1. Discovery of Obesity-Induced Inflammation
1.1. Early Observations
The link between obesity and inflammation was initially observed in the early 1990s when researchers began to unravel the complex interactions within adipose tissue. One pivotal moment came with the study by Hotamisligil et al., who identified the increased expression of tumor necrosis factor-alpha (TNF-α) by adipocytes in adipose tissues of obese mice and humans; moreover, the increased TNF-α expression was associated with insulin resistance in the obese mice; injection of TNF-α antibody into the mice to neutralize the TNF-α was found to improve the insulin sensitivity [
1]. In another early study, infusion of TNF-α into rats was found to reduce insulin sensitivity by induction of hepatic glucose production and inhibition of muscle glucose uptake in rats [
2]. These findings set up the foundation for a couple of popular concepts, including obesity-associated inflammation, immunometabolism, and adipose tissue inflammation in systemic insulin resistance. However, another activity of TNF-α has been ignored for a long time. TNF-α was reported to stimulate glucose uptake in the liver, muscle, and many other tissues in the rats [
2], which may be responsible for the reduction in insulin-stimulated glucose uptake and insulin-trigged inhibition of hepatic glucose production. In support, TNF-α was found to inhibit the signaling activity of insulin receptors, which provided a molecular mechanism for insulin resistance [
3]. Therefore, as an inflammatory cytokine, TNF-α is a representative model for the concept of inflammation in insulin resistance ().
. Supportive (black) and challenging (red) studies for the inflammation concept. Major studies are listed here to give a overview of evidence that either supports or challenges the inflammation concept. These are original studies for each important step in the evoluation of the concept. The confirmative studies are not listed here. Abbreviation: WF, White Fat; IR, Insulin Resistance; InR, Insulin Receptor; TNFR, TNF receptor; KO, Knockout; ATM, Adipose Tissue Macrophages; P65, NF-κB p65; OE, Over-expression.
These early studies, together with many subsequent studies, consistently suggest that TNF-α may serve as a signal of immune cells in the regulation of energy metabolism. Glucose, fatty acids, and amino acids are the major energy substrates that are used to generate ATP and synthesize biomaterials. Immune cells are the most active cells in the secretion of TNF-α, especially macrophages. In the adipose tissue, mouse adipocytes may express TNF-α protein, but human adipocytes do not express TNF-α protein. Later studies found that in obese mice, TNF-α protein came from macrophages instead of adipocytes in the year 2003 [
4,
5], which has been well-accepted in the field since then (). Macrophages are immune cells that play an important role in the organization of immune responses through the cytokine secretion to manage energy supply to the immune cells. TNF-α induces glucose uptake in target cells and, at the same time, inhibits insulin action in the cells, which are examples of cytokine activities in the regulation of energy metabolism.
In the traditional view, adipose tissue was considered an inert energy storage depot. Now, adipose tissue is generally accepted as an active endocrine organ for its secretion of adipokines, such as leptin, adiponectin, etc. [
6]. The finding that TNF-α levels were elevated in adipose tissue of obese individuals and correlated with insulin resistance provided the first direct evidence linking obesity-associated inflammation to metabolic disorders, which is the basis of the concept of immunometabolism. In this subfield of immunology, energy metabolism in immune cells and its impact on other types of cells have been the focus [
7]. TNF-α from macrophages regulates energy metabolism in immune cells as well as in non-immune cells, including hepatocytes and muscle cells [
3]. These effects of TNF-α were observed
in vivo with TNF-α infusion and
in vitro with TNF-α treatment at concentrations above nM, as reported by many groups, including us [
8,
9]. However, TNF-α concentration only reaches this level locally in the fat tissue under obesity. This may explain why TNF-α antibody treatment could not improve insulin sensitivity in obese patients. In obese patients, TNF-α concentration cannot reach the level in the circulation, which may explain why the anti-TNF therapy could not improve insulin sensitivity in T2DM patients.
In addition to TNF-α, other pro-inflammatory cytokines are elevated in obesity. Elevations in other pro-inflammatory cytokines are extensively reported, such as interleukin-6 (IL-6), IL-1β, and C-reactive protein (CRP) in the circulation of obese individuals [
10]. These markers of inflammation were persistently higher in the blood of obese patients compared to lean counterparts, suggesting a chronic and systemic low-grade inflammatory state associated with excess body fat. The association of these inflammatory mediators and metabolic disorders sparked interest in exploring the causes and impact of inflammation on human health.
1.2. Key Concepts
Systemic low-grade inflammation in obesity represents a mild and chronic inflammatory state, which is commonly characterized by elevated blood levels of C– reactive protein ranging approximately from 2–10 mg/L [
11]. Distinct from acute inflammation, it entails a subtle yet persistent activation of the immune system with a strong elevation of pro-inflammatory cytokines. Despite often being symptomless, chronic inflammation can play a significant role in the development and progression of various diseases and aging, such as autoimmune diseases, cancers, and cardiovascular diseases.
Adipose tissue inflammation represents a part of systemic low-grade inflammation. The initiation of adipose inflammation is related to several factors, including local hypoxia response from adipose tissue expansion [
12,
13], an imbalanced extracellular matrix and fibrosis [
14], as well as the overloading of cells by intermediate metabolites that disrupts cellular homeostasis [
15]. This type of inflammation is marked by an elevation in M1-like macrophage phenotype, more production of reactive oxygen species, endoplasmic reticulum stress, endothelial dysfunction, cell death, and senescence. The adipose inflammation is induced to repair the stress-induced damages from tissue expansion in favor of maintenance of tissue homeostasis. It may contribute to the systemic low-grade inflammation in obesity. The inflammation was considered a target in the treatment of type 2 diabetes, but the possibility has been challenged by the results of most clinical trials in the past 30 years [
16].
The resolution of inflammation is an actively regulated physiological process that commences at the peak of inflammation [
17]. Its purpose is to re-establish homoeostasis and clear cellular debris. The effectors of this resolution are specialized pro-resolving mediators synthesized through transcellular processes by immune, epithelial, and endothelial cells.
