Intercellular Targetable Mechanistic Interface for Cardiac Fibrosis

Intercellular Targetable Mechanistic Interface for Cardiac Fibrosis
Received: 30 March 2026 Revised: 29 April 2026 Accepted: 04 June 2026 Published: 25 June 2026

© 2026 The authors. This is an open access article under the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/).
1. Introduction
Heart failure (HF) remains a leading cause of morbidity and mortality worldwide, with a lifetime risk approaching 20–25% in many populations [1]. Despite major therapeutic advances targeting neurohormonal signaling and hemodynamic load, long-term outcomes remain poor, particularly in HF with preserved ejection fraction (HFpEF). A consistent pathological feature across diverse HF etiologies is cardiac fibrosis, a maladaptive remodeling process characterized by expansion of extracellular matrix, increased myocardial stiffness, impaired electrical coupling, and progressive contractile dysfunction [2]. Rather than a passive byproduct of injury, fibrosis is increasingly recognized as an active, regulated process that both reflects and drives disease progression.
Cardiac fibrosis develops in both ischemic and non-ischemic settings, yet the biological context differs substantially. In ischemic heart disease, particularly following myocardial infarction, cardiomyocyte necrosis triggers a well-orchestrated inflammatory cascade that transitions toward scar formation in order to preserve structural integrity [3,4]. In this setting, fibrosis initially serves as a reparative function. However, excessive or dysregulated scar formation contributes to chamber dilation, arrhythmogenesis, and long-term functional decline. In contrast, non-ischemic heart disease, such as that associated with hypertension, metabolic syndrome, cardiomyopathies, HFpEF, and atrial fibrillation, often exhibits diffuse interstitial and perivascular fibrosis in the absence of overt cardiomyocyte death [4,5]. In these conditions, inflammatory and fibrotic pathways are activated in parallel and may precede detectable structural damage. The temporal coupling of metabolic stress, inflammation, and matrix remodeling in non-ischemic HF suggests that upstream cellular signaling events, rather than cell death alone, play a central role in the initiation of fibrosis. Importantly, therapeutic strategies aimed at targeting downstream fibrosis have demonstrated limited clinical efficacy, emphasizing the need to better define the initiating mechanisms of pathological remodeling.
Cardiac fibrosis does not arise from the action of a single cell type. Instead, it reflects a coordinated response among multiple resident and infiltrating cells within the myocardium. While cardiac fibroblasts ultimately execute extracellular matrix deposition, fibrotic remodeling reflects dynamic communication between cardiomyocytes, endothelial cells, pericytes, and immune cells within a stress-responsive microenvironment (Figure 1). Increasingly, evidence supports a hierarchical model in which upstream stress signaling, particularly from cardiomyocytes, shapes inflammatory amplification and fibroblast activation [6,7]. We discuss next the role of various cell types in the heart and its communication with fibroblasts to promote or dampen fibrosis.
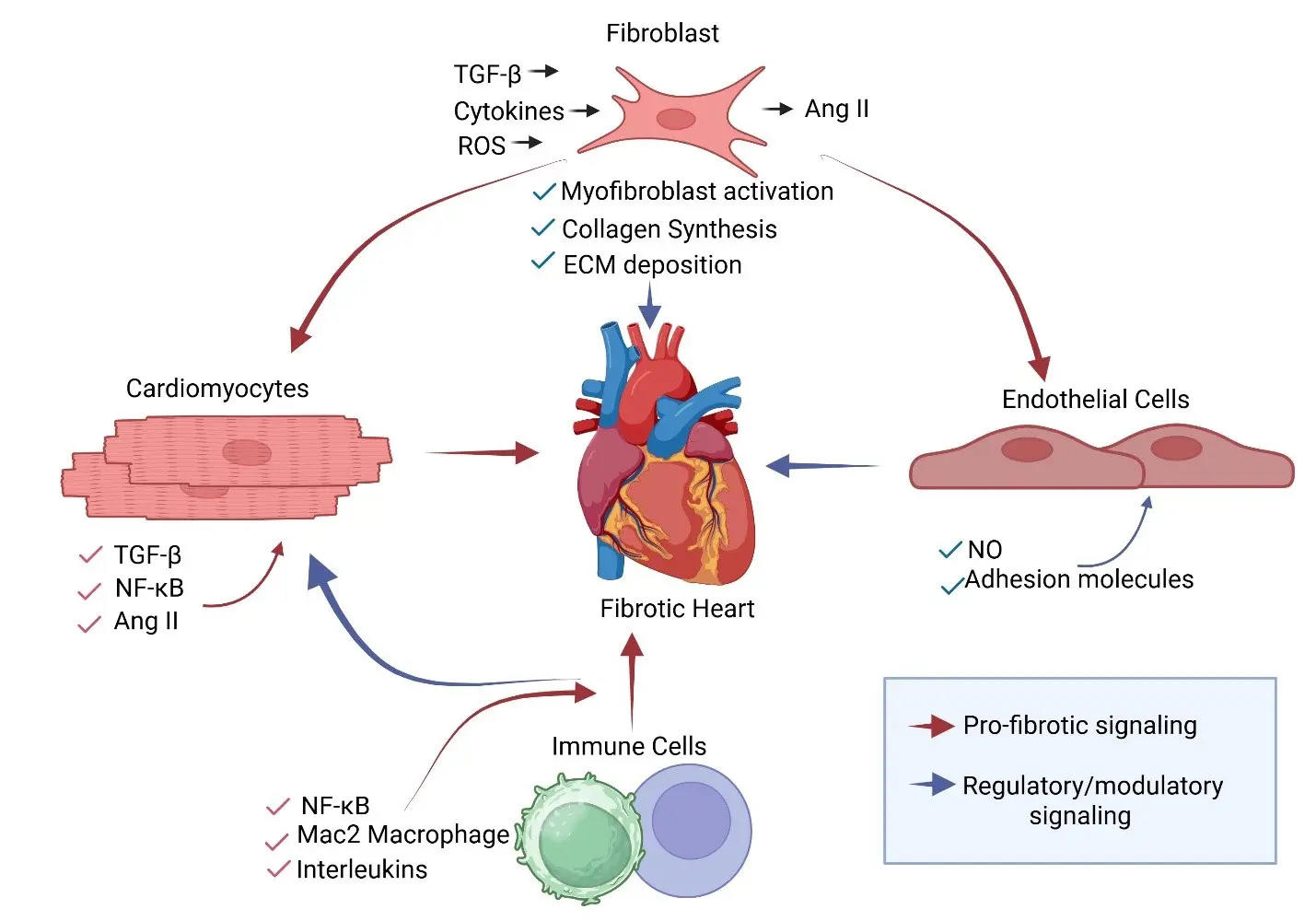
Figure 1. Cellular Crosstalk Driving Cardiac Fibrosis. Coordinated interactions among cardiomyocytes, fibroblasts, immune cells, and endothelial cells drive cardiac fibrosis. Cardiomyocytes subjected to stress (mechanical, metabolic, or neurohormonal) produce profibrotic mediators, including TGF-β, angiotensin II (Ang II), and inflammatory cytokines. These signals promote immune cell recruitment and fibroblast activation. Immune cells further amplify profibrotic signaling through the secretion of interleukins and TGF-β. Activated fibroblasts differentiate into myofibroblasts, leading to increased collagen synthesis and extracellular matrix (ECM) deposition. Endothelial cells modulate inflammatory responses through nitric oxide (NO) and adhesion molecule expression. Red arrows indicate profibrotic signaling; blue arrows indicate regulatory or modulatory signaling. Biorender was used to generate the figure.
2. Cell Types Associated with Cardiac Fibrosis
2.1. Cardiac Fibroblasts: Central Effectors with Regional and Phenotypic Diversity
Cardiac fibroblasts constitute the principal source of fibrillar collagens and extracellular matrix proteins during pathological remodeling. In the healthy adult heart, fibroblasts contribute to basal matrix turnover, mechanical coupling, and provide structural support for cardiomyocytes [8,9,10,11]. Following biomechanical stress, ischemia, or neurohormonal activation, fibroblasts undergo activation and differentiate into myofibroblasts characterized by α-smooth muscle actin (α-SMA or ACTA2) expression, enhanced contractility, resistance to apoptosis, and increased synthesis of collagen types I and III. Transforming growth factor-β (TGF-β)-Smad2/3 signaling is considered the canonical driver of this transition [12,13,14], with strong evidence supporting its role in pressure overload, myocardial infarction, and genetic cardiomyopathy models [12,13,15]. Additional pathways, including Wnt/β-catenin, Platelet-derived growth factor (PDGF), angiotensin II (Ang II), and mechanotransduction through integrin signaling, also cooperate in sustaining fibroblast activation (Figure 1). Our focus in this review is primarily on TGF-β-driven cardiac fibrosis. For other signaling pathways and their implications in cardiac fibrosis and therapy, we refer the reader to other reviews [16,17,18].
Recent lineage tracing and single-cell transcriptomic studies of the adult mammalian heart have revealed substantial fibroblast heterogeneity, including chamber-specific transcriptional differences between atrial and ventricular fibroblasts that may contribute to regional susceptibility to fibrosis [19,20,21,22,23,24,25]. Distinct fibroblast subsets have been identified that emerge during ischemic versus non-ischemic remodeling, including inflammatory-like, matrifibrocyte, and proliferative populations. In pressure overload and chronic human HF settings, Cartilage Intermediate Layer Protein (CILP)+ or Thromobospondin-4 (THBS4)+ fibroblast clusters are observed that remain ACTA2− but produce abundant ECM [26,27,28]. In post infarction models, ACTA2 expressing activated fibroblasts is upregulated, which is the classical myofibroblast phenotype. Thus, fibroblast signatures vary widely depending on the temporal and spatial characteristics of injury.
Importantly, fibroblasts exhibit chamber-specific transcriptional identities even under basal conditions. Atrial fibroblasts differ from ventricular fibroblasts in gene expression programs related to extracellular matrix composition, ion channel expression, and profibrotic sensitivity [29]. These intrinsic differences may help explain the pronounced susceptibility of the atrium to diffuse fibrosis in atrial fibrillation and HFpEF, where remodeling frequently precedes overt ventricular dysfunction. Atrial fibroblasts have also been reported to exhibit heightened responsiveness to TGF-β and Ang II stimulation, potentially lowering the threshold for fibrotic expansion in atrial tissue [30].
In addition to genetic programming, the mechanical environment further contributes to the regional diversity of fibroblasts. The atrium experiences distinct wall stress patterns and stretch dynamics compared with the ventricle, which may shape fibroblast mechano-responsive signaling and matrix composition [31,32]. Additionally, differences in extracellular matrix architecture between chambers can influence fibroblast behavior through integrin-mediated mechanotransduction [3,33]. In addition to genetic program and mechanical forces, epigenetics regulation of cardiac fibrosis is important. For example, DNA methylation [34] and demethylation [35] signatures, histone modifications [36], and chromatin remodeling play a critical role in cardiac fibrosis. There are various mechanisms that are involved in initiating and maintaining fibrotic signaling, eventually leading to fibrosis. Cell-cell communication in fibrosis is gaining increasing attention. Fibroblasts in general are highly responsive to cytokines, chemokines, and growth factors derived from neighboring cardiomyocytes, immune cells, and endothelial cells [10,37]. Their activation state reflects integration of these instructive cues within the myocardial microenvironment. Thus, while fibroblasts serve as indispensable executors of extracellular matrix deposition, the signals that initiate and sustain their activation frequently originate from other cell types, especially in the heart, which are discussed next.
