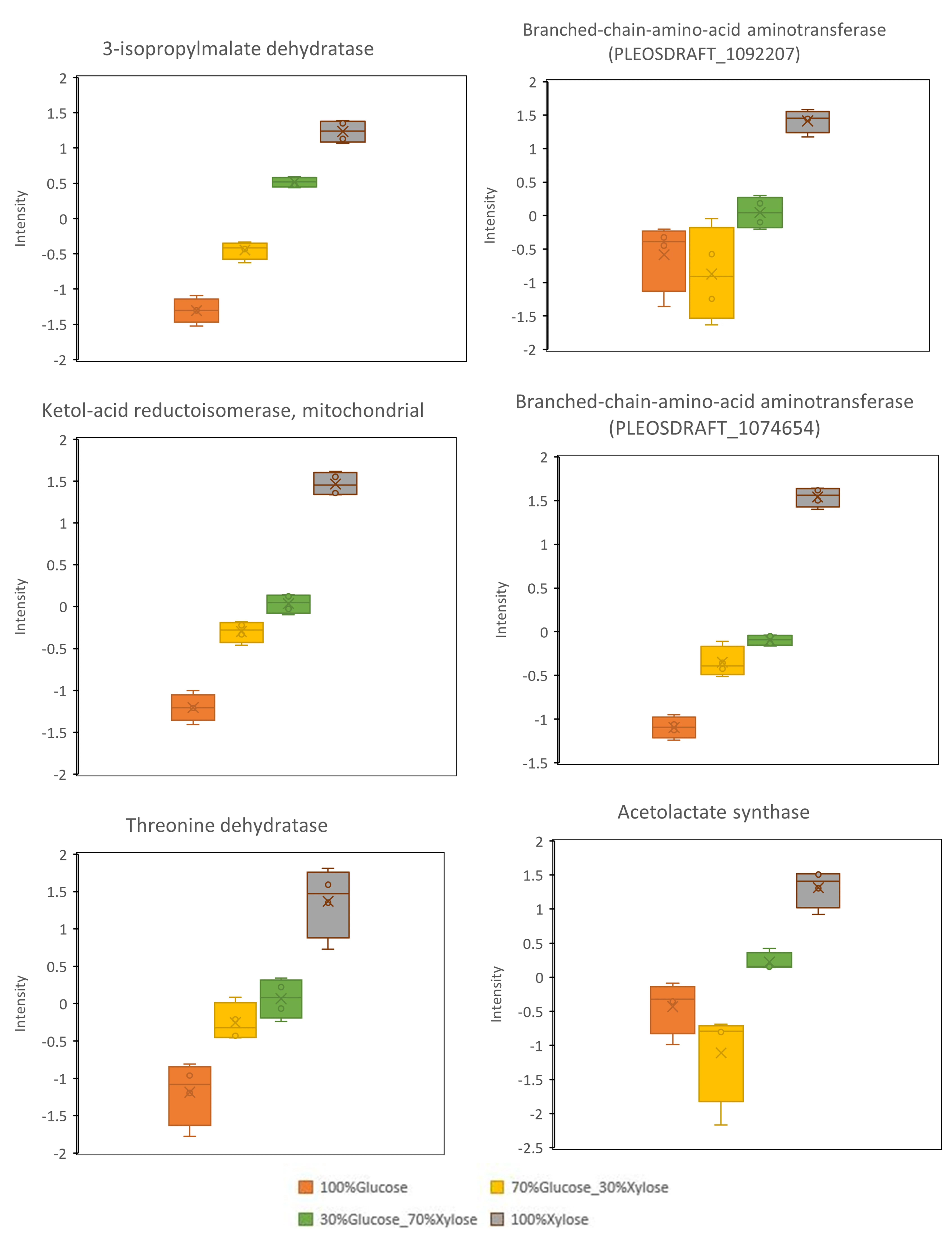
As we can see from an analysis of the fundamental enzymes that were upregulated in Valine, Leucine and Isoleucine biosynthesis pathway, as shown in the above KEGG map was conducted to see the different enzyme expression in all the tested conditions (100%_Glucose, 70%Glucose_30%Xylose, 30%Glucose_70%Xylose, 100%_Xylose). From the Box and Whisker plots is obvious that for all the tested enzymes, 3-isopropylmalate dehydratase (1.1.1.85), Branched-chain-amino-acid aminotransferase, genes: PLEOSDRAFT_1092207 and PLEOSDRAFT_1074654 (2.6.1.42) Ketol-acid reductoisomerase (1.1.1.86), Threonine dehydratase (4.3.1.19) and acetolactate synthase (2.2.1.6), there is an increase in enzyme regulation intensity when the xylose concentration is increased in growth medium. Specifically, all the enzymes were downregulated when sole glucose was used in the growth medium, whereas higher intensity values but still negative were observed for the 70%Glucose_30%Xylose condition. As concerns all the enzymes except for 3-isopropylmalate dehydratase, zero intensity values were presented in 30%Glucose_70%Xylose condition, while 3-isopropylmalate dehydratase has an intensity value of 0.5. All the tested enzymes were upregulated with an intensity value close to 1.5 when the 100%_Xylose condition was used.