The NLRP3 inflammasome is a multiprotein complex that participates in the innate immune response through inflammation. It consists of three main components: NLRP3 (NOD-, LRR- and pyrin domain- containing 3), ASC (apoptosis-associated speck-like protein containing a CARD), and procaspase 1. The NLRP3 sensor can detect signals of endogenous danger-associated molecular patterns (DAMPs) released during cellular stress and tissue damage, as well as signals of pathogen-associated molecular patterns (PAMPs) released during infection. The ASC adapter protein connects NLRP3 to pro-caspase 1, facilitating the cleavage of the inactive enzyme into active caspase 1. Subsequently, active caspase 1 activates cytokine precursors such as IL-1β and IL-18. In this way, the NLRP3 inflammasome initiates a cascade of inflammatory responses, leading to the production and release of pro-inflammatory cytokines, recruitment of immune cells, and amplification of the inflammatory process. The pathway was considered to contribute to obesity-associated chronic inflammation through bacteria-derived endotoxin from gut microbiota. While blood levels of IL-1β and IL-18 in obese patients do not support the possibility.
1.3. Subsequent Key Findings
Building on the early observations, subsequent research has been conducted to understand the impact of inflammation in T2DM patients. Those subsequent studies led to several key findings that have reshaped our understanding of the metabolic effects of obesity-induced inflammation. In this line, TNF-α was a dominant model and extensively investigated in both animals and humans as being reviewed [
18]. In the clinical studies, Bernstein et al. reported that anti-TNF drugs like Etanercept were effective in improving inflammation parameters in T2DM patients, but the drug could not improve insulin sensitivity [
19]. For IL-1, Larsen et al., demonstrated that IL-1 receptor antagonist (IL-1Ra) was able to improve glycemic control in T2DM patients but could not improve insulin sensitivity [
20]. In the CANTOS trial, IL-1β inhibition with Canakinumab for 3.5 years did not reduce the incidence of new-onset T2DM [
21]. These clinical studies challenged the expectations derived from the classical inflammation concept. Meta-analysis of numerous studies leads to a conclusion that inhibition of IL-1β activity by the receptor antagonist reduced HbA1c effectively in T2DM patients with shorter duration but not in patients with longer duration (>3 years) [
22]. However, the therapeutic effect was not a result of the improvement of insulin sensitivity. Those anti-inflammatory studies consistently suggest that inhibition of TNF-α or IL-1β could not improve insulin sensitivity in the treatment of T2DM patients.
IL-1 secretion is increased in macrophages by activation of NLRP3 inflammasome. Colchicine is considered a safe and affordable NLRP3 inflammasome inhibitor [
23]. Its main activity involves disrupting microtubule dynamics inhibiting immune cell recruitment/division [
24]. Colchicine also blocks oligomerization of NLRP3 inflammasome and prevents active IL-1β processing/release. Colchicine decreased cardiovascular event risk (cardiovascular-related death, myocardial infarction, stroke, and urgent coronary revascularizations) in a study [
23], but there is no evidence for its activity in the prevention of T2DM by reducing the disease incidence.
In addition to these clinical trials, laboratory research has elucidated the cellular and molecular mechanisms of obesity-related inflammation. It was demonstrated that induction of chronic inflammation in adipose tissue by NF-κB p65 overexpression could enhance energy expenditure, uncoupling inflammation from insulin resistance, offering new insights into the relationship between inflammation and metabolism [
25]. A later study of the effect of NF-κB inhibition in adipocytes by Sherer’s group suggests that NF-κB inhibition led to adipocyte dysfunction in lipid storage and made the transgenic mice more susceptible to insulin resistance [
26]. Consistently, Zhu et al. reported that suppressing adipocyte inflammation using adenoviral protein RIDα/β paradoxically led to insulin resistance, further highlighting the beneficial effect of inflammatory response in the adipose tissues [
27]. In a review article, adipose tissue inflammation in obesity is proposed as a protective mechanism in the maintenance of energy balance [
10]. Recently, type 2 diabetes has been considered a “sacrifice program” that handles energy surplus through adaptive mechanisms, including insulin resistance and inflammation [
28]. Inflammatory cytokines are able to induce mobilization of energy like triglycerides and glycogens. The cytokines promote lipolysis of triglycerides in adipocytes to release free fatty acids and glycerol into the blood. They may induce hydrolysis of glycogen in liver and muscle cells to induce cell consumption of glucose. At the same time, the cytokines may promote energy expenditure to reduce energy surplus in obesity. When the cytokine level is returned to normal after inflammation, the cells should exhibit a higher insulin sensitivity due to the reduction in energy storage. The primary activity of insulin is to stimulate energy storage in the form of glycogen and triglycerides by consumption of blood glucose, which results in blood glucose reduction as a secondary effect. Energy surplus in cells is an important trigger of insulin resistance in both physiological and pathological conditions [
28].
2. Characteristics of Obesity-Related Inflammation
2.1. Cellular Markers
Obesity-related inflammation is characterized by distinct cellular markers that play crucial roles in progression and manifestation [
29]. Among these, adipose tissue macrophages (ATMs) are particularly noteworthy. ATMs are often classified into two major types: M1 and M2. M1 macrophages are pro-inflammatory, and exhibit increased activity during obesity, secreting cytokines such as TNF-α and interleukin-6 (IL-6). These inflammatory signals may contribute to insulin resistance by disrupting normal metabolic pathways within adipocytes and muscle cells. Conversely, M2 macrophages have an anti-inflammatory function and help maintain homeostasis in lean adipose tissue. However, their numbers decrease during the development of obesity.
In addition to macrophages, other immune cells also participate in the inflammatory process associated with obesity [
30]. T lymphocytes, for instance, infiltrate adipose tissues during obesity and produce interferon-gamma (IFN-γ), which exacerbates inflammation. Furthermore, B lymphocytes contribute through antibody production, potentially targeting adipocyte antigens and amplifying local inflammatory responses. The balance between regulatory T cells (Tregs) and effector T cells is critical; a shift towards more effector T cells promotes chronic inflammation in obese individuals.
