Quartz-Based Castables with Calcium Silicate Cement as Binder-Mineralizer: Replacing Shaped Product for Large-Scale Fabrication

Quartz-Based Castables with Calcium Silicate Cement as Binder-Mineralizer: Replacing Shaped Product for Large-Scale Fabrication
Zuling Chen 1 Yawei Li 1,2,* Ning Liao 1,2,* Wenjing Liu 1 Shengli Jin 1,2,* Wanneng Yuan 3 Baoshan Tian 3 Wenxian Bai 4 Dayan Xu 4
Received: 06 February 2026 Revised: 11 March 2026 Accepted: 16 March 2026 Published: 25 March 2026

© 2026 The authors. This is an open access article under the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/).
1. Introduction
Silica bricks are acidic refractory materials primarily composed of silicon dioxide (SiO2). Owing to their high refractoriness, excellent resistance to acid corrosion, and good volume stability at elevated temperatures [1], they are widely employed in critical sections of high-temperature industrial furnaces, such as coke ovens, hot blast stoves, and glass kilns [2,3]. With the advancement of hydrogen-enriched smelting technology in the iron and steel industry, quartz-based refractories now face new challenges associated with phase transformations and structural stability under reducing atmospheres [4]. Besides, in response to meet the growing demand for intelligent construction, silica bricks are increasingly developed into large and complex-shaped components.
In conventional silica bricks production, CaO-FeOx-based mineralizers are employed to promote the conversion of quartz into cristobalite and tridymite [5,6], thereby reducing thermal expansion and enhancing high-temperature stability. Under reducing atmospheres, carbon monoxide (CO), hydrogen (H2), and metallic oxides within the furnace jointly promote the evolution of a SiO2-CaO-FeOx ternary liquid phase with a lower Fe2+/Fe3+ ratio into a SiO2-CaO-Al2O3-FeOx quaternary liquid phase exhibiting an elevated Fe2+/Fe3+ ratio. The latter phase exhibits lower viscosity and stronger dissolution capacity toward SiO2. Repeated dissolution-precipitation cycles refine SiO2 grains and eventually lead to structural collapse, which is considered the primary degradation mechanism of silica bricks during service [7]. To address these limitations, a variety of novel mineralizer strategies have been developed. For instance, Rauta et al. [8] incorporated nano-TiO2 into silica bricks, which increased cristobalite content and cold crushing strength while significantly reducing thermal expansion, creep rate, and apparent porosity. Zhang et al. [9] reported that CaCO3 as a mineralizer promoted cristobalite formation up to 62% with a markedly reduced glassy phase content. Jia et al. [2] demonstrated that the addition of 2% ferrosilicon nitride enhanced creep resistance, albeit at the expense of a slightly lower softening temperature under load. In addition, Wang et al. [10] prepared silica bricks with excellent comprehensive properties using the CaO-FeOx-MnO2-SiC/TiO2 quaternary mineralization system and via high-temperature firing.
Although these mineralizers have been proven to be effective, their applications are limited by the reliance on the “semi-dry” pressing process, which is not suitable for producing large-sized silica bricks. To overcome this limitation, the development of monolithic castables has emerged as a highly promising direction. Calcium silicate cement exhibits excellent chemical compatibility with silica, making it an ideal binder for quartz-based castables. During curing and drying, hydrates nano- to submicron-sized C-S-H gels and Ca(OH)2 crystals [11], which in turn facilitate a more uniform distribution of the mineralizer and confer excellent early strength to quartz-based castables. During the high-temperature firing stage, the hydrates of calcium silicate cement decompose, generating CaO [12], which serves as an active mineralizer and significantly promotes the transformation of quartz into cristobalite and tridymite. Therefore, calcium silicate cement should have the application potential for preparing quartz-based castables, although its dual binding-mineralization mechanism remains to be fully elucidated.
In this study, calcium silicate cement, as both a binder and a mineralizer, was synthesized via solid-state reaction and employed in quartz-based castables, replacing conventional CaO-FeOx mineralizers. The effects of cement addition on the early-stage binding performance, mineralization behavior, refractoriness under load, and high-temperature creep of quartz-based castables were systematically investigated. The objective of this work is to elucidate the dual functional mechanism of calcium silicate cement and to evaluate its potential for the efficient fabrication of large or complex-shaped silica bricks.
2. Experimental
2.1. Materials
Tricalcium silicate cement was synthesized using high-purity CaCO3 (Sinopharm Chemical Reagent Co., Ltd., Shanghai, China) and SiO2 (Sinopharm Chemical Reagent Co., Ltd., China) as raw materials. The preparation procedure was as follows: the raw powders were weighed according to a Ca/Si molar ratio of 3:1 and homogenized in an Eirich mixer at 2500 r/min for 20 min. The blended powder was then placed in alumina crucibles and calcined at 1500 °C for 3−9 h. The heating schedule was as follows: the mixture was heated in air at 5 °C/min to 1000 °C with an isothermal hold at 800 °C for 1 h to ensure complete decomposition of CaCO3, followed by heating at 3 °C/min to 1400 °C and then at 2 °C/min to 1500 °C, with soaking times of 3 h, 6 h, and 9 h. Correspondingly, calcium silicate cements synthesized at 1500 °C with different holding times were designated as: 1500 °C−3 h; 1500 °C−6 h; 1500 °C−9 h, respectively. The synthesized clinker was subsequently ground and sieved through a 325−mesh (<0.044 mm) screen to obtain fine calcium silicate cement powders. Laser particle size analysis revealed that the average particle size of the cement held at 1500 °C for 9 h was 6.6 μm.
Quartz-based castables were prepared using quartz aggregates and fine powders (Sinosteel Luonai Materials Technology Co., Ltd., Luoyang, China) as the primary raw materials, with quartz as the dominant phase. Chemical analysis results indicated that the SiO2 content of the quartz exceeds 97.9 wt%, and the fusion index (wAl2O3 (%) + 2wK2O (%) + wNa2O (%)) was below 0.6%. The synthesized calcium silicate cement (calcined for 9 h) was used as a binder, while FS65 (Wuhan Shanda Chemical Co., Ltd., Wuhan, China) served as a dispersant, and monohydrate citric acid (Sinopharm Chemical Reagent Co., Ltd., China) was used as a retarder.
Based on the Modified Anderson’s model, the q−value of the quartz-based castable was adjusted to 0.26 to ensure an optimal particle packing. The experimental mix designs with different cement additions are listed in Table 1. The preparation procedure of the quartz-based castables is as follows: Firstly, quartz aggregates and powders were sequentially added into a mortar mixer and were dry-mixed for 3 min. During the mixing, 7.85 wt% deionized water was added, and wet mixing continued for another 3 min to achieve the required flowability for casting. To improve homogeneity and reduce the risk of particle segregation, particular attention was paid to the mixing sequence and mixing time. The homogenized wet mixtures were then poured into different molds for shaping: prismatic molds (40 mm × 40 mm × 160 mm), cylindrical molds (Φ50 mm × h50 mm), and cylindrical molds with a central through-hole (Φ13 mm) in Φ50 mm × h50 mm bodies. Samples were consolidated by vibration for 1 min to eliminate entrapped air and to promote better particle packing within the castables.
After casting, samples were cured in a chamber at 25 °C and 75% relative humidity for 24 h. Subsequently, samples were dried at 110 °C for 48 h and then pre-fired in a muffle furnace, where samples were heated 1 °C/min up to 500 °C with intermediate holding steps at 200 °C, 300 °C, 400 °C, and 500 °C for 2 h each to remove chemically bound water and organic additives. Such a stepwise heating schedule was adopted to minimize thermal gradients and mitigate cracking caused by uneven dehydration. Sintering was conducted in a high-temperature furnace under the following regime: samples were heated at 5 °C/min to 500 °C, then at 2 °C/min to 1000 °C with an intermediate dwell at 600 °C for 1 h. The temperature was increased at 1 °C/min to 1400 or 1430 °C, with a final soaking time of 20 h, facilitating completion of the transformation and enhancing bulk densification. Afterwards, cooling was carried out at 1 °C/min down to 600 °C, followed by furnace cooling to room temperature.
Table 1. Batch compositions of quartz-based castables (wt/%).
|
Raw Materials |
Particle Size |
C3 |
C4 |
C5 |
|---|---|---|---|---|
|
Quartz aggregate |
2.6−1.5 mm |
25 |
25 |
25 |
|
1.5−0 mm |
45 |
45 |
45 |
|
|
Quartz powder |
≤0.074 mm |
19 |
18 |
17 |
|
Microsilica |
<1 μm |
8 |
8 |
8 |
|
Calcium silicate cement |
≤0.044 mm |
3 |
4 |
5 |
|
Dispersant: FS65 |
≤0.074 mm |
+0.2 |
+0.2 |
+0.2 |
|
Citric acid |
− |
+0.015 |
+0.015 |
+0.015 |
|
Deionized water |
− |
+7.85 |
+7.85 |
+7.85 |
Note: “+” denotes supplementary addition.
2.2. Methods
Phase identification was carried out by X-ray diffraction (XRD) on a Philips X’Pert PRO diffractometer (Philips, Amsterdam, The Netherlands) with Cu Kα radiation (40 kV, 40 mA) over a 2θ range of 7−90°. Quantitative phase analysis of the castables was performed using the Rietveld refinement method. The hydrated samples of tricalcium silicate cement were examined by scanning electron microscopy (SEM, EVO−10, Zeiss, Oberkochen Germany) equipped with an energy-dispersive X-ray spectrometer (EDS, X-MAX50, Oxford Instruments, Abingdon, UK) for compositional analysis. The relative contents of hydrates in calcium silicate cement are determined by thermogravimetric (TG) analysis. The early setting behavior of the castables was monitored in real time using an ultrasonic pulse velocity tester (IP−8 ultrasonic measurement system, Germany). The cold crushing strength (CCS) of the specimens was measured according to GB/T 5072-2023 [13]. The cold modulus of rupture (CMOR) was determined according to GB/T 3001-2017 [14]. The bulk density and apparent porosity are measured using water as the immersion medium following GB/T 2997-2015 [15]. The creep behavior under load was tested according to GB/T 5073-2005 [16]. The refractoriness under load (RUL) was determined in accordance with GB/T 5989-2023 [17]. The PLC (Permanent Linear Change) test method in accordance with GB/T 5988-2022 [18].
