Found 10 results
Open Access
Commentary
06 February 2026Novel Therapeutic Targets of Endothelial Inflammation in Acute Lung Injury and Acute Respiratory Distress Syndrome
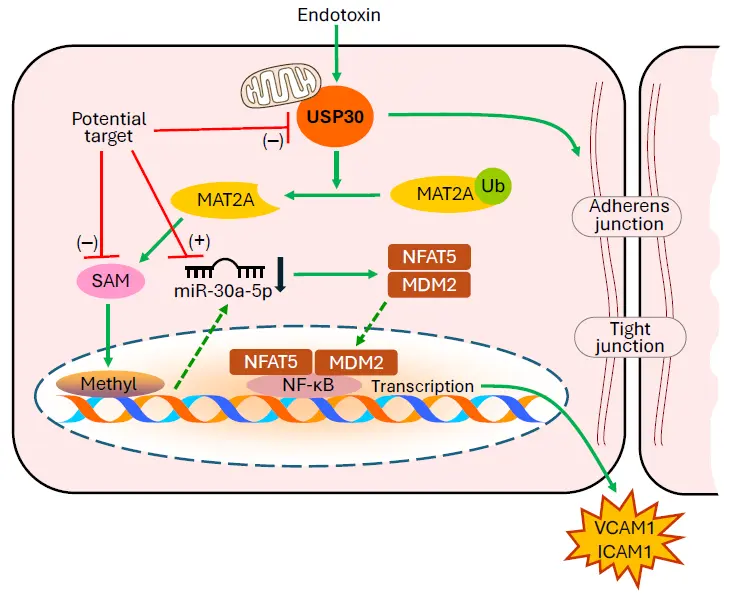
Lung microvascular endothelial inflammation and barrier dysfunction play critical roles in the pathogenesis of acute lung injury (ALI)/acute respiratory distress syndrome (ARDS). Despite recent scientific advances, the mortality of ALI/ARDS is still extremely high because the molecular mechanisms involved in ALI/ARDS remain unclear. In a recent issue of the journal Advanced Science, Baoyinna and colleagues reported that deubiquitinase USP30 induces lung microvascular inflammation and endothelial barrier disruption through the S-adenosylmethionine (SAM) cycle, DNA methylation, and miR-30a-5p down-regulation in ALI/ARDS. Their findings provide a strong rationale for targeting microRNAs, S-adenosylmethionine, DNA methylation, and deubiquitinating enzymes as potential therapeutic strategies for the treatment of ALI/ARDS.
Open Access
Review
10 December 2025Intracellular Chloride Channels: A Rising Target in Lung Disease Research
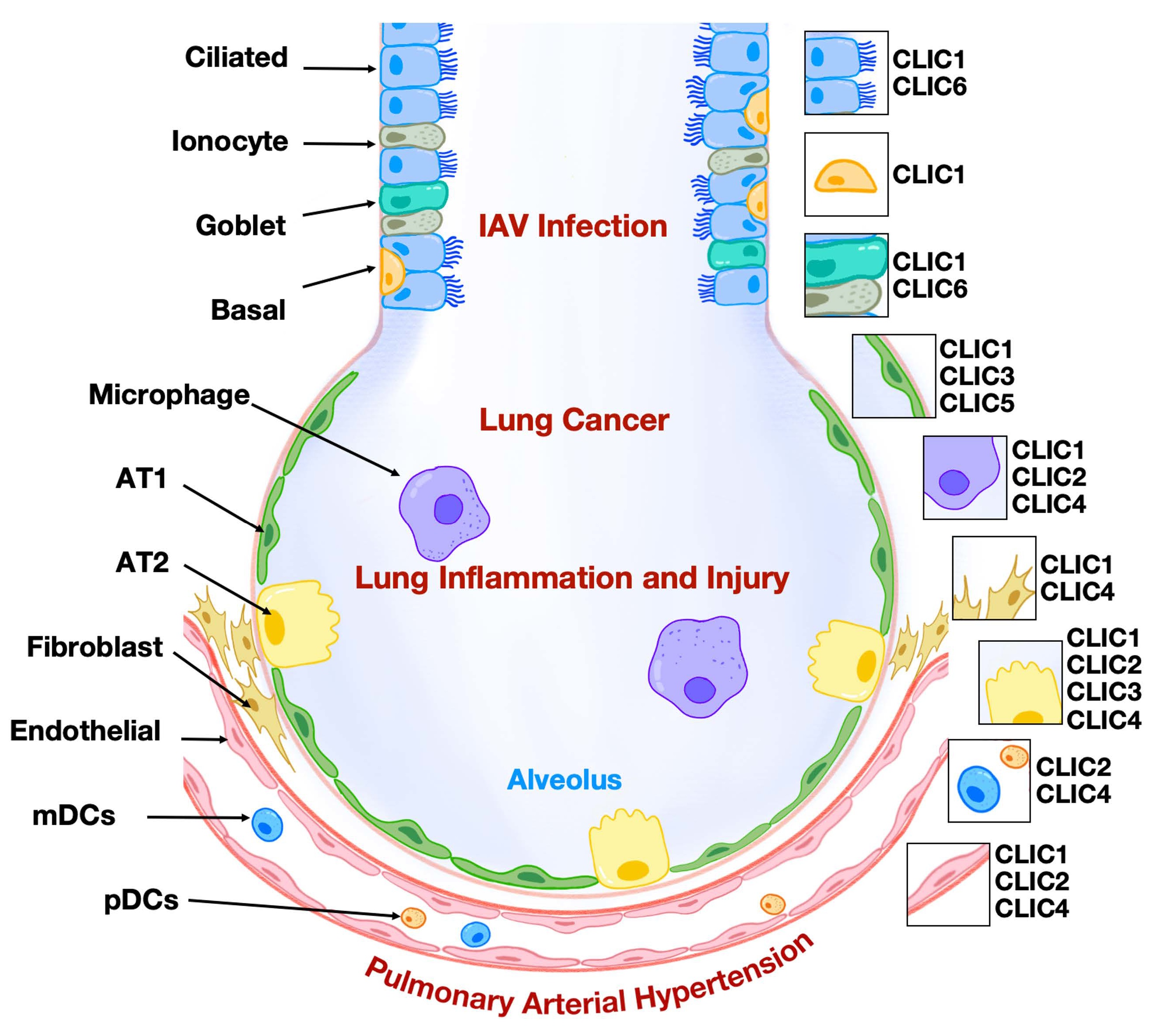