2.2. Cardiomyocytes: Initiators of Stress, Inflammation, and Fibrosis
Cardiomyocytes occupy a structurally and metabolically dominant position in the myocardium. They are also one of the major cell types in the heart, constituting roughly 23% of the total population [9,25,38,39]. As excitable energy-demanding cells, they are the first to sense hemodynamic overload, metabolic shifts, oxidative injury, and neurohormonal stimulation [40]. Importantly, stress-induced signaling within cardiomyocytes extends beyond contractile adaptation and includes activation of inflammatory pathways and secretion of paracrine mediators that influence surrounding cell populations. Cardiomyocytes are increasingly recognized as active regulators of inflammatory and fibrotic signaling [41,42]. In response to stressors (biomechanical stress, metabolic imbalance, oxidative injury, neurohormonal activation), cardiomyocytes activate intracellular pathways, including NF-κB signaling, oxidative stress cascades, and metabolic sensors, that influence chemokine production, immune cell recruitment, and fibroblast activation (Figure 1) [43,44,45,46]. Further, metabolic disruption and mitochondrial dysfunction within cardiomyocytes amplifies inflammatory signaling and matrix deposition [47]. These studies collectively inform the role of cardiomyocytes in promoting fibrosis more directly than purely secondary to fibroblast-autonomous activation or upon cardiomyocyte death. Within this emerging paradigm, metabolic kinases have gained attention as upstream regulators of stress signaling. Previously, we identified a member of the AMP-activated protein kinase (AMPK) family kinase, sucrose nonfermenting-related kinase (SNRK), as a cardiomyocyte-enriched metabolic regulator that is responsible for cardiac metabolism and function, also constrains inflammatory activation [48,49]. Cardiomyocyte-specific deletion of SNRK enhances NF-κB signaling, increases inflammatory mediator expression, promotes macrophage accumulation, and accelerates fibrotic remodeling under non-ischemic cardiac remodeling conditions (Ang II-induced stress) [43]. Importantly, endothelial-specific deletion does not recapitulate this fibrotic phenotype, underscoring the central role of cardiomyocyte-intrinsic signaling in governing pathological remodeling.
Beyond SNRK, stressed cardiomyocytes secrete various profibrotic mediators that act directly on fibroblasts, immune cells, and the ECM. These include TGF-β isoforms, connective tissue growth factor (CTGF/CCN2), interleukin-11 (IL-11), angiotensin II (Ang II), chemokines such as CCL2, and extracellular vesicles carrying fibrosis-regulatory microRNAs (e.g., miR-21-5p, miR-208a). IL-11 has received recent attention as a cardiomyocyte-derived fibrogenic cytokine that drives myofibroblast transition via ERK signaling in a Smad-independent manner, amplifying the fibrotic response downstream of TGF-β stimulation [50]. Cardiomyocyte-derived exosomes represent a complementary, non-soluble paracrine mode of intercellular communication, transferring miRNA cargo and chaperone proteins directly to fibroblasts to promote activation state. It is important to note that SNRK mechanistically is distinct from these secreted effectors. For example, SNRK is not released from cardiomyocytes but functions as an intracellular metabolic kinase that constrains NF-κB activation and thereby limits the production and release of profibrotic mediators, including TGF-β1, chemokines, and inflammatory cytokines, under stress conditions [43,44,48,51]. In that context, SNRK occupies a regulatory position upstream of the cardiomyocyte secretory program, functioning as a metabolic checkpoint that determines the threshold at which stressed cardiomyocytes transition to a profibrotic state, while the paracrine factors listed above represent the downstream effector outputs of that transition.
2.3. Endothelial Cells: Vascular Sensors, and Context-Dependent Regulators
Endothelial cells (EC) regulate vascular tone, barrier integrity, and immune cell trafficking, positioning them at the interface between systemic hemodynamic and myocardial signaling [52]. Single cell analysis of EC in the heart revealed an endocardial EC population, a coronary vascular EC population, and an aorta-specific EC population [52]. These anatomically distinct EC clusters provide a sense that each cluster may have a unique role as it relates to fibrosis. For example, aorta-specific EC will experience distinct shear stress patterns compared to coronary vascular EC, and thus may have a distinct role in their cell surface inflammatory responses to ischemic and non-ischemic cardiac events. EC from these distinct vascular beds may secrete different types of angiocrine factors that can shape cardiomyocyte and fibroblast behavior. It is well known that endothelial dysfunction, characterized by reduced nitric oxide (NO) bioavailability, increases adhesion molecule expression, impairs barrier function, and alters permeability (Figure 1). These disparate phenotypes can amplify inflammatory signaling and contribute to fibrotic progression [53]. EC are unique from other cardiac cell types in that they act as mechanosensors of blood flow. Shear stress-responsive transcriptional programs (e.g., KLF2/KLF4-associated pathways) and endothelial glycocalyx integrity help maintain an anti-inflammatory, anti-thrombotic state [54]. When this homeostatic program is disrupted, for example, by hypertension, diabetes, obesity, or aging, EC shift toward a pro-inflammatory phenotype that facilitates leukocyte adhesion and transmigration, which indirectly promotes fibroblast activation. In HFpEF, microvascular inflammation and capillary dysfunction are closely linked to interstitial fibrosis, supporting the idea that endothelial health strongly influences fibrotic susceptibility even when EC are not the primary matrix-producing cells [55,56]. Besides ECs role to indirectly contribute to fibrosis, in certain pathological contexts, EC are known to transition to mesenchymal phenotype, a process called endothelial-to-mesenchymal transition (EndoMT), which has been proposed as a direct contributor to fibroblast-like populations and matrix deposition [57]. However, the quantitative contribution of EndoMT to cardiac fibrosis may vary by model and injury type and is yet to be fully determined [9,11,58,59]. Further, in the adult, the role of EndoMT is unsettled, as previous mouse lineage tracing studies, where one third fibroblasts were suggested to arise from EndoMT [60], have been recently questioned with newer tools that can continuously track endothelial cell fate. Using EndoMTracer system that constitutes three mouse lines combined (UBC-CreER; α-sma-LSL-Dre; R26R-rox-tdT) to achieve CRE-induced constitutively active Dre to trace α-SMA activation specifically in ECs [61]. Using this system, the authors showed that adult ECs expressed α-SMA transiently in the developing cardiac valves during EndoMT. However, they did not observe the expression of α-SMA in a transient or reversible fashion during initiation and progression of cardiac fibrosis. Thus, they concluded that ECs do not transdifferentiate into myofibroblasts or transiently expressed mesenchymal markers in MI and TAC models post myocardial infarction [61]. However, additional studies [62] did identify transient endothelial-mesenchymal activation, which is a reversible state and thus may be targetable for fibrotic remodeling.
Intact EC signaling in the heart can also be protective and limit pro-inflammatory pathway activation, which then influences fibrosis. We observed this inflammatory protective effect in the heart endothelium in our studies. Endothelial-specific disruption of SNRK enhances NF-κB signaling (inflammation) but does not induce fibrosis or cardiac dysfunction [43,48]. On the other hand, cardiomyocyte-specific disruption of SNRK enhances NF-κB signaling (inflammation), promotes fibrosis, and affects cardiac function [43,48,49]. Taken together, this suggests that cardiomyocyte SNRK is more protective of fibrosis while inflammation upregulation seems to be a response to SNRK deletion in tissue. In fact, investigators have observed that SNRK deletion in other tissues, for example, adipocytes, also prevents inflammation activation [63]. These finding aligns with broader observations that endothelial inflammation can increase “background signals” in the myocardium, yet robust fibrosis typically requires an upstream instructive program, often arising from cardiomyocyte stress signaling, together with immune amplification and fibroblast responsiveness.
2.4. Immune Cells: Translators of Inflammatory Stress into Fibrosis
Immune cells play a central role in coordinating inflammatory and fibrotic responses in the heart. Among these populations, macrophages have emerged as key regulators of tissue remodeling. Macrophages contribute to cardiac modeling and fibrosis by directly transitioning to myofibroblasts, which was demonstrated in a zebrafish model [64]. This direct transition of macrophage to myofibroblast has also been observed in pressure-induced cardiac fibrosis mouse models [35]. In addition to direct transition, both resident cardiac macrophages and recruited monocyte-derived macrophages contribute to paracrine mechanisms involved in fibrotic remodeling through the production of cytokines, growth factors, and extracellular matrix-modifying enzymes [65,66]. Studies from several groups have demonstrated that macrophage-derived mediators, including TGF-β, interleukins, and chemokines, promote fibroblast activation and extracellular matrix deposition following cardiac injury and pressure overload [67,68,69]. The balance between pro-inflammatory and reparative macrophage subsets is therefore a major determinant of fibrotic progression and resolution. Beyond fibroblasts, macrophages also influence cardiomyocyte survival, angiogenesis, and extracellular matrix turnover during cardiac repair. Recent work has highlighted functional heterogeneity among cardiac macrophage populations, including resident c-c motif chemokine receptor type 2 (CCR2) macrophages involved in tissue homeostasis and monocyte-derived CCR2+ macrophages that expand during inflammatory remodeling [70]. These immune populations integrate signals from damaged or stressed cardiomyocytes and contribute to shaping the local inflammatory milieu (Figure 1). Immune cells beyond macrophages, such as Chimeric Antigen Receptor T cells (CAR-T) cells, can also directly communicate with fibroblasts to influence fibrosis [71,72]. Immune cell recruitment often reflects upstream stress signaling from cardiomyocytes. Several studies have demonstrated that cardiomyocyte activation of inflammatory pathways, such as NF-κB signaling, can drive chemokine production, including c-c motif ligand 2 (CCL2), thereby promoting monocyte recruitment and macrophage accumulation in the myocardium [73,74,75]. Consistent with this broader framework, cardiomyocyte-specific disruption of metabolic signaling in our models leads to increased Mac2+ macrophage infiltration even under basal conditions, with further expansion under angiotensin II-induced stress [43]. These findings support the concept that cardiomyocyte-derived inflammatory signals can shape immune cell recruitment during pathological remodeling. Together, these observations from multiple experimental systems indicate that immune cells function as key intermediaries linking cardiomyocyte stress signaling to fibroblast activation and matrix deposition. In this model, macrophages amplify inflammatory cues originating from stressed cardiomyocytes and translate them into sustained profibrotic signaling within the myocardial microenvironment.
2.5. Pericytes in Cardiac Fibrosis
Recent transcriptomic studies have revealed that pericytes, which are specialized mural cells that are critical for vascular stability and regulation of blood flow [76] are also involved in cardiac fibrosis post myocardial infarction [24,77]. Pericytes are known to acquire fibroblast-like phenotype, and post myocardial infarction, these cells maintain expression of pericyte markers and acquire fibroblast markers such as PDGFRA and TCF21 [77,78]. The profibrotic program is activated in a progressive manner, which includes increased expression of growth factors TGF-β1, VEGFA, PDGF, matrix-remodeling enzymes MMP2 and TIMP3, matrix proteins (COL1A2 and COL3A1) and integrins involved in fibrosis (ITGA1, ITGA2, ITGAV, ITGB1). In human MI samples, fibroblasts at the infarct region come from NG2+ pericytes [79]. They comprise 4.3% of the infarct fibroblasts. These studies collectively suggest that the complex interaction among multiple cell types in the heart facilitates cardiac fibrosis.