3. Results and Discussion
3.1. Preparation of Tricalcium Silicate and Its Hydration
Figure 1a shows the XRD patterns of the synthesized calcium silicate cement with different soaking times. At 1500 °C for 3 h, the main phases were tricalcium silicate (C3S) and dicalcium silicate (C2S), with a weak CaO peak at 2θ ≈ 37.5°. With extended holding time (6−9 h), C2S peaks decreased or disappeared, while C3S and CaO became dominant, indicating progressive conversion of C2S to C3S. The residual CaO is likely due to incomplete reaction or limited decomposition of C3S during cooling [19].
The hydrates of prepared cements were analyzed by XRD, and the results are shown in Figure 1b−d. The main phases of the 1500 °C−3 h sample after hydration for 6−24 h were C2S and Ca(OH)2, with no detectable diffraction peaks of C3S. This absence of diffraction peaks of C3S is presumably due to its partial dissolution during the hydration process, which diminishes its diffraction signal. In contrast, the 1500 °C−6 h sample still exhibited pronounced diffraction peaks of C3S after hydration for 6 h, alongside detectable diffraction peaks of Ca(OH)2. With further hydration up to 12 h and 24 h, the intensity of C3S diffraction peaks slightly decreased, while that of Ca(OH)2 increased significantly, indicating ongoing hydration of C3S to form Ca(OH)2. For the 1500 °C−9 h sample, no diffraction peaks of Ca(OH)2 were observed after hydration for 6 h, and C3S remained the dominant phase. After 12 h, diffraction peaks of Ca(OH)2 appeared, and their intensity increased markedly by 24 h, suggesting progressive hydration. It should be noted that the hydration of C2S and C3S produces Ca(OH)2 and amorphous C-S-H gel at 25 °C. Since the C-S-H gel lacks a well-defined crystalline structure, it does not produce characteristic diffraction peaks in the XRD patterns and is therefore not shown [20]. A comparative analysis of the hydration behavior of the three samples revealed that the 1500 °C−3 h sample exhibited only weak Ca(OH)2 diffraction peaks after 24 h of hydration, suggesting relatively low hydration reactivity. In contrast, the 1500 °C−6 h and 1500 °C−9 h samples exhibited distinct diffraction peaks of Ca(OH)2 after hydration for 6 h and 12 h, respectively, indicating higher hydration reactivity.
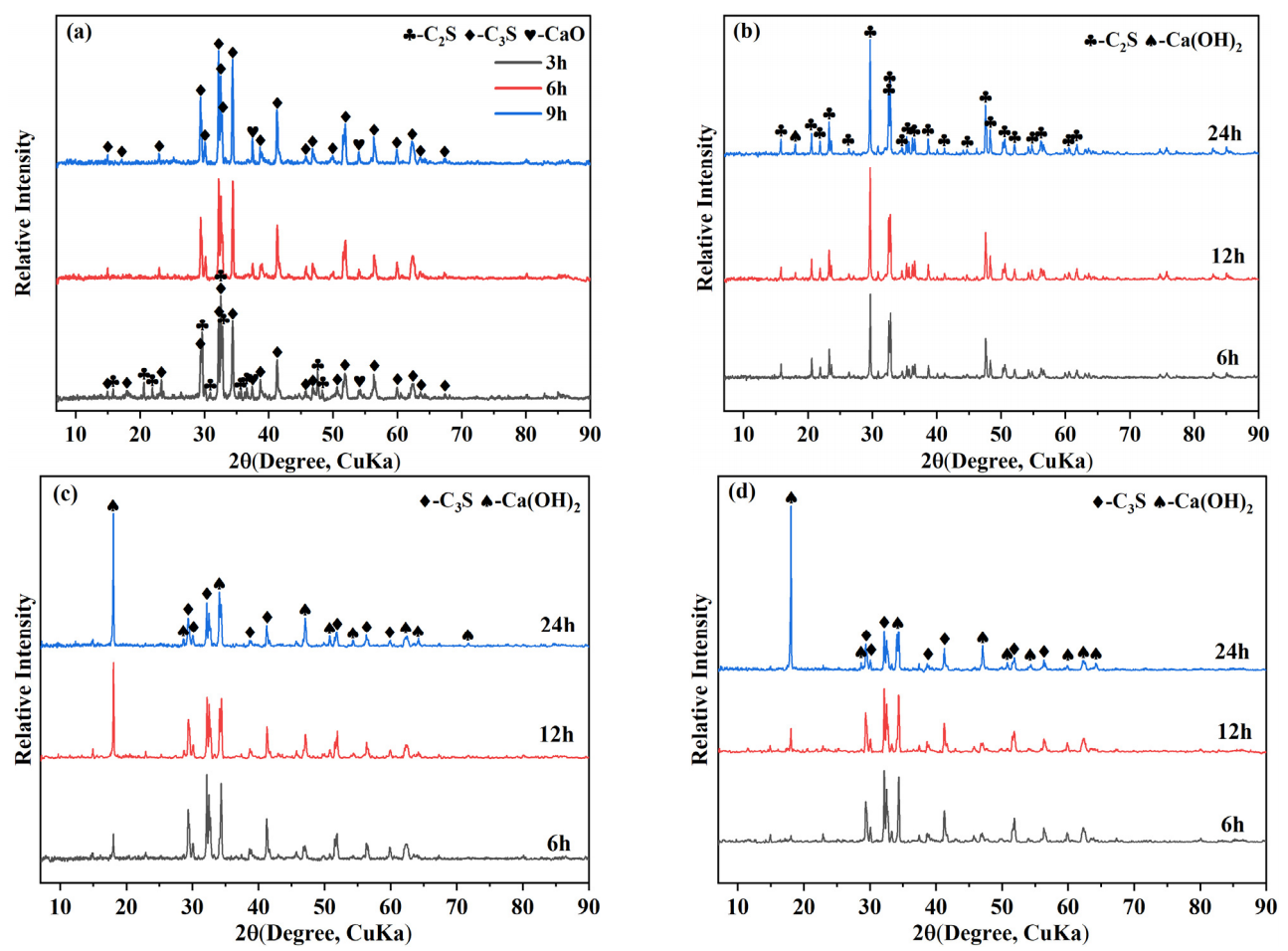
Figure 1. XRD patterns of synthesized calcium silicate cement at different holding times (a) and hydration at various curing times: (b) 1500 °C−3 h; (c) 1500 °C−6 h; (d) 1500 °C−9 h.
To gain deeper insights into the structural evolution of cement samples during hydration, SEM was employed to observe their microstructures after different hydration times, as shown in Figure 2. For the 1500 °C−3 h sample, no significant hydrates were observed after hydration for 6 h, with C2S particles still dominating the microstructure. After 12 h, although no diffraction peaks of hydrates were detected in XRD, SEM images revealed the formation of a small amount of fibrous nanoscale structures on the particle surfaces, presumed to be C-S-H gel, indicating the initiation of hydration [21]; Further extending the hydration time to 24 h, the number of un-hydrated cement particles decreased substantially and was replaced by abundant fibrous C-S-H gel attached to the particle surfaces, forming a continuous network, which demonstrates a significantly increased degree of hydration. For the 1500 °C−6 h sample, after hydration for 6 h, a small amount of C-S-H gel was observed on the cement particles, alongside hexagonal plate-like Ca(OH)2 crystals approximately 8−10 μm in size, interspersed within the hydrates. At 12 h, C-S-H gel formed extensively and densely accumulated together with Ca(OH)2. After 24 h, the C-S-H gel network further developed, covering a wider area. For the 1500 °C−9 h sample, after hydration for 6 h, only a small amount of C-S-H gel was formed, with minor Ca(OH)2 crystals embedded between particles. Compared to the 1500 °C−6 h sample, the crystal size was smaller (~1−2 μm). At 12 h, C-S-H gel formation became extensive, and Ca(OH)2 crystals grew to 5−6 μm. Extending hydration time to 24 h, the C-S-H gel exhibited typical fibrous and nanosheet morphologies [22], indicating a well-progressed hydration process and characteristic evolution of hydrates.
A comparative analysis of the SEM observations for the three samples revealed that the 1500 °C−6 h and 1500 °C−9 h samples exhibited more pronounced early-stage formation of C-S-H gel and development of Ca(OH)2 crystals, which was in good agreement with the higher hydration activity indicated by XRD analyses. This difference can be attributed to the influence of sintering regimes on the mineral composition and crystal structure of the cement, which in turn affects the nucleation and growth kinetics of hydrates.

