The Role of ILC2s in Tissue Injury and Repair

The Role of ILC2s in Tissue Injury and Repair
Jun Shang
1
Ruifeng Wang
1
Qi Cao
2,*

Received: 31 December 2025 Revised: 02 February 2026 Accepted: 20 March 2026 Published: 26 March 2026

© 2026 The authors. This is an open access article under the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/).
1. Introduction
Group 2 innate lymphoid cells (ILC2s) are tissue-resident innate lymphocytes that lack antigen-specific receptors. Their development and maintenance depend on the transcription factors GATA binding protein 3 (GATA3) and retinoic acid receptor-related orphan receptor alpha (RORα), with GATA3 acting as the master regulator and RORα essential for lineage stability [1,2,3]. First formally identified around 2010, ILC2s exhibit a lymphoid morphology and lack lineage markers characteristic of T cells, B cells, or myeloid cells [4,5]. Prior to the unified nomenclature established in 2013 [6], these cells were described under various names, including natural helper cells [4] and innate type 2 helper cells [5]. They are predominantly located at barrier surfaces, where they function as rapid response “immune sentinels” [7] and exhibit distinct tissue-specific phenotypic and functional adaptations.
The tissue repair process is a conserved, multi-phase cascade that begins immediately after injury and unfolds over several weeks. This orderly progression includes hemostasis, acute inflammation, inflammation resolution with debris clearance, and finally tissue regeneration and remodeling [8,9]. Within this cascade, alarmins such as interleukin-33 (IL-33), IL-25, and thymic stromal lymphopoietin (TSLP) are released from damaged or stressed cells and serve as critical early signals, bridging initial damage detection with the orchestration of subsequent immune and regenerative responses [10]. ILC2s are pivotal cellular targets of alarmins, initially characterized at mucosal barrier surfaces, but research has progressively revealed their equally important and complex roles within parenchymal organs, where they govern the delicate balance between regeneration and fibrosis.
In summary, ILC2s act as crucial immune sentinels across diverse tissues, balancing protective functions in host defense and acute repair against potential pathogenic contributions to chronic inflammation and fibrosis when their activity becomes dysregulated. To visually summarize this context-dependent duality, the following sections will systematically detail the dual roles of ILC2s in major organ systems—including the respiratory, skin, intestinal, hepatic, renal, cardiovascular, and nervous systems—highlighting their function as critical contextual regulators of tissue fate.
Before discussing organ-specific contexts, it is important to clarify the “dual” role of ILC2s. We have integrated their core mechanisms in Figure 1. The immunological function and fate of ILC2s are not fixed, but are dynamically shaped by the nature and duration of tissue injury, the local cytokine milieu, and tissue-specific niche signals. In acute damage, alarmins such as IL-33, IL-25, and TSLP, together with neural, microbial, and metabolic cues, generally drive ILC2s toward a reparative program characterized by amphiregulin production, type 2 cytokine release, and crosstalk with eosinophils, macrophages, and Tregs to promote inflammation resolution and tissue regeneration. By contrast, under persistent inflammation or fibrotic conditions, prolonged alarmin exposure, altered stromal signals, and subset imbalance can redirect ILC2s toward pathogenic states marked by sustained IL-5/IL-13 production, fibroblast activation, mucus hypersecretion, or even phenotypic plasticity toward ILC1-like cells. Thus, the “duality” of ILC2s refers to their context-dependent capacity to either preserve tissue integrity and repair or to amplify chronic inflammation and remodeling, depending on the signals that determine their activation state, subset composition, and differentiation trajectory.
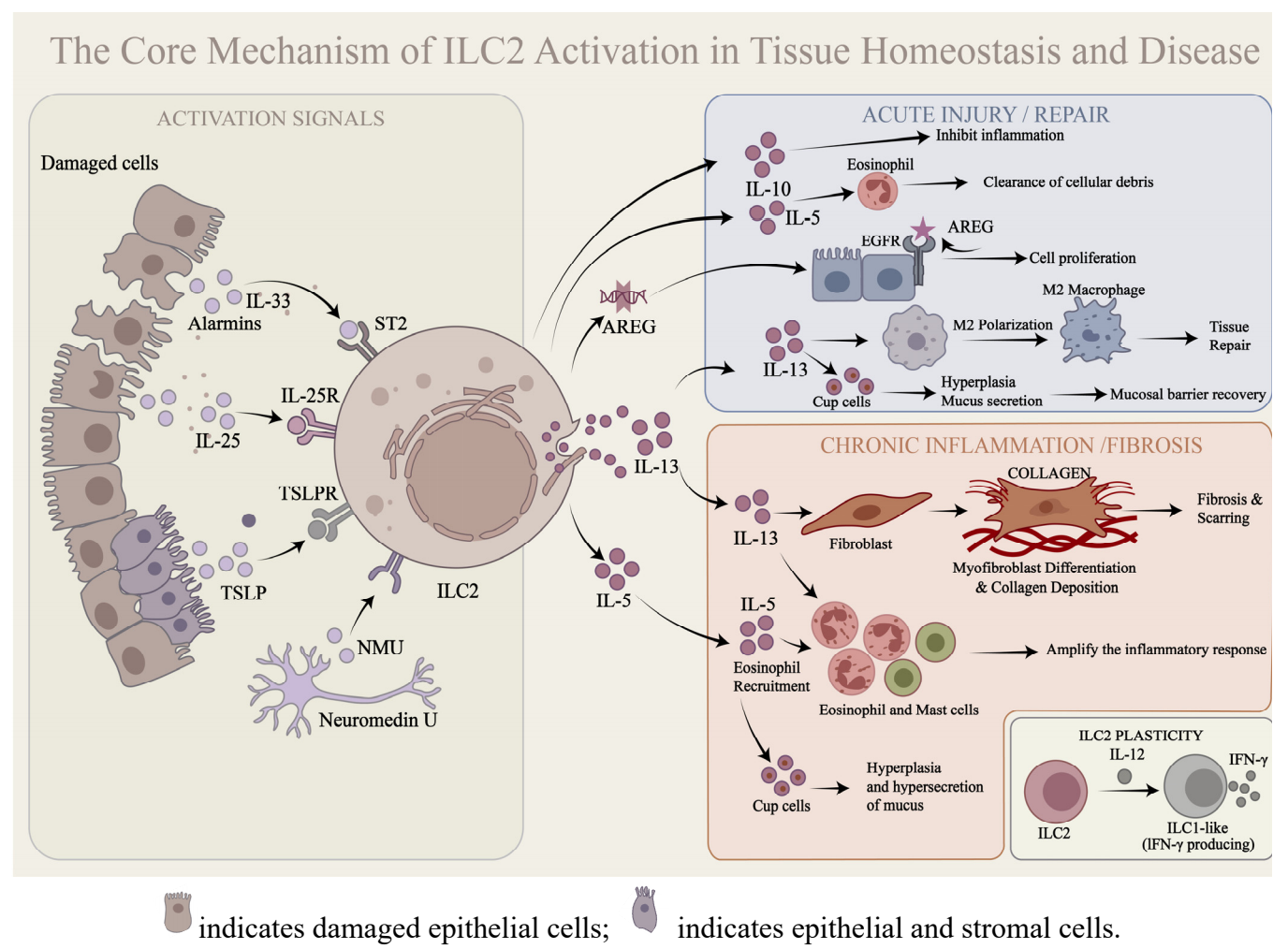 |
Figure 1. The dual roles of ILC2s in tissue injury, repair, and fibrosis. Upon acute injury, damaged cells release alarmins such as IL-33 and IL-25, which bind to receptors including ST2 and IL-25R on ILC2s, leading to their activation. Additional signals such as TSLP and the neuropeptide NMU can also regulate ILC2 function. Activated ILC2s promote tissue repair through a coordinated secretion of mediators: amphiregulin (AREG) stimulates epithelial proliferation; IL-13 induces goblet-cell hyperplasia, mucus secretion, and polarization of macrophages toward an anti-inflammatory M2 phenotype; IL-5 recruits eosinophils that help clear cellular debris; and IL-10 suppresses inflammation, collectively supporting tissue restoration. Under chronic inflammatory conditions, ILC2 function becomes dysregulated. Excessive IL-13 drives fibroblast-to-myofibroblast differentiation and collagen deposition, promoting fibrosis. IL-5-mediated recruitment of eosinophils and mast cells amplifies inflammatory responses, while goblet-cell-derived mucus becomes hypersecreted. Furthermore, in the presence of cytokines such as IL-12, ILC2s can undergo phenotypic plasticity and convert into IFN-γ-producing ILC1-like cells, further exacerbating tissue damage and pathological remodeling. TSLP, thymic stromal lymphopoietin; NMU, neuromedin U.
2. The Respiratory System: Coordinator of Acute Repair and Driver of Chronic Remodeling
The respiratory system, consisting of both conducting (trachea, bronchi) and respiratory (bronchioles, alveoli) zones, provides distinct microenvironments for resident ILC2s. Under homeostatic conditions, murine studies have identified ILC2s as being primarily localized to two key niches: the adventitial connective tissue surrounding large airways and the interstitial spaces of the alveolar parenchyma [11,12]. This strategic positioning places ILC2s at the forefront of detecting airborne insults. Their activation is primarily triggered by alarmins, such as IL-33, IL-25, and TSLP, released from damaged airway and alveolar epithelial cells [13,14]. In addition to tissue-resident populations, the pulmonary ILC2 pool exhibits considerable dynamic plasticity during inflammation. Following influenza virus infection or allergen exposure, circulating inflammatory ILC2s (iILC2s) are recruited to the lungs, where they collaborate with tissue-resident natural ILC2s (nILC2s) to orchestrate the immune response [15,16]. This recruitment emphasizes that ILC2-mediated immunity in the lung is not a static function of resident cells, but rather a dynamic process involving both local expansion and systemic mobilization.
Upon activation, ILC2s orchestrate a multi-layered repair response [17,18]. The dominant reparative pathway is the secretion of amphiregulin (AREG), which binds to the epidermal growth factor receptor (EGFR) on alveolar type II and airway epithelial cells, directly driving their proliferation and migration to restore barrier integrity. A secondary, immunomodulatory mechanism involves ILC2-derived IL-13, which polarizes alveolar macrophages toward an anti-inflammatory M2 phenotype, contributing to the resolution of inflammation. Additionally, IL-5-driven eosinophil recruitment plays an auxiliary role. These eosinophils do not directly clear debris; rather, they secrete IL-4 and IL-13, which enhance macrophage-mediated efferocytosis, a more plausible mechanism for tissue cleanup [19].