. Box and Whisker plots for the analysis of the expression of the fundamental enzymes of Valine Leucine Isoleucine biosynthesis map, in each tested condition of P. ostreatus LGAM 1123 in glucose, xylose and mixtures of them.
Table 3. STRING Database analysis for the up-regulated proteins that participate in amino acid biosynthesis process when xylose used for P. ostreatus LGAM 1123 submerged cultivation.
4. Discussion
In this study,
P. ostreatus LGAM 1123 was submerged cultivated in the fundamental sugars of lignocellulosic biomass, glucose, xylose and mixtures of them. At the exponential phase cells were withdrawn from the cultures and biochemical compositions were determined. From the biochemical analysis of the cells, we observed that xylose favors macromolecule production, except for lipids which did not have statistically different values, but reduced biomass production in contrast to glucose which favors biomass production. A study for
Pleurotus pulmonarius using different carbon sources has shown that glucose favored biomass production and EPS production whereas xylose favored protein production. Lipids were also not statistically different between these two carbon sources [
31]. Concerning IPS production a study for
Mortierella (
Umbelopsis)
isabelline has shown that glucose as a carbon source favored the production of endopolysaccharides in contrast to xylose [
32]. Also, a proteomic analysis was conducted to reveal the differentially regulated expression of proteins in each case. Cultivation in glucose/xylose mixtures has potential as they constitute the fundamental sugars in lignocellulosic biomass and most lignocellulosic substrates contain glucose and xylose in a ratio of 60–70% Glucose to30–40% xylose [
24]. In addition, some studies suggest that the presence of glucose suppresses genes that are related to alternative carbon sources [
33]. Also, a study has shown that using different lignocellulosic substrates in
P. ostreatus enhances the ability to transform sugars via different metabolic pathways and improves adaptability in the environment [
17,
24].
The proteomic analysis of
P. ostreatus LGAM 1123 has revealed that a different cluster was upregulated when glucose and xylose were used as carbon source. Specifically, 128 proteins responsible for amino acid biosynthesis had a differential expression, of which 104 are significantly different. When xylose was used as a carbon source, 78 of these proteins were up-regulated, and the other 28 were up-regulated by glucose. For carbohydrate biosynthesis 16 proteins were identified of which 11 were significantly different, 8 were up-regulated when glucose was used and 3 were up-regulated when xylose was used. For lipid biosynthesis 27 proteins were responsible with 20 of them being significantly different. 13 were upregulated when glucose was used and 7 when xylose was used. Furthermore, when filtering of proteins as concerns the two fundamental pathways for energy production in cells, glycolysis, and TCA cycle, were used, it seems that xylose upregulates TCA cycle whereas the existence of even small concentrations of xylose in the mixture conditions up-regulates some of the related proteins. In contrast, glycolysis seems to be upregulated when sole glucose was used in the medium and similar results were observed for 70%Glucose_30%Xylose mixture. In a study for
P. ostreatus proteomic analysis in response to lignocellulosic components 531 ± 23 were detected when control Kirk medium was used, 496 ± 19 when lignin was used, 567 ± 38 in xylan-supplemented medium, and 601 ± 27 for CMC medium. Xylan is composed of pentoses and especially xylose, therefore this condition could be compared to the xylose one of this study. CMC was used as a substitute for cellulase which is a polymer of glucose units and could be compared to glucose conditions in this study. Similar to our results, this study confirms that glucose, or the respective polymer CMC activates glycolysis while xylose, or the respective polymer, xylan activates TCA cycle. Johnston 1999 and Kim et al. 2013 have confirmed that for yeast cells that grow on high levels of sugars the metabolism of them is completed through glycolysis [
34,
35]. In our study, the presence of glucose does not seem to suppress genes that are related to alternative carbon sources as stated before [
33], while the mixture conditions seem to upregulate proteins from both the clusters of sole glucose and xylose. The carbon-dependent expression of metabolic enzymes has also been confirmed by proteomic analysis of
Trichoderma reesei, grown on different carbon sources [
8]. In addition, a study of the proteome of
C. acetobutylicum using glucose and xylose has shown that in total 826 proteins upregulated when xylose was used as a carbon source whereas glucose upregulated 717 proteins. From them, the number of proteins related to carbohydrate metabolism identified only in glucose was 7 and in xylose only 24. For lipid metabolism, most proteins were both presented in glucose and xylose. For amino acid metabolism, 4 proteins were identified only in glucose and 9 when xylose was used [
9].