Adipocytes themselves undergo phenotypic changes under conditions of obesity, becoming hypertrophic and dysfunctional [
31,
32]. Hypertrophic adipocytes release altered profiles of adipokines like leptin and adiponectin, which influence both systemic metabolism and local inflammatory processes. Leptin levels rise significantly in obesity, contributing to increased inflammation via activation of various signaling pathways, including JAK/STAT and NF-κB. On the other hand, adiponectin levels fall, reducing its protective effects against inflammation and insulin resistance.
2.2. Cytokine Involvement
Cytokines serve as key mediators of the inflammatory response in obesity, linking adipose tissue dysfunction to systemic metabolic disorders [
33]. TNF-α was the first cytokine identified in adipose tissue during obesity. Its role in promoting insulin resistance has been extensively studied in cellular models, demonstrating how it interferes with insulin signaling by activating serine kinases that phosphorylate IRS proteins, thereby impairing downstream glucose uptake mechanisms. The TNF activities
in vitro include the induction of lipolysis and the release of fatty acids by adipocytes. However, the observations may not apply to the systemic insulin resistance in obesity as the cytokine concentrations in the cellular models were very high and are only observed
in vivo under acute infection by bacteria or viruses.
Interleukins constitute another important group of cytokines involved in obesity-induced inflammation. IL-6, produced not only by immune cells but also by other cell types, including adipocytes, induces hepatic acute-phase protein synthesis and affects insulin sensitivity negatively [
34]. Another interleukin, IL-1β, activates the NLRP3 inflammasome, leading to further secretion of pro-inflammatory cytokines and perpetuating the cycle of inflammation. Additionally, IL-10, an anti-inflammatory cytokine, tends to be downregulated in obese states, failing to counterbalance the heightened inflammatory environment effectively.
Beyond traditional cytokines, resistin and visfatin are found in obesity-associated inflammation. Resistin expression increases in obesity and correlates with insulin resistance, although its exact mechanisms remain partially unclear. Visfatin, originally thought to mimic insulin actions, paradoxically shows elevated levels in obese subjects and may promote inflammatory responses rather than alleviate them.
The interplay among those cytokines forms complex networks that govern the dynamics of obesity-associated inflammation. Understanding these interactions at a molecular level offers potential therapeutic targets for mitigating adverse health outcomes related to obesity and type 2 diabetes mellitus. For example, blocking specific cytokine pathways could reduce inflammation without compromising essential physiological functions mediated by others. This approach underscores the importance of personalized medicine strategies tailored to individual patient profiles based on detailed assessments of cytokine profiles and other biomarkers.
3. Failure of Anti-Inflammatory Drugs in Clinical Trials
The efficacy of anti-inflammatory drugs on the improvement of insulin resistance has been the subject of intense investigation. Etanercept, a TNF-α inhibitor (Neutralization antibody), was tested in patients with metabolic syndrome. Bernstein et al. reported that such an intervention did not improve insulin sensitivity and increased triglyceride accumulation in the skeletal muscle in the patients [
35]. These findings suggest that inhibiting TNF-α had no beneficial effects on individuals with metabolic syndrome. This may explain why anti-TNF therapies have never been tested in a large clinical trial for their efficacy in the treatment of T2DM [
11].
IL-1 activity was tested in the treatment of T2DM in a couple of clinical trials. Blocking IL-1 activity with receptor antagonists (IL-1Ra, Anakinra) led to a modest improvement in inflammation, such as a reduction in C-peptide, but no improvement was observed in insulin sensitivity [
36]. In another study, Larsen et al. conducted a trial of IL-1Ra (Anakinra) impact in T2DM patients. They observed a reduction in glycated hemoglobin (HbA1c) and inflammatory markers, such as IL-6 and C-reactive protein (CRP) [
20]. However, the patients did not show an improvement in insulin sensitivity. The clinical significance of these results remains under debate, as the observed benefits were not observed in a subsequent large-scale clinical trial [
15]. Diacerein, an indirect inhibitor of IL-1, was evaluated in a randomized clinical trial involving T2DM patients. Tres et al. noted limited improvements in metabolic control and inflammatory markers [
37]. In a long-term clinical study of IL-1β in the cardiovascular system, inhibition of IL-1β with neutralization antibody (Canakinumab) reduced cardiovascular events in the patients over a median period of 3.7 years but did not reduce the incidence of diabetes [
21]. These studies consistently suggest that IL-1β may not play a major role in the pathogenesis of T2DM.
IL-6 activities have been extensively studied in obesity, as documented in several excellent review articles [
34,
38]. In non-obese conditions, IL-6 is secreted by skeletal muscle during exercise to the blood circulation as a myokine, which coordinates energy metabolism. IL-6 stimulates adipocytes to release fatty acids in energy mobilization for energy supply to the muscle. IL-6 also stimulates the liver for an increase in glucose production in the maintenance of blood glucose. In the obese condition, IL-6 is elevated in the bloodstream, and the adipose tissue is a source of the circulating IL-6. This elevation contributes to liver insulin resistance as glucose production by the liver is not inhibited by the physiological level of insulin. In this condition, inhibition of IL-6 activity with receptor antagonist (Tocilizumab) improved insulin sensitivity in patients with rheumatoid diseases [
39]. However, the long-term effect remains to be determined.
In addition to the cytokine-specific anti-inflammatory therapies, broad-spectrum anti-inflammation medicines have also been tested for the improvement of insulin sensitivity. The conclusion is that the treatment may improve blood glucose in T2Dm patients, but the activity is not due to an improvement in insulin sensitivity. Instead, the effect is from an increase in insulin secretion [
28]. The point is supported by a meta-analysis of anti-inflammatory therapies in T2DM [
22].
4. Beneficial Activities of Obesity-Associated Inflammation
The translation of preclinical studies on inflammation into T2DM therapy fails in almost all clinical trials. Emerging data suggest that we have to change the view about inflammation in T2DM. Inflammation during obesity is not entirely detrimental but can serve protective roles under certain conditions, as proposed in 2013 [
10]. Both animal and human studies suggest the existence of low-grade chronic inflammation in obesity, but the biological significance of the inflammation has been under debate for decades. We propose that obesity-associated inflammation might function as an adaptive mechanism to handle excessive energy storage in adipocytes [
28], which induces energy mobilization and energy consumption in the control of energy surplus. However, when this process becomes prolonged or exacerbated, it may contribute to tissue damage or metabolic disorders. The detrimental effects of inflammation have been well documented in the cardiovascular system in a recent review [
11].