However, under chronic pathological conditions, persistent exposure to alarmins disrupts these reparative functions, causing ILC2s to drive pathological remodeling. In such conditions, the role of IL-13 shifts from immune modulation to direct pro-fibrotic activity. Murine models of asthma and idiopathic pulmonary fibrosis have demonstrated that excessive and sustained IL-13 production by ILC2s directly activates lung fibroblasts, promoting collagen deposition and airway remodeling [20,21,22]. Beyond this functional shift, chronic inflammation can also induce ILC2 plasticity. For example, in a murine model of silicosis, a mechanosensitive fibroblast niche drives ILC2 transdifferentiation into IFN-γ-producing, ILC1-like cells via the Notch3-IL-18 axis, a conversion that exacerbates tissue damage and fibrotic progression [23]. The critical role of ILC2 dysregulation in human disease is exemplified by chronic rhinosinusitis with nasal polyps (CRSwNP). Patient-derived nasal polyp tissues are highly enriched with activated ILC2s persistently producing IL-5 and IL-13, which drive eosinophil inflammation and tissue remodeling [24]. Allergen immunotherapy demonstrates the inducibility of ILC2 function by promoting the appearance of IL-10-producing ILC2s in the nasal mucosa. This is correlated with improved epithelial barrier integrity and better clinical outcomes [25].
In addition to these clinical observations, emerging evidence from murine studies reveals that ILC2 activity is regulated by broader systemic cues. Neural regulation has been supported by findings showing that agonists of the α7 nicotinic acetylcholine receptor attenuate ILC2 activation and reduce Alternaria-induced airway inflammation, suggesting the involvement of a cholinergic anti-inflammatory pathway [14]. Regulation by the microbiota is also evident through metabolites such as butyrate, which suppresses ILC2 function via HDAC inhibition. This suggests a gut-lung axis [26]. The functional significance of this crosstalk is clear: vancomycin-induced intestinal dysbiosis primes lung ILC2s for enhanced cytokine production, an effect that can be reversed by butyrate supplementation, demonstrating that microbial signals from distant sites can influence pulmonary ILC2 responsiveness [27]. Hormonal signals within the lung microenvironment also play a role in fine-tuning ILC2 activity. Studies in mice have shown that testosterone, a sex hormone present in the pulmonary niche, directly inhibits ILC2 proliferation and suppresses their secretion of IL-5 and IL-13 [28,29]. This finding offers a potential mechanistic explanation for sex-based differences in the prevalence of type 2 inflammatory diseases, such as asthma, and highlights how the local milieu, including structural cells, microbial products, and endocrine signals, shapes ILC2 functional output.
In summary, ILC2s in the respiratory system illustrate a context-dependent duality closely tied to tissue architecture. In their perivascular and interstitial niches, they act as first responders, with AREG-driven epithelial repair as the dominant pathway for restoring barrier integrity. However, in the chronically inflamed lung, persistent alarmin signaling within a fibrotic niche can alter this function, leading ILC2s to skew toward IL-13-driven fibroblast activation and even transdifferentiation into pro-inflammatory ILC1-like cells. It is important to note that this mechanistic understanding is primarily derived from murine models. Although human data confirm the presence and activation of ILC2s in diseases such as CRSwNP, these findings remain largely correlative [24]. The emerging evidence for inducible IL-10-producing ILC2s in humans [25] provides a promising avenue for future therapeutic exploration, though it requires further functional validation.
3. The Skin: From Acute Wound Healing to Chronic Inflammation and Fibrosis
The skin, as the body’s outermost barrier, possesses unique anatomical structures, including hair follicles and sebaceous glands, which extend deep into the dermis and create specialized immune niches. In murine skin, ILC2s are not uniformly distributed; they predominantly reside in the dermis, comprising 5–10% of CD45+ leukocytes, and are strategically localized around hair follicles and near sebaceous glands and blood vessels [30,31,32]. This perifollicular niche is significant as it exposes ILC2s to hair cycle-dependent epithelial signals and places them at the interface with the skin microbiota [31,32]. Human studies have confirmed these findings, demonstrating ILC2 presence in healthy skin and marked enrichment in the dermis of atopic dermatitis (AD) lesions, where they co-localize with basophils [33].
During homeostasis, dermal ILC2s can interact with local mast cells, suppressing their activity through IL-13 [31]. Beyond this direct immunoregulatory role, skin ILCs also contribute to microbiota homeostasis through two distinct mechanisms. First, a specialized subset of RORγt+ ILCs, distinct from classical ILC2s, resides near sebaceous glands within hair follicles, where they constitutively produce TNF and lymphotoxins to negatively regulate sebaceous gland function via Notch repression, thereby controlling antimicrobial lipid production and shaping Gram-positive bacterial communities [34]. Second, classical ILC2s directly sense bacteria through NOD2, responding to skin-resident Staphylococcus species by inducing IL-6 and IL-8, cytokines involved in barrier maintenance [30].
In response to skin injury, ILC2s transition from homeostatic sentinels to active mediators of repair. Upon injury, IL-33 released from damaged keratinocytes activates dermal ILC2s in an ST2-dependent manner, prompting AREG secretion that directly promotes keratinocyte proliferation and migration, accelerating re-epithelialization and wound closure [32,35]. This AREG-driven pathway represents a primary, non-redundant role for ILC2s in restoring skin barrier integrity. Following injury, ILC2s dynamically accumulate in the granulation tissue at wound edges, positioning them to influence keratinocyte function during re-epithelialization [35] directly.
In chronic inflammatory skin diseases, these protective functions are subverted, leading to tissue damage. In AD, keratinocyte-derived IL-25 activates ILC2s via IL-25R, driving IL-13 production that directly downregulates key epidermal barrier proteins such as filaggrin, establishing a vicious cycle of barrier disruption and sustained inflammation [36]. ILC2-derived IL-5 serves as a secondary amplifier by recruiting and activating eosinophils and mast cells [31,32,36]. Human studies confirm that ILC2 enrichment in AD lesions correlates with disease severity [33]. In fibrotic diseases such as systemic sclerosis (SSc), a distinct TGFβ-driven mechanism operates: TGFβ downregulates the inhibitory receptor KLRG1 on ILC2s, reducing their IL-10 production while enabling sustained IL-13 secretion that directly activates dermal myofibroblasts, driving excessive collagen deposition and fibrosis [37].
The critical importance of ILC-microbiota dialogue is further underscored by genetic lesions disrupting this axis. In mice with epidermal ADAM10 deletion, spontaneous dysbiosis drives inflammation, with distinct species dictating specific pathologies [38]. In hair follicles, ADAM10-Notch disruption downregulates β-defensin 6, allowing C. mastitidis dominance, which triggers IL-7R-/S1PR1-/CCR6-dependent recruitment of inflammatory ILC2s (co-expressing GATA3/RORγt and producing IL-17/type 2 cytokines) that mediate pyroptotic destruction of hair follicle stem cells, causing irreversible alopecia [38]. These models establish a direct causal chain linking epithelial defects, impaired antimicrobial defense, dysbiosis, and pathogenic ILC2 activation in the process of tissue destruction.
In summary, skin ILC2s exemplify a context-dependent duality rooted in tissue architecture. In acute wound healing, AREG-driven epithelial repair constitutes the dominant protective pathway. However, in chronic inflammatory diseases such as AD and SSc, this function is subverted toward IL-13-driven barrier disruption and fibrosis. Importantly, skin ILCs, including RORγt+ subsets, also play a homeostatic role in shaping the microbiota through regulation of the sebaceous glands, and disruptions to this epithelial-ILC-microbiota dialogue can drive severe inflammatory pathology.
4. The Intestine: A Multi-Signal Integrator in Barrier Maintenance and Inflammatory Regulation
In the intestinal tract, ILC2s are strategically positioned at the frontline of host-environment interactions. They predominantly reside in the lamina propria of both the small intestine and colon, where they localize adjacent to intestinal epithelial cells and in close contact with crypt progenitors [39,40]. In the small intestine, ILC2s are found near tuft cells, forming a functional unit referred to as the “tuft cell-ILC2 circuit”, which serves as a dedicated sensor for helminth infection [41,42]. In addition to the lamina propria, ILC2s also populate intestinal lymphoid structures such as Peyer’s patches and mesenteric lymph nodes, where they contribute to the broader mucosal immune network [39].
The intestinal microenvironment exposes ILC2s to a remarkable diversity of signals; the neuronal peptide neuromedin U (NMU) binds to NMUR1 on intestinal ILC2s, activating them to secrete type 2 cytokines. This neuro-immune circuit allows ILC2s to respond to local neuronal cues independently of epithelial alarmins [43]. Additionally, dietary and microbial signals play a critical role in regulating ILC2 function. Murine studies have shown that dietary components, such as inulin fiber, modulate ILC2 function indirectly via microbiota-derived bile acids [42]. In contrast, microbial metabolites such as butyrate can directly suppress ILC2 activation, demonstrating a gut microbiota-ILC2 axis that translates nutritional inputs into immune responses [26].
In response to acute challenges, intestinal ILC2s orchestrate a coordinated, multi-layered response. The dominant mechanism for helminth defense is the tuft cell-ILC2 circuit. Upon parasite infection, tuft cell-derived IL-25 activates ILC2s, which in turn secrete IL-13. This ILC2-derived IL-13 drives tuft cell and goblet cell hyperplasia, creating a feedback loop that amplifies the type 2 response and promotes effective pathogen expulsion [41,44]. This circuit represents a specialized adaptation of the small intestine to parasitic threats. Additionally, ILC2s contribute to tissue repair through AREG secretion, which directly stimulates intestinal epithelial proliferation and supports wound closure, complementing their pathogen-clearing function by restoring the barrier [45,46].
Beyond epithelial interactions, ILC2s engage with other immune cells to fine-tune the mucosal environment. ILC2s contribute to macrophage polarization toward an anti-inflammatory M2 phenotype, creating a pro-repair microenvironment that supports tissue regeneration [45]. Through IL-9 production, ILC2s may also potentiate regulatory T cell (Treg) function, forming an auxiliary immunoregulatory circuit that complements epithelial repair pathways [25].
When the finely tuned signal integration is disrupted, intestinal ILC2s can transition from protective mediators to drivers of pathology. In chronic intestinal inflammation, the “diet-microbiota-metabolite” axis emerges as a key programming mechanism. Murine studies have demonstrated that inulin fiber, through modulation of the microbiota-bile acid axis, promotes a pro-inflammatory ILC2 phenotype characterized by elevated IL-5 and reduced AREG production, exacerbating colitis [42]. In the context of polymicrobial sepsis, ILC2s’ protective functions can be subverted toward immunosuppression. Both human and murine studies have shown that intestinal ILC2s activated via the IL-33/ST2 axis can exacerbate injury by promoting IL-13 release. This IL-13, in turn, stimulates IL-10 production from local immune cells and may also induce an IL-10-producing ILC2 subset, leading to immunosuppression that impairs bacterial clearance and worsens outcomes [47].