Volcano plot analysis of combinations between groups of all the tested conditions and categorization of proteins related to amino acid, carbohydrate and lipid biosynthesis, has revealed that the most differentiated biosynthesis process was that of amino acid biosynthesis. For that reason, a filtering of proteins related to amino acid biosynthesis has shown that xylose condition seems to favor the upregulation of a wide range of proteins related to amino acid biosynthesis. The mixture conditions are clustered together with some distinct proteins to be upregulated in comparison to glucose and xylose. The 100% Glucose condition upregulated the fewest proteins among all the tested conditions. This is also confirmed by our previous study for amino acid analysis through an HPLC unit which showed that eight amino acids were clustered with sole xylose condition whereas only four amino acids clustered with sole glucose condition [
24]. The different expression of essential and nonessential amino acids was also confirmed when
P. ostreatus was grown in black poplar wood logs, WS, and lignocellulosic byproducts, LcS after a multivariate statistical models analysis [
18]. It has been suggested that fungi can catabolize glucose into amino acids using at first glycolysis and proceeding to TCA cycle. About xylose, there are three potential ways. In the two of them xylose catabolism is linked with glucose one through the pentose phosphate pathway and only one way has been suggested that connects xylose directly to the TCA cycle (Weimberg pathway) [
24,
36,
37].
The STRING Database analysis of proteins related to amino acid biosynthesis and overexpressed in xylose medium has shown that the first upregulated KEGG pathway was that of Leucine, Valine, and Isoleucine biosynthesis. In this study, the enzymes 3-isopropylmalate dehydrogenase (1.1.1.85), threonine dehydratase (4.3.1.19), acetolactate synthase (2.2.1.6), Ketol-acid reductoisomerase (1.1.1.86) and Branched-chain-amino-acid aminotransferase (2.6.1.42) are overexpressed. To see how these enzymes are expressed in all the tested conditions, Box and Whisker plots were constructed. For all the enzymes the intensities were gradually increased when xylose concentration was increased in the growth medium. From the upregulation of the specific enzymes and taking into account , we can suggest that for
P. ostreatus LGAM 1123 isoleucine is synthesized mainly from threonine and not from pyruvate, valine from pyruvate, while leucine is synthesized from 3-isopropylmalate. Leucine, Valine and Isoleucine amino acids also referred to as Branched-chain amino acids (BCAAs) are required for ribosomal protein and peptide-based secondary metabolites biosynthesis and cannot be synthesized by humans [
38,
39]. BCAAs are synthesized by fungi and are essential for the human diet as they are key components of human and animal nutrition and need to be obtained from daily nutrition. Also, they play a role in protein synthesis and muscle metabolism, with increased BCAAs concentrations to indicatingdecreased muscle soreness and prevention of muscle breakdown during exercise [
40] Their applications are related to food, feed, cosmetic and pharmaceutical industries [
41]. The BCAA metabolic pathway is well characterized in Saccharomyces cerevisiae, in which the transcriptional regulation is conducted by the Zn(II)2Cys6 transcription factors LEU3. In
A. nidulans and
A. fumigatus biosynthesis occurs by LeuB, 3-isopropylmalate dehydrogenase, similar to our results. The activity of these transcription factors is modulated by the intermediate α-isopropylmalate [
38]. In a study for the regulation of Leucine metabolism in Mortierella alpina the key regulators were found to be α-isopropylmalate synthase LeuA1, α-ketoisocaproate, and propionyl-CoA [
42]. Concerning
P. ostreatus metabolism Pellegrino et al. after a metabolic pathway analysis for growth in black poplar wood logs, and lignocellulosic byproducts have revealed that the two highest differentially expressed pathways were glycine, serine, and threonine metabolism, followed by valine, leucine, and isoleucine biosynthesis, alike to our study, and D-glutamine and D-glutamate metabolism [
18]. Glycine, serine and threonine metabolism as well as valine, leucine and isoleucine biosynthesis were also reported in a study for