In support, several studies in mouse models suggest that suppressing adipocyte inflammation can impair insulin sensitivity [
27,
40] (). In an early study, insulin sensitivity was examined in mice of TNF-a receptor knockout mice [
40]. Inhibition of TNF-a signaling activity in the global receptor KO mice led to more severe insulin resistance fed on a high-fat diet with a four-fold elevation in the fasting insulin. The TNF-a effect was tested in adipose tissue by over-expression of TNF-α in adipocytes in mice [
41]. The result was that the increased activity of TNF-α significantly reduced obesity, but this could not improve systemic insulin sensitivity. This RIDα/β, an adenoviral protein complex, inhibits inflammatory pathways of TLR4, TNF-α, and IL-1β. Inhibition of the inflammation by over-expression of RIDα/β in an adipocyte-specific manner reduced the adiposity and adipose chronic inflammation in the diet-induced obese (DIO) mice but promoted insulin resistance [
27]. In another mouse study, suppression of the inflammation by blocking IL-6 trans-signaling reduced adipose tissue inflammation and prevented adipose tissue macrophage recruitment in DIO mice [
42,
43], but this intervention did not improve insulin resistance. Inhibition of TNF-α activity by its receptor gene deletion was reported to enhance liver steatosis and insulin resistance in TNFRα-KO mice [
44]. These findings underscore the beneficial roles of adipose tissue inflammation in the control of insulin sensitivity.
The concept of a beneficial effect of inflammation in adipose tissue is supported by a study of induction of inflammation. The evidence comes from the induction of inflammation by over-expression of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), a transcription factor central to inflammatory processes. Tang et al.'s work revealed that up-regulation of adipose tissue inflammation by overexpression of NF-κB p65 subunit generated chronic inflammation in transgenic mice by failing to induce insulin resistance [
25] (). The mechanism is that energy expenditure was enhanced in the mice by NF-κB activation. In this line, induction of the inflammation by over-expression of IKKβ in adipocytes, an upstream kinase of NF-κB, generated a similar impact in the transgenic mice for improvement of insulin sensitivity [
45]. In contrast, inhibition of inflammation by adipose IKKβ gene knockout impaired insulin sensitivity in mice [
46]. IL-2, a cytokine from T cells with an activity in the promotion of inflammation, was found to improve insulin sensitivity by induction of sympathetic nerve activities in mice [
47]. This group of studies by this and other laboratories consistently suggests that obesity-associated inflammation may protect systemic insulin sensitivity by induction of energy expenditure, which was originally proposed by us in 2012 [
48] and 2013 [
10], respectively.
The beneficial activity of obesity-associated inflammation was then extended into physiological activities of inflammatory cytokines in a review in 2015 [
49] (). In the review, we examined activities of several cytokines (TNF-α, IL-1, IL-6, IL-7, IL-15, IL-18) in the regulation of energy metabolism in both physiological and pathological conditions. The cytokine activities are discussed in obesity (energy surplus condition), calorie restriction (energy deficient condition), physical exercise (energy consumption condition), and cancer cachexia (high energy demand condition). The conclusion is that the inflammatory cytokines are signaling molecules in the induction of energy mobilization and energy consumption. Inhibition of the cytokine activities may increase the risk of obesity from a deficiency in energy mobilization and consumption. These views received support from review articles in the immunology field, in which the cytokine activities are discussed in the evolution of immunometabolism and regulation of tissue homeostasis by Medzhitov’s group in 2019 [
50] and 2021 [
17,
51]. In a recent perspective, Rosen and Kajimura proposed that the conventional view linking adipose inflammation to insulin resistance requires reevaluation [
52] (). Their analysis indicates that inflammation might sometimes function as a protective mechanism rather than solely as a pathological process. This perspective is in line with our view to challenge the traditional concept of inflammation in T2DM. In extension to these views, we proposed that obesity-associated inflammation and insulin resistance both are “sacrifice programs” handling energy surplus in obesity to take care of the energy surplus in a recent perspective [
28].
5. Potential Strategies for Future Research
The exploration of new targets in the treatment of obesity-associated chronic inflammation and type 2 diabetes mellitus (T2DM) is essential for advancing therapeutic strategies. In the past, most studies focus on the inflammatory cytokine network, particularly on TNF-α, IL-6, and IL-1β. Clinical studies have shown that targeting these cytokines has no benefit on the control of insulin resistance, a hallmark of T2DM. Several new drugs for T2DM have led us to focus on the role of energy surplus in the pathogenesis. In this line, GLP-1 receptor activators (such as Semaglutide) improved insulin sensitivity by reducing food intake through satiety. SGLT2 (sodium-dependent glucose transporters 2) inhibitors (such as Empagliflozin, Dapagliflozin, and Canagliflozin) have shown reliable therapeutic effects on T2DM through energy (glucose) discharge in urine. These medicines both resolve the energy surplus problem and reduce obesity in T2DM patients. In addition, the medicines also reduce inflammatory status in the patients from energy metabolism activities [
53,
54]. Clinical data from these new medicines consistently suggest that obesity-associated inflammation and insulin resistance are results of energy surplus. Control of energy surplus is a strategy for the control of chronic inflammation and insulin resistance in obese subjects. In this way, GLP-1R and SGLT2 are new targets in the control of obesity-associated chronic inflammation.
Another emerging area of interest involves the identification of signals of energy surplus in cells. In this aspect, intracellular ATP has been a new potential candidate for the signal in addition to ER stress, lipokines, oxidative stress, etc [
11]. Lee et al. found that over-supply of intracellular ATP promoted the expression of inflammatory cytokines in adipose tissue [
55].