In summary, intestinal ILC2s serve as central orchestrators, integrating neural, dietary, microbial, and epithelial signals to regulate mucosal immune balance. In acute settings, they coordinate repair through the tuft cell-ILC2 circuit and AREG-mediated epithelial regeneration. In chronic conditions, however, they become hubs where environmental signals—particularly those from diet and microbiota—dictate immunological outcomes, driving either inflammation or immunosuppression depending on context.
5. The Liver and Biliary Tract: A Dual-Pronged Role in Repair and Fibrosis
Murine studies have shown that ILC2s localize to the non-parenchymal cell fraction, specifically in the subcapsular region and around portal tracts [48,49,50,51]. Under steady-state conditions, ILC2s make up a small proportion of hepatic lymphocytes and maintain a naive, non-activated phenotype, with low expression of KLRG1, CD25, and GATA3 [52]. Following liver injury, ILC2s are recruited to the affected areas and accumulate in regions of damage [53,54]. In the biliary system, ILC2s infiltrate the subepithelial connective tissue of the extrahepatic bile ducts, where they localize in close proximity to peribiliary glands [51]. Human studies have confirmed the presence of ILC2s in liver tissue, where they make up 0.03–0.89% of CD45+ lymphocytes, a frequency significantly higher than in peripheral blood. Their activation status (as indicated by CD69 expression) correlates with the severity of liver fibrosis, suggesting clinical relevance [50]. In patients with biliary atresia, the balance between nILC2 and iILC2 within the biliary tree is predictive of clinical outcomes, highlighting the prognostic value of ILC2 subset composition [51].
In immune-mediated hepatitis, such as concanavalin A-induced hepatitis, murine studies have consistently demonstrated that ILC2s primarily exert a pro-damage effect. Hepatocyte damage releases IL-33, which activates ILC2s and drives them to produce IL-5 and IL-13. These cytokines subsequently recruit and activate inflammatory cells, including eosinophils, which in this pro-inflammatory context may contribute to tissue damage rather than repair. This highlights the context-dependent role of eosinophils in inflammation [52,53]. The dominant mechanism in this model is IL-13-driven amplification of the T cell-mediated inflammatory response. A secondary, protective mechanism involves ILC2-derived AREG, which suppresses ILC2 activation and promotes the induction of ST2+ Tregs that produce IL-10, a cytokine more prominently associated with Tregs than with ILC2s under homeostatic conditions. This offers a negative feedback loop that modulates inflammation and provides protection [55].
In contrast, in ischemia-reperfusion injury (IRI), a model characterized by innate immune responses, murine studies have shown that the IL-33/ILC2 axis promotes tissue protection and repair. In this context, the dominant mechanism shifts to IL-13-mediated macrophage reprogramming toward an anti-inflammatory M2 phenotype. IL-5-driven eosinophil pathways further contribute to this protective effect, as eosinophils produce IL-13 and promote anti-inflammatory macrophage polarization [56].
A parallel functional duality is evident in the biliary system, where ILC2s orchestrate epithelial repair in acute settings but drive pathological remodeling under chronic conditions. In response to biliary injury, such as in models of biliary atresia, murine studies have shown that ILC2s are recruited to the periductal region via chemokine cues such as CCL20 [51]. Once localized, they orchestrate epithelial repair through two complementary mechanisms: AREG directly stimulates cholangiocyte proliferation via EGFR signaling, while IL-13 modulates the local immune milieu to support regeneration [48,51].
However, within the sustained inflammatory milieu of chronic injury, this reparative program is often derailed. Persistently activated ILC2s, through sustained IL-13 secretion, not only drive excessive peribiliary glandular hyperplasia but also directly activate hepatic stellate cells and portal fibroblasts, leading to pathological fibrosis. In severe cases, this dysregulated repair and proliferative response can even create a tumor-promoting microenvironment, contributing to the development of cholangiocarcinoma [48].
The functional duality of hepatic ILC2s is underpinned by the plasticity between two distinct subsets: nILC2s and iILC2s. Murine studies have defined nILC2s by high ST2 expression and responsiveness to IL-33, while iILC2s are characterized by high expression of IL-25R and KLRG1 [51]. These subsets show functional specialization: nILC2s are primarily responsible for AREG-dependent epithelial repair, while iILC2s are biased toward inflammatory and pro-fibrotic responses. The relative abundance and stability of the nILC2 subset are regulated by IL-4Rα/STAT6 signaling. Both human and murine studies have demonstrated that the balance between nILC2 and iILC2 subsets predicts clinical outcomes. In biliary atresia, a higher nILC2-to-iILC2 ratio correlates with better repair outcomes and a more favorable clinical prognosis, while predominance of iILC2s is associated with progressive fibrosis and worse outcomes [51].
In addition to their immune functions, hepatic ILC2s may also play a role in metabolic regulation. Murine studies have demonstrated that liver ILC2s contribute to blood glucose homeostasis through IL-13 signaling, suggesting an endocrine-related function that extends beyond tissue repair and inflammation [57]. This discovery opens new avenues for understanding ILC2s as integrators of both immune and metabolic homeostasis in the liver.
In summary, ILC2s are pivotal regulators of hepato-biliary homeostasis, with their dual capacity for regeneration and fibrosis governed by the type of injury, the chronicity of the disease, and the plasticity between subsets. In acute settings, ILC2s can either exacerbate damage or promote repair, depending on the dominant immune response. In the biliary system, ILC2s promote epithelial repair following acute injury but drive fibrosis and tumorigenesis under chronic inflammatory conditions. This functional diversity is further compartmentalized at the subset level, with nILC2s mediating repair and iILC2s driving pathology, and the balance between these subsets predicts clinical outcomes in biliary atresia.
These findings must be interpreted in light of several key experimental variables: model specificity, as the roles of ILC2s differ in Con A hepatitis versus IRI; injury kinetics; and subset definitions, which are well-defined in murine models but less established in human liver disease. Tissue specificity also plays a role, as ILC2 functions differ between the liver parenchyma and the biliary system, highlighting the importance of considering the anatomical context. The mechanistic understanding of hepatic ILC2 function is largely based on murine models, while human studies have provided critical validation of subset imbalance and its clinical correlation in biliary atresia [50,51]. Future studies should aim to functionally validate the roles of specific subsets in human tissues and explore whether therapeutic modulation of the nILC2/iILC2 balance can shift outcomes from fibrosis toward regeneration.
6. The Kidney: A Model of Acute Repair and Therapeutic Targeting
In the kidney, ILC2s are positioned at the corticomedullary junction and outer medulla. These regions are particularly vulnerable to ischemic injury due to their high metabolic demand and limited oxygen supply [58,59]. Parabiosis models have confirmed their tissue-resident nature, showing minimal exchange between the circulation and the kidney under steady-state conditions, distinguishing them from circulating immune cells that only infiltrate during injury [59]. Single-cell analysis has identified renal ILC2s through their expression profile, including CD45+, lineage marker-negative, and GATA3+ [60]. This anatomical positioning at the corticomedullary junction places them at the frontline of sensing ischemic damage, enabling rapid responses to tubular epithelial injury.
Renal ischemia-reperfusion injury (IRI) is an ideal paradigm for delineating the protective roles of ILC2s in acute kidney injury. Murine studies have demonstrated that renal IRI induces tubular epithelial necrosis and the rapid release of the alarmin IL-33 [59,60]. IL-33, acting predominantly via its receptor ST2, recruits and activates both tissue-resident and circulating ILC2s, initiating a reparative program [59,60]. Once activated, ILC2s promote repair through two complementary mechanisms: direct epithelial support and immune microenvironment modulation. First, ILC2s secrete AREG, which binds to EGFR on tubular epithelial cells, promoting their survival and proliferation [58,60,61]. Second, ILC2s release IL-13, which polarizes macrophages toward an anti-inflammatory M2 phenotype. These M2 macrophages contribute to inflammation resolution and create a pro-repair environment that supports tubular regeneration [59,60]. This dual-action mechanism, combining direct epithelial support with indirect immune modulation, positions ILC2s as central orchestrators of the acute repair response.
This core mechanism underscores the therapeutic potential of targeting ILC2s, with strategies that aim to amplify or supplement this endogenous response. One approach involves using biomaterial carriers for localized IL-33 administration. Murine studies have shown that using biomaterial carriers, such as DNA nanorafts, enables precise and sustained release of IL-33 at the injury site, effectively expanding the local ILC2 pool while circumventing the risks associated with systemic administration. The targeted IL-33 delivery activates ILC2s, which in turn promote M2 macrophage polarization and increase local IL-10 production, likely from multiple cellular sources, including macrophages and potentially a subset of ILC2s induced in this therapeutic context, contributing to the suppression of inflammation and acceleration of tubular epithelial replacement and functional recovery [62]. An alternative strategy is the adoptive transfer of ex vivo expanded ILC2s. Murine studies have demonstrated that these pre-activated cells home to the kidneys and efficiently execute reparative functions [60,62]. Both interventions have been shown to significantly accelerate renal function recovery, evidenced by rapid declines in serum creatinine and blood urea nitrogen. They also markedly mitigate histological damage, including reduced tubular injury scores, diminished tubular epithelial cell apoptosis, and attenuated inflammatory cell infiltration [60,62].
In contrast, during the progression of chronic kidney disease to fibrosis, murine studies have suggested that persistently activated ILC2s may undergo a maladaptive shift. Through sustained secretion of pro-fibrotic factors such as IL-13, they can contribute to pathological remodeling and interstitial fibrosis [7].
In summary, the kidney serves as an instructive model for understanding both the therapeutic potential and inherent risks of ILC2 modulation. Preclinical studies have successfully harnessed this biology through targeted IL-33 delivery and adoptive ILC2 transfer, demonstrating accelerated functional recovery in murine models of acute kidney injury [60,62]. However, the transition from acute repair to chronic fibrosis reveals a critical duality: the same IL-13 that promotes regeneration in acute settings can, when persistently produced, drive fibroblast activation and interstitial fibrosis [7].
The findings described in this section are derived from murine studies and have not been directly validated in humans to date. The therapeutic strategies validated in murine models, DNA nanoraft-mediated IL-33 delivery, and adoptive ILC2 transfer, have not been tested in humans, and their translational relevance remains uncertain. While direct evidence for butyrate-mediated modulation of renal ILC2s is currently lacking, the established role of butyrate in suppressing ILC2 function at other barrier sites raises the possibility of similar regulatory circuits in the kidney.