P. ostreatus mycelia growth under heat-stress conditions [
20]. In a previous study of our group, concerning amino acid concentration in
P. ostreatus submerged cultivation in different glucose/xylose combinations, valine and leucine were observed to be one of the sixth most abundant amino acids in the biomass, whereas for all the tested conditions valine, leucine and isoleucine concentrations found to be over the minimum recommended patterns according to the report of FAO Expert Consultation for Dietary Protein Quality Evaluation in Human Nutrition [
24]. Also, a study on free amino acids in Pleurotus species reported that leucine, together with valine and lysine were the predominant essential free amino acids [
43]. We can conclude that the importance of valine, leucine, and isoleucine pathways in the metabolism of
P. ostreatus fungi is evident. Further research into the specific biosynthetic pathways and regulatory mechanisms would provide a more comprehensive understanding of the biosynthesis of these amino acids in this
P. ostreatus.
5. Conclusions
In this study, P. ostreatus was submerged cultivated in the fundamental sugars of agro-industrial biomass, glucose and xylose, and mixtures of them, to see the different proteomic response in each case. The produced biomass was analyzed in their biochemical compounds and a proteomic analysis was conducted to reveal the different upregulated proteins in each condition. The proteomic analysis revealed that glucose and xylose upregulate different clusters of proteins whereas the mixtures present similarities with the proteome of sole glucose condition. Xylose is found to up-regulate TCA cycle whereas glucose upregulates glycolysis cycle. Between the basic biosynthetic processes of carbohydrates, lipids and amino acids the most significantly different process for all the tested conditions were found to be the amino acid biosynthesis. Xylose upregulates mostly the proteins related to amino acid biosynthesis, and the enriched pathway was confirmed by the analysis of the upregulated proteins in the STRING Database. Leucine, Valine and Isoleucine biosynthesis pathways were found to be the most triggered pathway. All the BCAAs-related enzymes intensities were gradually increased when xylose concentration was increased in the growth medium. BCAAs play an important role in the human diet so the enhancement of BCAAs pathway for P. ostreatus could convert it to a very remarkable protein substitute in human diet. These findings provide new insights into the proteomic and metabolic response of the fungus to the major sugars of lignocellulosic biomass, and could contribute to the production of high nutritional value mycelial protein by the edible fungus Pleurotus ostreatus in submerged fermentation.
Supplementary Materials
The following supporting information can be found at: https://www.sciepublish.com/article/pii/153; Table S1: Enrichment analysis of Gene Ontology in biological processes that upregulate the different clusters of the HeatMap for proteomic analysis of P. ostreatus LGAM 1123 submerged cultivation using glucose, xylose and mixtures of them.; Figure S1. Enrichment analysis of Gene Ontology in biological processes that upregulate the different clusters of the HeatMap upregulated from glucose and xylose for proteomic analysis of P. ostreatus LGAM 1123 submerged cultivation using glucose, xylose and mixtures of them. Table S2: Gene Ontology biological process categorization of up and down-regulated proteins when glucose condition compared to xylose one.
Acknowledgments
We acknowledge support of this work by the project “The Greek Research Infrastructure for Personalised Medicine (pMED-GR)” (MIS 5002802) which is implemented under the Action “Reinforcement of the Research and Innovation Infra-structure”, funded by the Operational Programme “Competitiveness, Entrepreneurship and Innovation” (NSRF 2014-2020) and co-financed by Greece and the European Union (European Regional Development Fund). We gratefully acknowledge Prof. Georgios I. Zervakis from the Laboratory of General and Agricultural Microbiology, Agricultural University of Athens, Iera Odos 75, 11855 Athens, Greece; for providing the fungal strain.