Chloride intracellular ion channels (CLICs) represent a relatively underexplored class of chloride channels and are included in a research initiative that focuses on druggable genes that have not been well studied yet. As a unique family, CLICs exist in membrane and soluble forms and play a role in regulating chloride flux and modulating various aspects of cellular biology. To date, six mammalian CLICs have been cloned and characterized at molecular and physiological levels. The respiratory system, responsible for gas exchange between the atmosphere and the human body, has recently been shown to express CLICs with functional relevance in lung pathophysiology, including lung carcinoma, inflammation, and endothelial dysfunction. Notably, the expression patterns of CLIC isoforms in lung cell types are distinct. Among them, CLIC1, CLIC3, and CLIC4 have been investigated more extensively, particularly in the context of lung cancer, inflammatory diseases, and pulmonary arterial hypertension. A deeper understanding of the role of CLICs in regulating lung cellular function may pave the way for developing novel therapeutic strategies to treat pulmonary disorders. In this review, we summarize the expression and functional roles of CLICs in lung pathophysiology, with particular emphasis on CLIC1, CLIC3, and CLIC4.
Open Access
Review
20 August 2025On the Utility of Nailfold Capillaroscopy in Detecting the Effects of Fibrinaloid Microclots in Diseases Involving Blood Stasis
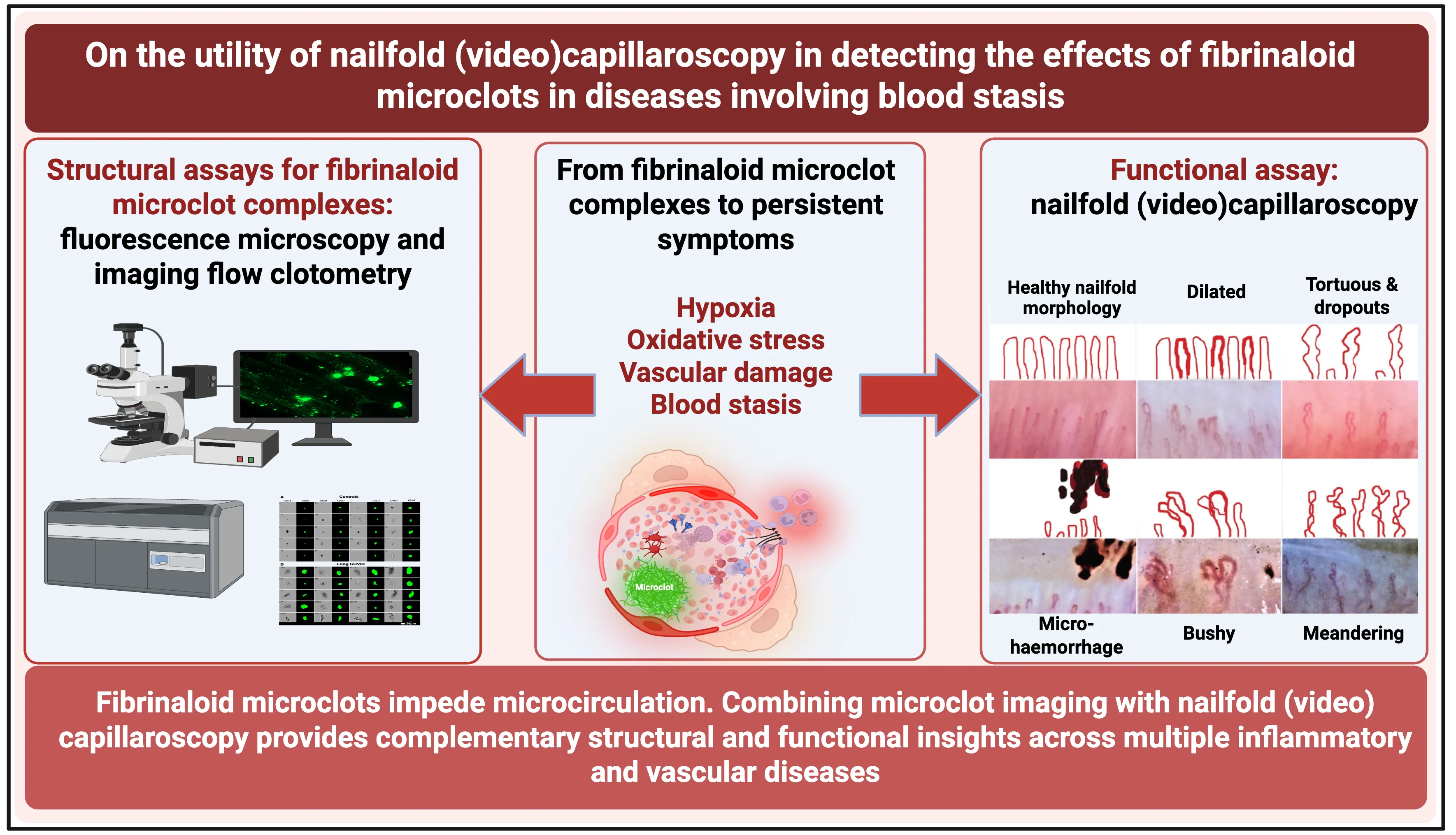
A variety of chronic, inflammatory vascular and autoimmune diseases are accompanied by fibrinaloid microclots. Such diseases reflect endothelial dysfunction and may be detected using a ‘structural’ assay in the form of the fluorescence microscopic or flow ‘clotometry’ analysis of suitably stained platelet-poor plasma. Their amyloid nature and the presence of anti-fibrinolytic molecules therein make the fibrinaloid microclots comparatively resistant to the normal processes of clot degradation. By inhibiting the free flow of blood, the many effects of fibrinaloid microclots include those causing hypoxia, oxidative stress, and ‘blood stasis’ in the microcirculation. Nailfold capillaroscopy is an established observational technique (with both ‘structural’ and ‘functional’ elements) for assessing the microcirculation, and it is thus of interest to establish whether it too demonstrates changes when these syndromes are diagnosed. All diseases in which both methods have been applied show both the presence of fibrinaloid microclots and changes in capillary properties, indicating the complementary value of the structural and functional assays. This also suggests the potential value of nailfold capillaroscopy in a variety of other diseases involving coagulopathies or a deficient microcirculation, which has been little studied to date.