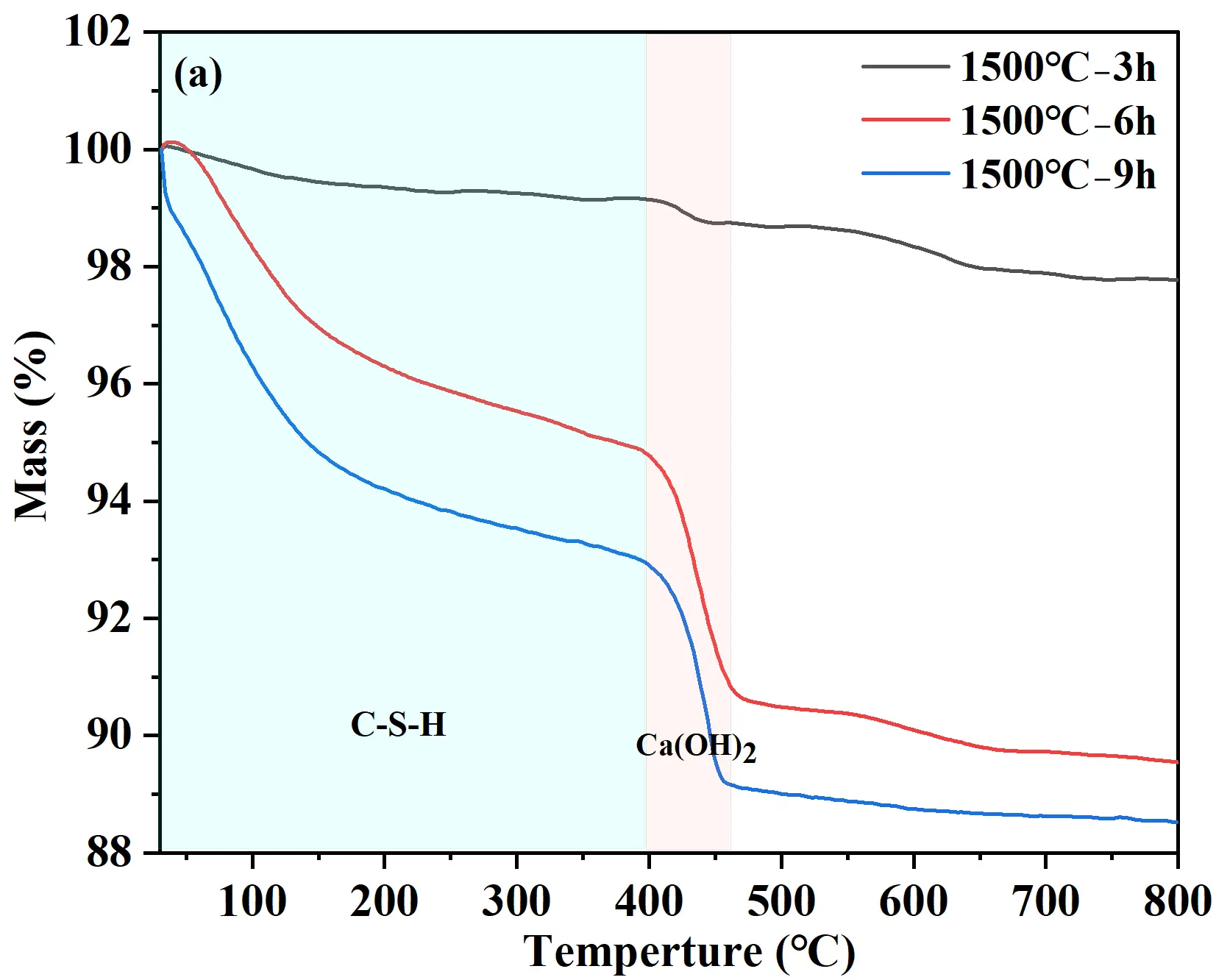
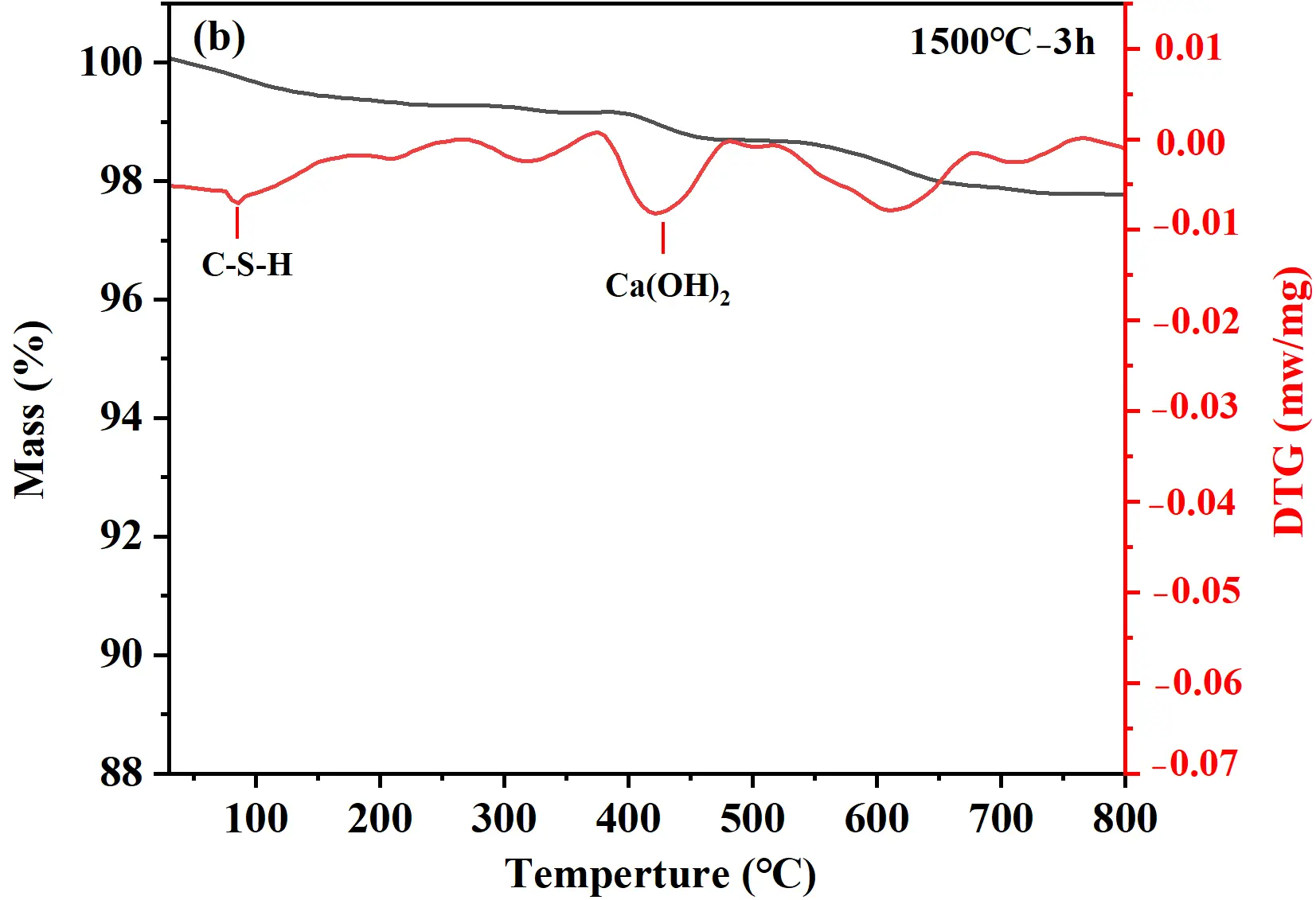
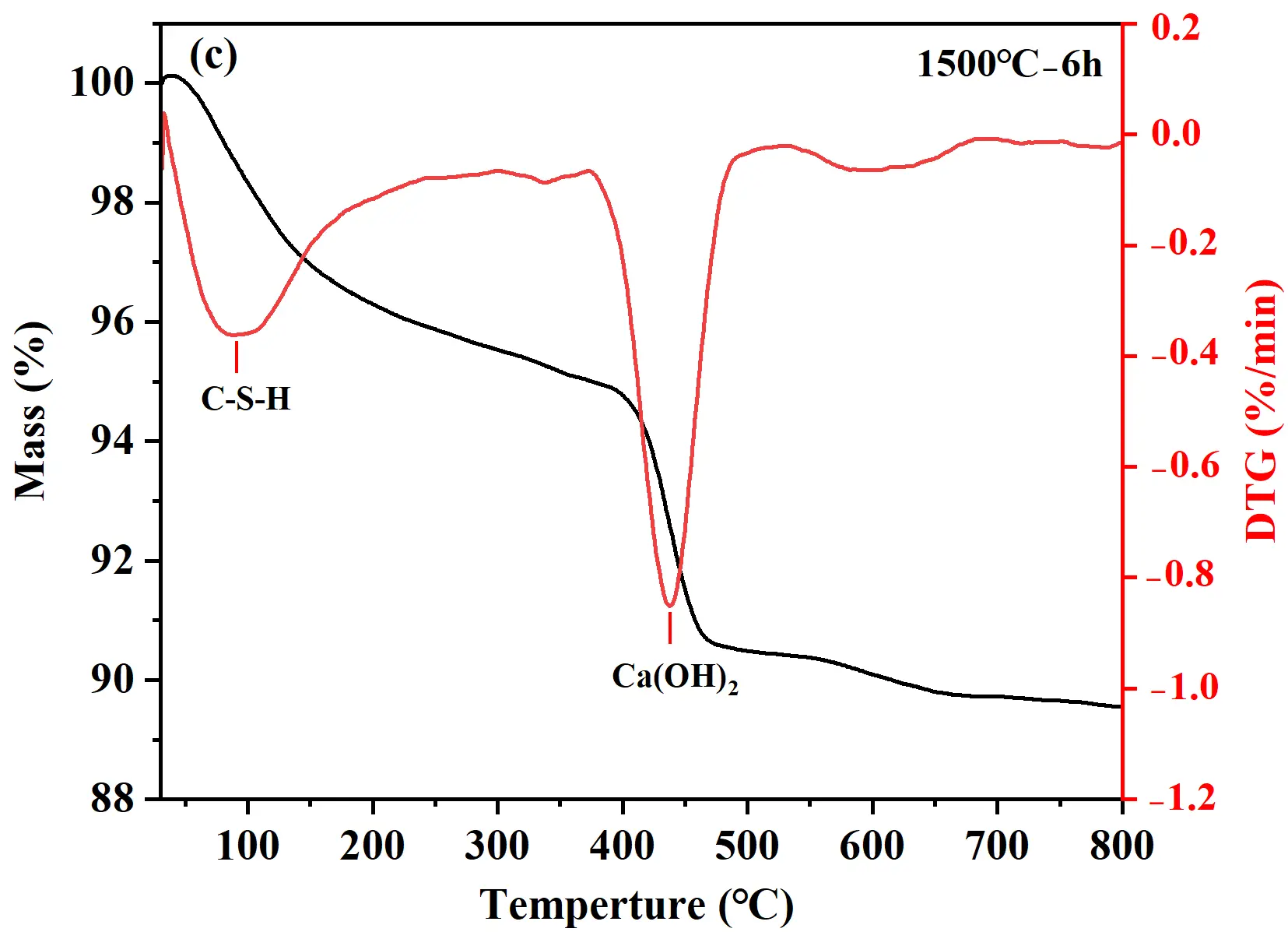
TG analyses were conducted to investigate the hydrates and hydration degree of cement samples after hydration at 25 °C for 24 h, with the TG-DTG curves presented in Figure 3. The weight loss of samples occurred mainly in two characteristic stages: 30−400 °C and 400−550 °C, corresponding to the dehydration of C-S-H gel and the decomposition of Ca(OH)2, respectively [23], as shown in Figure 3a. With increasing holding time, the overall weight loss of the hydrated cement samples increased. The 1500 °C−3 h sample exhibited the lowest total weight loss (2.5%), indicating limited formation of hydrates and relatively low hydration activity (Figure 3b). For the 1500 °C−6 h sample, the total weight loss after hydration of 24 h reached 10.5%. Within the 30−400 °C range, the weight loss attributed to C-S-H gel dehydration was 5.3% (Figure 3c). Although C-S-H gel diffraction peaks were not detected in XRD, TG-DTG analysis confirmed its presence and provided a quantitative measure of its formation. The weight loss corresponding to the thermal decomposition of Ca(OH)2 (400−550 °C) was 4.0%. For the 1500 °C−9 h sample (Figure 3d), the TG curve showed a weight loss of 7.1% owing to C-S-H gel dehydration and 3.7% owing to Ca(OH)2 decomposition. The C-S-H gel-related weight loss of the 1500°C−9 h sample (7.1%) was significantly higher than that of the 1500 °C−6 h sample (5.3%), demonstrating a more complete hydration reaction and the formation of a large amount of gel-like hydrates. Therefore, the sample held at 1500 °C for 9 h was selected to serve as the binder for quartz-based castables.