Author Contributions
Conceptualization, P.K.; Methodology, M.S., P.K.; Software, M.S.; Validation, G.B, P.K.; Formal Analysis, G.B.; M.S.; Investigation, G.B.; Resources, H.S., P.K.; Data Curation, G.B.; Writing – Original Draft Preparation, G.B; Writing – Review & Editing, P.K. M.S. and H.S.; Visualization, G.B.; M.S; Supervision, P.K., and H.S.; Project Administration, H.S. and P.K.; Funding Acquisition, H.S.
Ethics Statement
Not applicable.
Informed Consent Statement
Not applicable.
Funding
This research was funded by “Synthetic Biology: from omics technologies to genomic engineering (OMIC-ENGINE)” (MIS 5002636), which is implemented under the Action “Reinforcement of the Research and Innovation Infrastructure,” funded by the Operational Program “Competitiveness, Entrepreneurship and Innovation” (NSRF 2014–2020) and co-financed by Greece and the European Union (European Regional Development Fund).
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
-
1.
Kim Y, Nandakumar MP, Marten MR. Proteomics of Filamentous Fungi.
Trends. Biotechnol. 2007,
25, 395–400.
[Google Scholar]
-
2.
Jagadeesh DS, Kannegundla U, Reddy RK. Application of Proteomic Tools in Food Quality and Safety.
Adv. Anim. Vet. Sci. 2017,
5, 213–225.
[Google Scholar]
-
3.
Ortea I, O’Connor G, Maquet A. Review on Proteomics for Food Authentication.
J. Proteomics 2016,
147, 212–225.
[Google Scholar]
-
4.
Duracova M, Klimentova J, Fucikova A, Dresler J. Proteomic Methods of Detection and Quantification of Protein Toxins.
Toxins 2018,
10, 1–30.
[Google Scholar]
-
5.
Fayyaz K, Nawaz A, Olaimat AN, Akram K, Farooq U, Fatima M, et al. Microbial Toxins in Fermented Foods: Health Implications and Analytical Techniques for Detection.
J. Food Drug Anal. 2022,
30, 523–537.
[Google Scholar]
-
6.
López-Pedrouso M, Lorenzo JM, Alché JD, Moreira R, Franco D. Advanced Proteomic and Bioinformatic Tools for Predictive Analysis of Allergens in Novel Foods.
Biology 2023,
12, 1–14.
[Google Scholar]
-
7.
Sivamaruthi BS, Kesika P, Chaiyasut C. Toxins in Fermented Foods: Prevalence and Preventions—A Mini Review.
Toxins 2019,
11, 4.
[Google Scholar]
-
8.
Jun, H Guangye H, Daiwen C. Insights into Enzyme Secretion by Filamentous Fungi : Comparative Proteome Analysis of Trichoderma Reesei Grown on Different Carbon Sources.
J. Proteomics 2013,
89, 191–201.
[Google Scholar]
-
9.
Sivagnanam K, Raghavan VGS, Shah M, Hettich RL, Verberkmoes NC, Lefsrud MG. Comparative Shotgun Proteomic Analysis of Clostridium Acetobutylicum from Butanol Fermentation Using Glucose and Xylose.
Proteome Sci. 2011,
9, 66.
[Google Scholar]
-
10.
Liu D, Wang S, Xu B, Guo Y, Zhao J, Liu W, et al. Proteomics Analysis of Bifidobacterium Longum NCC2705 Growing on Glucose, Fructose, Mannose, Xylose, Ribose, and Galactose.
Proteomics 2011,
11, 2628–2638.
[Google Scholar]
-
11.
Bellettini MB, Fiorda FA, Maieves HA, Teixeira GL, Ávila S, Hornung PS, et al. Factors Affecting Mushroom
Pleurotus Spp.
Saudi. J. Biol. Sci. 2019,
26, 633–646.
[Google Scholar]
-
12.
Bakratsas G, Polydera A, Katapodis P, Stamatis H. Recent Trends in Submerged Cultivation of Mushrooms and Their Application as a Source of Nutraceuticals and Food Additives.
Future Foods 2021,
4, 100086.
[Google Scholar]
-
13.
Bakratsas G, Polydera A, Nilson O, Κossatz L, Xiros C, Katapodis P, et al. Single-Cell Protein Production by Pleurotus Ostreatus in Submerged Fermentation.