3. Metabolism as a Regulator of Fibrotic Remodeling
Cardiomyocytes are among the most metabolically demanding cells in the body, continuously adjusting substrate utilization to maintain ATP production and contractile function. In the healthy adult heart, fatty acid oxidation (FAO) accounts for the majority of ATP generation, while glucose oxidation and glycolysis provide additional metabolic flexibility (Figure 2). This metabolic profile is tightly regulated to sustain mitochondrial efficiency and redox balance. Under pathological stress, including pressure overload, neurohormonal activation, ischemia, and metabolic disease, the heart undergoes metabolic remodeling characterized by a partial shift away from FAO toward greater reliance on glycolysis and glucose utilization [80,81]. Although this metabolic reprogramming may initially support energy production under stress, sustained shifts in substrate utilization can impair mitochondrial function, increase reactive oxygen species (ROS) generation, and activate stress-responsive signaling pathways that contribute to inflammatory and fibrotic remodeling.
A growing body of work indicates that cardiomyocyte metabolic imbalance is not merely a consequence of cardiac disease but can actively influence inflammatory signaling and tissue remodeling [82,83]. Mitochondrial dysfunction altered NAD+/NADH balance, and ROS accumulation activates redox-sensitive pathways such as NF-κB and MAP kinase signaling, which in turn promote cytokine production, immune cell recruitment, and fibroblast activation (Figure 2) [84]. Beyond ROS and redox-sensitive kinase pathways, emerging evidence implicates lactylation, a post-translational modification in which lactate-derived lactyl groups are added to lysine residues of histones. This novel mechanism links cardiomyocyte metabolic reprogramming to fibrotic remodeling. For example, during pathological glycolytic shift, intracellular and extracellular lactate accumulation drives histone lactylation (e.g., H3K18la) in cardiac fibroblasts, activating profibrotic transcriptional programs and promoting myofibroblast differentiation [85]. This pathway provides a direct epigenetic link between cardiomyocyte metabolic state and fibroblast activation, extending the concept of metabolite-driven intercellular communication in cardiac fibrosis. Thus, cardiomyocyte metabolic state represents an important upstream regulator of the inflammatory environment that precedes fibrosis. When discussing cardiac metabolism and its effect on fibrosis, it would be remiss if we did not discuss the AMPK family kinases, which serve as central sensors of cellular energetic stress, integrating ATP depletion and metabolic imbalance with downstream transcriptional and signaling responses. The reader is directed to many reviews on AMPK and its role in cardiac metabolism, repair, and fibrosis [86,87]. The broad-spectrum nature of AMPK in cardiac function makes it a challenging target for drug discovery campaigns [88]. We posit that cardiac-specific AMPK family members are an alternative. For example, SNRK has emerged as a cardiomyocyte-enriched metabolic regulator that links cardiac metabolism to cardiac function. Further, SNRK is linked to preserving mitochondrial coupling efficiency to improve cardiac function [89]. In our own work, SNRK has been linked to Ang II non-ischemic heart failure conditions [43,48]. During embryonic development, global deletion of Snrk causes disruption of the embryonic heart substrate utilization and metabolic homeostasis [49] resulting in embryonic death at E17.5. Knocking out Snrk in cardiomyocytes specifically also leads to death at 9 months of age. These mice show defects in cardiac metabolism and cardiac functional parameters measured by ECHO (HPrEF) [48,49]. However, in these studies, we disrupted Snrk in embryonic cardiomyocytes at the start of development. So, whether adult cardiomyocyte-specific Snrk knockout models exhibit a similar cardiac metabolism and function phenotype is not known and represents the current limitation of these studies. Despite this gap, SNRK’s function as a protector of various metabolic processes in multiple tissues, including the heart, warrants further investigation into its potential as a therapeutic target for cardiac metabolism-induced heart failure. We support a model in which cardiomyocyte metabolic sensing pathways act upstream of inflammatory and fibrotic signaling networks. In this framework, disruption of metabolic checkpoints, such as those mediated by SNRK and related AMPK family kinases, can shift the myocardium toward a pro-inflammatory and profibrotic state, linking energetic stress to pathological tissue remodeling.
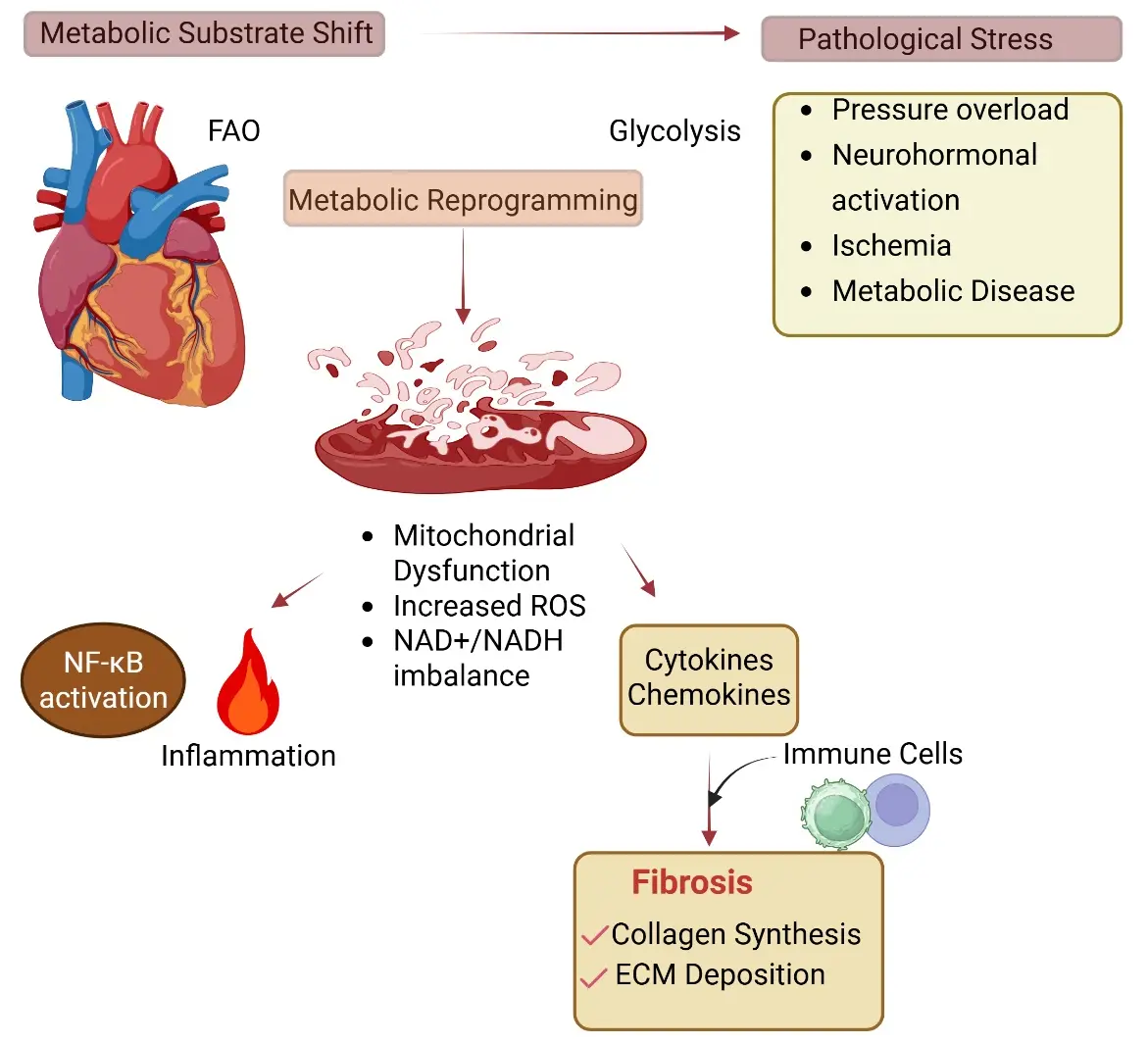
Figure 2. Cardiomyocyte Metabolism as a Regulator of Fibrotic Remodeling. Cardiomyocytes undergo metabolic reprogramming under pathological stress (pressure overload, neurohormonal activation, ischemia, and metabolic disease), characterized by a shift from fatty acid oxidation (FAO) toward glycolysis. This metabolic imbalance results in mitochondrial dysfunction, increased reactive oxygen species (ROS), and altered NAD+/NADH balance. These changes activate inflammatory signaling pathways, including NF-κB, leading to the production of cytokines and chemokines. Subsequent immune cell recruitment (arrow) and inflammatory amplification contribute to fibroblast activation and extracellular matrix deposition, culminating in cardiac fibrosis. Biorender was used to generate the figure.
4. TGF-β and Its Role in Cardiac Fibrosis
Among the signaling pathways involved, TGF-β remains the most extensively studied profibrotic mediator in the heart [14]. TGF-β family of ligands binds to specific transmembrane serine threonine kinase receptors (type I and type II). Upon ligand binding, heterotrimeric complexes of type I and II receptors form, leading to transphosphorylation of type 2 receptors, which, in turn, activate type I receptors. This phosphorylation cascades down to regulated (R)-SMAD transcription factor at two C-terminal serine residues, which activates it and leads to complex formation with a related Co-Smad molecule (SMAD4). This complex transports into the nucleus to activate downstream target genes. Although TGF-β family members have distinct functions, the canonical signaling converges into 5 type II receptors and 7 type I (also called activin receptor-like kinases—ALKs) receptors, and two main intracellular SMAD pathways (SMAD2/3 and SMAD1/5/8). All three TGF-β isoforms (TGF-β1, TGF-β2, and TGF-β3) are secreted in latent forms and need activation prior to interaction with signaling receptors. For a more comprehensive analysis of TGF-β signaling pathway, the reader is guided to other expansive reviews on this topic [14,90].