 |
 |
 |
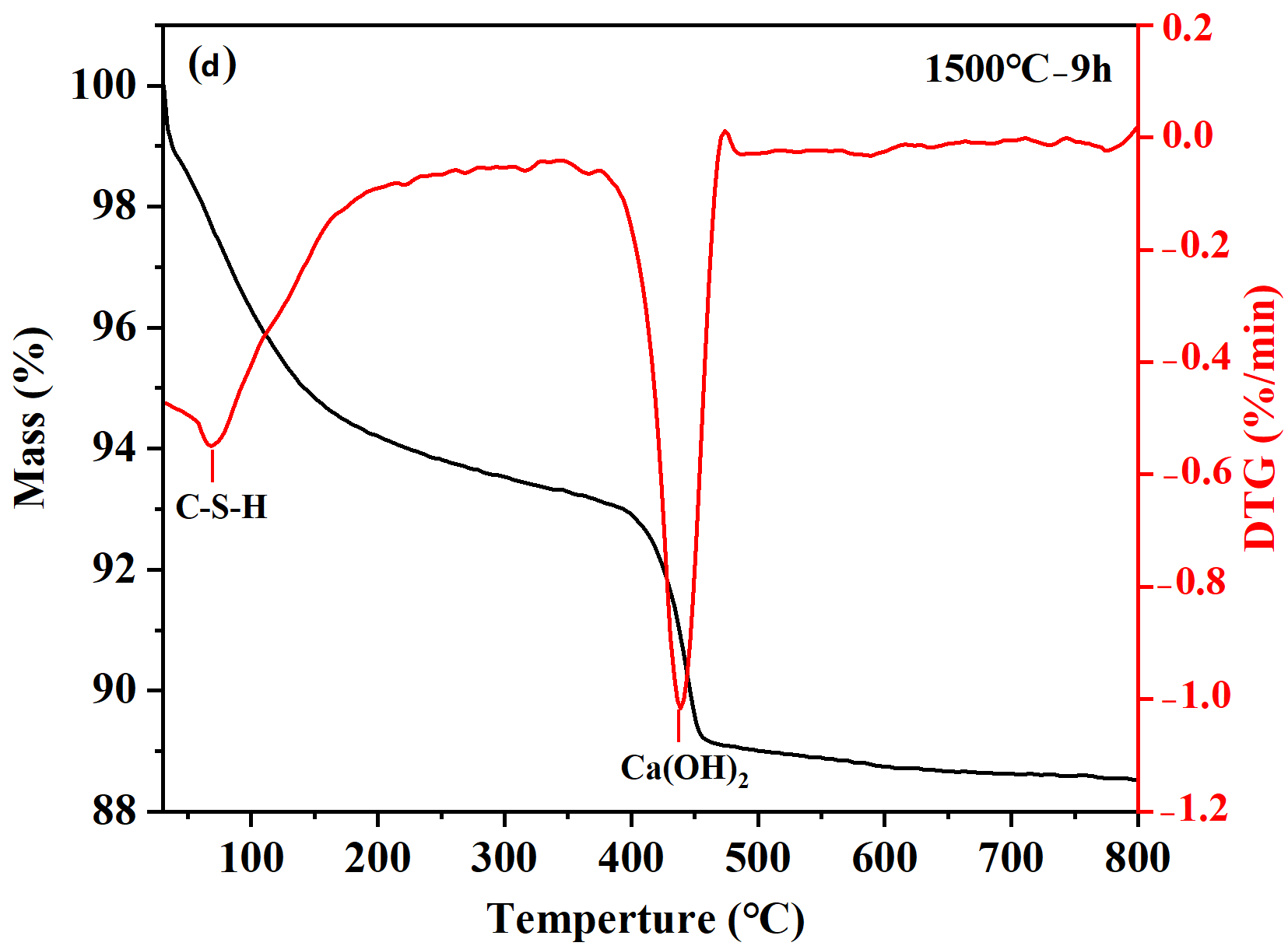 |
Figure 3. TG analysis of hydrated calcium silicate cement samples: (a) TG curves; (b−d) TG-DTG curves of samples synthesized at 1500 °C with different holding times: (b) 3 h; (c) 6 h; (d) 9 h.
3.2. Setting Behavior and Green Strength of Quartz-Based Castables
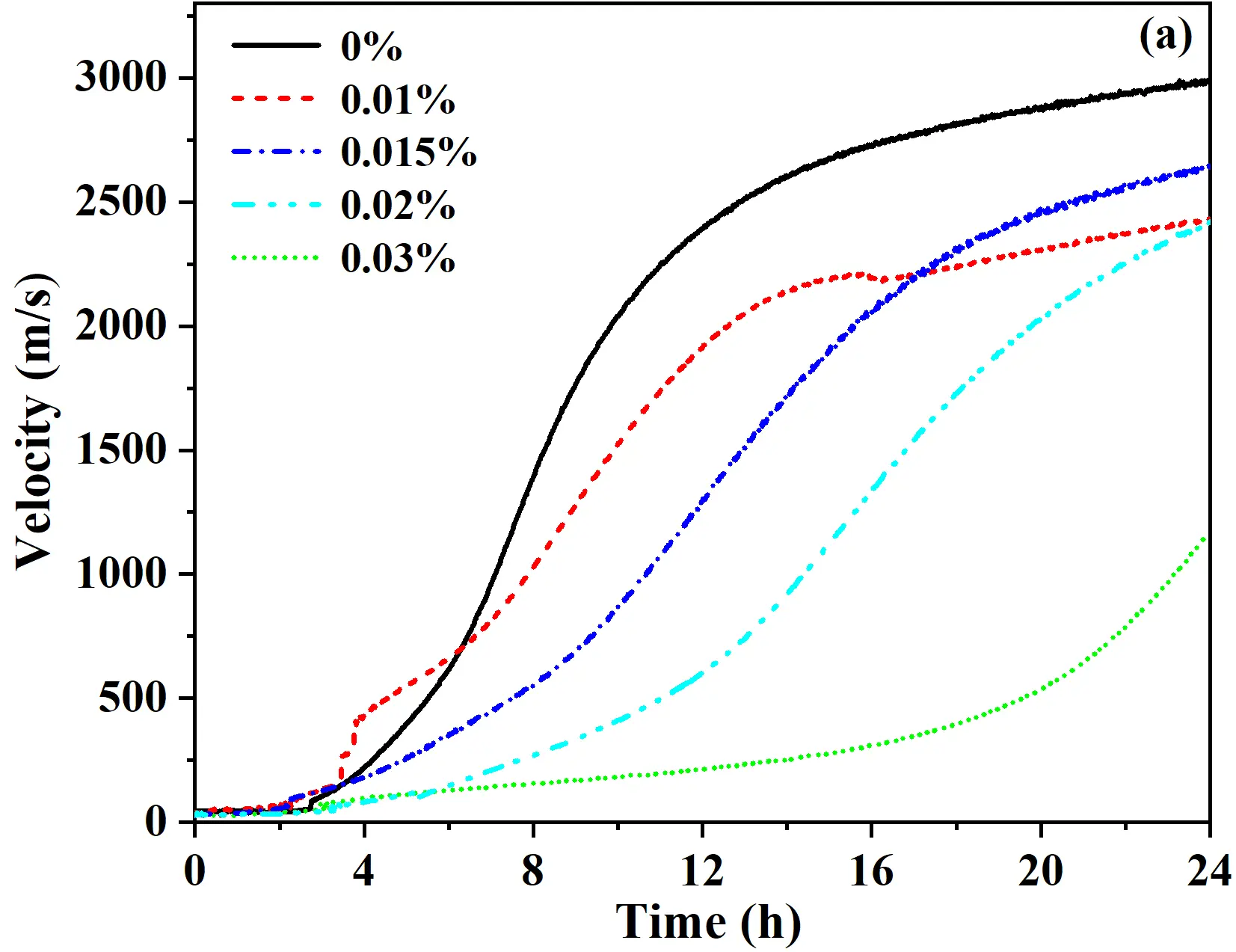
To achieve an acceptable setting behavior of the quartz-based castables, varying mass fractions of citric acid monohydrate were incorporated as a retarder. The ultrasonic setting apparatus was employed to monitor the real-time variation of ultrasonic velocity during curing, and the ultrasonic velocity versus time curves are presented in Figure 4a. The results indicated that the setting process of the castables could be divided into three stages: 0−4 h, 4−18 h, and 18−24 h. During the initial 0−4 h period, the ultrasonic velocity remained low without a significant increase, indicating a slow hydration rate at 25 °C and insufficient formation of hydrates, leaving the system in a flocculated-deflocculated state [24]. In the 4−18 h stage, the ultrasonic velocity of all samples increased markedly, primarily due to the combined effect of C-S-H gel and Ca(OH)2 formation during the hydration of calcium silicate cement. Compared with the reference sample without a retarder, the introduction of citric acid monohydrate significantly reduced the rate of velocity increase. The retarding effect strengthened with higher retarder dosage, with the most pronounced inhibition observed at 0.03 wt%. In the 18−24 h stage, the rate of ultrasonic velocity increased slowly or plateaued. After curing for 24 h, the final ultrasonic velocities were approximately 3000 m/s for the reference sample, ~2500 m/s for samples with 0.01−0.02 wt% retarder, and only ~1200 m/s for the sample with 0.03 wt% retarder. Rapid early setting can cause volumetric expansion of the quartz-based castables, mainly due to the expansion associated with the hydration of free lime (f-CaO) [25]. Therefore, a retarder dosage of 0.015 wt% was selected to control the early setting behavior.