Sustain. Food Technol. 2023,
1, 377–389.
[Google Scholar]
-
14.
Bakratsas G, Polydera A, Nilson O, Chatzikonstantinou AV, Xiros C, Katapodis P, et al. Mycoprotein Production by Submerged Fermentation of the Ed- Ible Mushroom Pleurotus Ostreatus in a Batch Stirred Tank Bio- Reactor Using Agro-Industrial Hydrolysate.
Foods 2023,
12, 2295.
[Google Scholar]
-
15.
Bakratsas G, Antoniadis K, Athanasiou PE, Katapodis P, Stamatis H. Laccase and Biomass Production via Submerged Cultivation of Pleurotus ostreatus Using Wine Lees.
Biomass 2024,
4, 1–22.
[Google Scholar]
-
16.
Zerva A, Papaspyridi LM, Christakopoulos P, Topakas E. Valorization of Olive Mill Wastewater for the Production of β-Glucans from Selected Basidiomycetes.
Waste Biomass Valoriz. 2017,
8, 1721–1731.
[Google Scholar]
-
17.
Xiao Q, Ma F, Li Y, Yu H, Li C, Zhang X. Differential Proteomic Profiles of Pleurotus Ostreatus in Response to Lignocellulosic Components Provide Insights into Divergent Adaptive Mechanisms.
Front. Microbiol. 2017,
8, 480.
[Google Scholar]
-
18.
Pellegrino RM, Blasi F, Angelini P, Ianni F, Alabed HBR, Emiliani, C, et al. LC/MS Q-TOF Metabolomic Investigation of Amino Acids and Dipeptides in Pleurotus Ostreatus Grown on Different Substrates.
J. Agric. Food Chem. 2022,
70, 10371–10382.
[Google Scholar]
-
19.
Fernández-Fueyo E, Ruiz-Dueñas FJ, López-Lucendo MF, Pérez-Boada M, Rencoret J, Gutiérrez A, et al. A Secretomic View of Woody and Nonwoody Lignocellulose Degradation by Pleurotus Ostreatus.
Biotechnol. Biofuels 2016,
9, 1–18.
[Google Scholar]
-
20.
Yan Z, Zhao M, Wu X, Zhang J. Metabolic Response of Pleurotus Ostreatus to Continuous Heat Stress.
Front. Microbiol. 2020,
10, 1–13.
[Google Scholar]
-
21.
Zhu W, Hu J, Li Y, Yang B, Guan Y, Xu C, et al. Comparative Proteomic Analysis of Pleurotus Ostreatus Reveals Great Metabolic Differences in the Cap and Stipe Development and the Potential Role of Ca
2+ in the Primordium Differentiation.
Int. J. Mol. Sci. 2019,
20, 6317.
[Google Scholar]
-
22.
Lin R, Zhang L, Yang X, Li Q, Zhang C, Guo L, et al. Responses of the Mushroom Pleurotus Ostreatus under Different CO
2 Concentration by Comparative Proteomic Analyses.
J. Fungi 2022,
8, 1–14.
[Google Scholar]
-
23.
Zhu W, Hu J, Chi J, Li Y, Yang B, Hu W, et al. Label-Free Proteomics Reveals the Molecular Mechanism of Subculture Induced Strain Degeneration and Discovery of Indicative Index for Degeneration in Pleurotus Ostreatus.
Molecules 2020,
25, 4920.
[Google Scholar]
-
24.
Bakratsas G, Polydera A, Nilson O, Chatzikonstantinou AV, Xiros C, Katapodis P, et al. Mycoprotein Production by Submerged Fermentation of the Edible Mushroom Pleurotus Ostreatus in a Batch Stirred Tank Bioreactor Using Agro-Industrial Hydrolysate.
Foods 2023,
12, 2295.
[Google Scholar]
-
25.
Miller GL. Use of Dinitrosalicylic Acid Reagent for Determination of Reducing Sugar.
Anal. Chem. 1959,
31, 426–428.
[Google Scholar]
-
26.
Folch J, Lees M, Sloane Stanley GH. A Simple Method for the Isolation and Purification of Total Lipides from Animal Tissues.
J. Biol. Chem. 1957,
226, 497–509.
[Google Scholar]
-
27.
Dubois M, Gilles KA, Hamilton JK, Rebers PA, Smith F. Colorimetric Method for Determination of Sugars and Related Substances.