In vitro, induction of ATP production in macrophages with lauric acid treatment induced expression of pro-inflammatory cytokines. Inhibition of ATP production with β-oxidation inhibitor (Etomoxir) or mitochondrial uncoupler (2,4-dinitrophenol, DNP) suppressed the cytokine expression. ATP is able to activate inflammasome in the induction of pro-inflammatory response. AMPK (AMP-activated protein kinase) and SIRT1 (Sirtuin 1) are intracellular energy sensors that regulate ATP production through an impact on metabolic pathways, including glycolysis and mitochondrial respiration. This suggests that metabolic regulators such as AMPK or SIRT1 are potential targets in the control of obesity-related inflammation and improving insulin sensitivity.
Additionally, recent research has focused on the role of macrophages in adipose tissue homeostasis. Chavakis et al. highlighted how macrophage function within adipose tissue can promote or inhibit inflammation [
56]. Specifically, M1 macrophages contribute to pro-inflammatory responses, while M2 macrophages exhibit anti-inflammatory properties. Modulating the polarization state of these cells presents another avenue for developing targeted therapies against obesity-induced inflammation and its associated metabolic disorders.
6. Improved Methodologies
Improving methodologies for studying obesity-associated chronic inflammation requires integrating multidisciplinary approaches. Advanced imaging techniques, such as positron emission tomography (PET) combined with computed tomography (CT), offer non-invasive ways to monitor inflammatory processes
in vivo [
57]. These tools enable researchers to track changes in adipose tissue composition and immune cell infiltration over time, providing valuable insights into disease progression and response to interventions.
Moreover, advancements in omics technologies, including genomics, transcriptomics, proteomics, and metabolomics, facilitate comprehensive profiling of biological systems affected by obesity and T2DM. By employing systems biology approaches, it becomes possible to identify key nodes within complex networks that drive pathological conditions. For example, Wang et al.'s work on immunometabolism from an evolutionary perspective underscores the importance of considering both innate immunity and metabolism when designing experiments [
50].
Animal models remain crucial for preclinical testing but must be refined to reflect human physiology better. Farooqi and Xu emphasized the translational potential of mouse models in understanding human metabolic diseases [
33]. However, discrepancies between species necessitate careful consideration during experimental design. Developing more accurate animal models that capture genetic diversity and environmental factors influencing human obesity will enhance their predictive power.
7. Bridging the Gap between Bench and Bedside
7.1. Lessons from Clinical Trials
Clinical trials have been instrumental in shaping our understanding of the relationship between obesity-associated inflammation and T2DM. The early discovery that adipose tissue expresses TNF-α highlighted the potential role of inflammation in insulin resistance. Subsequent clinical studies aimed at targeting inflammatory pathways with drugs demonstrated negative results, underscoring the complexity of translating bench findings into effective bedside treatments.
One significant lesson learned from the clinical trials is the disassociation of inflammation and insulin resistance in T2DM patients under anti-inflammatory therapies. Bernstein et al. (2006) reported that while patients with metabolic syndrome experienced improvements in inflammation markers after treatment with TNF inhibitor (etanercept), they did not show any benefit in insulin sensitivity [
19]. This observation challenged the concept of inflammation in the pathogenesis of insulin resistance.
The clinical trials on other anti-inflammation therapies gave similar challenges. Larsen et al. (2007) demonstrated that interleukin-1 receptor antagonist therapy improved glycemic control in patients with T2DM over a six-month period [
20]. However, the effect was not from the improvement of insulin sensitivity. Furthermore, Tres et al.'s randomized controlled trial using IL-1 inhibitor (diacerein) illustrated that inflammatory markers were decreased together with the improvement of blood glucose, and there was no significant improvement in insulin sensitivity [
37], suggesting that inflammation might not be the sole driver of insulin resistance.
The failure of anti-inflammatory strategies to consistently improve insulin sensitivity raises questions about the underlying mechanisms linking inflammation and metabolic disorders. It was proposed that anti-inflammatory therapies fail due to an incomplete understanding of the physiology of inflammatory response within the adipose microenvironment [
49]. Future approaches should consider both pro- and anti-inflammatory cytokines, as well as their balance, which could better reflect the nuanced nature of inflammation in obesity and diabetes.
Moreover, clinical trials have underscored the importance of personalized medicine. Not all patients respond equally to the same treatment, necessitating tailored therapeutic regimens based on individual characteristics such as age, sex, ethnicity, and specific biomarkers. These lessons highlight the need for more comprehensive studies that incorporate diverse populations and evaluate multiple endpoints to ensure robust conclusions regarding efficacy and safety.
7.2. Integrating Multidisciplinary Approaches
To bridge the gap between basic research and clinical application, it is essential to adopt multidisciplinary approaches that integrate insights from immunology, metabolism, genetics, and systems biology. The intricate interplay between immune cells and adipocytes requires a holistic perspective that considers how different components interact under physiological and pathological conditions. Farooqi and Xu (2024) emphasized the translational potential of mouse models in human metabolic diseases, but they also cautioned against over-reliance on animal data without corroborating evidence in humans.
Systems biology offers a powerful framework for analyzing complex networks involved in obesity-induced inflammation and T2DM. By leveraging advanced computational tools and high-throughput technologies, researchers can identify key nodes and pathways that are most amenable to therapeutic modulation. Wang et al. (2015) reviewed the regulatory role of energy balance in inflammation, suggesting that integrating metabolic flux analysis with inflammatory signaling could yield novel targets for intervention.
Genomics and epigenomics provide further avenues for investigation. Genetic variations influencing cytokine production or receptor responsiveness may explain why certain individuals develop severe insulin resistance despite similar levels of obesity-related inflammation. Epigenetic modifications triggered by environmental exposures, such as diet and physical activity, add another layer of complexity that must be considered in designing effective interventions.
Immunometabolism represents another promising area where multidisciplinary collaboration holds great promise. Lee et al. (2018) presented an integrated view of immunometabolism, highlighting the bidirectional relationship between immune function and metabolic processes. Understanding how immune cells adapt their metabolism during activation could lead to innovative strategies for mitigating inflammation without compromising overall health.