Open Access
Article
24 July 2025Spermidine Dampens Inflammation by Directly Inhibiting Th17 Cytokine Production through a PRDX1 Associated Antioxidant Pathway
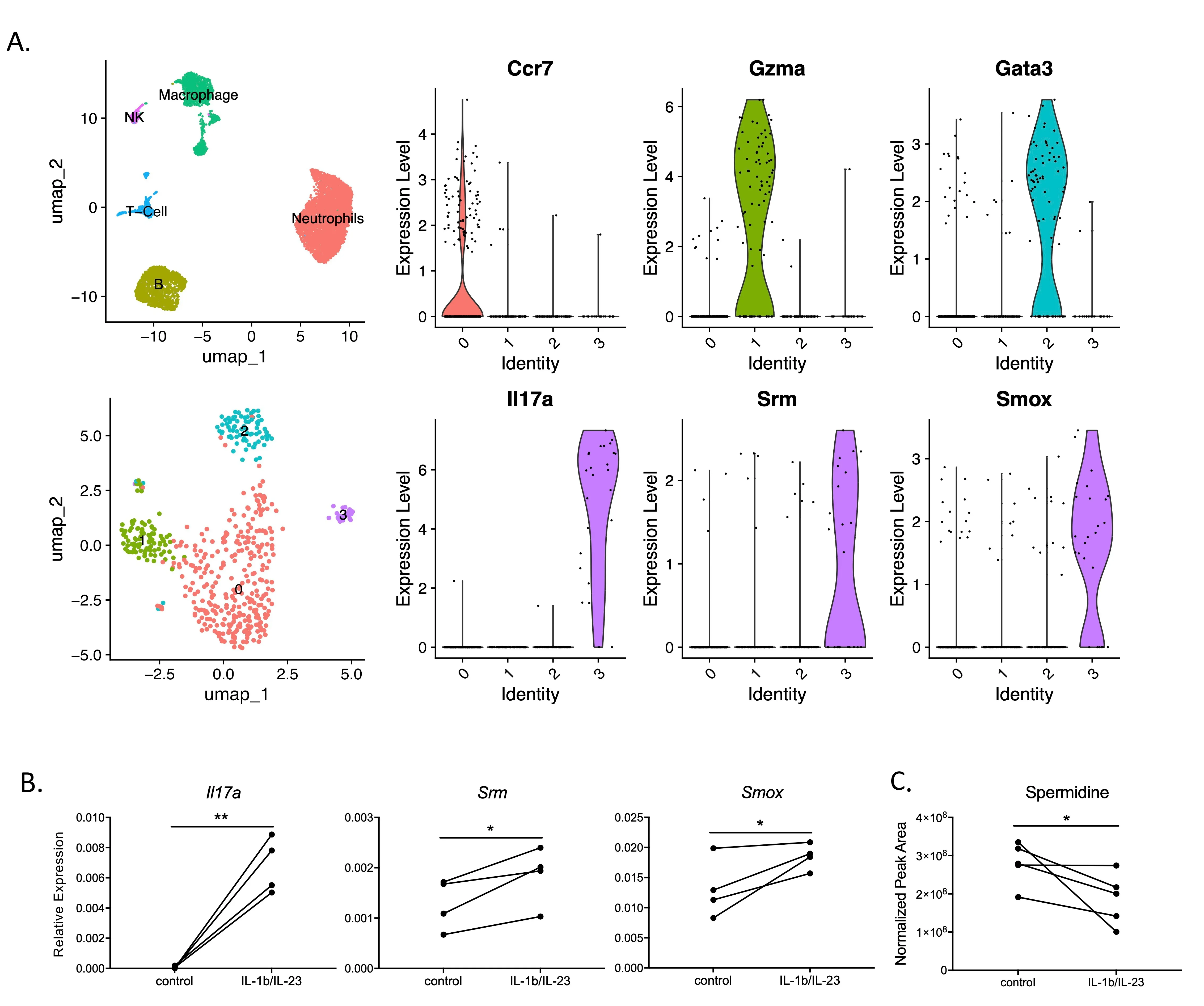
The activation of IL-17 signaling has been linked to the pathogenesis of many chronic, inflammatory lung diseases including cystic fibrosis. Through unbiased single-cell RNAseq screening, we found that IL-17+ T cells highly express Srm and Smox, which encode two key enzymes involved in spermidine synthesis, spermidine synthase and spermine oxidase respectively. Spermidine has been shown to reduce inflammation by regulating macrophage activation and balancing Th17/Treg differentiation; however its direct effects on Th17 cytokine production have not been carefully investigated. Here, using already differentiated Th17 cells from cultured mouse splenocytes, we found that exogenous spermidine directly inhibits IL-1β/IL-23-induced IL-17 production. Blockade of endogenous spermidine synthesis enhanced IL-17 production above native levels, further supporting the notion that spermidine is a direct regulator of cytokine secretion independent of differentiation. In vivo, spermidine alleviates lung inflammation in both Pseudomonas aeruginosa (PA) and LPS induced acute lung injury models. Further RNA-seq analysis suggests that spermidine suppression of Th17 cytokine production is mediated through its PRDX1-dependent antioxidant activity. Our data suggests that spermidine is a direct regulator of Type-17 T cell cytokine production and has potent anti-inflammatory effects against lung inflammation.