Anal. Chem. 1956,
28, 350–356.
[Google Scholar]
-
28.
Hughes CS, Moggridge S, Müller T, Sorensen PH, Morin GB, Krijgsveld J. Single-Pot, Solid-Phase-Enhanced Sample Preparation for Proteomics Experiments.
Nat. Protoc. 2019,
14, 68–85.
[Google Scholar]
-
29.
Demichev V, Messner CB, Vernardis SI, Lilley KS, Ralser M. DIA-NN: Neural Networks and Interference Correction Enable Deep Proteome Coverage in High Throughput.
Nat. Methods 2020,
17, 41–44.
[Google Scholar]
-
30.
Szklarczyk D, Franceschini A, Kuhn M, Simonovic M, Roth A, Minguez P, et al. The STRING Database in 2011: Functional Interaction Networks of Proteins, Globally Integrated and Scored.
Nucleic Acids Res. 2011,
39, 561–568.
[Google Scholar]
-
31.
Kanehisa M, Furumichi M, Sato Y, Kawashima M, Ishiguro-Watanabe M. KEGG for Taxonomy-Based Analysis of Pathways and Genomes.
Nucleic Acids Res. 2023,
51, D587–D592.
[Google Scholar]
-
32.
Perez-Riverol Y, Bai J, Bandla C, García-Seisdedos D, Hewapathirana S, Kamatchinathan S, et al. The PRIDE Database Resources in 2022: A Hub for Mass Spectrometry-Based Proteomics Evidences.
Nucleic Acids Res. 2022,
50, D543–D552.
[Google Scholar]
-
33.
Bahn YS, Xue C, Idnurm A, Rutherford JC, Heitman J, Cardenas ME. Sensing the Environment: Lessons from Fungi.
Nat. Rev. Microbiol. 2007,
5, 57–69.
[Google Scholar]
-
34.
Johnston M. Feasting, Fasting and Fermenting: Glucose Sensing in Yeast and Other Cells.
Trends Gene 1999,
15, 29–33.
[Google Scholar]
-
35.
Kim JH, Roy A, Jouandot D, Cho KH. The Glucose Signaling Network in Yeast.
Biochim. Biophys. Acta Gen. Subj. 2013,
1830, 5204–5210.
[Google Scholar]
-
36.
Hou J, Suo F, Wang C, Li X, Shen Y, Bao X. Fine-Tuning of NADH Oxidase Decreases Byproduct Accumulation in Respiration Deficient Xylose Metabolic
Saccharomyces cerevisiae.
BMC Biotechnol. 2014,
14, 1–10.
[Google Scholar]
-
37.
Liu D, Zhang Y, Li J, Sun W, Yao Y, Tian C. The Weimberg Pathway: An Alternative for Myceliophthora Thermophila to Utilize d-Xylose.
Biotechnol. Biofuels Bioproducts 2023,
16, 1–16.
[Google Scholar]
-
38.
Steyer JT, Todd RB. Branched-Chain Amino Acid Biosynthesis in Fungi.
Essays Biochem. 2023,
67, 865–876.
[Google Scholar]
-
39.
Neinast M, Murashige D, Arany Z. Branched Chain Amino Acids.
Annu. Rev. Physiol. 2019,
81, 139–164.
[Google Scholar]
-
40.
Wolfe RR. Branched-Chain Amino Acids and Muscle Protein Synthesis in Humans: Myth or Reality?
J. Int. Soc. Sports Nutr. 2017,
14, 1–7.
[Google Scholar]
-
41.
Gao H, Tuyishime P, Zhang X, Yang T, Xu M, Rao Z. Engineering of Microbial Cells for L-Valine Production: Challenges and Opportunities.
Microb. Cell Fact. 2021,
20, 1–16.
[Google Scholar]
-
42.
Sonnabend R, Seiler L, Gressler M. Regulation of the Leucine Metabolism in Mortierella Alpina.
J. Fungi 2022,
8, 196.
[Google Scholar]
-
43.
Tagkouli D, Kaliora A, Bekiaris G, Koutrotsios G, Christea M, Zervakis GI, et al. Free Amino Acids in Three Pleurotus Species Cultivated on Agricultural and Agro-Industrial By-Products.
Molecules 2020,
25, 4015.
[Google Scholar]