7. The Cardiovascular System: Disease-Specific Roles—From Plaque Stabilization to Myocardial Repair
In the cardiovascular system, ILC2s occupy multiple distinct anatomical niches, reflecting their functional specialization in response to various types of insult. In models of atherosclerosis, murine studies have localized ILC2s to the aorta, periaortic adipose tissue, and periaortic lymph nodes [63,64,65]. Within the aorta itself, ILC2s are found in the adventitia and within tertiary lymphoid structures that form around atherosclerotic plaques, especially in advanced disease, positioning them to directly influence plaque inflammation and stability [65]. In periaortic adipose tissue, ILC2s are present in fat-associated lymphoid clusters (FALCs), which also contain T cells, B cells, and plasma cells, suggesting that they play a role in coordinating local immune responses [64]. Additionally, gonadal white adipose tissue harbors ILC2s with a natural ILC2 phenotype (KLRG1+ST2+), where they contribute to metabolic homeostasis [66,67]. After myocardial infarction, murine studies have shown that cardiac ILC2s accumulate specifically in the infarcted area and border zone during the first week after injury [68,69]. These cells are found in the myocardial interstitium, not within blood vessels, and also in pericardial adipose tissue, where they reside within fat-associated lymphoid clusters adjacent to the epicardium [69]. This dual localization, both within the damaged myocardium and in the adjacent pericardial fat, positions ILC2s to integrate signals from both the injured tissue and the surrounding adipose microenvironment. While human studies have detected ILC2s in cardiac tissue, detailed localization data remain limited [70].
In atherosclerosis, ILC2s play a defined protective role through a dual immunomodulatory strategy that targets lipid clearance and plaque stability. The primary mechanism involves IL-5-mediated stimulation of B1a cells. ILC2-derived IL-5 promotes B1a cell production of natural IgM antibodies, which recognize and facilitate the clearance of oxidized LDL, thereby reducing lipid accumulation and inflammation within plaques [63,71]. This pathway links ILC and humoral immunity in atherosclerosis. A secondary, reinforcing mechanism involves IL-13-mediated macrophage modulation. ILC2-derived IL-13 polarizes plaque macrophages toward a stabilizing M2 phenotype, enhancing collagen deposition and fibrous cap integrity [65,71]. This protection is dynamically regulated, as seen with high-fat diets, which suppress ILC2 numbers and function [65].
In contrast to the consistent protective role in atherosclerosis, ILC2 function in myocardial injury shows marked phase dependence, shifting from protection in acute settings to potential harm in chronic disease. Following acute insults like myocardial infarction (MI), murine studies demonstrate that cardiac ILC2s help mitigate damage. Core mechanisms include IL-13 secretion to modulate macrophage polarization and suppress inflammation [72,73], alongside synergistic effects from factors such as bone morphogenetic protein-7 (BMP-7) or an IL-5-eosinophil axis, in which recruited eosinophils secrete IL-4 and IL-13 to enhance macrophage-mediated repair processes [19,68,74]. This acute protective role is evident in sepsis-induced cardiac injury, where IL-33-activated ILC2s improve function, a finding supported by preclinical and preliminary clinical studies [70].
Conversely, the long-term role of ILC2s in chronic cardiac pathologies remains ambiguous and may become detrimental. In models like doxorubicin-induced cardiotoxicity, increased ILC2s post-injury suggest a potential pro-fibrotic risk, raising the question of whether chronic ILC2 activation drives maladaptive remodeling [75]. The same IL-13 that promotes macrophage-mediated repair in acute settings may, when chronically produced, activate cardiac fibroblasts and promote pathological fibrosis, a mechanism well-established in other organs but requiring further validation in the heart.
The divergent roles of ILC2s in cardiovascular disease—protective in atherosclerosis and acute myocardial injury, potentially pathogenic in chronic cardiac conditions—can be attributed to a central mechanistic paradox: IL-13, the key effector cytokine of ILC2s, exerts context-dependent effects. In protective contexts, IL-13 promotes M2 macrophage polarization, supporting inflammation resolution, tissue repair, and plaque stabilization [65,71,72,73]. In pathogenic contexts, sustained IL-13 production can activate fibroblasts, driving excessive collagen deposition and maladaptive fibrosis [75]. This fibrotic potential, well documented in other organs such as the liver, lungs, and skin, likely contributes to adverse remodeling in chronic cardiac conditions, although direct evidence in the heart remains limited.
In summary, the divergent roles of ILC2s in acute MI versus chronic cardiomyopathy highlight the influence of injury duration and disease stage [72,73,74,75]. Short-term, controlled ILC2 activation appears beneficial, while persistent activation may promote pathological remodeling. Second, all mechanistic insights into cardiovascular ILC2 function are derived from murine studies [63,64,65,66,67,68,69,70,71,72,73,74,75], with human data limited to correlative observations such as detection of ILC2s in cardiac tissue and associations with clinical outcomes [70] without functional validation. Whether the IL-5/IgM axis or IL-13/fibroblast pathways operate similarly in humans with atherosclerosis or heart failure remains unknown.
8. The Central Nervous System: Emerging Neuro-Immune Regulators and Protectors
In the central nervous system (CNS), ILC2s are strategically localized to areas that enable immune surveillance while respecting the blood-brain barrier. Studies in mice have shown that ILC2s are primarily found in the meninges and choroid plexus, which are interfaces between the CNS and the periphery that are accessible to immune cells [76,77]. In the meninges, ILC2s are concentrated around the dural sinuses and distributed sparsely throughout the dura mater and spinal meninges [76]. This positioning adjacent to cerebrospinal fluid (CSF) compartments allows ILC2s to rapidly detect damage-associated molecular patterns (DAMPs) released from injured brain tissue [78]. Following traumatic brain injury, ILC2s accumulate in the meninges and CSF, with evidence suggesting bidirectional communication with peripheral immune organs through meningeal lymphatic vessels [79]. In the choroid plexus, ILC2s localize within the stromal compartment, close to the blood-CSF barrier [77]. These cells make up about 50% of the lymphocytes in the choroid plexus of both aged mice and humans, suggesting their role in lifelong immune surveillance [77].
Murine studies have now confirmed the existence of resident group 2 innate lymphoid cells within the meninges and choroid plexus, where they form a unique barrier for immune surveillance [76,78]. In response to acute CNS insults—such as spinal cord injury or cerebral ischemia DAMPs, including the alarmin IL-33, promptly activate these tissue-resident ILC2s [78,79].
Activated ILC2s mediate neuroprotection through three complementary mechanisms, with their relative contributions varying by injury type and stage. First, ILC2-derived IL-13 drives microglial reprogramming: it suppresses pro-inflammatory microglial activation (reducing TNF-α and IL-1β) while promoting anti-inflammatory factors, such as IL-10—which may be produced by both microglia and infiltrating ILC2s [80,81]. This limits secondary tissue damage and creates a pro-repair microenvironment. Second, ILC2s recruit eosinophils via IL-5; these eosinophils secrete IL-4 and IL-13, further supporting anti-inflammatory polarization and enhancing efferocytosis, contributing to debris clearance and tissue recovery [19,78]. Third, ILC2-derived CGRP directly protects neurons from apoptosis and modulates local vascular tone, improving blood flow to injured areas [78,79]. Beyond these core mechanisms, additional regulatory pathways operate in specific injury contexts. Following traumatic brain injury, for instance, AMPK activation promotes the expansion of an IL-10-producing regulatory ILC2 subset that inhibits pro-inflammatory ILC1/ILC3 responses, further suppressing neuroinflammation and improving long-term neurological outcomes [82]. Importantly, these protective mechanisms exhibit injury-specific nuances. For example, in intracerebral hemorrhage, the IL-13/microglial axis appears particularly dominant [81], whereas in spinal cord injury, CGRP-mediated neuroprotection may play a more prominent role [78]. This context-dependence highlights the importance of considering injury type when designing ILC2-targeted therapies.
The role of ILC2s in the CNS extends beyond acute injury to include fundamental functions in neurodevelopment and cognitive maintenance throughout life. During early postnatal stages, ILC2s play a key role in shaping hippocampal synaptic development. Mice deficient in ILC2s exhibit selective reductions in inhibitory synapse frequency, which leads to long-lasting impairments in learning and memory tasks [83]. This suggests a previously unrecognized role for ILC2s in neuronal network maturation, likely mediated by local cytokine production that influences synaptic pruning and stabilization. Additionally, during aging, ILC2s residing in the choroid plexus help maintain brain homeostasis by suppressing neuroinflammation and promoting hippocampal neurogenesis in an IL-5-dependent manner [84]. This IL-5-mediated effect recruits eosinophils and promotes a pro-regenerative environment, mitigating age-associated cognitive decline. These homeostatic functions position ILC2s as lifelong regulators of CNS health, and their dysfunction may contribute to neurodevelopmental disorders and accelerated cognitive aging.
In aging and neurodegenerative diseases, ILC2s exhibit a dual trajectory, maintaining resilience in normal aging but showing deficits in pathological conditions. During normal aging, ILC2s in the brain resist senescence and retain self-renewal capacity, unlike many other immune cells [84]. Choroid plexus ILC2s continue to produce IL-5, supporting eosinophil recruitment and hippocampal neurogenesis, which may explain their role in preserving cognitive function in aged mice [83,84]. In contrast, in models of Alzheimer’s disease, ILC2s exhibit numerical and functional deficits, potentially exacerbating neuroinflammation and cognitive decline [84]. The mechanisms underlying this dysfunction remain unclear, but they may involve chronic amyloid-β-induced inflammation that impairs ILC2 survival or their responsiveness to IL-33. Whether similar deficits occur in human Alzheimer’s disease patients remains an unresolved issue, representing a critical gap in translational knowledge. These observations suggest that while ILC2s are intrinsically equipped to support CNS health throughout life, their dysfunction in disease contexts may contribute to pathology, opening the possibility of ILC2-targeted therapies for neurodegenerative conditions.
In summary, ILC2s in the CNS play protective roles in response to acute injury and contribute to neurodevelopment and cognitive maintenance. They mediate neuroprotection through IL-13-driven microglial modulation, IL-5-dependent eosinophil recruitment, and CGRP secretion, with mechanisms varying depending on injury type. In aging, ILC2s maintain brain health by suppressing neuroinflammation and promoting neurogenesis, while their dysfunction in neurodegenerative diseases such as Alzheimer’s disease may contribute to pathology. These findings underscore the potential of ILC2-targeted therapies for neurodegenerative diseases, although further studies are needed to understand the underlying mechanisms and translate these findings to human conditions.