Open Access
Review
24 June 2025Obesity-Associated Chronic Inflammation: A Protective Mechanism against Type 2 Diabetes
Chronic inflammation is widely considered a risk factor for T2DM by inducing insulin resistance, but all attempts to translate the concept into clinical therapies have failed in the past 30 years. Anti-inflammatory medicines, including anti-TNF-α antibody (Etanercept), anti-IL1 antibody (Anakinra), anti-IL6 (Ziltivekimab), and NLRP3 inflammasome inhibitor (Colchicine) have excellent activities in the control of inflammation in arthritis. They reduced inflammation in T2DM patients in the clinical trials, but none improved insulin sensitivity. Some of them exhibited a mild and transient activity in the control of blood glucose, but the activities were related to the improvement of insulin secretion by β-cells. The failure may be related to followings: over-interpretation of TNF-α activity; ignoring the role of anti-inflammatory cytokines; differences between mice and humans. However, the species difference cannot fully explain the failure as these therapies did not work in the animal models as well. Moreover, genome-wide association studies (GWAS) show that T2DM is not associated with proinflammatory cytokine genes, including TNF-α, IL-1β, IL-6, and CCL2(MCP1). More studies suggest that inflammation has beneficial activities in the mobilization of energy stores and promotion of energy expenditure to prevent energy surplus, a risk factor of obesity-associated T2DM. Inflammatory cytokines induce lipolysis, thermogenesis, and satiety. In this regard, the inflammatory response is a compensatory event to obesity-associated stress with beneficial effects on energy metabolism. It is time to reconsider inflammation activity in obesity for protective activities.

Open Access
Review
20 March 2025The Fate and Dynamics of Neural Stem Cells (NSCs) and Their Neurogenic Decline in Alzheimer’s Disease
Neural stem cells (NSCs) are crucial for neurogenesis in the mammalian brain, supporting the generation of neurons and glial cells during both development and adulthood. However, aging—driven by factors such as reduced growth factors, heightened inflammation, oxidative stress, and epigenetic modifications—leads to a decline in NSC activity, which is closely associated with cognitive decline. This article explores the significant reduction in neurogenesis observed in Alzheimer’s disease (AD), where amyloid-beta (Aβ) accumulation, tau pathology, mitochondrial dysfunction, and chronic neuroinflammation disrupt NSC