According to elastic theory, ultrasonic velocity could serve as an indicator for the development of strength, suggesting that the addition of a retarder effectively guarantees the working time of castables. Since early setting was closely related to cement hydration, the influence of calcium silicate cement content on early setting was further investigated. As was shown in Figure 4b, during the initial 0−4 h period, the ultrasonic velocity of all samples increased slowly. The C3 sample showed only a minor increase of ~50 m/s in the first 4 h, indicating insufficient C-S-H gel and Ca(OH)2 formation due to the low cement content, with the system remaining in a near-wet state. In contrast, C4 and C5 samples entered a slow growth phase within the first 2 h, followed by a rapid increase in ultrasonic velocity between 2−4 h, demonstrating that higher cement content accelerated hydrate accumulation, promoting early structural development. During the 4−18 h period, all samples exhibited a rapid and continuous increase in ultrasonic velocity, reflecting the accelerated hydration stage. Extending the curing to 24 h, the ultrasonic velocities of C3 and C4 samples reached ~2150 m/s and ~2270 m/s, respectively, whereas the C5 sample achieved the highest velocity of 2650 m/s. Considering both the setting rate and early strength development, the optimal dosage of retarder and calcium silicate cement in quartz-based castables was 0.015% and 5%, respectively.
The physical and mechanical properties of the specimens after demolding and drying are summarized in Table 2. The demolding strength primarily arose from the bonding and filling effects of hydrates during curing. After 24 h of curing, the CMOR of all samples was approximately 1.30 MPa, and the CCS was ~7.0 MPa. C3 sample exhibited the highest porosity (4.86%), while C4 and C5 samples showed similar porosity of 4.79%. The cement content had a minor effect on demolding strength, mainly because the retarder delayed both the rate of cement hydration and early strength development of the castables. The strength of samples after drying primarily originated from the further hydration of cement and the densification of C-S-H gel through dehydration-condensation [26], which enhanced inter-particle bonding. After drying at 110 °C for 24 h, both CMOR and CCS increased significantly. The CMOR of C3, C4, and C5 samples increased from an initial 1.28 MPa to 7.10 MPa, 8.73 MPa, and 7.72 MPa, respectively, while CCS increased to 47.1 MPa, 63.2 MPa, and 52.2 MPa. Following drying, the apparent porosity gradually increased with higher cement content, likely due to the dehydration of C-S-H gel [27].
 |
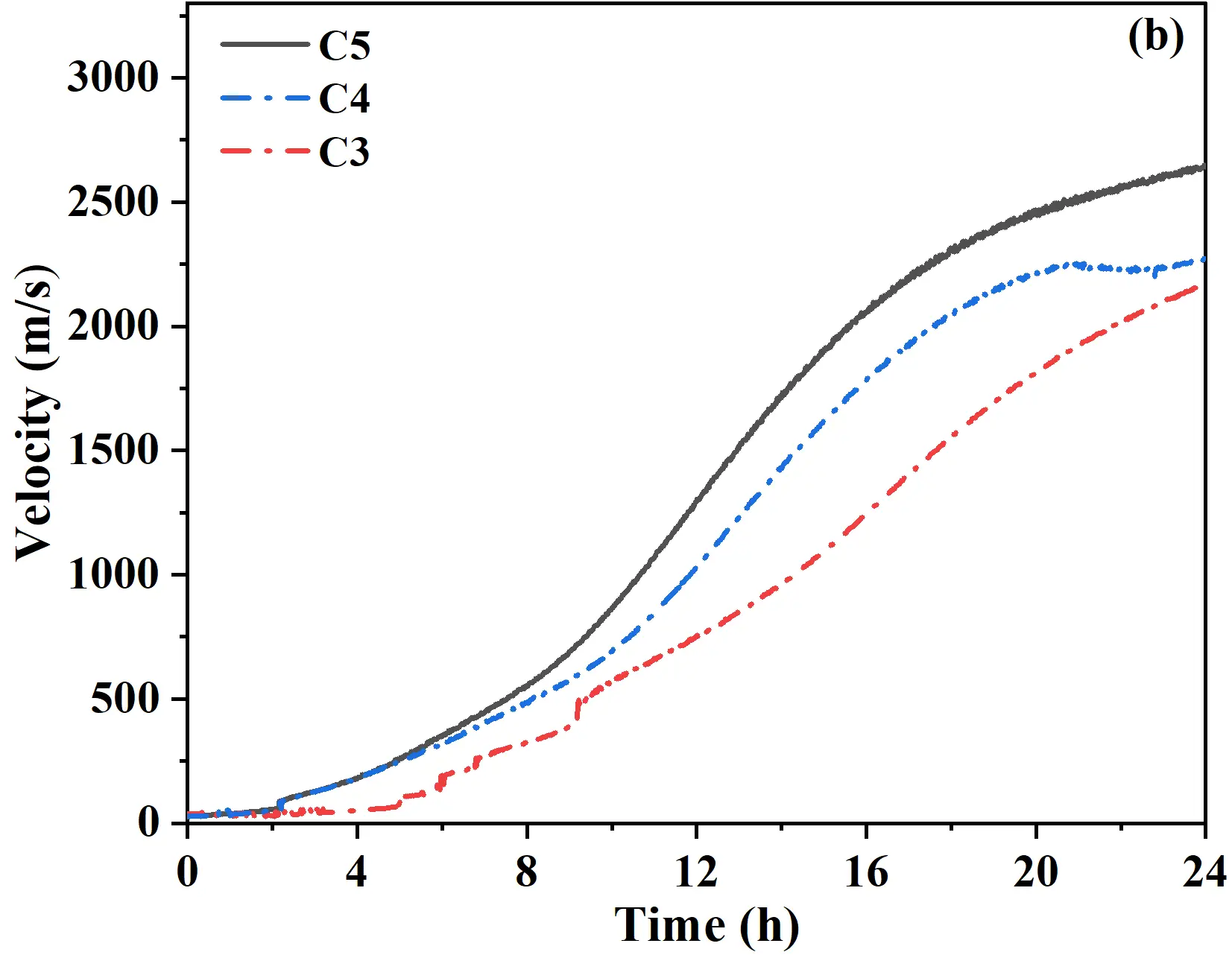 |
Figure 4. Variation of ultrasonic velocity versus time curves of castables: (a) effect of retarder content; (b) effect of calcium silicate cement content.
Table 2. Physical properties of quartz-based castables.