A large body of work from multiple laboratories has demonstrated that activation of the TGF-β1-Smad2/3 pathway promotes fibroblast-to-myofibroblast transition (Figure 3), characterized by induction of α-smooth muscle actin (α-SMA), enhanced extracellular matrix synthesis, and increased contractile capacity of fibroblasts [7,33,91,92]. TGF-β within the myocardium can originate from multiple cellular sources, including fibroblasts, infiltrating immune cells, endothelial cells, and cardiomyocytes [14,93]. Increasing evidence indicates that stressed cardiomyocytes contribute to TGF-β1 availability within the tissue microenvironment [94]. In models of pressure overload, neurohormonal stimulation, and atrial remodeling, stressed cardiomyocytes have been shown to release cytokines, growth factors, and matricellular proteins capable of activating neighboring fibroblasts [95]. Further, cardiomyocyte-derived mediators, including TGF-β family ligands, connective tissue growth factor (CTGF), and other secreted factors, can directly stimulate fibroblast activation (Figure 3). TGF-β can also influence cardiac inflammation directly by modulating immune and stromal cell behavior and indirectly by remodeling ECM and activating FBs [93,96]. Depending on the isoform secreted, TGF-β can also be protective in cardiac fibrosis. For example, in a pressure load situation, cardiomyocyte-derived TGF-β3 attenuates cardiac fibrosis and preserves cardiac function in HF [97]. TGF-β2 also plays a prominent role in inducing EndMT in cultured ECs [98,99]. Further, TGF-β1 and TGF-β3 were shown to induce TGF-β2 in ECs indicating the importance of TGF-β2 to EndMT [98,100]. Upon exposure to TGF-β, ECs transdifferentiate into myofibroblast-like cells (lose VE-cadherin, gain α-SMA and collagen), which becomes a source of fibrosis in the heart [101]. Previous work from our team has implicated Rho-associated kinase (ROCK) as a substrate of SNRK and is involved in SNRK-mediated cardiac function in mice [48]. ROCK in CMs acts as a critical amplifier of TGF-β signaling [102] by enhancing Smad nuclear activity, cytoskeletal tension, and ECM stiffening [103,104]. This promotes fibrosis (via collagen and ECM production) and inflammation (via NF-κB–mediated cytokine secretion).
In atrial remodeling models, cardiomyocyte stress has been associated with increased TGF-β expression and paracrine activation of fibroblast Smad signaling, contributing to diffuse interstitial fibrosis commonly observed in atrial fibrillation and HFpEF [105,106,107]. For example, mitogen-activated protein kinase kinase 4 (Mkk4), a critical component of the stress-activated mitogen-activated protein kinase family, when selectively inactivated in atrial cardiomyocyte Mkk4 [Mkk4(ACKO)], upregulates TGF-β1-mediated fibrosis. Thus, Mkk4 is a negative regulator of the TGF-β1 signaling associated with atrial remodeling [94]. Consistent with this concept, we also observed that SNRK, an AMPK family kinase enzyme, restrains cardiomyocyte-derived TGF-β1 secretion, thereby limiting fibroblast Smad2/3 activation and myofibroblast transition in non-ischemic remodeling models [44]. Conversely, TGF-β ligand family or specific TGF-β isoform [97] secreted by cardiomyocytes can protect fibrosis activation. For example, bone-morphogenetic protein-2 (BMP-2), when secreted from cardiomyocytes, has a potential beneficial effect in attenuating pressure overload-induced cardiac fibrosis [102]. In this mechanism, BMP-2 antagonizes TGF-β1-mediated Rho-associated kinase (ROCK) signaling in cardiac fibroblasts through Smurf1/Smad6 complex [102]. Collectively, emerging data from multiple studies suggest that cardiomyocytes are not merely reactive to fibrosis deposition but play a critical role in preventing fibrosis activation to protect the heart from further injury. These findings also suggest that cardiomyocyte metabolic stress can amplify established profibrotic pathways and position cardiomyocytes as active regulators of fibroblast behavior. Together, these observations support a model in which cardiomyocyte stress signals, both inflammatory and metabolic, are translated into fibrotic remodeling through paracrine communication with fibroblasts.
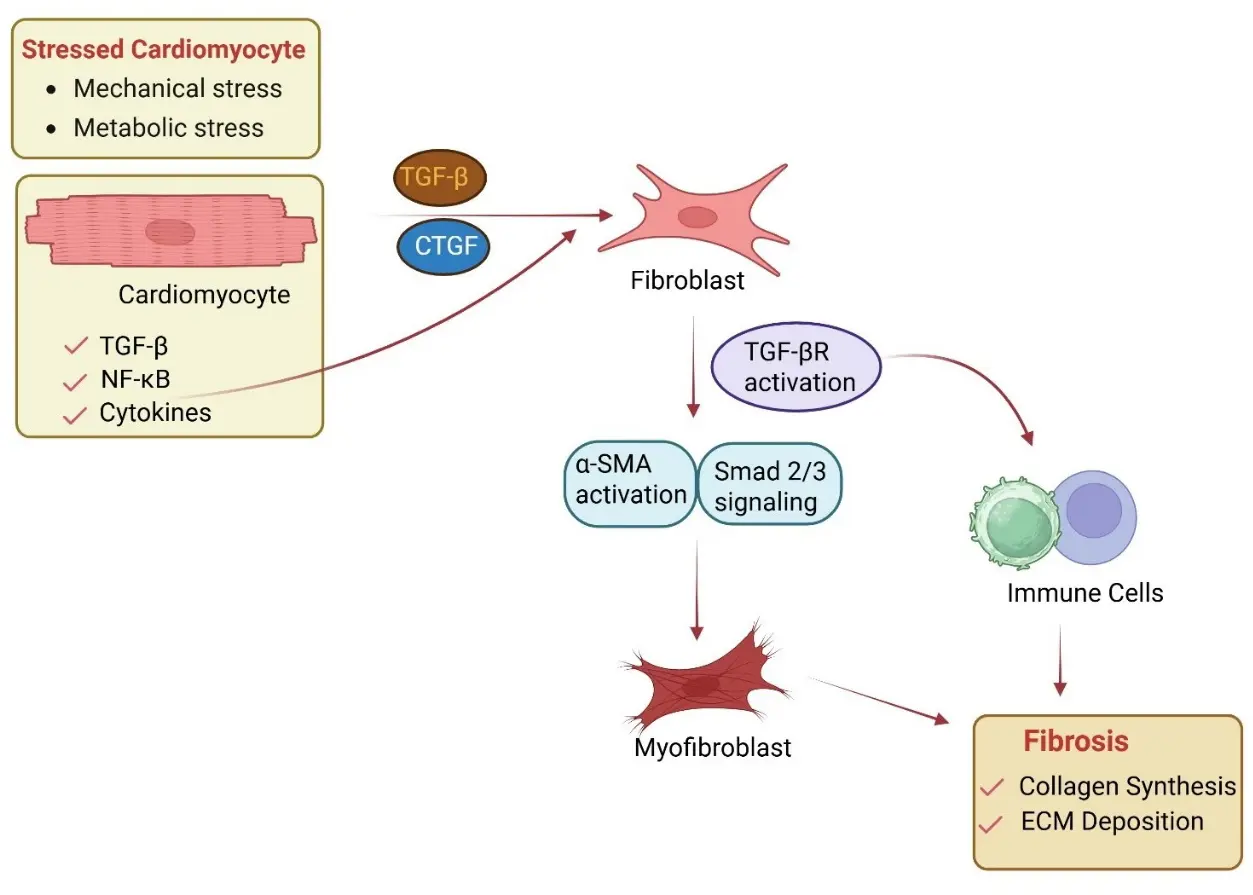
Figure 3. Paracrine Signaling Between Cardiomyocytes and Fibroblasts. Stressed cardiomyocytes release profibrotic mediators, including TGF-β, connective tissue growth factor (CTGF), and cytokines, which act on adjacent fibroblasts. These signals activate TGF-β receptor (TGF-βR) signaling and downstream Smad2/3 pathways, promoting α-smooth muscle actin (α-SMA) expression and myofibroblast differentiation. Myofibroblasts increase extracellular matrix (ECM) production, leading to fibrosis. Immune cells contribute to this process by enhancing profibrotic signaling through cytokine release. Biorender was used to generate the figure.
5. Ischemic Versus Non-Ischemic Fibrosis: Similarities and Differences
Cardiac fibrosis can arise through distinct biological pathways depending on the underlying disease context. In ischemic heart disease, fibrosis typically follows cardiomyocyte necrosis resulting from myocardial infarction. This process involves a well-defined sequence of events beginning with acute inflammation, followed by immune cell infiltration, fibroblast activation, and ultimately scar formation that preserves the structural integrity of the injured myocardium [4]. In contrast, non-ischemic fibrosis often develops without overt cardiomyocyte death. Instead, chronic cardiomyocyte stress, including pressure overload, metabolic dysfunction, neurohormonal activation, and oxidative injury, can drive sustained inflammatory signaling and fibroblast activation. In these conditions, inflammation and fibrosis frequently evolve in parallel rather than sequentially [4]. These distinctions are particularly evident in conditions such as hypertensive heart disease, HFpEF, and atrial fibrillation, where diffuse interstitial fibrosis emerges gradually and may precede measurable systolic dysfunction. Atrial fibrosis represents a notable example of this pattern. Compared with ventricular myocardium, atrial tissue appears more susceptible to metabolic and inflammatory perturbations, potentially reflecting differences in fibroblast populations, extracellular matrix composition, and mechanical stress. For example, overexpression of a mutant form of constitutively active TGF-β1 expression under cardiac-restricted α-myosin heavy chain promoter resulted in selective atrial chamber-specific fibrosis but not ventricular chamber fibrosis. This result is particularly intriguing given that TGF-β1 expression was expressed constitutively in both chambers [108]. Why this selectivity in fibrosis in atria vs. ventricle remains an interesting question. In the clinic, atrial fibrosis has been proposed as a precursor for promoting atrial fibrillation, a common arrhythmia associated with HF [109,110]. A recent single cell study of human heart revealed that atrium consists of 30.1% cardiomyocytes, 24.3% fibroblasts, while in ventricle, the proportions were 49.2% cardiomyocytes and 15.5% fibroblasts [24]. Thus, the higher density of fibroblasts in atria may contribute to this susceptibility. In the end, irrespective of the initial triggers for ischemic and non-ischemic fibrosis, which may differ, the downstream pathways that govern fibroblast activation, including inflammatory signaling, TGF-β activation, and extracellular matrix deposition, extensively overlap. Understanding how cardiomyocyte stress signaling interacts with these pathways may therefore provide insight into shared mechanisms that contribute to fibrotic remodeling in ischemic- and non-ischemic-driven heart failure.
6. Cardiac Fibrosis Targeting Strategies
The identification of genetic drivers of cardiac fibrosis, and the emergence of multi-omics studies (single-cell RNA and single-nucleus RNA-seq, proteomics, epigenetics, metabolomics), have all contributed to a rich source of targets and pathways for therapeutic intervention in cardiac fibrosis [111]. GWAS and whole exome sequencing studies have identified novel genetic variants in CRTAC1, CAPN1, UNC45A, UNC45B, and SMARCB1 in patients with sudden cardiac death, in which cardiac fibrosis observed by imaging is the key driving event [112,113,114,115]. Similarly, numerous fibrosis-related proteomic biomarkers in blood in HF have been discovered, especially those related to specific HF subtypes—HFrEF and HFpEF. A comprehensive list of proteomic biomarkers in cardiac fibrosis is available elsewhere [111]. As metabolic substrate alterations drive fibrotic remodeling, metabolomic profiling is yet another rich source of targets for cardiac fibrosis. For example, branched-chain amino acids (BCAAs), which play a significant role in cardiac remodeling and fibrosis progression, are one such target [116,117]. Similarly, the Framingham Heart study metabolomic profiling revealed elevated levels of kynurenine and aminoadipate that correlate with adverse structural remodeling [118]. These metabolite levels, along with protein levels in blood, serve as excellent surrogates for anti-fibrotic therapy responses in patients.