function in the hippocampus and subventricular zone (SVZ). These disruptions impair NSC proliferation, differentiation, and migration, contributing to the progression of cognitive deficits. Additionally, this article examines experimental studies suggesting that deficits in neurogenesis often precede amyloid plaque formation in animal models, positioning impaired neurogenesis as a potential early biomarker for AD. Therapeutic strategies targeting neurogenesis, epigenetics, and inflammation—such as anti-inflammatory treatments, environmental enrichment, and modulation of systemic factors—hold promise for reversing neurogenic deficits and enhancing cognitive function. Furthermore, this article discusses both pharmacological agents and non-pharmacological strategies that show potential in promoting neurogenesis, though further research is needed to evaluate their safety and efficacy. The decline of NSC is driven by many interconnected factors, making it challenging to understand and address fully. This highlights the need for ongoing research.

Open Access
Review
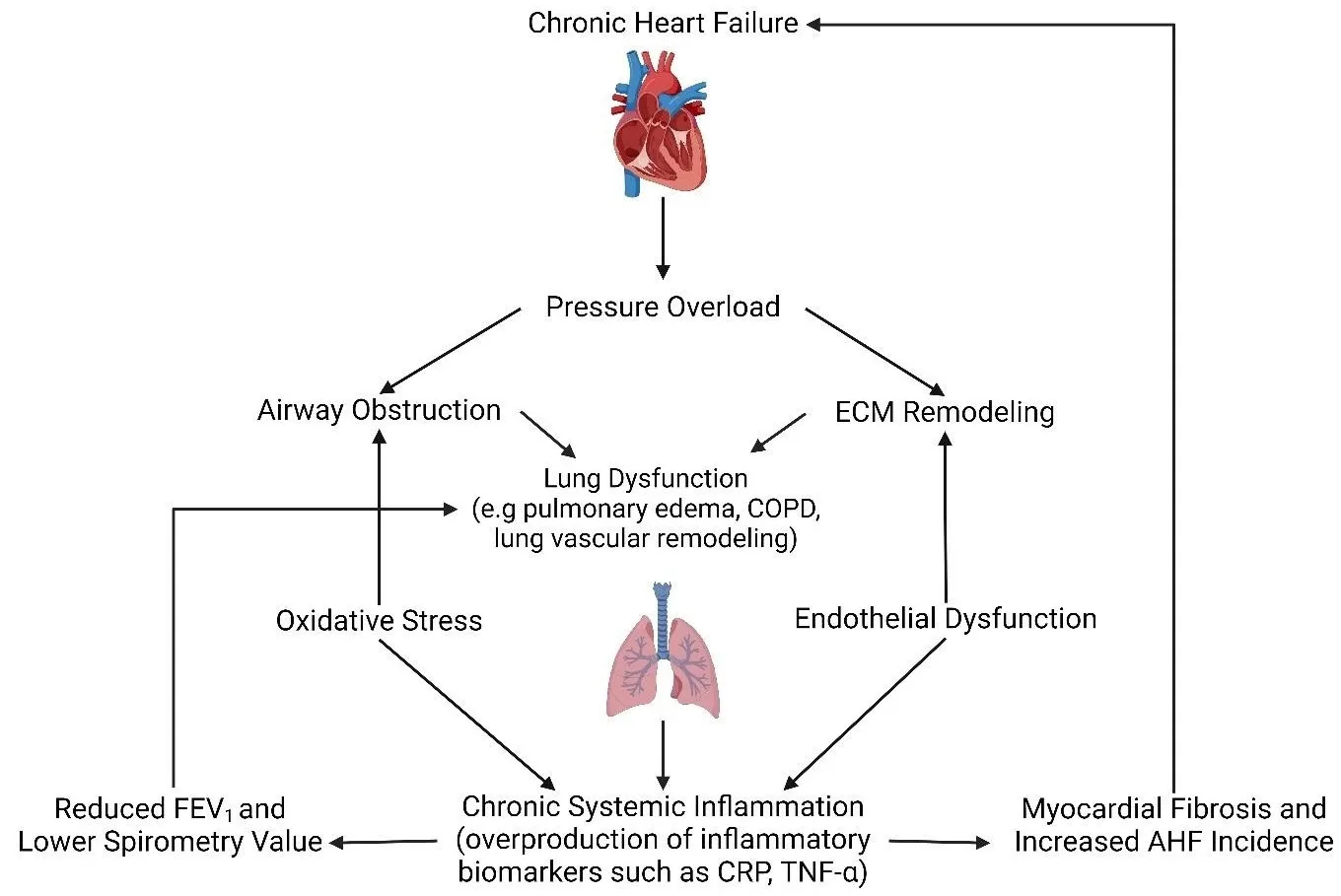
02 December 2024The Interplay of Heart Failure and Lung Disease: Clinical Correlations, Mechanisms, and Therapeutic Implications
Heart failure (HF) is a common clinical syndrome marked by reduced cardiac output, elevated intracardiac pressures, and heart dysfunction. Chronic HF (CHF) is a syndrome characterized by a lack of blood flow and impaired pumping ability to the heart over time, while acute HF (AHF) arises suddenly due to incidents like myocardial infarction or cardiac arrest. HF has a significant impact on pulmonary health and function, leading to conditions such as pulmonary edema and restrictive lung patterns. Clinical evidence highlights the bidirectional relationship between HF and lung dysfunction. Declining lung function serves as a predictor for HF progression and severity, while HF contributes to worsening lung health. Animal models that induce HF through surgical methods further demonstrate the connection between heart and lung pathology. The main mechanisms linking HF and lung dysfunction are pressure overload and chronic systemic inflammation, with changes in the extracellular matrix (ECM) also playing a role. Additionally, environmental factors like air pollution exacerbate lung inflammation, increasing the risk of both HF and chronic obstructive pulmonary disease (COPD) incidence. Combined treatment approaches involving pharmaceutical drugs such as statins, Angiotensin-converting enzyme (ACE) inhibitors, and Angiotensin receptor blockers (ARBs) may benefit by reducing inflammation. This review will explore the complex interplay between HF and lung function, emphasizing their interconnected pathophysiology and potential integrated treatment strategies.