9. ILC2s in Other Tissues: Diverse Roles in Homeostasis and Repair
Beyond major organ systems, ILC2s contribute critically to tissue homeostasis and post-injury repair in several specialized local niches. Murine studies have demonstrated that a resident population of ILC2s is found in the limbal region. Upon corneal epithelial injury, alarmins, including IL-25, IL-33, and TSLP, released from epithelial cells and macrophages activate ILC2s, stimulating their local expansion. Activated ILC2s then secrete amphiregulin, which directly promotes the proliferation and migration of corneal epithelial cells, thereby accelerating wound re-epithelialization. Conversely, depletion of ILC2s markedly impairs this repair process [85]. Within the bone marrow, murine studies have shown that ILC2s can be stimulated by IL-33 to secrete IL-4 and IL-13. These type 2 cytokines potently inhibit osteoclast differentiation. In a model of postmenopausal osteoporosis, ILC2s utilize this pathway to exert a net bone-protective effect by restraining excessive bone resorption [86]. Following acute thymic insults such as radiotherapy or chemotherapy, murine studies have revealed that intrathymic ILC2s are activated by IL-33 and IL-25. They promote the regeneration of thymic epithelial cells (TECs) and have a notable effect on medullary TEC differentiation by secreting AREG and IL-13. This ILC2-mediated repair is vital for restoring thymic architecture and function, including the re-establishment of central tolerance [87]. Moreover, in the context of islet transplantation for type 1 diabetes, murine studies have demonstrated that IL-33-induced IL-10-producing ILC2s [88]. These cells migrate to the graft site, where they suppress CD4+ T cell-mediated rejection by secreting IL-10, prolonging allograft survival, and demonstrating a repair function in an immunologically mediated chronic injury setting [88]. All findings in this section are derived from murine studies, highlighting the need for translational research in humans.
The Functional Division of ILC2 and Tregs in IL-10 Production
Tregs constitutively produce IL-10, maintaining immune tolerance under steady-state conditions. In contrast, ILC2s produce IL-10 only in specific contexts—such as following allergen immunotherapy in the nasal mucosa [25], AMPK activation after traumatic brain injury [82], or IL-33 stimulation during islet transplantation [88]—forming an inducible, context dependent IL-10 producing subset. In conventional inflammation models lacking such inductive signals, ILC2-derived IL-10 is undetectable. For instance, in immune-mediated hepatitis, ST2+ Tregs are the major source of protective IL-10, whereas ILC2s do not directly produce IL-10 but instead regulate inflammation through other mechanisms [55]. Thus, the functional dichotomy in IL-10 production, constitutive and systemic for Tregs versus inducible and tissue-specific for ILC2s underpins their distinct roles in immune responses.
10. Integrating ILC2s into the Cellular Network: Lessons from Eosinophils and Macrophages
Eosinophils as a Bridge Between ILC2 and Macrophages. ILC2-derived IL-5 recruits eosinophils to sites of injury, where they secrete IL-4 and IL-13 to enhance macrophage phagocytic activity and tissue-remodeling functions, thereby indirectly promoting repair [17,18]. This IL-5-eosinophil axis operates across multiple organs: after myocardial infarction, it protects the heart [68,74]; during hepatic ischemia-reperfusion injury, eosinophils contribute to anti-inflammatory macrophage polarization [56]; yet in immune-mediated hepatitis, the same axis may exacerbate hepatocyte damage [52,53]. These contrasting outcomes highlight that eosinophils, as intermediaries, execute functions dictated by the local inflammatory microenvironment, not solely by ILC2 signals.
Macrophages as Key Effector Partners of ILC2. ILC2-derived IL-13 is a critical signal for polarizing macrophages toward a pro-repair M2 phenotype [17,18,60]. However, for macrophages to fully execute repair functions—phagocytosis, growth factor production, and matrix remodeling—they require a second signal: apoptotic cells. A study demonstrated that IL-4/IL-13 alone induces genes related to pattern recognition, chemotaxis, and adhesion, but the induction of repair-associated genes requires co-stimulation with apoptotic cells, a process dependent on the AXL and MERTK receptors [19]. Thus, ILC2-provided IL-13 is necessary but not sufficient for macrophage-mediated repair; the outcome hinges on the availability of apoptotic cells in the microenvironment. This may explain variability in observed ILC2 pro-repair effects across studies: in settings rich in apoptotic cells, ILC2 signals translate effectively into repair; when apoptotic cells are scarce or rapidly cleared, the same signals may fail to elicit equivalent repair.
11. Translational Prospects
The dual role of ILC2s, protective in acute injury and potentially pathogenic in chronic inflammation, presents both significant opportunities and challenges for therapeutic development, as summarized in Figure 2. Current strategies are increasingly focusing on precision targeting. One approach aims to inhibit pathogenic ILC2 responses by using antagonists that target alarmin pathways, such as IL-33, TSLP, and IL-25, or the CRTH2 receptor, all of which are relevant in conditions like asthma, atopic dermatitis, and fibrosis [89]. The complementary strategy seeks to enhance reparative ILC2 functions through local administration of IL-33 or amphiregulin, metabolic interventions, induction of regulatory ILC210 subsets, or adoptive ILC2 transfer. These methods are designed to augment endogenous repair mechanisms following acute injury [60,62].
However, several critical limitations must be considered. The dual nature of ILC2 function inherently carries risks: systemic inhibition may hinder essential tissue repair, while non-specific activation could exacerbate fibrosis or allergic inflammation through sustained IL-13 secretion [7,20,21,22]. Achieving precise spatiotemporal control to balance these opposing outcomes remains a major technical challenge. Moreover, each therapeutic approach has its own safety concerns. While IL-33 delivery has shown protective effects in acute kidney injury, chronic administration may promote fibrosis [59,60]. Adoptive ILC2 transfer, although effective in preclinical models of renal injury and islet transplantation, faces challenges regarding cell survival, phenotypic stability, and the risk of maladaptive differentiation within inflammatory microenvironments [60,62,88]. Similarly, metabolic interventions such as butyrate, which are effective in suppressing ILC2-driven inflammation, may have off-target effects on other immune populations, raising concerns about broader immunosuppression [26,27].
Furthermore, significant gaps remain in translating preclinical findings into clinical practice. The majority of mechanistic insights and therapeutic strategies are based on murine models, with human data still limited to correlative observations [70]. Key challenges include the lack of functional validation in human tissues, the absence of standardized protocols for ILC2 identification and manipulation, and a limited understanding of human ILC2 biology across different tissues and disease states [22,24,55]. To bridge these gaps, it will be essential to integrate advanced technologies such as single-cell multi-omics and spatial transcriptomics. These tools will aid in unraveling ILC2 heterogeneity and tissue-specific regulatory circuits. A deeper understanding of these processes will be crucial for developing precision immunotherapies that maximize the protective potential of ILC2s while minimizing their pathogenic risks.
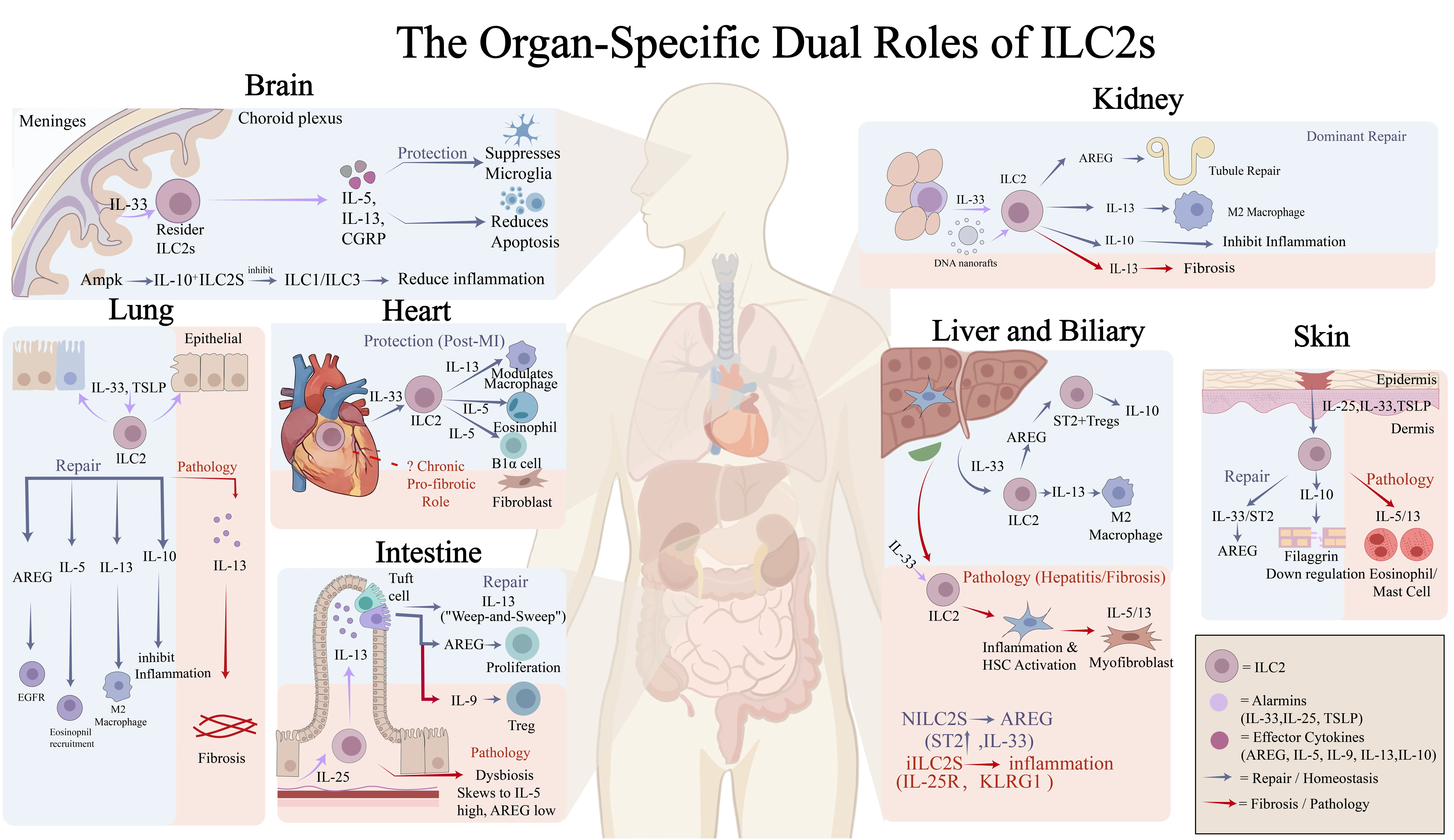
Figure 2. Organ-specific dual roles of ILC2s. Brain: IL-33-activated ILC2s secrete IL-5 and IL-13, suppressing microglial activation and reducing neuronal apoptosis. AMPK-driven expansion of IL-10-producing regulatory ILC2s (IL-10+ ILC2s) further dampens neuroinflammation by inhibiting pro-inflammatory ILC1/ILC3 responses. Lung: ILC2s promote repair through secretion of AREG, IL-13, IL-5, and IL-10, which drive epithelial regeneration, M2 macrophage polarization, and eosinophil-mediated debris clearance. In chronic settings, excessive IL-13 leads to mucus hypersecretion and fibroblast activation, contributing to airway remodeling and fibrosis. Heart: After myocardial infarction, ILC2s exert acute protection by modulating macrophage polarization, recruiting eosinophils, and stimulating B1a-cell-derived natural IgM. However, persistent ILC2 activation may promote maladaptive remodeling and chronic fibrosis. Intestine: ILC2-derived IL-13 facilitates pathogen clearance (“weep-and-sweep”), while AREG enhances epithelial proliferation. IL-9 potentiates regulatory T-cell function, supporting mucosal homeostasis. Dysbiosis skews ILC2s toward a pro-inflammatory phenotype (high IL-5, low AREG), exacerbating inflammation. Kidney: IL-33 recruits and activates ILC2s, which secrete AREG to support tubular epithelial survival and release IL-13 to polarize macrophages toward an M2 phenotype. IL-10 further suppresses inflammation. Therapeutic strategies such as DNA-nanoraft-mediated IL-33 delivery accelerate functional recovery, whereas chronic IL-13 secretion may drive interstitial fibrosis. Liver & Biliary Tract: The ST2+ Treg-nILC2 axis promotes repair via IL-10 and AREG. Conversely, excessive ILC2 activation triggers inflammatory cell recruitment and hepatic stellate-cell activation, leading to hepatitis or fibrosis. Skin: ILC2s maintain homeostasis through AREG and IL-10. In diseases such as atopic dermatitis, ILC2-derived IL-5 and IL-13 activate eosinophils and mast cells and downregulate barrier proteins (e.g., filaggrin), driving chronic inflammation and barrier dysfunction.