The next generation of targets will emerge from the integration of the multi-omics data. Some druggable candidates have already been revealed from the integration of GWAS and proteomic data. For example, CAMK2D, PRKD1, APOC3, TNFSF12, and MAPK3 have been identified as associated with fibroblast activation and fibrotic remodeling pathways [119]. More such integrations of datasets are likely to reveal additional targets for fibrosis, which will eventually lead to clinical trials. One recent example includes the identification of miR-132 through integration of transcriptomic and proteomic profiling of failing hearts. CDR132L, a locked nucleic-acid based antisense oligonucleotide inhibitor of miR-132, has shown efficacy in preclinical models of ischemic and non-ischemic HF [120,121], and is known to modulate the fibrotic and hypertrophic pathways via MEF2 and GATA3 transcription factors, respectively. A phase Ib clinical study with CDR132L has been initiated, demonstrating a good tolerance profile in chronic HF patients and an observed dose-dependent reduction in circulating miR-132 levels in plasma [122]. Antisense oligonucleotides (ASOs) and siRNA-based platforms offer sequence-specific silencing of fibrosis-promoting genes. Preclinical studies have demonstrated the efficacy of siRNA targeting HSP47 (SERPINH1) and CTGF/CCN2 in reducing myocardial collagen deposition [123,124]. ASOs targeting disease-relevant miRNAs, exemplified by CDR132L (anti-miR-132), currently in clinical development, have shown favorable safety and dose-dependent target reduction in HF patients. The therapeutic landscape for cardiac fibrosis continues to broaden with advances in nucleic acid therapeutics and small-molecule pharmacology. mRNA-based strategies, leveraging lipid nanoparticle delivery systems refined during the COVID-19 vaccine era, are being explored to transiently restore anti-fibrotic protein expression in the heart. Small molecule inhibitors targeting ALK5 (TGFβRI kinase), and ROCK1/2 proteins have shown anti-fibrotic activity in pressure overload and ischemic models [125,126]. Collectively, these emerging modalities complement the upstream cellular targeting strategies discussed above and point toward a multi-pronged therapeutic approach for cardiac fibrosis, one that may combine pathway-selective small molecules with nucleic acid therapeutics tailored to specific fibrotic pathways. Thus, there is great promise in the next generation of therapeutics for cardiac fibrosis.
While such approaches are promising, we think that strategies targeting fibrosis prior to its initiation, i.e., upstream drivers of pathological remodeling, are equally important. For example, cardiomyocyte stress signaling may represent an earlier intervention point in the fibrotic cascade. In this strategy, we are targeting an indirect source of fibrosis, which is the cardiomyocyte. Similarly, metabolic dysregulation, mitochondrial dysfunction, and inflammatory activation within cardiomyocytes can generate signals that subsequently recruit immune cells and activate fibroblasts. Intervening at this level could potentially prevent amplification of the fibrotic response. For example, an engineered chimeric antigen receptor T cells that target FAP can effectively reduce fibroblast activation [72]. IL-1β is a similar target that functions at the immune-fibroblast junction [71]. This strategy offers the benefit of not broadly suppressing fibroblast activity, which may be essential for other normal physiological repair processes, but rather selectively targets the cardiomyocyte-immune interface or immune-fibroblast interface or other cell-cell interface that influences cardiac fibrosis.
7. Knowledge Gaps and Future Directions
Despite significant advances in understanding the cellular and molecular drivers of cardiac fibrosis, several important questions remain. First, the temporal window during which cardiomyocyte-driven inflammatory and fibrotic signaling remains reversible is not well defined. Identifying early metabolic or inflammatory markers of fibrotic initiation may help determine when therapeutic intervention is most effective. Second, emerging evidence suggests that atrial and ventricular cardiomyocytes may differ in their responses to metabolic and inflammatory stress. Understanding how chamber-specific cardiomyocyte signaling contributes to regional susceptibility to fibrosis will be particularly important in conditions such as atrial fibrillation and HFpEF. Third, the influence of biological variables, including sex, aging, obesity, and metabolic disease, on cardiomyocyte inflammatory signaling remains incompletely understood. These factors likely modulate the threshold at which cardiomyocyte stress transitions into chronic inflammatory and fibrotic remodeling. Fourth, translating insights from experimental models to human disease remains a major challenge. Integrating emerging technologies (cardiac organoids with vasculature and flow) with mechanistic studies will be critical for identifying new therapeutic targets and developing strategies to prevent or reverse cardiac fibrosis. Finally, the era of AI is upon us. How AI can be effectively used to guide experimental and translational studies or to integrate into current workflows to make cardiac fibrosis clinical management more effective is still to be determined and remains the next frontier.
Author Contributions
K.T.: Conceptualization, writing, and editing. R.R.: Writing and editing, funding acquisition, supervision, and resources.
Ethics Statement
Not applicable.
Informed Consent Statement
All studies references in this article have adhered to best practices for human and animal work, and were performed under approved protocols. Informed consent was obtained from all subjects involved in the referenced studies.
Data Availability Statement
All data references in this article are from the original papers that were cited. Data availability is included in the original papers.
Funding
R.R. and K.T. are partly funded through NIH grants R33 HL154254 and HL179583. R.R. and K.T. are also supported by program funds provided by Children’s Research Institute, and Department of Pediatrics, MCW.
Declaration of Competing Interest
Both authors in this manuscript do not have competing financial interest associated with the content in this manuscript.
References
- Fonarow GC, Ahmad FS, Ahmad T, Albert NM, Alexander KM, Baker WL, et al. HF STATS 2025: Heart Failure Epidemiology and Outcomes Statistics An Updated 2025 Report from the Heart Failure Society of America. J. Card. Fail. 2026, 32, 439–498. DOI:10.1016/j.cardfail.2025.07.007 [Google Scholar]
- BaniHani HA, Khaled LH, Al Sharaa NM, Al Saleh RA, Bin Ghalaita AK, Bin Sulaiman AS, et al. Causes, Diagnosis, Treatment, and Prognosis of Cardiac Fibrosis: A Systematic Review. Cureus 2025, 17, e81264. DOI:10.7759/cureus.81264 [Google Scholar]
- Frangogiannis NG. The Extracellular Matrix in Ischemic and Nonischemic Heart Failure. Circ. Res. 2019, 125, 117–146. DOI:10.1161/CIRCRESAHA.119.311148 [Google Scholar]
- Bacmeister L, Schwarzl M, Warnke S, Stoffers B, Blankenberg S, Westermann D, et al. Inflammation and fibrosis in murine models of heart failure. Basic Res. Cardiol. 2019, 114, 19. DOI:10.1007/s00395-019-0722-5 [Google Scholar]
- Zhang YZ, Wu Y, Li MJ, Mijiti A, Cheng LF. Identification of macrophage driver genes in fibrosis caused by different heart diseases based on omics integration. J. Transl. Med. 2024, 22, 839. DOI:10.1186/s12967-024-05624-7 [Google Scholar]
- Dolmatova E, Spagnol G, Boassa D, Baum JR, Keith K, Ambrosi C, et al. Cardiomyocyte ATP release through pannexin 1 aids in early fibroblast activation. Am. J. Physiol. Heart Circ. Physiol. 2012, 303, H1208–H1218. DOI:10.1152/ajpheart.00251.2012 [Google Scholar]
- Koitabashi N, Danner T, Zaiman AL, Pinto YM, Rowell J, Mankowski J, et al. Pivotal role of cardiomyocyte TGF-beta signaling in the murine pathological response to sustained pressure overload. J. Clin. Investig. 2011, 121, 2301–2312. DOI:10.1172/JCI44824 [Google Scholar]
- Camelliti P, Borg TK, Kohl P. Structural and functional characterisation of cardiac fibroblasts. Cardiovasc. Res. 2005, 65, 40–51. DOI:10.1016/j.cardiores.2004.08.020 [Google Scholar]
- Pinto AR, Ilinykh A, Ivey MJ, Kuwabara JT, D’Antoni ML, Debuque R, et al. Revisiting Cardiac Cellular Composition. Circ. Res. 2016, 118, 400–409. DOI:10.1161/CIRCRESAHA.115.307778 [Google Scholar]
- Tallquist MD. Cardiac fibroblasts: From origin to injury. Curr. Opin. Physiol. 2018, 1, 75–79. DOI:10.1016/j.cophys.2017.08.002 [Google Scholar]
- Tallquist MD, Molkentin JD. Redefining the identity of cardiac fibroblasts. Nat. Rev. Cardiol. 2017, 14, 484–491. DOI:10.1038/nrcardio.2017.57 [Google Scholar]
- Souders CA, Bowers SL, Baudino TA. Cardiac fibroblast: The renaissance cell. Circ. Res. 2009, 105, 1164–1176. DOI:10.1161/CIRCRESAHA.109.209809 [Google Scholar]
- Li P, Wang D, Lucas J, Oparil S, Xing D, Cao X, et al. Atrial natriuretic peptide inhibits transforming growth factor beta-induced Smad signaling and myofibroblast transformation in mouse cardiac fibroblasts. Circ. Res. 2008, 102, 185–192. DOI:10.1161/CIRCRESAHA.107.157677 [Google Scholar]
- Massague J, Sheppard D. TGF-beta signaling in health and disease. Cell 2023, 186, 4007–4037. DOI:10.1016/j.cell.2023.07.036 [Google Scholar]
- Chen F, Lyu L, Xing C, Chen Y, Hu S, Wang M, et al. The pivotal role of TGF-beta/Smad pathway in fibrosis pathogenesis and treatment. Front. Oncol. 2025, 15, 1649179. DOI:10.3389/fonc.2025.1649179 [Google Scholar]
- Bertaud A, Joshkon A, Heim X, Bachelier R, Bardin N, Leroyer AS, et al. Signaling Pathways and Potential Therapeutic Strategies in Cardiac Fibrosis. Int. J. Mol. Sci. 2023, 24, 1756. DOI:10.3390/ijms24021756 [Google Scholar]
- Improta-Caria AC, Rodrigues LF, Joaquim VHA, De Sousa RAL, Fernandes T, Oliveira EM. MicroRNAs regulating signaling pathways in cardiac fibrosis: Potential role of the exercise training. Am. J. Physiol. Heart Circ. Physiol. 2024, 326, H497–H510. DOI:10.1152/ajpheart.00410.2023 [Google Scholar]