Open Access
Review
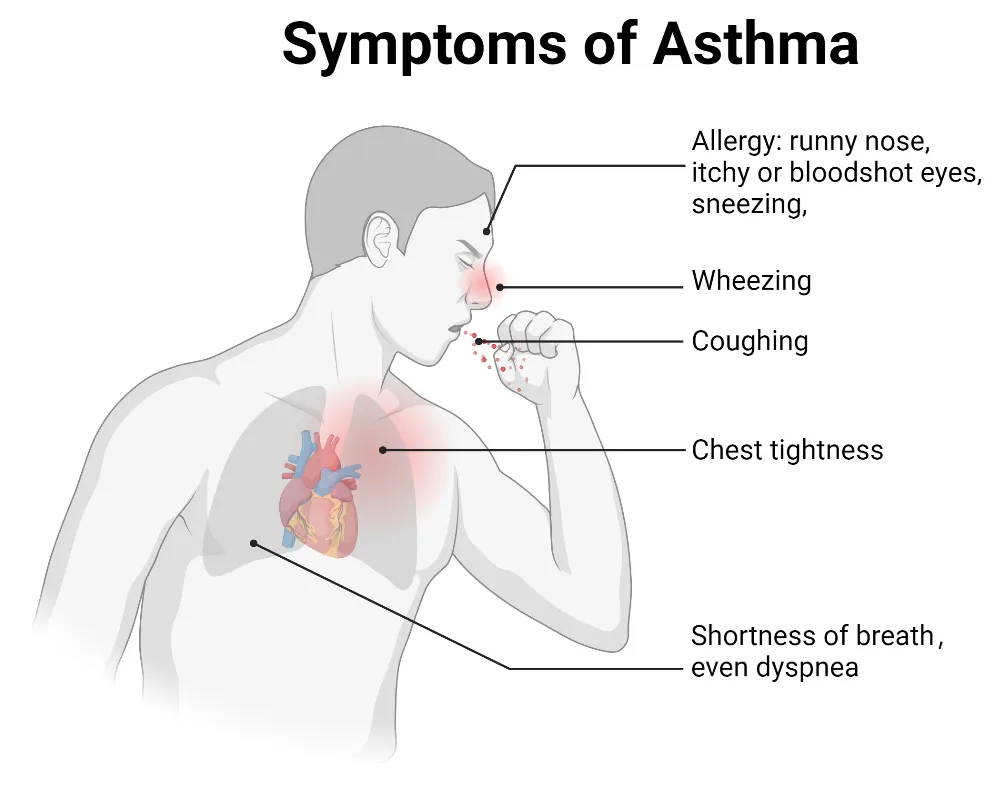
15 November 2024Ion Channels in the Immune Response of Asthma
Asthma is a common respiratory disorder characterized by chronic inflammation of the lower airways, contributing to significant morbidity, mortality, and a substantial global economic burden. It is now understood as a heterogeneous condition, with ongoing research shedding light on its complex immunological underpinnings. Ion channels, which are specialized transmembrane proteins that facilitate ion movement based on electrochemical gradients, play a crucial role in the pathophysiology of asthma. Ion channels regulate essential processes like maintaining epithelial hydroelectrolyte balance and also play a role in modulating immune responses involved in asthma. We discuss the connection between ion channel activity and immune regulation in asthma, focusing on ion channel regulation of immune cell behavior, airway hyperresponsiveness, and inflammation in asthma. Understanding ion channels in asthma could lead to the development of targeted therapies modulating their activity, thereby enhancing disease management and patient outcomes.
Open Access
Article
31 March 2024
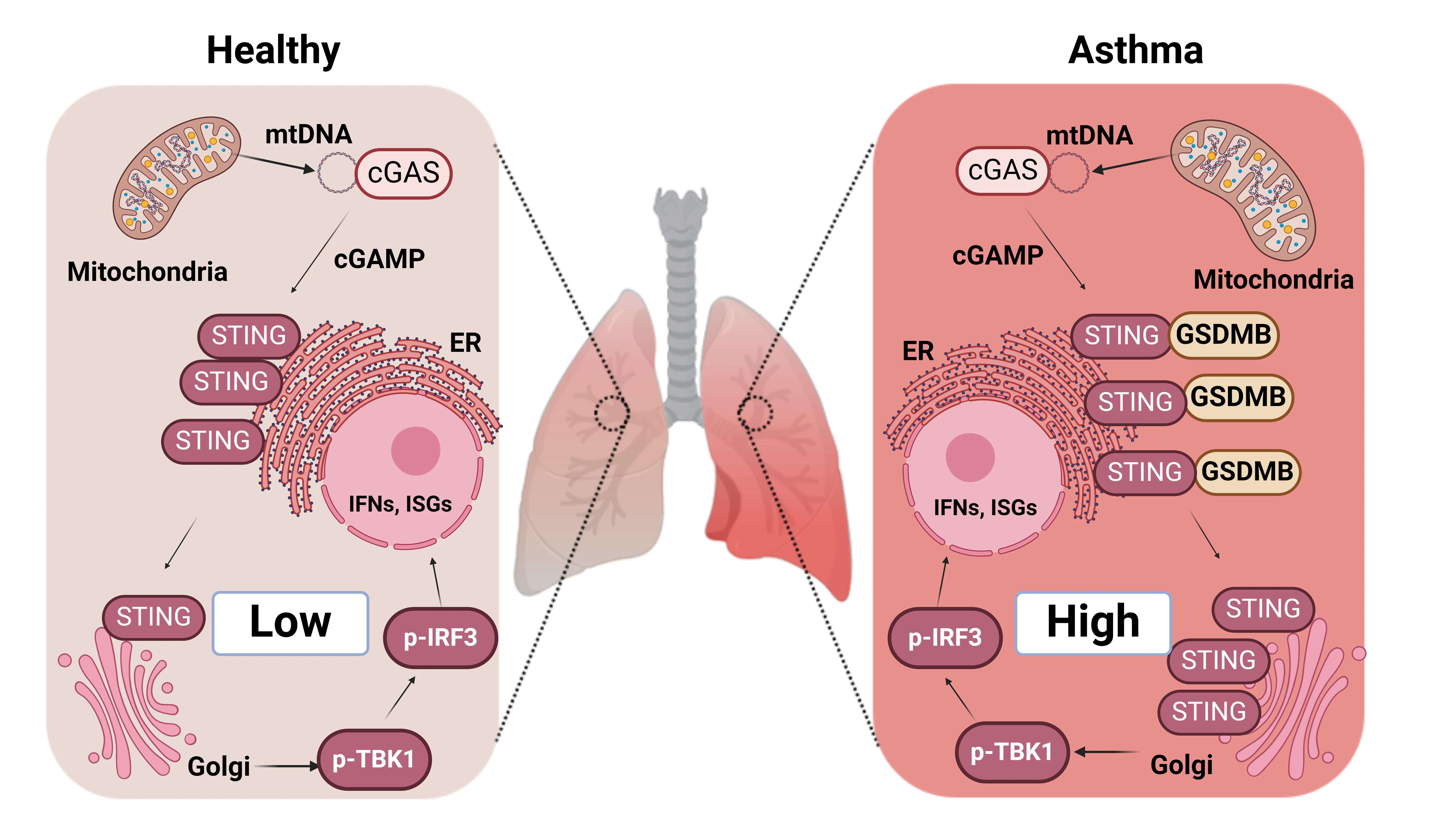
The Asthma Risk Gene, GSDMB, Promotes Mitochondrial DNA-induced ISGs Expression
Released mitochondrial DNA (mtDNA) in cells activates cGAS-STING pathway, which induces expression of interferon-stimulated genes (ISGs) and thereby promotes inflammation, as frequently seen in asthmatic airways. However, whether the genetic determinant, Gasdermin B (GSDMB), the most replicated asthma risk gene, regulates this pathway remains unknown. We set out to determine whether and how GSDMB regulates mtDNA-activated cGAS-STING pathway and subsequent ISGs induction in human airway epithelial cells. Using qPCR, ELISA, native polyacrylamide gel electrophoresis, co-immunoprecipitation and immunofluorescence assays, we evaluated the regulation of GSDMB on cGAS-STING pathway in both BEAS-2B cells and primary normal human bronchial epithelial cells (nHBEs). mtDNA was extracted in plasma samples from human asthmatics and the correlation between mtDNA levels and eosinophil counts was analyzed. GSDMB is significantly associated with RANTES expression