Statement of the Use of Generative AI and AI-Assisted Technologies in the Writing Process
During the preparation of this manuscript, the authors used ChatGPT-5 for language polishing. The authors subsequently reviewed and edited the content as needed and take full responsibility for the accuracy and integrity of the published article.
Author Contributions
Conceptualization, Q.C.; Methodology, J.S. and R.W.; Investigation, J.S. and R.W.; Writing—Original Draft Preparation, J.S.; Writing—Review & Editing, Q.C., R.W. and J.S.; Visualization, J.S.; Supervision, Q.C.; Project Administration, Q.C. All authors have read and agreed to the published version of the manuscript.
Ethics Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were generated or analyzed in support of this review. All data discussed or cited are available from the original publications provided in the reference list.
Funding
This work was supported by the National Health and Medical Research Council of Australia (2008347 and 2012351), the National Natural Science Foundation of China (82370748), and the Anhui Provincial Natural Science Foundation (2208085MH207). The APC for this invited article was covered by the journal.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Mjösberg J, Bernink J, Golebski K, Karrich JJ, Peters CP, Blom B, et al. The transcription factor GATA3 is essential for the function of human type 2 innate lymphoid cells. Immunity 2012, 37, 649–659. DOI:10.1016/j.immuni.2012.08.015 [Google Scholar]
- Wong SH, Walker JA, Jolin HE, Drynan LF, Hams E, Camelo A, et al. Transcription factor RORα is critical for nuocyte development. Nat. Immunol. 2012, 13, 229–236. DOI:10.1038/ni.2208 [Google Scholar]
- Yagi R, Zhong C, Northrup DL, Yu F, Bouladoux N, Spencer S, et al. The transcription factor GATA3 is critical for the development of all IL-7Rα-expressing innate lymphoid cells. Immunity 2014, 40, 378–388. DOI:10.1016/j.immuni.2014.01.012 [Google Scholar]
- Moro K, Yamada T, Tanabe M, Takeuchi T, Ikawa T, Kawamoto H, et al. Innate production of T(H)2 cytokines by adipose tissue-associated c-Kit(+)Sca-1(+) lymphoid cells. Nature 2010, 463, 540–544. DOI:10.1038/nature08636 [Google Scholar]
- Neill DR, Wong SH, Bellosi A, Flynn RJ, Daly M, Langford TK, et al. Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature 2010, 464, 1367–1370. DOI:10.1038/nature08900 [Google Scholar]
- Klose CS, Kiss EA, Schwierzeck V, Ebert K, Hoyler T, d’Hargues Y, et al. A T-bet gradient controls the fate and function of CCR6-RORγt+ innate lymphoid cells. Nature 2013, 494, 261–265. DOI:10.1038/nature11813 [Google Scholar]
- Eberl G, Colonna M, Di Santo JP, McKenzie AN. Innate lymphoid cells. Innate lymphoid cells: A new paradigm in immunology. Science 2015, 348, aaa6566. DOI:10.1126/science.aaa6566 [Google Scholar]
- Rodrigues M, Kosaric N, Bonham CA, Gurtner GC. Wound healing: A cellular perspective. Physiol. Rev. 2019, 99, 665–706. DOI:10.1152/physrev.00067.2017 [Google Scholar]
- Almadani YH, Vorstenbosch J, Davison PG, Murphy AM. Wound healing: A comprehensive review. Semin. Plast. Surg. 2021, 35, 141–144. DOI:10.1055/s-0041-1731791 [Google Scholar]
- Shi X, Mekis G, Simms KJ, Gao B, Zhang P. Cell phenotypic conversion and fate change during the host defense response, tissue injury and repair. Pharmacol. Ther. 2026, 277, 108959. DOI:10.1016/j.pharmthera.2025.108959 [Google Scholar]
- Dahlgren MW, Jones SW, Cautivo KM, Dubinin A, Ortiz-Carpena JF, Farhat S, et al. Adventitial stromal cells define group 2 innate lymphoid cell tissue niches. Immunity 2019, 50, 707–722. DOI:10.1016/j.immuni.2019.02.002 [Google Scholar]
- Monticelli LA, Sonnenberg GF, Abt MC, Alenghat T, Ziegler CG, Doering TA, et al. Innate lymphoid cells promote lung-tissue homeostasis after infection with influenza virus. Nat. Immunol. 2011, 12, 1045–1054. DOI:10.1038/ni.2131 [Google Scholar]
- Rajput C, Han M, Ishikawa T, Lei J, Goldsmith AM, Jazaeri S, et al. Rhinovirus C infection induces type 2 innate lymphoid cell expansion and eosinophilic airway inflammation. Front. Immunol. 2021, 12, 668699. DOI:10.3389/fimmu.2021.649520 [Google Scholar]
- Yuan F, Jiang L, Li Q, Sokulsky L, Wanyan Y, Wang L, et al. A selective α7 nicotinic acetylcholine receptor agonist, PNU-282987, attenuates ILC2s activation and Alternaria-induced airway inflammation. Front. Immunol. 2021, 11, 598165. DOI:10.3389/fimmu.2020.598165 [Google Scholar]
- Huang Y, Guo L, Qiu J, Chen X, Hu-Li J, Siebenlist U, et al. IL-25-responsive, lineage-negative KLRG1hi cells are multipotential 'inflammatory' type 2 innate lymphoid cells. Nat. Immunol. 2015, 16, 161–169. DOI:10.1038/ni.3078 [Google Scholar]
- Huang Y, Mao K, Chen X, Sun MA, Kawabe T, Li W, et al. S1P-dependent interorgan trafficking of group 2 innate lymphoid cells supports host defense. Science 2018, 359, 114–119. DOI:10.1126/science.aam5809 [Google Scholar]
- Chang Y, Kang JS, Jung K, Chung DH, Ha SJ, Kim YJ, et al. OASL1-mediated inhibition of type I IFN reduces influenza A infection-induced airway inflammation by regulating ILC2s. Allergy Asthma Immunol. Res. 2022, 14, 99–116. DOI:10.4168/aair.2022.14.1.99 [Google Scholar]
- Sheikh A, Lu J, Melese E, Seo JH, Abraham N. IL-7 induces type 2 cytokine response in lung ILC2s and regulates GATA3 and CD25 expression. J. Leukoc. Biol. 2022, 112, 1105–1113. DOI:10.1002/JLB.3AB1220-819RRR [Google Scholar]
- Bosurgi L, Cao YG, Cabeza-Cabrerizo M, Tucci A, Hughes LD, Kong Y, et al. Macrophage function in tissue repair and remodeling requires IL-4 or IL-13 with apoptotic cells. Science 2017, 356, 1072–1076. DOI:10.1126/science.aai8132 [Google Scholar]
- Liu B, Jiang Q, Chen R, Zhang H, Xia Q, Shao C, et al. Tacrolimus alleviates pulmonary fibrosis progression through inhibiting the activation and interaction of ILC2 and monocytes. Int. Immunopharmacol. 2024, 132, 111999. DOI:10.1016/j.intimp.2024.111999 [Google Scholar]
- Nakatsuka Y, Yaku A, Handa T, Vandenbon A, Hikichi Y, Motomura Y, et al. Pro-fibrotic function of pulmonary group 2 innate lymphoid cells is controlled by regnase-1. Eur. Respir. J. 2020, 57, 2000018. DOI:10.1183/13993003.00018-2020 [Google Scholar]
- Zhang J, Qiu J, Zhou W, Cao J, Hu X, Mi W, et al. Neuropilin-1 mediates lung tissue-specific control of ILC2 function in type 2 immunity. Nat. Immunol. 2022, 23, 237–250. DOI:10.1038/s41590-021-01097-8 [Google Scholar]
- He Y, Yang F, Yang L, Yuan H, You Y, Chen Y, et al. Mechanics-activated fibroblasts promote pulmonary group 2 innate lymphoid cell plasticity propelling silicosis progression. Nat. Commun. 2024, 15, 4128. DOI:10.1038/s41467-024-54174-5 [Google Scholar]
- Poposki JA, Klingler AI, Tan BK, Soroosh P, Banie H, Lewis G, et al. Group 2 innate lymphoid cells are elevated and activated in chronic rhinosinusitis with nasal polyps. Immun. Inflamm. Dis. 2017, 5, 233–243. DOI:10.1002/iid3.161 [Google Scholar]
- Golebski K, Layhadi JA, Sahiner U, Steveling-Klein EH, Lenormand MM, Li RCY, et al. Induction of IL-10-producing type 2 innate lymphoid cells by allergen immunotherapy is associated with clinical response. Immunity 2021, 54, 291–307.e7. DOI:10.1016/j.immuni.2020.12.013 [Google Scholar]
- Thio CLP, Chi PY, Lai ACY, Chang YJ. Regulation of type 2 innate lymphoid cell–dependent airway hyperreactivity by butyrate. J. Allergy Clin. Immunol. 2018, 142, 1867–1883.e12. DOI:10.1016/j.jaci.2018.02.032 [Google Scholar]
- Jiang M, Li Z, Zhang F, Li Z, Xu D, Jing J, et al. Butyrate inhibits iILC2-mediated lung inflammation via lung-gut axis in chronic obstructive pulmonary disease (COPD). BMC Pulm. Med. 2023, 23, 104. DOI:10.1186/s12890-023-02438-z [Google Scholar]