|
Sample |
CMOR/MPa |
CCS/MPa |
AP/% |
BD/(g/cm3) |
|
|---|---|---|---|---|---|
|
Demolding |
C3 |
1.28 ± 0.15 |
6.9 ± 0.1 |
4.9 ± 0.0 |
2.22 ± 0.01 |
|
C4 |
1.30 ± 0.14 |
7.4 ± 0.1 |
4.8 ± 0.0 |
2.22 ± 0.01 |
|
|
C5 |
1.31 ± 0.12 |
7.1 ± 0.1 |
4.8 ± 0.0 |
2.22 ± 0.01 |
|
|
110 °C drying |
C3 |
7.10 ± 0.30 |
47.1 ± 3.0 |
7.0 ± 0.2 |
2.15 ± 0.01 |
|
C4 |
8.73 ± 0.90 |
63.2 ± 0.3 |
8.4 ± 0.3 |
2.15 ± 0.01 |
|
|
C5 |
7.72 ± 0.50 |
52.2 ± 0.6 |
10.2 ± 0.1 |
2.15 ± 0.01 |
3.3. Mineralization Behavior
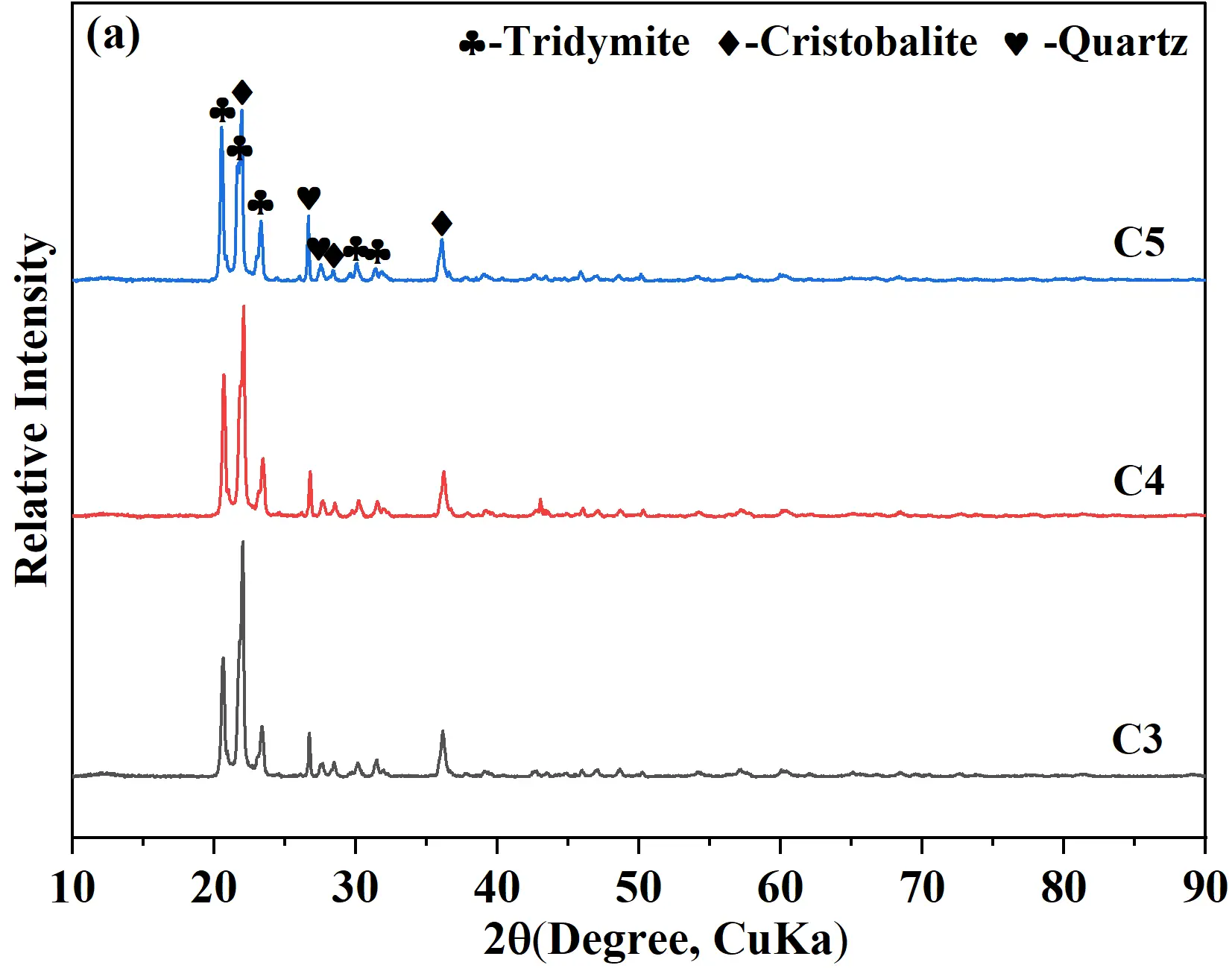
To clarify the effect of calcium silicate cement content on the phase composition of quartz-based castables and to elucidate the relationship between mineralization and high-temperature properties, XRD analysis was performed on the sintered samples, as shown in Figure 5a. Based on the XRD results, a semi-quantitative analysis was carried out to determine the relative contents of the main crystalline phases, and the permanent linear changes were also recorded, with all the relevant results summarized in Table 3. After firing at 1400 °C for 20 h, the main crystalline phases identified were tridymite (PDF#01−086−0680), cristobalite (PDF#01−082−0680), and quartz (PDF#01−079−1906). Cristobalite presents typical cubic-to-tetragonal phase transformation cracking with a characteristic cauliflower/scaly crack structure and is mainly distributed in the large quartz aggregate region with a blocky morphology; residual quartz retains the original dense granular morphology of quartz aggregates without obvious phase transformation cracks and is mostly concentrated in the core of large aggregates with insufficient contact with mineralizers; tridymite exhibits a flaky/scaly loose morphology, is predominantly distributed in the matrix region, and closely combines with the calcium silicate glass phase without obvious phase transformation cracks. Indicating polymorphic transformation of quartz under the mineralizing effect of calcium silicate. The data showed that with increasing cement, the relative amount of tridymite continuously increased, whereas the contents of cristobalite and quartz decreased. Notably, for sample C5 (5 wt% cement), the tridymite content reached a maximum of 65%, while quartz decreased to 8%. This suggested that the well−dispersed Ca(OH)2 crystals in the hydrates synergistically interacted with the C-S-H gel. During hydration, a substantial amount of C-S-H gel was generated, which, upon subsequent high-temperature firing, decomposed to form reactive CaO that could fully react with the SiO2 in matrices, promoting the formation of tridymite. The higher the cement content, the more pronounced the mineralization effect, attributable to the increased availability of CaO for reaction, which facilitated the quartz-to-tridymite transformation during firing. Although samples C4 and C5 showed favorable mineralization, residual quartz remained, likely due to limited phase transformation driving force (high temperature or enough surrounding liquids).
To deal with this issue, comparative experiments were conducted at an elevated firing temperature of 1430 °C for 20 h. The XRD patterns of the samples are shown in Figure 5b, where the main phases remained tridymite, cristobalite, and quartz. With increasing firing temperature, the intensity of quartz diffraction peaks gradually decreased, indicating enhanced transformation of quartz to tridymite and cristobalite. Compared with firing at 1400 °C, the 1430 °C heat treatment significantly reduced the residual quartz content, demonstrating a more favorable condition for phase transformation. Obviously, the residual quartz content in samples C3 and C4 decreased, while that in the C5 sample was further reduced to 1%, which was close to that of conventional silica bricks (R sample) treated at 1450 °C for at least 40 h. The present work confirmed that the nano-micro CaO and SiO2 containing hydrates provided sufficient possibilities for the uniformity of the mineralizer during the high-temperature treatment process and for reducing the formation energy of the liquid phase.
 |
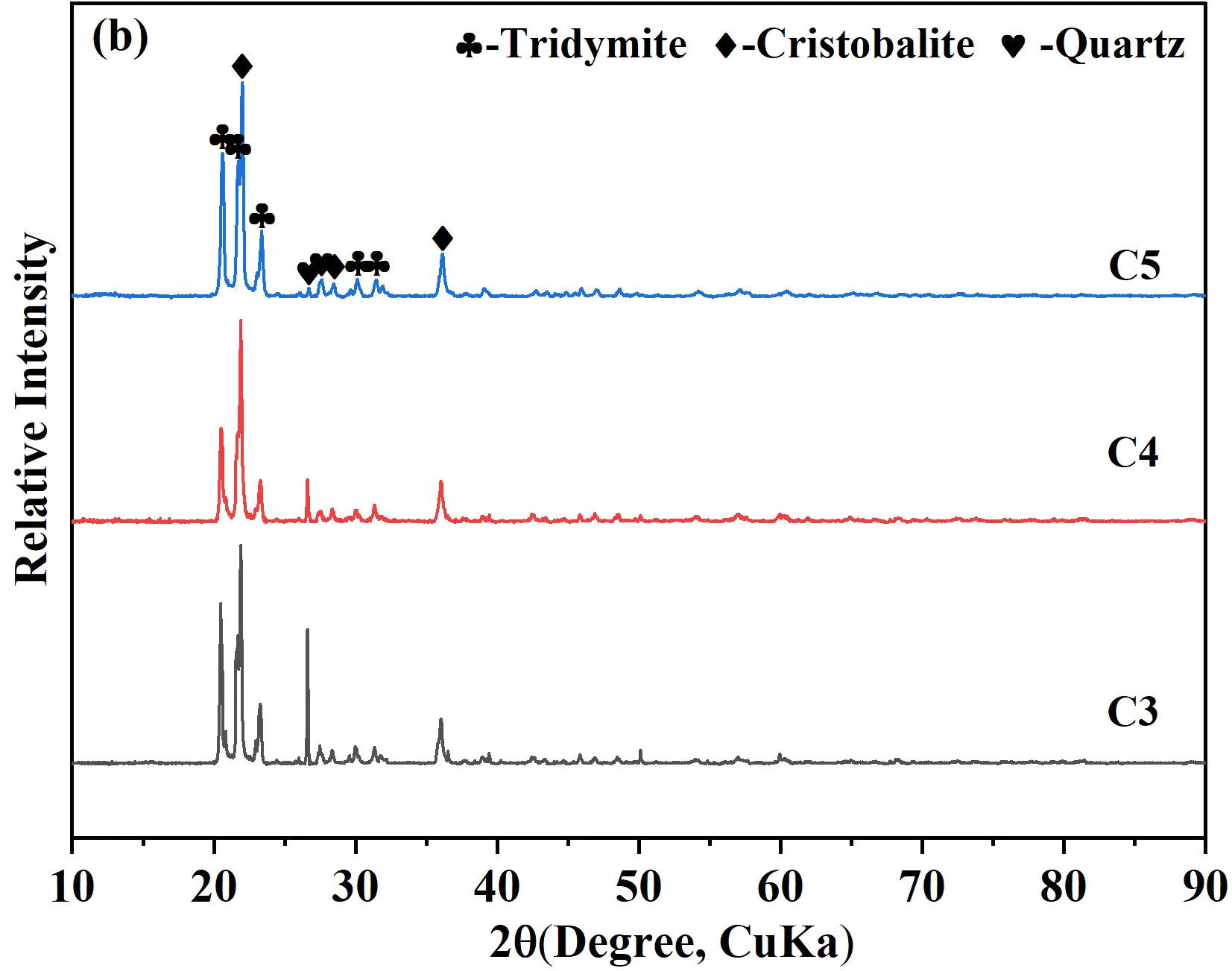 |
Figure 5. XRD patterns of quartz-based castables with different calcium silicate cement contents and firing temperatures: (a) Fired at 1400 °C × 20 h; (b) Fired 1430 °C × 20 h at 1430 °C.