8. Conclusions
Obesity-associated chronic inflammation has been extensively studied over the past 30 years. There is no debate about the existence of inflammation, but there is a debate about the impact of inflammation on insulin resistance. The cause/effect relationship could not be approved for inflammation/insulin resistance, especially in the human body. The debate is supported by the failure of anti-inflammatory therapies in the improvement of insulin sensitivity in all clinical studies. Targeting TNF-α with etanercept does not lead to improvements in insulin resistance or other metabolic benefits in the human body. Similarly, interventions involving interleukin-1 receptor antagonists failed to improve insulin sensitivity in the human body, although a mild improvement of blood glucose was observed through induction of insulin secretion. Non-steroid anti-inflammation medicines such as salsalate also failed to improve insulin resistance in T2DM patients [
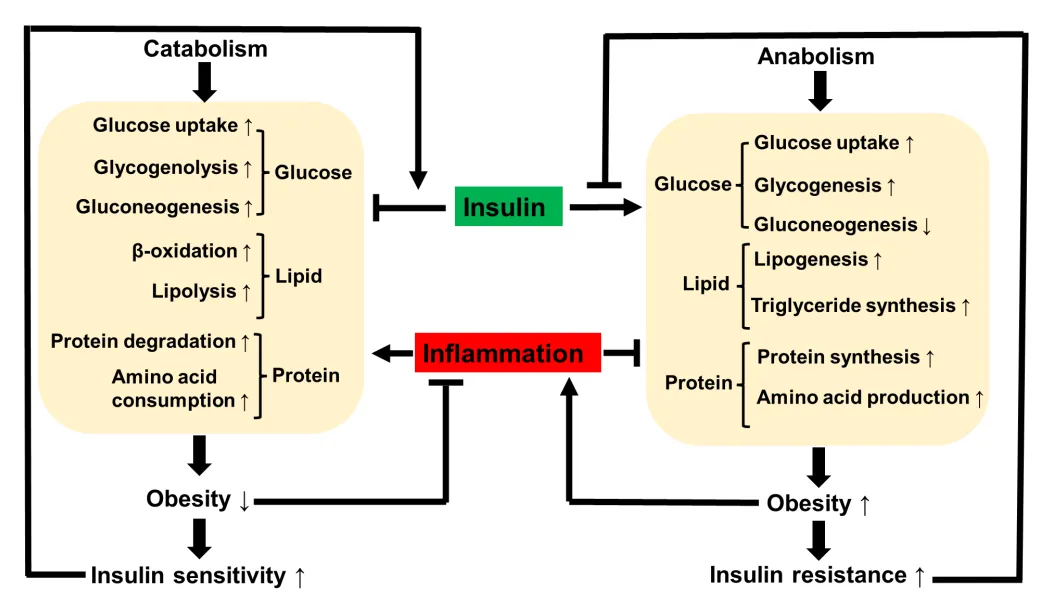
58]. Evidence from animal and human studies consistently suggests that inflammatory response may protect insulin sensitivity by induction of energy expenditure through taking care of energy surplus. In this line, the inflammatory response is considered a feedback mechanism to the energy surplus condition in obesity to prevent more severe metabolic disorders. The feedback effects include induction of lipolysis, inhibiting adipocyte expansion, and induction of energy expenditure to promote energy consumption (). Therefore, induction of inflammation under certain conditions may favor control of blood glucose in a long run by reducing obesity. Energy surplus is the primary cause of obesity, inflammation, and insulin resistance.
. Impact of insulin and inflammation in anabolism and catabolism. Insulin is the main driving force for anabolism that promotes storage of glucose, lipids and proteins through biosynthesis. The storage under conditions of ecessive substrate supply leads to expansion of adipsoe tissue for obesity, which in turn causes insulin resistance. Insuin resistance is able to inhibit the insulin activity in a feedback manner to attenuate the energy storage process in fight against sverer obesity. Obesity may trigger inflammation to slow down the anabolism to decrease energy storage, and at the same time to promote catabolism for an increase in energy mobilization and consumption. The inflammation effect contributes to loss of energy storage and body weight in the control of obesity. Weight loss eventually increase insulin sensitivity and reduce inflammation to attenuate the catabolism to prevent body weight over loss. These events keep runing in the physiological condition to maintain energy balance in the body.
Ethics Statement
Not applicable.
Informed Consent Statement
Not applicable.
Funding
The study is supported by the National Natural Science Foundation of China (Grant No. 32271220 ) to Jianping Ye.
Declaration of Competing Interest
The author declares that he has no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
-
1.
Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-α: direct role in obesity-linked insulin resistance.
Science 1993,
259, 87–91.
[Google Scholar]
-
2.
Lang CH, Dobrescu C, Bagby GJ. Tumor necrosis factor impairs insulin action on peripheral glucose disposal and hepatic glucose output.
Endocrinology 1992,
130, 43–52.
[Google Scholar]
-
3.
Feinstein R, Kanety H, Papa M, Lunenfeld B, Karasik A. Tumor necrosis factor-alpha suppresses insulin-induced tyrosine phosphorylation of insulin receptor and its substrates..
J. Biol. Chem. 1993,
268, 26055–26058.
[Google Scholar]
-
4.
Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW. Obesity is associated with macrophage accumulation in adipose tissue.
J. Clin. Investig. 2003,
112, 1796–1808.
[Google Scholar]
-
5.
Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance.
J. Clin. Investig. 2003,
112, 1821–1830.
[Google Scholar]
-
6.
Kershaw EE, Flier JS. Adipose tissue as an endocrine organ.
J. Clin. Endo. Metab. 2004,
89, 2548–2556.
[Google Scholar]
-
7.
Hu T, Liu C, Lei M, Zeng Q, Li L, Tang H, et al. Metabolic regulation of the immune system in health and diseases: mechanisms and interventions.
Sign. Transduct. Target. Ther. 2024,
9, 268.
[Google Scholar]
-
8.
Gao Z, Zuberi A, Quon MJ, Dong Z, Ye J. Aspirin inhibits serine phosphorylation of insulin receptor substrate 1 in tumor necrosis factor-treated cells through targeting multiple serine kinases.
J. Biol. Chem. 2003,
278, 24944–24950.
[Google Scholar]
-
9.
Rui L, Aguirre V, Kim JK, Shulman GI, Lee A, Corbould A, et al. Insulin/IGF-1 and TNF-α stimulate phosphorylation of IRS-1 at inhibitory Ser 307 via distinct pathways.
J. Clin. Investig. 2001,
107, 181–189.