- Kittana N. Calcium Signaling in Cardiac Fibroblasts: Roles in Fibrosis and Therapeutic Implications. Cardiovasc. Drugs Ther. 2026, 40, 393–410. DOI:10.1007/s10557-025-07699-w [Google Scholar]
- Ali SR, Ranjbarvaziri S, Talkhabi M, Zhao P, Subat A, Hojjat A, et al. Developmental heterogeneity of cardiac fibroblasts does not predict pathological proliferation and activation. Circ. Res. 2014, 115, 625–635. DOI:10.1161/CIRCRESAHA.115.303794 [Google Scholar]
- Cui Y, Zheng Y, Liu X, Yan L, Fan X, Yong J, et al. Single-Cell Transcriptome Analysis Maps the Developmental Track of the Human Heart. Cell Rep. 2019, 26, 1934–1950.e5. DOI:10.1016/j.celrep.2019.01.079 [Google Scholar]
- DeLaughter DM, Bick AG, Wakimoto H, McKean D, Gorham JM, Kathiriya IS, et al. Single-Cell Resolution of Temporal Gene Expression during Heart Development. Dev. Cell 2016, 39, 480–490. DOI:10.1016/j.devcel.2016.10.001 [Google Scholar]
- Hernandez SC, Ainciburu M, Sudupe L, Planell N, Lopez-Moreno M, Vilas-Zornoza A, et al. Single-cell and spatial transcriptomic profiling of cardiac fibroblasts following myocardial infarction. Sci. Data 2026, 13, 216. DOI:10.1038/s41597-025-06533-0 [Google Scholar]
- Lanzer JD, Wienecke LM, Ramirez Flores RO, Zylla MM, Kley C, Hartmann N, et al. Single-cell transcriptomics reveal distinctive patterns of fibroblast activation in heart failure with preserved ejection fraction. Basic Res. Cardiol. 2024, 119, 1001–1028. DOI:10.1007/s00395-024-01074-w [Google Scholar]
- Litviňuková M, Talavera-López C, Maatz H, Reichart D, Worth CL, Lindberg EL, et al. Cells of the adult human heart. Nature 2020, 588, 466–472. DOI:10.1038/s41586-020-2797-4 [Google Scholar]
- Wolfien M, Galow AM, Muller P, Bartsch M, Brunner RM, Goldammer T, et al. Single-Nucleus Sequencing of an Entire Mammalian Heart: Cell Type Composition and Velocity. Cells 2020, 9, 318. DOI:10.3390/cells9020318 [Google Scholar]
- McLellan MA, Skelly DA, Dona MSI, Squiers GT, Farrugia GE, Gaynor TL, et al. High-Resolution Transcriptomic Profiling of the Heart During Chronic Stress Reveals Cellular Drivers of Cardiac Fibrosis and Hypertrophy. Circulation 2020, 142, 1448–1463. DOI:10.1161/CIRCULATIONAHA.119.045115 [Google Scholar]
- Rahman MT, Muppala S, Wu J, Krukovets I, Solovjev D, Verbovetskiy D, et al. Effects of thrombospondin-4 on pro-inflammatory phenotype differentiation and apoptosis in macrophages. Cell Death Dis. 2020, 11, 53. DOI:10.1038/s41419-020-2237-2 [Google Scholar]
- Peisker F, Halder M, Nagai J, Ziegler S, Kaesler N, Hoeft K, et al. Mapping the cardiac vascular niche in heart failure. Nat. Commun. 2022, 13, 3027. DOI:10.1038/s41467-022-30682-0 [Google Scholar]
- Burstein B, Libby E, Calderone A, Nattel S. Differential behaviors of atrial versus ventricular fibroblasts: A potential role for platelet-derived growth factor in atrial-ventricular remodeling differences. Circulation 2008, 117, 1630–1641. DOI:10.1161/CIRCULATIONAHA.107.748053 [Google Scholar]
- Saljic A, Grandi E, Dobrev D. TGF-β1-induced endothelial-mesenchymal transition: A potential contributor to fibrotic remodeling in atrial fibrillation? J. Clin. Investig. 2022, 132, e161070. DOI:10.1172/JCI161070 [Google Scholar]
- Tsai CT, Chiang FT, Tseng CD, Yu CC, Wang YC, Lai LP, et al. Mechanical stretch of atrial myocyte monolayer decreases sarcoplasmic reticulum calcium adenosine triphosphatase expression and increases susceptibility to repolarization alternans. J. Am. Coll. Cardiol. 2011, 58, 2106–2115. DOI:10.1016/j.jacc.2011.07.039 [Google Scholar]
- Wyndham CRC. Atrial fibrillation—The most common arrhythmia. Tex. Heart Inst. J. 2000, 27, 257–267. Available online: https://pmc.ncbi.nlm.nih.gov/articles/PMC101077/ (accessed on 1 January 2026).
- Sygitowicz G, Maciejak-Jastrzebska A, Sitkiewicz D. A Review of the Molecular Mechanisms Underlying Cardiac Fibrosis and Atrial Fibrillation. J. Clin. Med. 2021, 10, 4430. DOI:10.3390/jcm10194430 [Google Scholar]
- Glezeva N, Moran B, Collier P, Moravec CS, Phelan D, Donnellan E, et al. Targeted DNA Methylation Profiling of Human Cardiac Tissue Reveals Novel Epigenetic Traits and Gene Deregulation Across Different Heart Failure Patient Subtypes. Circ. Heart Fail. 2019, 12, e005765. DOI:10.1161/CIRCHEARTFAILURE.118.005765 [Google Scholar]
- Zhuang T, Chen MH, Wu RX, Wang J, Hu XD, Meng T, et al. ALKBH5-mediated m6A modification of IL-11 drives macrophage-to-myofibroblast transition and pathological cardiac fibrosis in mice. Nat. Commun. 2024, 15, 1995. DOI:10.1038/s41467-024-46357-x [Google Scholar]
- Zhang QJ, Tran TAT, Wang M, Ranek MJ, Kokkonen-Simon KM, Gao J, et al. Histone lysine dimethyl-demethylase KDM3A controls pathological cardiac hypertrophy and fibrosis. Nat. Commun. 2018, 9, 5230. DOI:10.1038/s41467-018-07173-2 [Google Scholar]
- Bowers SLK, Meng Q, Molkentin JD. Fibroblasts orchestrate cellular crosstalk in the heart through the ECM. Nat. Cardiovasc. Res. 2022, 1, 312–321. DOI:10.1038/s44161-022-00043-7 [Google Scholar]
- Farbehi N, Patrick R, Dorison A, Xaymardan M, Janbandhu V, Wystub-Lis K, et al. Single-cell expression profiling reveals dynamic flux of cardiac stromal, vascular and immune cells in health and injury. Elife 2019, 8, e43882. DOI:10.7554/eLife.43882 [Google Scholar]
- Zhou P, Pu WT. Recounting Cardiac Cellular Composition. Circ. Res. 2016, 118, 368–370. DOI:10.1161/CIRCRESAHA.116.308139 [Google Scholar]
- Munch J, Abdelilah-Seyfried S. Sensing and Responding of Cardiomyocytes to Changes of Tissue Stiffness in the Diseased Heart. Front. Cell Dev. Biol. 2021, 9, 642840. DOI:10.3389/fcell.2021.642840 [Google Scholar]
- Venugopal H, Frangogiannis NG. Cardiomyocytes regulate Inflammation in the failing heart. Am. J. Physiol. Heart Circ. Physiol. 2025, 329, H1013–H1015. DOI:10.1152/ajpheart.00529.2025 [Google Scholar]
- Ninh VK, Barlow M, Aydin S, Brand CS, Yu J, Smith J, et al. Cardiomyocyte YAP represses myocardial inflammation and fibrosis and restrains MEF2-regulated gene expression. Am. J. Physiol. Heart Circ. Physiol. 2025, 329, H774–H787. DOI:10.1152/ajpheart.00799.2024 [Google Scholar]
- Thirugnanam K, Cossette SM, Lu Q, Chowdhury SR, Harmann LM, Gupta A, et al. Cardiomyocyte-Specific Snrk Prevents Inflammation in the Heart. J. Am. Heart Assoc. 2019, 8, e012792. DOI:10.1161/JAHA.119.012792 [Google Scholar]
- Thirugnanam K, Rizvi F, Jahangir A, Homar P, Shabnam F, Palecek SP, et al. SNRK facilitates cardiac repair associated with nonischemic fibrosis: Regulating transforming growth factor-beta1 levels in atrial cardiomyocytes. Regen. Med. Rep. 2025, 2, 45–52. DOI:10.4103/regenmed.regenmed-d-25-00009 [Google Scholar]
- Zhang H, Dhalla NS. The Role of Pro-Inflammatory Cytokines in the Pathogenesis of Cardiovascular Disease. Int. J. Mol. Sci. 2024, 25, 1082. DOI:10.3390/ijms25021082 [Google Scholar]
- Haque ZK, Wang DZ. How cardiomyocytes sense pathophysiological stresses for cardiac remodeling. Cell Mol. Life Sci. 2017, 74, 983–1000. DOI:10.1007/s00018-016-2373-0 [Google Scholar]
- Pietrangelo D, Lopa C, Litterio M, Cotugno M, Rubattu S, Lombardi A. Metabolic Disturbances Involved in Cardiovascular Diseases: The Role of Mitochondrial Dysfunction, Altered Bioenergetics and Oxidative Stress. Int. J. Mol. Sci. 2025, 26, 6791. DOI:10.3390/ijms26146791 [Google Scholar]
- Cossette SM, Bhute VJ, Bao X, Harmann LM, Horswill MA, Sinha I, et al. Sucrose Nonfermenting-Related Kinase Enzyme-Mediated Rho-Associated Kinase Signaling is Responsible for Cardiac Function. Circ. Cardiovasc. Genet. 2016, 9, 474–486. DOI:10.1161/CIRCGENETICS.116.001515 [Google Scholar]
- Cossette SM, Gastonguay AJ, Bao X, Lerch-Gaggl A, Zhong L, Harmann LM, et al. Sucrose non-fermenting related kinase enzyme is essential for cardiac metabolism. Biol. Open 2014, 4, 48–61. DOI:10.1242/bio.20149811 [Google Scholar]
-
Adami E, Viswanathan S, Widjaja AA, Ng B, Chothani S, Zhihao N, et al. IL11 is elevated in systemic sclerosis and IL11-dependent ERK signalling underlies TGFbeta-mediated activation of dermal fibroblasts. Rheumatology 2021, 60, 5820–5826. DOI:10.1093/rheumatology/keab168 [Google Scholar]
-
Thirugnanam K, Ramchandran R. SNRK: A metabolic regulator with multifaceted role in development and disease. Vessel Plus 2020, 4, 26. DOI:10.20517/2574-1209.2020.18 [Google Scholar]
- Feng W, Chen L, Nguyen PK, Wu SM, Li G. Single Cell Analysis of Endothelial Cells Identified Organ-Specific Molecular Signatures and Heart-Specific Cell Populations and Molecular Features. Front. Cardiovasc. Med. 2019, 6, 165. DOI:10.3389/fcvm.2019.00165 [Google Scholar]
- Murdoch CE, Chaubey S, Zeng L, Yu B, Ivetic A, Walker SJ, et al. Endothelial NADPH oxidase-2 promotes interstitial cardiac fibrosis and diastolic dysfunction through proinflammatory effects and endothelial-mesenchymal transition. J. Am. Coll. Cardiol. 2014, 63, 2734–2741. DOI:10.1016/j.jacc.2014.02.572 [Google Scholar]
- Joshi D, Coon BG, Chakraborty R, Deng H, Yang Z, Babar MU, et al. Endothelial gamma-protocadherins inhibit KLF2 and KLF4 to promote atherosclerosis. Nat. Cardiovasc. Res. 2024, 3, 1035–1048. DOI:10.1038/s44161-024-00522-z [Google Scholar]
- Yan X, Wang R, Xu H, Tao Z, Ling J. The Mechanisms Associated with Inflammation and Coronary Microvascular Dysfunction in Heart Failure with Preserved Ejection Fraction. Med. Princ. Pract. 2025, 35, 101–113. DOI:10.1159/000548233 [Google Scholar]
- Mohammed SF, Hussain S, Mirzoyev SA, Edwards WD, Maleszewski JJ, Redfield MM. Coronary microvascular rarefaction and myocardial fibrosis in heart failure with preserved ejection fraction. Circulation 2015, 131, 550–559. DOI:10.1161/CIRCULATIONAHA.114.009625 [Google Scholar]
- Zeisberg EM, Tarnavski O, Zeisberg M, Dorfman AL, McMullen JR, Gustafsson E, et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat. Med. 2007, 13, 952–961. DOI:10.1038/nm1613 [Google Scholar]
- Le D. Endothelial-to-Mesenchymal Transition in Post-Myocardial Infarction Fibrosis: A Maladaptive but Targetable Pathway. EJIFCC 2026, 37, 26–41. Available online: https://pmc.ncbi.nlm.nih.gov/articles/PMC12882090/ (accessed on 1 January 2026).