Table 3. Relative contents of crystalline phases and permanent linear changes of samples after firing at different temperatures.
|
Sample |
Mass Fraction/% |
PLC/% |
|||
|---|---|---|---|---|---|
|
Residual Quartz |
Cristobalite |
Tridymite |
|||
|
1400 °C × 20 h |
C3 |
13 |
39 |
48 |
2.67 |
|
C4 |
7 |
36 |
58 |
3.64 |
|
|
C5 |
8 |
32 |
60 |
4.29 |
|
|
1430 °C × 20 h |
C3 |
10 |
41 |
49 |
1.20 |
|
C4 |
6 |
46 |
48 |
3.28 |
|
|
C5 |
1 |
34 |
65 |
4.10 |
|
|
1450 °C × 40 h |
R |
1 |
33 |
66 |
− |
SEM images of conventional silica bricks and quartz aggregates are shown in Figure 6. Conventional silica bricks were composed of aggregates, matrices, and pores, exhibiting a typical microstructure of refractory materials. In terms of distribution characteristics, the mineralizer was mainly concentrated in the matrices; meanwhile, cracks and pores generated by phase transformation could be observed inside the aggregates, resulting in a relatively loose overall structure (Figure 6a). In contrast, for quartz aggregates (Figure 6b), the internal pores were extremely scarce and scattered, mostly intrinsic tiny pores inherent in the raw material. By comparison, the quartz aggregates exhibited significantly higher compactness than conventional silica bricks, with no obvious concentrated pores. Figure 7 presents SEM images of quartz-based castable samples after heat treatments at 1400 °C and 1430 °C for 20 h. Dark regions corresponded to pores filled with epoxy resin; gray areas represented aggregates; and white and yellow boxes indicated regions of cristobalite and quartz, respectively. It was observed that the aggregate remained partially intact in sample C3, with no obvious cracks or pores after firing at 1400 °C. In contrast, aggregates in samples C4 and C5 exhibited internal cracks and pore structures, indicating phase transformations within the aggregates. The higher firing temperature (1430 °C) led to a significant reduction in internal porosity compared to the 1400 °C samples and conventional silica bricks. The bonding between matrices and aggregates became more compact, with the mineralizer uniformly distributed within the matrices. The white dashed lines in the images indicate regions of cristobalite and the yellow dotted lines in the image indicate the areas with larger residual quartz. The “scaly” crack structures within cristobalite particles resulted from the high-temperature cubic-to-tetragonal phase transformation of cristobalite, which was closely related to the polymorphic behavior of quartz [28]. In the microstructure, blocky cristobalite was mainly distributed within larger aggregates, whereas scaly tridymite was concentrated in the matrices. Additionally, cristobalite