- Laffont S, Blanquart E, Savignac M, Cénac C, Laverny G, Metzger D, et al. Androgen signaling negatively controls group 2 innate lymphoid cells. J. Exp. Med. 2017, 214, 1581–1592. DOI:10.1084/jem.20161807 [Google Scholar]
- Cephus JY, Stier MT, Fuseini H, Yung JA, Toki S, Bloodworth MH, et al. Testosterone attenuates group 2 innate lymphoid cell-mediated airway inflammation. Cell Rep. 2017, 21, 2487–2499. DOI:10.1016/j.celrep.2017.10.110 [Google Scholar]
- Hardman CS, Chen YL, Salimi M, Nahler J, Corridoni D, Jagielowicz M, et al. IL-6 effector function of group 2 innate lymphoid cells (ILC2) is NOD2 dependent. Sci. Immunol. 2021, 6, eabe5084. DOI:10.1126/sciimmunol.abe5084 [Google Scholar]
- Roediger B, Kyle R, Yip KH, Sumaria N, Guy TV, Kim BS, et al. Cutaneous immunosurveillance and regulation of inflammation by group 2 innate lymphoid cells. Nat. Immunol. 2013, 14, 564–573. DOI:10.1038/ni.2584 [Google Scholar]
- Imai Y. Interleukin-33 in atopic dermatitis. J. Dermatol. Sci. 2019, 96, 2–7. DOI:10.1016/j.jdermsci.2019.08.006 [Google Scholar]
- Kim BS, Wang K, Siracusa MC, Saenz SA, Brestoff JR, Monticelli LA, et al. Basophils promote innate lymphoid cell responses in inflamed skin. J. Immunol. 2014, 193, 3717–3725. DOI:10.4049/jimmunol.1401307 [Google Scholar]
- Kobayashi T, Voisin B, Kim DY, Kennedy EA, Jo JH, Shih HY, et al. Homeostatic Control of Sebaceous Glands by Innate Lymphoid Cells Regulates Commensal Bacteria Equilibrium. Cell 2019, 176, 982–997.e16. DOI:10.1016/j.cell.2018.12.031 [Google Scholar]
- Rak GD, Osborne LC, Siracusa MC, Kim BS, Wang K, Bayat A, et al. IL-33-dependent group 2 innate lymphoid cells promote cutaneous wound healing. J. Investig. Dermatol. 2016, 136, 487–496. DOI:10.1038/JID.2015.406 [Google Scholar]
- Leyva-Castillo JM, Galand C, Mashiko S, Bissonnette R, McGurk A, Ziegler SF, et al. ILC2 activation by keratinocyte-derived IL-25 drives IL-13 production at sites of allergic skin inflammation. J. Allergy Clin. Immunol. 2020, 145, 1606–1614.e4. DOI:10.1016/j.jaci.2020.02.026 [Google Scholar]
- Wohlfahrt T, Usherenko S, Englbrecht M, Dees C, Weber S, Beyer C, et al. Type 2 innate lymphoid cell counts are increased in patients with systemic sclerosis and correlate with the extent of fibrosis. Ann. Rheum. Dis. 2016, 75, 623–626. DOI:10.1136/annrheumdis-2015-207388 [Google Scholar]
- Sakamoto K, Jin SP, Goel S, Jo JH, Voisin B, Kim D, et al. Disruption of the endopeptidase ADAM10-Notch signaling axis leads to skin dysbiosis and innate lymphoid cell-mediated hair follicle destruction. Immunity 2021, 54, 2321–2337.e10. DOI:10.1016/j.immuni.2021.09.001 [Google Scholar]
- Deng F, Hu JJ, Yang X, Sun QS, Lin ZB, Zhao BC, et al. Gut microbial metabolite pravastatin attenuates intestinal ischemia/reperfusion injury through promoting IL-13 release from type II innate lymphoid cells via IL-33/ST2 signaling. Front. Immunol. 2021, 12, 704836. DOI:10.3389/fimmu.2021.704836 [Google Scholar]
- von Moltke J, Ji M, Liang HE, Locksley RM. Tuft-cell-derived IL-25 regulates an intestinal ILC2-epithelial response circuit. Nature 2016, 529, 221–225. DOI:10.1038/nature16161 [Google Scholar]
- Schneider C, O’Leary CE, von Moltke J, Liang HE, Ang QY, Turnbaugh PJ, et al. A metabolite-triggered tuft cell-ILC2 circuit drives small intestinal remodeling. Cell 2018, 174, 271–284.e14. DOI:10.1016/j.cell.2018.05.014 [Google Scholar]
- Arifuzzaman M, Won TH, Yano H, Uddin J, Emanuel ER, Hu E, et al. Dietary fiber is a critical determinant of pathologic ILC2 responses and intestinal inflammation. J. Exp. Med. 2024, 221, e20232148. DOI:10.1084/jem.20232148 [Google Scholar]
- Klose CSN, Mahlakõiv T, Moeller JB, Rankin LC, Flamar AL, Kabata H, et al. The neuropeptide neuromedin U stimulates innate lymphoid cells and type 2 inflammation. Nature 2017, 549, 282–286. DOI:10.1038/nature23676 [Google Scholar]
- McGinty JW, Ting HA, Billipp TE, Nadjsombati MS, Khan DM, Barrett NA, et al. Tuft-cell-derived leukotrienes drive rapid anti-helminth immunity in the small intestine but are dispensable for anti-protist immunity. Immunity 2020, 52, 528–541.e7. DOI:10.1016/j.immuni.2020.02.005 [Google Scholar]
- Irie E, Ishihara R, Mizushima I, Hatai S, Hagihara Y, Takada Y, et al. Enrichment of type I interferon signaling in colonic group 2 innate lymphoid cells in experimental colitis. Front. Immunol. 2022, 13, 982827. DOI:10.3389/fimmu.2022.982827 [Google Scholar]
- Uddin MJ, Thompson B, Leslie JL, Fishman C, Sol-Church K, Kumar P, et al. Investigating the impact of antibiotic-induced dysbiosis on protection from Clostridium difficile colitis by mouse colonic innate lymphoid cells. mBio 2024, 15, e0333823. DOI:10.1128/mbio.03338-23 [Google Scholar]
- Chun TT, Chung CS, Fallon EA, Hutchins NA, Clarke E, Rossi AL, et al. Group 2 Innate Lymphoid Cells (ILC2s) Are Key Mediators of the Inflammatory Response in Polymicrobial Sepsis. Am. J. Pathol. 2018, 188, 2097–2108. DOI:10.1016/j.ajpath.2018.05.009 [Google Scholar]
- Nakagawa H, Suzuki N, Hirata Y, Hikiba Y, Hayakawa Y, Kinoshita H, et al. Biliary epithelial injury-induced regenerative response by IL-33 promotes cholangiocarcinogenesis from peribiliary glands. Proc. Natl. Acad. Sci. USA 2017, 114, E3806–E3815. DOI:10.1073/pnas.1619416114 [Google Scholar]
- Loh Z, Fitzsimmons RL, Reid RC, Ramnath D, Clouston A, Gupta PK, et al. Inhibitors of class I histone deacetylases attenuate thioacetamide-induced liver fibrosis in mice by suppressing hepatic type 2 inflammation. Br. J. Pharmacol. 2019, 176, 3775–3790. DOI:10.1111/bph.14768 [Google Scholar]
- Gonzalez-Polo V, Pucci-Molineris M, Cervera V, Gambaro S, Yantorno SE, Descalzi V, et al. Group 2 innate lymphoid cells exhibit progressively higher levels of activation during worsening of liver fibrosis. Ann. Hepatol. 2019, 18, 366–372. DOI:10.1016/j.aohep.2018.12.001 [Google Scholar]
- Russi AE, Shivakumar P, Luo Z, Bezerra JA. Plasticity between type 2 innate lymphoid cell subsets and amphiregulin expression regulates epithelial repair in biliary atresia. Hepatology 2023, 78, 1035–1049. DOI:10.1097/HEP.0000000000000418 [Google Scholar]
- Neumann K, Karimi K, Meiners J, Voetlause R, Steinmann S, Dammermann W, et al. A pro-inflammatory role of type 2 innate lymphoid cells in murine immune-mediated hepatitis. J. Immunol. 2017, 198, 128–137. DOI:10.4049/jimmunol.1600418 [Google Scholar]
- Zhang Y, Qi C, Li L, Hua S, Zheng F, Gong F, et al. CD8+ T cell/IL-33/ILC2 axis exacerbates the liver injury in Con A-induced hepatitis in T cell-transferred Rag2-deficient mice. Inflamm. Res. 2019, 68, 75–91. DOI:10.1007/s00011-018-1197-9 [Google Scholar]
- Steinmann S, Schoedsack M, Heinrich F, Breda PC, Ochel A, Tiegs G, et al. Hepatic ILC2 activity is regulated by liver inflammation-induced cytokines and effector CD4+ T cells. Sci. Rep. 2020, 10, 1071. DOI:10.1038/s41598-020-57985-w [Google Scholar]
- Wachtendorf S, Jonin F, Ochel A, Heinrich F, Westendorf AM, Tiegs G, et al. The ST2+ Treg/amphiregulin axis protects from immune-mediated hepatitis. Front. Immunol. 2024, 15, 1351405. DOI:10.3389/fimmu.2024.1351405 [Google Scholar]
- Cao Q, Wang R, Niu Z, Chen T, Azmi F, Read SA, et al. Type 2 innate lymphoid cells are protective against hepatic ischaemia/reperfusion injury. JHEP Rep. 2023, 5, 100837. DOI:10.1016/j.jhepr.2023.100837 [Google Scholar]
- Fujimoto M, Yokoyama M, Kiuchi M, Hosokawa H, Nakayama A, Hashimoto N, et al. Liver group 2 innate lymphoid cells regulate blood glucose levels through IL-13 signaling and suppression of gluconeogenesis. Nat Commun. 2022, 13, 5408. DOI:10.1038/s41467-022-33171-6 [Google Scholar]