- Ivey MJ, Kuwabara JT, Pai JT, Moore RE, Sun Z, Tallquist MD. Resident fibroblast expansion during cardiac growth and remodeling. J. Mol. Cell Cardiol. 2018, 114, 161–174. DOI:10.1016/j.yjmcc.2017.11.012 [Google Scholar]
- Kovacic JC, Dimmeler S, Harvey RP, Finkel T, Aikawa E, Krenning G, et al. Endothelial to Mesenchymal Transition in Cardiovascular Disease: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2019, 73, 190–209. DOI:10.1016/j.jacc.2018.09.089 [Google Scholar]
- Zhang S, Li Y, Huang X, Liu K, Wang QD, Chen AF, et al. Seamless Genetic Recording of Transiently Activated Mesenchymal Gene Expression in Endothelial Cells During Cardiac Fibrosis. Circulation 2021, 144, 2004–2020. DOI:10.1161/CIRCULATIONAHA.121.055417 [Google Scholar]
- Tombor LS, John D, Glaser SF, Luxan G, Forte E, Furtado M, et al. Single cell sequencing reveals endothelial plasticity with transient mesenchymal activation after myocardial infarction. Nat. Commun. 2021, 12, 681. DOI:10.1038/s41467-021-20905-1 [Google Scholar]
- Li Y, Nie Y, Helou Y, Ding G, Feng B, Xu G, et al. Identification of sucrose non-fermenting-related kinase (SNRK) as a suppressor of adipocyte inflammation. Diabetes 2013, 62, 2396–2409. DOI:10.2337/db12-1081 [Google Scholar]
- Simoes FC, Cahill TJ, Kenyon A, Gavriouchkina D, Vieira JM, Sun X, et al. Macrophages directly contribute collagen to scar formation during zebrafish heart regeneration and mouse heart repair. Nat. Commun. 2020, 11, 600. DOI:10.1038/s41467-019-14263-2 [Google Scholar]
- Toba H, Cannon PL, Yabluchanskiy A, Iyer RP, D’Armiento J, Lindsey ML. Transgenic overexpression of macrophage matrix metalloproteinase-9 exacerbates age-related cardiac hypertrophy, vessel rarefaction, inflammation, and fibrosis. Am. J. Physiol. Heart Circ. Physiol. 2017, 312, H375–H383. DOI:10.1152/ajpheart.00633.2016 [Google Scholar]
- Shen SC, Xu J, Cheng C, Xiang XJ, Hong BY, Zhang M, et al. Macrophages promote the transition from myocardial ischemia reperfusion injury to cardiac fibrosis in mice through GMCSF/CCL2/CCR2 and phenotype switching. Acta Pharmacol. Sin. 2024, 45, 959–974. DOI:10.1038/s41401-023-01222-3 [Google Scholar]
- Parthiban P, Barrow F, Wang H, Chalise U, Araujo N, Souza-Neto F, et al. Macrophage-Derived CCL24 Promotes Cardiac Fibrosis Via Fibroblast CCR3. Circ. Res. 2025, 137, 1140–1156. DOI:10.1161/CIRCRESAHA.125.326599 [Google Scholar]
- Zhang XZ, Li QL, Tang TT, Cheng X. Emerging Role of Macrophage-Fibroblast Interactions in Cardiac Homeostasis and Remodeling. JACC Basic Transl. Sci. 2025, 10, 113–127. DOI:10.1016/j.jacbts.2024.06.003 [Google Scholar]
- Frangogiannis N. Transforming growth factor-beta in tissue fibrosis. J. Exp. Med. 2020, 217, e20190103. DOI:10.1084/jem.20190103 [Google Scholar]
- Chen B, Brickshawana A, Frangogiannis NG. The Functional Heterogeneity of Resident Cardiac Macrophages in Myocardial Injury: CCR2+ Cells Promote Inflammation, Whereas CCR2− Cells Protect. Circ. Res. 2019, 124, 183–185. DOI:10.1161/CIRCRESAHA.118.314357 [Google Scholar]
- Amrute JM, Luo X, Penna V, Yang S, Yamawaki T, Hayat S, et al. Targeting immune-fibroblast cell communication in heart failure. Nature 2024, 635, 423–433. DOI:10.1038/s41586-024-08008-5 [Google Scholar]
- Rurik JG, Tombacz I, Yadegari A, Mendez Fernandez PO, Shewale SV, Li L, et al. CAR T cells produced in vivo to treat cardiac injury. Science 2022, 375, 91–96. DOI:10.1126/science.abm0594 [Google Scholar]
- Willeford A, Suetomi T, Nickle A, Hoffman HM, Miyamoto S, Heller Brown J. CaMKIIδ-mediated inflammatory gene expression and inflammasome activation in cardiomyocytes initiate inflammation and induce fibrosis. JCI Insight 2018, 3, e97054. DOI:10.1172/jci.insight.97054 [Google Scholar]
- Feng Y, Chen H, Cai J, Zou L, Yan D, Xu G, et al. Cardiac RNA induces inflammatory responses in cardiomyocytes and immune cells via Toll-like receptor 7 signaling. J. Biol. Chem. 2015, 290, 26688–26698. DOI:10.1074/jbc.M115.661835 [Google Scholar]
- Wang L, Zhang YL, Lin QY, Liu Y, Guan XM, Ma XL, et al. CXCL1-CXCR2 axis mediates angiotensin II-induced cardiac hypertrophy and remodelling through regulation of monocyte infiltration. Eur. Heart J. 2018, 39, 1818–1831. DOI:10.1093/eurheartj/ehy085 [Google Scholar]
- Bergers G, Song S. The role of pericytes in blood-vessel formation and maintenance. Neuro-Oncol. 2005, 7, 452–464. DOI:10.1215/S1152851705000232 [Google Scholar]
- Alex L, Tuleta I, Hernandez SC, Hanna A, Venugopal H, Astorkia M, et al. Cardiac Pericytes Acquire a Fibrogenic Phenotype and Contribute to Vascular Maturation After Myocardial Infarction. Circulation 2023, 148, 882–898. DOI:10.1161/CIRCULATIONAHA.123.064155 [Google Scholar]
- Alex L, Tuleta I, Harikrishnan V, Frangogiannis NG. Validation of Specific and Reliable Genetic Tools to Identify, Label, and Target Cardiac Pericytes in Mice. J. Am. Heart Assoc. 2022, 11, e023171. DOI:10.1161/JAHA.121.023171 [Google Scholar]
- Kuppe C, Ramirez Flores RO, Li Z, Hayat S, Levinson RT, Liao X, et al. Spatial multi-omic map of human myocardial infarction. Nature 2022, 608, 766–777. DOI:10.1038/s41586-022-05060-x [Google Scholar]
- Fonseka O, Gare SR, Chen X, Zhang J, Alatawi NH, Ross C, et al. Molecular Mechanisms Underlying Heart Failure and Their Therapeutic Potential. Cells 2025, 14, 324. DOI:10.3390/cells14050324 [Google Scholar]
- Umbarawan Y, Syamsunarno M, Koitabashi N, Yamaguchi A, Hanaoka H, Hishiki T, et al. Glucose is preferentially utilized for biomass synthesis in pressure-overloaded hearts: Evidence from fatty acid-binding protein-4 and -5 knockout mice. Cardiovasc. Res. 2018, 114, 1132–1144. DOI:10.1093/cvr/cvy063 [Google Scholar]
- He L, Zhang D, Zhang S, Ren Z, Huangfu Y, Cheng X, et al. Dapagliflozin mitigates myocardial inflammation and metabolic stress in heart failure through STAT1 inhibition: Evidence from multi-omics analyses and experimental exploration. PLoS ONE 2026, 21, e0343296. DOI:10.1371/journal.pone.0343296 [Google Scholar]
- Pliouta L, Lampsas S, Kountouri A, Korakas E, Thymis J, Kassi E, et al. Mitochondrial Dysfunction in the Development and Progression of Cardiometabolic Diseases: A Narrative Review. J. Clin. Med. 2025, 14, 3706. DOI:10.3390/jcm14113706 [Google Scholar]
- Yang HM. Mitochondrial Dysfunction in Cardiovascular Diseases. Int. J. Mol. Sci. 2025, 26, 1917. DOI:10.3390/ijms26051917 [Google Scholar]
-
Fang N, Zhang N, Jiang X, Yan S, Wang Z, Gao Q, et al. PFKM-Driven Lactate Overproduction Promotes Atrial Fibrillation via Triggering Cardiac Fibroblasts Histone Lactylation. Adv. Sci. 2025, 12, e00963. DOI:10.1002/advs.202500963 [Google Scholar]
- Daskalopoulos EP, Dufeys C, Bertrand L, Beauloye C, Horman S. AMPK in cardiac fibrosis and repair: Actions beyond metabolic regulation. J. Mol. Cell Cardiol. 2016, 91, 188–200. DOI:10.1016/j.yjmcc.2016.01.001 [Google Scholar]
- Heidrich F, Schotola H, Popov AF, Sohns C, Schuenemann J, Friedrich M, et al. AMPK—Activated Protein Kinase and its Role in Energy Metabolism of the Heart. Curr. Cardiol. Rev. 2010, 6, 337–342. DOI:10.2174/157340310793566073 [Google Scholar]
- Kim M, Tian R. Targeting AMPK for cardiac protection: Opportunities and challenges. J. Mol. Cell Cardiol. 2011, 51, 548–553. DOI:10.1016/j.yjmcc.2010.12.004 [Google Scholar]
- Rines AK, Chang HC, Wu R, Sato T, Khechaduri A, Kouzu H, et al. Snf1-related kinase improves cardiac mitochondrial efficiency and decreases mitochondrial uncoupling. Nat. Commun. 2017, 8, 14095. DOI:10.1038/ncomms14095 [Google Scholar]
- ten Dijke P, Arthur HM. Extracellular control of TGFbeta signalling in vascular development and disease. Nat. Rev. Mol. Cell Biol. 2007, 8, 857–869. DOI:10.1038/nrm2262 [Google Scholar]
- Rainer PP, Hao S, Vanhoutte D, Lee DI, Koitabashi N, Molkentin JD, et al. Cardiomyocyte-specific transforming growth factor beta suppression blocks neutrophil infiltration, augments multiple cytoprotective cascades, and reduces early mortality after myocardial infarction. Circ. Res. 2014, 114, 1246–1257. DOI:10.1161/CIRCRESAHA.114.302653 [Google Scholar]
- Tsai CT, Tseng CD, Hwang JJ, Wu CK, Yu CC, Wang YC, et al. Tachycardia of atrial myocytes induces collagen expression in atrial fibroblasts through transforming growth factor beta1. Cardiovasc. Res. 2011, 89, 805–815. DOI:10.1093/cvr/cvq322 [Google Scholar]