in asthmatic nasal epithelial brushing samples from the Genes-environments and Admixture in Latino Americans (GALA) II study. Over-expression of GSDMB promotes DNA-induced IFN and ISGs expression in bronchial epithelial BEAS-2B cells and nHBEs. Conversely, knockout of GSDMB led to weakened induction of interferon (IFNs) and ISGs in BEAS-2B cells. Mechanistically, GSDMB interacts with the C-terminus of STING, promoting the translocalization of STING to Golgi, leading to the phosphorylation of IRF3 and induction of IFNs and ISGs. mtDNA copy number in serum from asthmatics was significantly correlated with blood eosinophil counts especially in male subjects. GSDMB promotes the activation of mtDNA and poly (dA:dT)-induced activation of cGAS-STING pathway in airway epithelial cells, leading to enhanced induction of ISGs.
Open Access
Meeting Report
20 February 2024The Cellular and Metabolic Bases of Organ Fibrosis: UNIA Workshop 2023 in Baeza, Spain
Fibrosis is defined by scarring and tissue hardening caused by excess deposition of extracellular matrix components, mainly collagens. A fibrotic response can occur in any tissue of the body and is the final outcome of an unbalanced reaction to inflammation and wound healing induced by a variety of insults, including persistent infections, autoimmune reactions, allergic responses, chemical exposure, radiation, and tissue injury. The accumulation of extracellular matrix proteins replaces the living tissue and disrupts the architecture leading to organ malfunction. Fibrosis remains a major clinical and therapeutic challenge and has been estimated to account for 45% of deaths in the developed world. While major advances regarding mechanistic knowledge on the underlying cell biology alterations in fibrosis have helped to characterize the main phases and mediators involved, this knowledge has not yielded significant progress in treatment. Only recently, the metabolic features associated to fibrosis have begun to emerge. This information, likely representing only the tip of the iceberg, suggests that metabolic derangement is a key culprit in the pathophysiology of fibrogenesis. The Workshop on The Cellular and Metabolic Bases of Organ Fibrosis, International University of Andalusia, Baeza, Spain, October 8–11, 2023 aimed to discuss the current knowledge and novel perspectives on the mechanisms contributing to the development of fibrosis in different organs and tissues, with particular focus on new methodological developments in metabolomics and therapeutic strategies.