- Nagashima R, Ishikawa H, Kuno Y, Kohda C, Iyoda M. HIF-PHD inhibitor regulates the function of group2 innate lymphoid cells and polarization of M2 macrophages. Sci. Rep. 2023, 13, 1587. DOI:10.1038/s41598-023-29161-3 [Google Scholar]
- Andrade-Oliveira V, Amano MT, Correa-Costa M, Castoldi A, Felizardo RJ, de Almeida DC, et al. Gut Bacteria Products Prevent AKI Induced by Ischemia-Reperfusion. J. Am. Soc. Nephrol. 2015, 26, 1877–1888. DOI:10.1681/ASN.2014030288 [Google Scholar]
- Xu L, Xing Z, Yuan J, Han Y, Jiang Z, Han M, et al. Ultrasmall nanoparticles regulate immune microenvironment by activating IL-33/ST2 to alleviate renal ischemia-reperfusion injury. Adv. Healthc. Mater. 2024, 13, e2303276. DOI:10.1002/adhm.202303276 [Google Scholar]
- Stremska ME, Jose S, Sabapathy V, Huang L, Bajwa A, Kinsey GR, et al. IL233, a novel IL-2 and IL-33 hybrid cytokine, ameliorates renal injury. J. Am. Soc. Nephrol. 2017, 28, 2681–2693. DOI:10.1681/ASN.2016121272 [Google Scholar]
- Li W, Wang C, Lv H, Wang Z, Zhao M, Liu S, et al. A DNA nanoraft-based cytokine delivery platform for alleviation of acute kidney injury. ACS Nano 2021, 15, 18237–18249. DOI:10.1021/acsnano.1c07270 [Google Scholar]
- Mantani PT, Dunér P, Bengtsson E, Alm R, Ljungcrantz I, Söderberg I, et al. IL-25 inhibits atherosclerosis development in apolipoprotein E deficient mice. PLoS ONE 2015, 10, e0117255. DOI:10.1371/journal.pone.0117255 [Google Scholar]
- Mantani PT, Dunér P, Ljungcrantz I, Nilsson J, Björkbacka H, Fredrikson GN. ILC2 transfers to apolipoprotein E deficient mice reduce the lipid content of atherosclerotic lesions. BMC Immunol. 2019, 20, 47. DOI:10.1186/s12865-019-0330-z [Google Scholar]
- Newland SA, Mohanta S, Clément M, Taleb S, Walker JA, Nus M, et al. Type-2 innate lymphoid cells control the development of atherosclerosis in mice. Nat. Commun. 2017, 8, 15781. DOI:10.1038/ncomms15781 [Google Scholar]
- Brestoff JR, Kim BS, Saenz SA, Stine RR, Monticelli LA, Sonnenberg GF, et al. Group 2 innate lymphoid cells promote beiging of white adipose tissue and limit obesity. Nature 2015, 519, 242–246. DOI:10.1038/nature14115 [Google Scholar]
- Molofsky AB, Nussbaum JC, Liang HE, Van Dyken SJ, Cheng LE, Mohapatra A, et al. Innate lymphoid type 2 cells sustain visceral adipose tissue eosinophils and alternatively activated macrophages. J. Exp. Med. 2013, 210, 535–549. DOI:10.1084/jem.20121964 [Google Scholar]
- Liu T, Meng Z, Liu J, Li J, Zhang Y, Deng Z, et al. Group 2 innate lymphoid cells protect mouse heart from myocardial infarction injury via interleukin 5, eosinophils, and dendritic cells. Cardiovasc. Res. 2023, 119, 1046–1061. DOI:10.1093/cvr/cvac144 [Google Scholar]
- Yu X, Newland SA, Zhao TX, Lu Y, Sage AS, Sun Y, et al. Innate Lymphoid Cells Promote Recovery of Ventricular Function After Myocardial Infarction. J. Am. Coll. Cardiol. 2021, 78, 1127–1142. DOI:10.1016/j.jacc.2021.07.018 [Google Scholar]
- Ortega-Rodriguez AC, Guerra de Blas PDC, Ramírez-Torres R, Martínez-Shio EB, Monsiváis-Urenda AE. Quantitative analysis of innate lymphoid cells in patients with ST-segment elevation myocardial infarction. Immunol. Investig. 2024, 53, 586–603. DOI:10.1080/08820139.2024.2316052 [Google Scholar]
- Molofsky AB, Van Gool F, Liang HE, Van Dyken SJ, Nussbaum JC, Lee J, et al. Interleukin-33 and Interferon-γ Counter-Regulate Group 2 Innate Lymphoid Cell Activation during Immune Perturbation. Immunity 2015, 43, 161–174. DOI:10.1016/j.immuni.2015.05.019 [Google Scholar]
- Hong T, Li S, Guo X, Wei Y, Zhang J, Su X, et al. IL-13 derived type 2 innate lymphoid cells ameliorates cardiomyocyte apoptosis through STAT3 signaling pathway. Front. Cell Dev. Biol. 2021, 9, 714229. DOI:10.3389/fcell.2021.742662 [Google Scholar]
- She L, Alanazi HH, Xu Y, Yu Y, Gao Y, Guo S, et al. Direct activation of toll-like receptor 4 signaling in group 2 innate lymphoid cells contributes to inflammatory responses of allergic diseases. iScience 2024, 27, 111240. DOI:10.1016/j.isci.2024.111240 [Google Scholar]
- Chen WY, Wu YH, Tsai TH, Li RF, Lai AC, Li LC, et al. Group 2 innate lymphoid cells contribute to IL-33-mediated alleviation of cardiac fibrosis. Theranostics 2021, 11, 2594–2611. DOI:10.7150/thno.51648 [Google Scholar]
- Fung ITH, Sankar P, Zhang Y, Robison LS, Zhao X, D’Souza SS, et al. Activation of group 2 innate lymphoid cells alleviates aging-associated cognitive decline. J. Exp. Med. 2020, 217, e20190915. DOI:10.1084/jem.20190915 [Google Scholar]
- Gadani SP, Smirnov I, Wiltbank AT, Overall CC, Kipnis J. Characterization of meningeal type 2 innate lymphocytes and their response to CNS injury. J. Exp. Med. 2017, 214, 285–296. DOI:10.1084/jem.20161982 [Google Scholar]
- Korin B, Ben-Shaanan TL, Schiller M, Dubovik T, Azulay-Debby H, Boshnak NT, et al. High-dimensional, single-cell characterization of the brain's immune compartment. Nat. Neurosci. 2017, 20, 1300–1309. DOI:10.1038/nn.4610 [Google Scholar]
- Baruch K, Ron-Harel N, Gal H, Deczkowska A, Shifrut E, Ndifon W, et al. CNS-specific immunity at the choroid plexus shifts toward destructive Th2 inflammation in ageing. Proc. Natl. Acad. Sci. USA 2013, 110, 2264–2269. DOI:10.1073/pnas.1211270110 [Google Scholar]
- Zheng P, Xiu Y, Chen Z, Yuan M, Li Y, Wang N, et al. Group 2 innate lymphoid cells resolve neuroinflammation following cerebral ischaemia. Stroke Vasc. Neurol. 2023, 8, 424–434. DOI:10.1136/svn-2022-001919 [Google Scholar]
- Liu M, Wang D, Xu L, Pan Y, Huang H, Li M, et al. Group 2 innate lymphoid cells suppress neuroinflammation and brain injury following intracerebral hemorrhage. J. Cereb. Blood Flow. Metab. 2024, 44, 355–366. DOI:10.1177/0271678X231208168 [Google Scholar]
- Walsh JT, Zheng J, Smirnov I, Lorenz U, Tung K, Kipnis J. Regulatory T cells in central nervous system injury: A double-edged sword. J. Immunol. 2014, 193, 5013–5022. DOI:10.4049/jimmunol.1302401 [Google Scholar]
- Baban B, Braun M, Khodadadi H, Ward A, Alverson K, Malik A, et al. AMPK induces regulatory innate lymphoid cells after traumatic brain injury. JCI Insight 2021, 6, e126766. DOI:10.1172/jci.insight.126766 [Google Scholar]
- Liu J, Xiao C, Wang H, Xue Y, Dong D, Lin C, et al. Local group 2 innate lymphoid cells promote corneal regeneration after epithelial abrasion. Am. J. Pathol. 2017, 187, 1313–1326. DOI:10.1016/j.ajpath.2017.02.010 [Google Scholar]
- Omata Y, Frech M, Lucas S, Primbs T, Knipfer L, Wirtz S, et al. Type 2 innate lymphoid cells inhibit the differentiation of osteoclasts and protect from ovariectomy-induced bone loss. Bone 2020, 136, 115335. DOI:10.1016/j.bone.2020.115335 [Google Scholar]
- Cosway EJ, James KD, White AJ, Parnell SM, Bacon A, McKenzie ANJ, et al. The alarmin IL33 orchestrates type 2 immune-mediated control of thymus regeneration. Nat. Commun. 2023, 14, 4704. DOI:10.1038/s41467-023-43072-x [Google Scholar]
- Huang Q, Ma X, Wang Y, Niu Z, Wang R, Yang F, et al. IL-10 producing type 2 innate lymphoid cells prolong islet allograft survival. EMBO Mol. Med. 2020, 12, e12305. DOI:10.15252/emmm.202012305 [Google Scholar]
- Corren J, Parnes JR, Wang L, Mo M, Roseti SL, Griffiths JM, et al. Tezepelumab in Adults with Uncontrolled Asthma. N. Engl. J. Med. 2017, 377, 936–946. DOI:10.1056/NEJMoa1704064 [Google Scholar]
- Kabashima K, Matsumura T, Komazaki H, Kawashima M. Trial of nemolizumab and topical agents for atopic dermatitis with pruritus. N. Engl. J. Med. 2020, 383, 141–150. DOI:10.1056/NEJMoa1917006 [Google Scholar]
- Li Y, Wang W, Ying S, Lan F, Zhang L. A potential role of group 2 innate lymphoid cells in eosinophilic chronic rhinosinusitis with nasal polyps. Allergy Asthma Immunol. Res. 2021, 13, 363–374. DOI:10.4168/aair.2021.13.3.363 [Google Scholar]