- Vallee A, Lecarpentier Y. TGF-beta in fibrosis by acting as a conductor for contractile properties of myofibroblasts. Cell Biosci. 2019, 9, 98. DOI:10.1186/s13578-019-0362-3 [Google Scholar]
- Davies L, Jin J, Shen W, Tsui H, Shi Y, Wang Y, et al. Mkk4 is a negative regulator of the transforming growth factor beta 1 signaling associated with atrial remodeling and arrhythmogenesis with age. J. Am. Heart Assoc. 2014, 3, e000340. DOI:10.1161/JAHA.113.000340 [Google Scholar]
- Russo I, Cavalera M, Huang S, Su Y, Hanna A, Chen B, et al. Protective Effects of Activated Myofibroblasts in the Pressure-Overloaded Myocardium Are Mediated Through Smad-Dependent Activation of a Matrix-Preserving Program. Circ. Res. 2019, 124, 1214–1227. DOI:10.1161/CIRCRESAHA.118.314438 [Google Scholar]
- Saadat S, Noureddini M, Mahjoubin-Tehran M, Nazemi S, Shojaie L, Aschner M, et al. Pivotal Role of TGF-beta/Smad Signaling in Cardiac Fibrosis: Non-coding RNAs as Effectual Players. Front. Cardiovasc. Med. 2020, 7, 588347. DOI:10.3389/fcvm.2020.588347 [Google Scholar]
- Xuan J, Zhou J, Huang Y, Huang M, Pu S, Fang L, et al. Cardiomyocyte-derived TGFB3 attenuates cardiac fibrosis and preserves cardiac function in heart failure. Sci. Rep. 2026, 16, 11534. DOI:10.1038/s41598-026-42367-5 [Google Scholar]
- Sabbineni H, Verma A, Somanath PR. Isoform-specific effects of transforming growth factor beta on endothelial-to-mesenchymal transition. J. Cell. Physiol. 2018, 233, 8418–8428. DOI:10.1002/jcp.26801 [Google Scholar]
- Alvandi Z, Bischoff J. Endothelial-Mesenchymal Transition in Cardiovascular Disease. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 2357–2369. DOI:10.1161/ATVBAHA.121.313788 [Google Scholar]
- Bischoff J, Casanovas G, Wylie-Sears J, Kim DH, Bartko PE, Guerrero JL, et al. CD45 Expression in Mitral Valve Endothelial Cells After Myocardial Infarction. Circ. Res. 2016, 119, 1215–1225. DOI:10.1161/CIRCRESAHA.116.309598 [Google Scholar]
- Becerra A, Rojas M, Vallejos A, Villegas V, Perez L, Cabello-Verrugio C, et al. Endothelial fibrosis induced by suppressed STAT3 expression mediated by signaling involving the TGF-beta1/ALK5/Smad pathway. Lab. Investig. 2017, 97, 1033–1046. DOI:10.1038/labinvest.2017.61 [Google Scholar]
- Wang S, Sun A, Li L, Zhao G, Jia J, Wang K, et al. Up-regulation of BMP-2 antagonizes TGF-beta1/ROCK-enhanced cardiac fibrotic signalling through activation of Smurf1/Smad6 complex. J. Cell Mol. Med. 2012, 16, 2301–2310. DOI:10.1111/j.1582-4934.2012.01538.x [Google Scholar]
- Loirand G, Guerin P, Pacaud P. Rho kinases in cardiovascular physiology and pathophysiology. Circ. Res. 2006, 98, 322–334. DOI:10.1161/01.RES.0000201960.04223.3c [Google Scholar]
- Noma K, Oyama N, Liao JK. Physiological role of ROCKs in the cardiovascular system. Am. J. Physiol. Cell Physiol. 2006, 290, C661–C668. DOI:10.1152/ajpcell.00459.2005 [Google Scholar]
- Thompson NL, Bazoberry F, Speir EH, Casscells W, Ferrans VJ, Flanders KC, et al. Transforming growth factor beta-1 in acute myocardial infarction in rats. Growth Factors 1988, 1, 91–99. DOI:10.3109/08977198809000251 [Google Scholar]
- Li D, Fareh S, Leung TK, Nattel S. Promotion of atrial fibrillation by heart failure in dogs: Atrial remodeling of a different sort. Circulation 1999, 100, 87–95. DOI:10.1161/01.cir.100.1.87 [Google Scholar]
- Ramos-Mondragon R, Galindo CA, Avila G. Role of TGF-beta on cardiac structural and electrical remodeling. Vasc. Health Risk Manag. 2008, 4, 1289–1300. DOI:10.2147/vhrm.s3985 [Google Scholar]
- Nakajima H, Nakajima HO, Salcher O, Dittie AS, Dembowsky K, Jing S, et al. Atrial but not ventricular fibrosis in mice expressing a mutant transforming growth factor-beta(1) transgene in the heart. Circ. Res. 2000, 86, 571–579. DOI:10.1161/01.res.86.5.571 [Google Scholar]
- Anter E, Jessup M, Callans DJ. Atrial fibrillation and heart failure: Treatment considerations for a dual epidemic. Circulation 2009, 119, 2516–2525. DOI:10.1161/CIRCULATIONAHA.108.821306 [Google Scholar]
- Everett TH, Olgin JE. Atrial fibrosis and the mechanisms of atrial fibrillation. Heart Rhythm 2007, 4, S24–S27. DOI:10.1016/j.hrthm.2006.12.040 [Google Scholar]
- Ghazal R, Wang M, Liu D, Tschumperlin DJ, Pereira NL. Cardiac Fibrosis in the Multi-Omics Era: Implications for Heart Failure. Circ. Res. 2025, 136, 773–802. DOI:10.1161/CIRCRESAHA.124.325402 [Google Scholar]
- Skarp S, Doedens A, Holmstrom L, Izzi V, Saarimaki S, Sliz E, et al. Novel Genetic Variants Associated with Primary Myocardial Fibrosis in Sudden Cardiac Death Victims. J. Cardiovasc. Transl. Res. 2024, 17, 1229–1239. DOI:10.1007/s12265-024-10527-5 [Google Scholar]
- Pirruccello JP, Bick A, Wang M, Chaffin M, Friedman S, Yao J, et al. Analysis of cardiac magnetic resonance imaging in 36,000 individuals yields genetic insights into dilated cardiomyopathy. Nat. Commun. 2020, 11, 2254. DOI:10.1038/s41467-020-15823-7 [Google Scholar]
- Shabani M, Wang M, Jenkins GD, Rotter JI, Rich SS, Batzler A, et al. Myocardial Fibrosis and Cardiomyopathy Risk: A Genetic Link in the MESA. Circ. Heart Fail. 2023, 16, e010262. DOI:10.1161/CIRCHEARTFAILURE.122.010262 [Google Scholar]
- Wang X, Lee RS, Alver BH, Haswell JR, Wang S, Mieczkowski J, et al. SMARCB1-mediated SWI/SNF complex function is essential for enhancer regulation. Nat. Genet. 2017, 49, 289–295. DOI:10.1038/ng.3746 [Google Scholar]
- Lu QB, Fu X, Liu Y, Wang ZC, Liu SY, Li YC, et al. Disrupted cardiac fibroblast BCAA catabolism contributes to diabetic cardiomyopathy via a periostin/NAP1L2/SIRT3 axis. Cell Mol. Biol. Lett. 2023, 28, 93. DOI:10.1186/s11658-023-00510-4 [Google Scholar]
- Uddin GM, Zhang L, Shah S, Fukushima A, Wagg CS, Gopal K, et al. Impaired branched chain amino acid oxidation contributes to cardiac insulin resistance in heart failure. Cardiovasc. Diabetol. 2019, 18, 86. DOI:10.1186/s12933-019-0892-3 [Google Scholar]
- Andersson C, Liu C, Cheng S, Wang TJ, Gerszten RE, Larson MG, et al. Metabolomic signatures of cardiac remodelling and heart failure risk in the community. ESC Heart Fail. 2020, 7, 3707–3715. DOI:10.1002/ehf2.12923 [Google Scholar]
- Rasooly D, Peloso GM, Pereira AC, Dashti H, Giambartolomei C, Wheeler E, et al. Genome-wide association analysis and Mendelian randomization proteomics identify drug targets for heart failure. Nat. Commun. 2023, 14, 3826. DOI:10.1038/s41467-023-39253-3 [Google Scholar]
- Foinquinos A, Batkai S, Genschel C, Viereck J, Rump S, Gyongyosi M, et al. Preclinical development of a miR-132 inhibitor for heart failure treatment. Nat. Commun. 2020, 11, 633. DOI:10.1038/s41467-020-14349-2 [Google Scholar]
- Batkai S, Spannbauer A, Viereck J, Genschel C, Rump S, Traxler D, et al. MiR-132 inhibition improves myocardial strain in a large animal model of chronic left ventricular adverse remodelling. Eur. Heart J. Imaging Methods Pract. 2025, 3, qyaf088. DOI:10.1093/ehjimp/qyaf088 [Google Scholar]
- Taubel J, Hauke W, Rump S, Viereck J, Batkai S, Poetzsch J, et al. Novel antisense therapy targeting microRNA-132 in patients with heart failure: Results of a first-in-human Phase 1b randomized, double-blind, placebo-controlled study. Eur. Heart J. 2021, 42, 178–188. DOI:10.1093/eurheartj/ehaa898 [Google Scholar]
-
Ruigrok MJR, El Amasi KEM, Leeming DJ, Sand JMB, Frijlink HW, Hinrichs WLJ, et al. Silencing Heat Shock Protein 47 (HSP47) in Fibrogenic Precision-Cut Lung Slices: A Surprising Lack of Effects on Fibrogenesis? Front. Med. 2021, 8, 607962. DOI:10.3389/fmed.2021.607962 [Google Scholar]
-
Quan T, Shao Y, He T, Voorhees JJ, Fisher GJ. Reduced expression of connective tissue growth factor (CTGF/CCN2) mediates collagen loss in chronologically aged human skin. J. Investig. Dermatol. 2010, 130, 415–424. DOI:10.1038/jid.2009.224 [Google Scholar]
-
Wu X, Verschut V, Woest ME, Ng-Blichfeldt JP, Matias A, Villetti G, et al. Rho-Kinase 1/2 Inhibition Prevents Transforming Growth Factor-beta-Induced Effects on Pulmonary Remodeling and Repair. Front. Pharmacol. 2020, 11, 609509. DOI:10.3389/fphar.2020.609509 [Google Scholar]
-
Smoktunowicz N, Alexander RE, Franklin L, Williams AE, Holman B, Mercer PF, et al. The anti-fibrotic effect of inhibition of TGFbeta-ALK5 signalling in experimental pulmonary fibrosis in mice is attenuated in the presence of concurrent gamma-herpesvirus infection. Dis. Model. Mech. 2015, 8, 1129–1139. DOI:10.1242/dmm.019984 [Google Scholar]