[Google Scholar]
-
10.
Ye J, McGuinness OP. Inflammation during obesity is not all bad: evidence from animal and human studies.
Am. J. Physiol.-Endo. Metab. 2013,
304, E466–E477.
[Google Scholar]
-
11.
Soták M, Clark M, Suur BE, Börgeson E. Inflammation and resolution in obesity.
Nat. Rev. Endo. 2025,
21, 45–61.
[Google Scholar]
-
12.
Trayhurn P. Hypoxia and adipose tissue function and dysfunction in obesity.
Physiol. Rev. 2013,
93, 1–21.
[Google Scholar]
-
13.
Ye J. Emerging role of adipose tissue hypoxia in obesity and insulin resistance.
Int. J. Obes. 2009,
33, 54–66.
[Google Scholar]
-
14.
Crewe C, An YA, Scherer PE. The ominous triad of adipose tissue dysfunction: inflammation, fibrosis, and impaired angiogenesis.
J. Clin. Investig. 2017,
127, 74–82.
[Google Scholar]
-
15.
Ye J. Mechanism of insulin resistance in obesity: a role of ATP.
Fron. Med. 2021,
15, 372–382.
[Google Scholar]
-
16.
Ye J. Failure of inflammation hypothesis in the pathogenesis of type 2 diabetes.
Obesity Med. 2024,
52, 100565.
[Google Scholar]
-
17.
Meizlish ML, Franklin RA, Zhou X, Medzhitov R. Tissue Homeostasis and Inflammation.
Ann. Rev. Immunol. 2021,
39, 557–581.
[Google Scholar]
-
18.
Sethi JK, Hotamisligil GS. Metabolic messengers: tumour necrosis factor.
Nat. Metab. 2021,
3, 1302–1312.
[Google Scholar]
-
19.
Bernstein LE, Berry J, Kim S, Canavan B, Grinspoon SK. Effects of etanercept in patients with the metabolic syndrome.
Arch. Intern. Med. 2006,
166, 902–908.
[Google Scholar]
-
20.
Larsen CM, Faulenbach M, Vaag A, Vølund A, Ehses JA, Seifert B, et al. Interleukin-1–receptor antagonist in type 2 diabetes mellitus.
N. Engl. J. Med. 2007,
356, 1517–1526.
[Google Scholar]
-
21.
Everett BM, Donath MY, Pradhan AD, Thuren T, Pais P, Nicolau JC, et al. Anti-inflammatory therapy with canakinumab for the prevention and management of diabetes.
J. Am. Coll. Card. 2018,
71, 2392–2401.
[Google Scholar]
-
22.
Li D, Zhong J, Zhang Q, Zhang J. Effects of anti-inflammatory therapies on glycemic control in type 2 diabetes mellitus.
Fron. Immunol. 2023,
14, 1125116.
[Google Scholar]
-
23.
Sattar L, Memon RA, Ashfaq F, Hamdani SSQ, Rahim Vohra R, Ashraf J, et al. Efficacy and Safety of Colchicine in Prevention of Secondary Cardiovascular Outcomes Among Patients With Coronary Vessel Disease: A Meta-Analysis.
Cureus 2022,
14, e26680.
[Google Scholar]
-
24.
Cronstein BN, Molad Y, Reibman J, Balakhane E, Levin RI, Weissmann G. Colchicine alters the quantitative and qualitative display of selectins on endothelial cells and neutrophils..
J. Clin. Investig. 1995,
96, 994–1002.
[Google Scholar]
-
25.
Tang T, Zhang J, Yin J, Staszkiewicz J, Gawronska-Kozak B, Jung DY, et al. Uncoupling of inflammation and insulin resistance by NF-κB in transgenic mice through elevated energy expenditure 2.
J. Biol. Chem. 2010,
285, 4637–4644.
[Google Scholar]
-
26.
Asterholm IW, Tao C, Morley TS, Wang QA, Delgado-Lopez F, Wang ZV, et al. Adipocyte inflammation is essential for healthy adipose tissue expansion and remodeling.
Cell. Metab. 2014,
20, 103–118.
[Google Scholar]
-
27.
Zhu Q, An YA, Kim M, Zhang Z, Zhao S, Zhu Y, et al. Suppressing adipocyte inflammation promotes insulin resistance in mice.
Mol. Metab. 2020,
39, 101010.
[Google Scholar]
-
28.
Ye J, Yin J. Type 2 diabetes: a sacrifice program handling energy surplus.
Life. Metab. 2024,
3, loae033.
[Google Scholar]
-
29.
Lee YS, Wollam J, Olefsky JM. An integrated view of immunometabolism.
Cell 2018,
172, 22–40.
[Google Scholar]
-
30.
Kalathookunnel Antony A, Lian Z, Wu H. T Cells in Adipose Tissue in Aging.
Front. Immunol. 2018,
9, 2945.
[Google Scholar]
-
31.
Sárvári AK, Van Hauwaert EL, Markussen LK, Gammelmark E, Marcher A, Ebbesen MF, et al. Plasticity of epididymal adipose tissue in response to diet-induced obesity at single-nucleus resolution.
Cell. Metab. 2021,
33, 437
–453. e5.
[Google Scholar]
-
32.
Song A, Dai W, Jang MJ, Medrano L, Li Z, Zhao H, et al. Low-and high-thermogenic brown adipocyte subpopulations coexist in murine adipose tissue.
J. Clin. Investig. 2020,
130, 247–257.
[Google Scholar]
-
33.
Farooqi IS, Xu Y. Translational potential of mouse models of human metabolic disease.
Cell 2024,
187, 4129–4143.
[Google Scholar]
-
34.
Wueest S, Konrad D. The controversial role of IL-6 in adipose tissue on obesity-induced dysregulation of glucose metabolism.
Am. J. Physiol.-Endo. Metab. 2020,
319, E607–E613.
[Google Scholar]
-
35.
Lo J, Bernstein LE, Canavan B, Torriani M, Jackson MB, Ahima RS, et al. Effects of TNF-α neutralization on adipocytokines and skeletal muscle adiposity in the metabolic syndrome.
Am. J. Physiol.-Endo. Metab. 2007,
293, E102–E109.
[Google Scholar]
-
36.
Larsen CM, Faulenbach M, Vaag A, Ehses JA, Donath MY, Mandrup-Poulsen T. Sustained effects of interleukin-1 receptor antagonist treatment in type 2 diabetes.
Diabetes Care. 2009,
32, 1663–1668.
[Google Scholar]
-
37.
Tres GS, Fuchs SC, Piovesan F, Koehler-Santos P, Pereira FDS, Camey S, et al. Effect of diacerein on metabolic control and inflammatory markers in patients with type 2 diabetes using antidiabetic agents: a randomized controlled trial.
J. Diabetes Res. 2018,
2018, 4246521.
[Google Scholar]
-
38.
Garbers C, Heink S, Korn T, Rose-John S. Interleukin-6: designing specific therapeutics for a complex cytokine.
Nat. Rev. Drug. Discov. 2018,
17, 395–412.
[Google Scholar]
-
39.
Schultz O, Oberhauser F, Saech J, Rubbert-Roth A, Hahn M, Krone W, et al. Effects of inhibition of interleukin-6 signalling on insulin sensitivity and lipoprotein (a) levels in human subjects with rheumatoid diseases.
PLoS ONE 2010,
5, e14328.
[Google Scholar]
-
40.
Schreyer SA, Chua SC, LeBoeuf RC. Obesity and diabetes in TNF-alpha receptor-deficient mice.
J. Clin. Investig. 1998,
102, 402–411.
[Google Scholar]
-
41.
Xu H, Hirosumi J, Uysal KT, Guler AD, Hotamisligil GS. Exclusive action of transmembrane TNFα in adipose tissue leads to reduced adipose mass and local but not systemic insulin resistance.
Endocrinology 2002,
143, 1502–1511.
[Google Scholar]
-
42.
Kraakman MJ, Kammoun HL, Allen TL, Deswaerte V, Henstridge DC, Estevez E, et al. Blocking IL-6 trans-signaling prevents high-fat diet-induced adipose tissue macrophage recruitment but does not improve insulin resistance.
Cell. Metab. 2015,
21, 403–416.
[Google Scholar]
-
43.
Findeisen M, Allen TL, Henstridge DC, Kammoun H, Brandon AE, Baggio LL, et al. Treatment of type 2 diabetes with the designer cytokine IC7Fc.
Nature 2019,
574, 63–68.
[Google Scholar]
-
44.
Lambertucci F, Arboatti A, Sedlmeier MG, Motiño O, de Luján Alvarez M, Ceballos MP, et al. Disruption of tumor necrosis factor alpha receptor 1 signaling accelerates NAFLD progression in mice upon a high-fat diet.
J. Nutr. Biochem. 2018,
58, 17–27.
[Google Scholar]
-
45.
Jiao P, Feng B, Ma J, Nie Y, Paul E, Li Y, et al. Constitutive activation of IKKβ in adipose tissue prevents diet-induced obesity in mice.
Endocrinology 2012,
153, 154–165.
[Google Scholar]
-
46.
Kwon H, Laurent S, Tang Y, Zong H, Vemulapalli P, Pessin JE. Adipocyte-specific IKKβ signaling suppresses adipose tissue inflammation through an IL-13-dependent paracrine feedback pathway.
Cell. Repo. 2014,
9, 1574–1583.
[Google Scholar]
-
47.
Moon S, Park Y, Jang S, Kim S, Song D, Shin D, et al. Interleukin-2 improves insulin sensitivity through hypothalamic sympathetic activation in obese mice.
J. Neur. 2024,
21, 250.
[Google Scholar]
-
48.
Gao Z, Ye J. Why do anti-inflammatory therapies fail to improve insulin sensitivity?
Acta Pharm. Sinica B 2012,
33, 182–188.
[Google Scholar]
-
49.
Wang H, Ye J. Regulation of energy balance by inflammation: common theme in physiology and pathology.
Rev. Endo. Metab. Disord. 2015,
16, 47–54.
[Google Scholar]
-
50.
Wang A, Luan HH, Medzhitov R. An evolutionary perspective on immunometabolism.
Science 2019,
363, eaar3932.
[Google Scholar]
-
51.
Medzhitov R. The spectrum of inflammatory responses.
Science 2021,
374, 1070–1075.
[Google Scholar]
-
52.
Rosen ED, Kajimura S. Is it time to rethink the relationship between adipose inflammation and insulin resistance?
J. Clin. Investig. 2024,
134.
[Google Scholar]
-
53.
Maretty L, Gill D, Simonsen L, Soh K, Zagkos L, Galanakis M, et al. Proteomic changes upon treatment with semaglutide in individuals with obesity.
Nat. Med. 2025,
31, 266–277.
[Google Scholar]
-
54.
Yokose C, McCormick N, Abhishek A, Dalbeth N, Pascart T, Lioté F, et al. The clinical benefits of sodium–glucose cotransporter type 2 inhibitors in people with gout.
Nat. Rev. Rheumatol. 2024,
20, 216–231.
[Google Scholar]
-
55.
Lee JH, Zhang Y, Zhao Z, Ye X, Zhang X, Wang H, et al. Intracellular ATP in balance of pro-and anti-inflammatory cytokines in adipose tissue with and without tissue expansion.
Int. J. Obes. 2017,
41, 645–651.
[Google Scholar]
-
56.
Chavakis T, Alexaki VI, Ferrante AW,
Jr. Macrophage function in adipose tissue homeostasis and metabolic inflammation.
Nat. Immunol. 2023,
24, 757–766.
[Google Scholar]
-
57.
Salehi Farid A, Rowley JE, Allen HH, Kruger IG, Tavakolpour S, Neeley K, et al. CD45-PET is a robust, non-invasive tool for imaging inflammation.
Nature 2025,
639, 214–224.
[Google Scholar]
-
58.
Goldfine A, Conlin P, Halperin F, Koska J, Permana P, Schwenke D, et al. A randomised trial of salsalate for insulin resistance and cardiovascular risk factors in persons with abnormal glucose tolerance.
Diabetologia 2013,
56, 714–723.
[Google Scholar]