in the aggregates was often partially encapsulated by the surrounding tridymite grown in the matrices. These structural distribution characteristics primarily arose because the mineralizer was initially concentrated in the matrix region. Consequently, during high-temperature firing, the liquid phase formed preferentially in the matrices, promoting the transformation of quartz to tridymite. It should be noted that the tridymite-to-cristobalite transformation was accompanied by a volumetric expansion of approximately 5−8% [29], and the porous matrices provided buffer space for crystal growth and transformation, thereby facilitating reduced microcrack formation. In contrast, large aggregates, due to insufficient contact with the mineralizer, were less fully mineralized within a short firing time, leading to the retention of residual quartz and cristobalite within their interiors [30].


3.4. Physical and Mechanical Properties
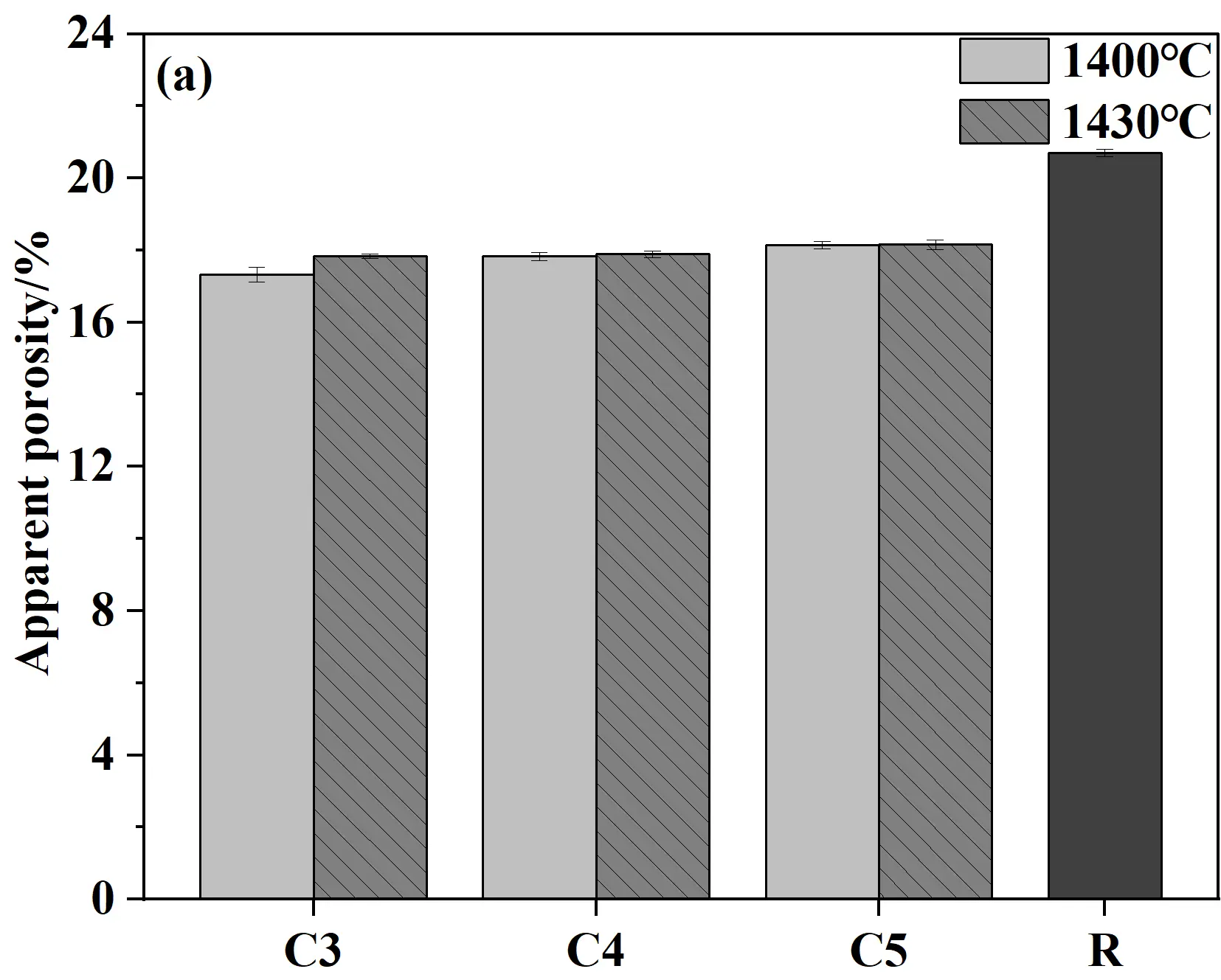
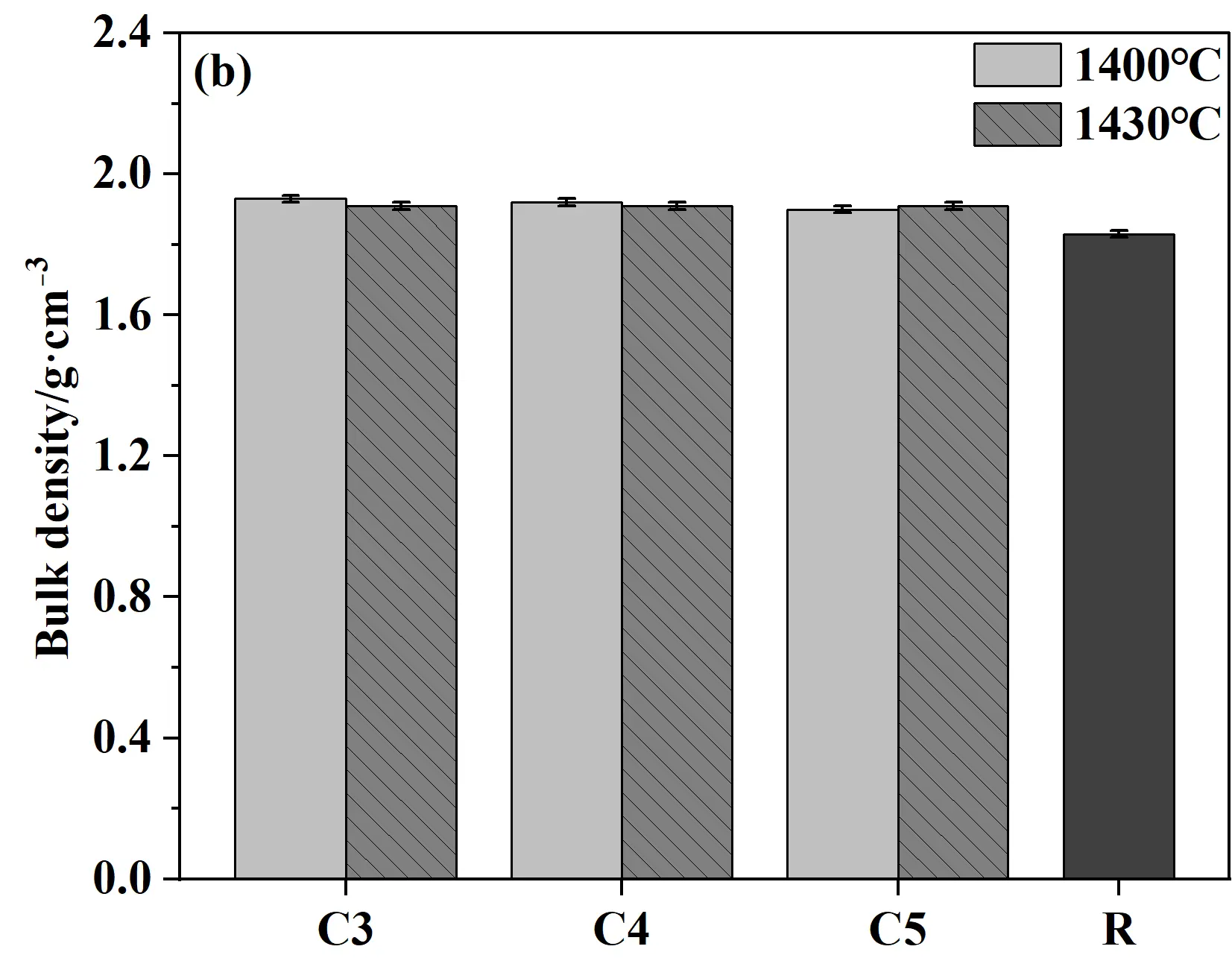
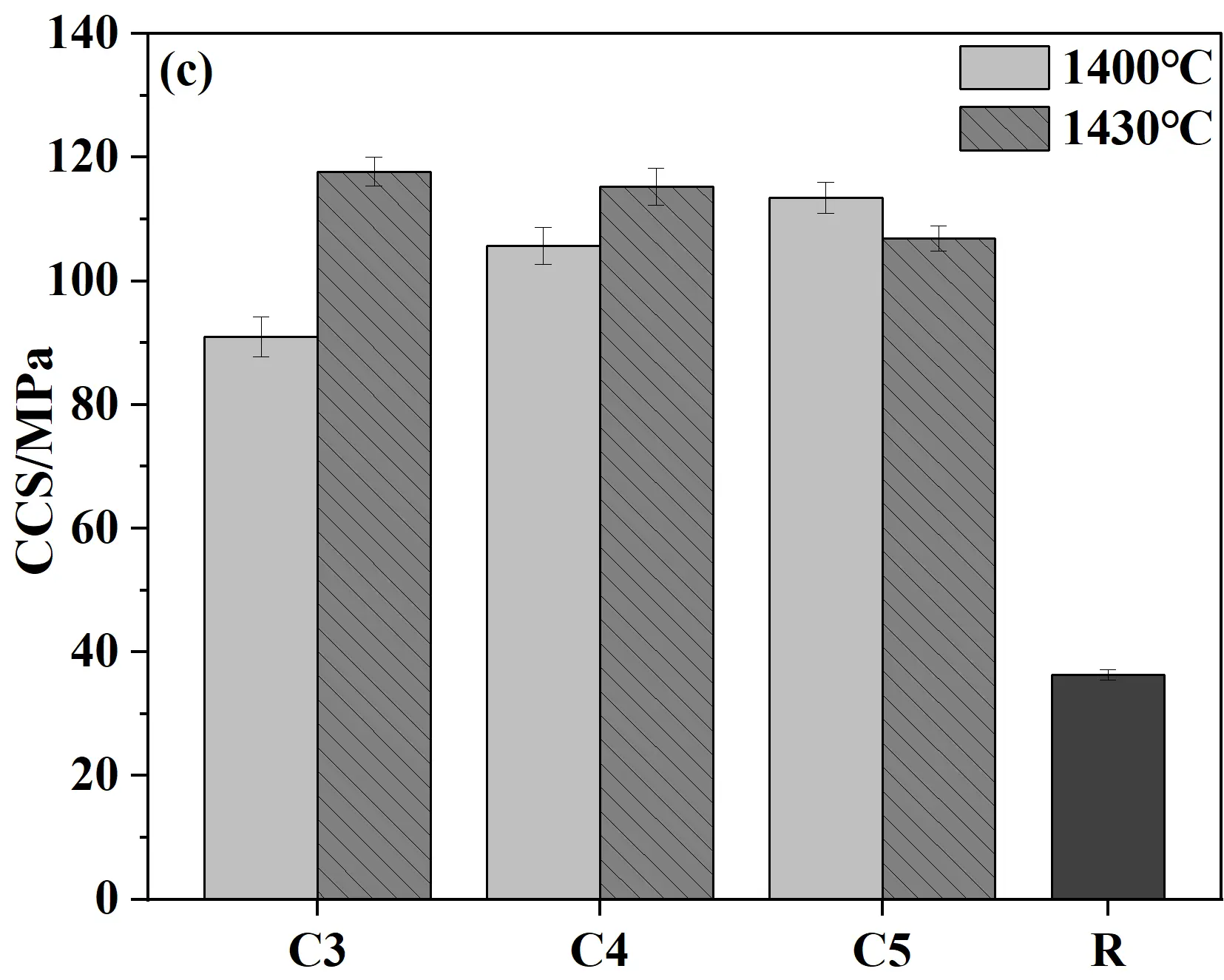
Figure 8 presents the apparent porosity, bulk density, cold modulus of rupture, and cold crushing strength of castables and conventional silica bricks (R sample). As shown in Figure 8a,b, when samples were fired at 1400 °C for 20 h, the porosity showed a gradual upward trend while the bulk density decreased correspondingly with the increase in cement content. This phenomenon was mainly attributed to the decomposition of the hydrates of calcium silicate cement (C-S-H and Ca(OH)2) at high temperatures, which in turn led to an increase in porosity. Meanwhile, the CMOR and CCS of samples generally increase with the increase in cement content. The key reason for this trend was the increased cement content, which introduced higher CaO content. CaO reacted with SiO2 at high temperatures to generate more liquid phase, which facilitated the phase transformation and ultimately improved the mechanical properties [31]. Compared to samples fired at 1400 °C, the CCS of specimens fired at 1430 °C decreased slightly (all above 100 MPa), which should be caused by the increased apparent porosity. Compared to conventional silica bricks, the CCS and CMOR of specimens from the present work were significantly improved, and the apparent porosity was correspondingly reduced, exhibiting distinct advantages in comprehensive performance.
 |
 |
 |
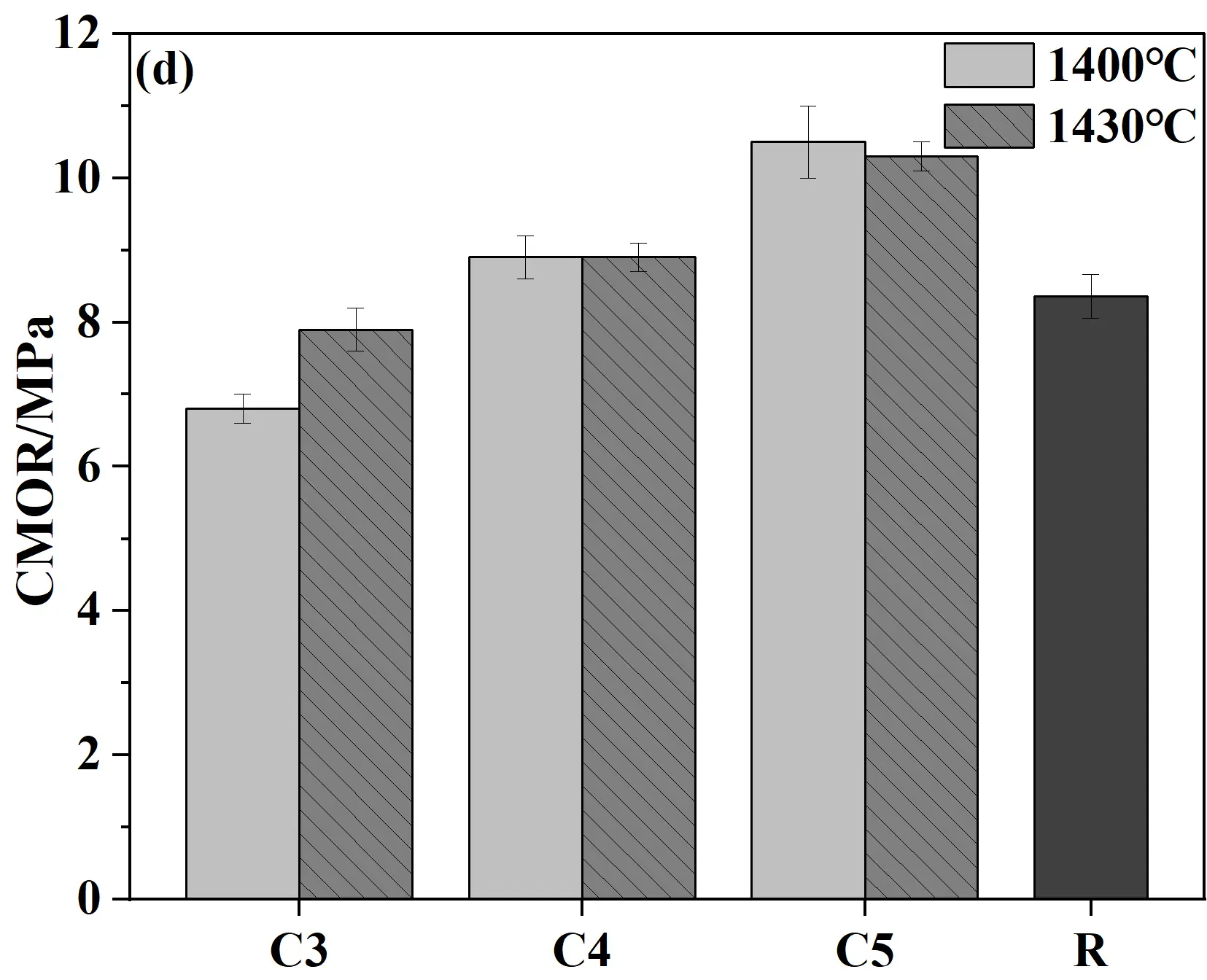 |
Figure 8. Physical and mechanical properties of quartz-based castables and conventional silica bricks (R): (a) Apparent porosity; (b) Bulk density; (c) Cold crushing strength (CCS); (d) Cold modulus of rupture (CMOR).
3.5. High-Temperature Properties
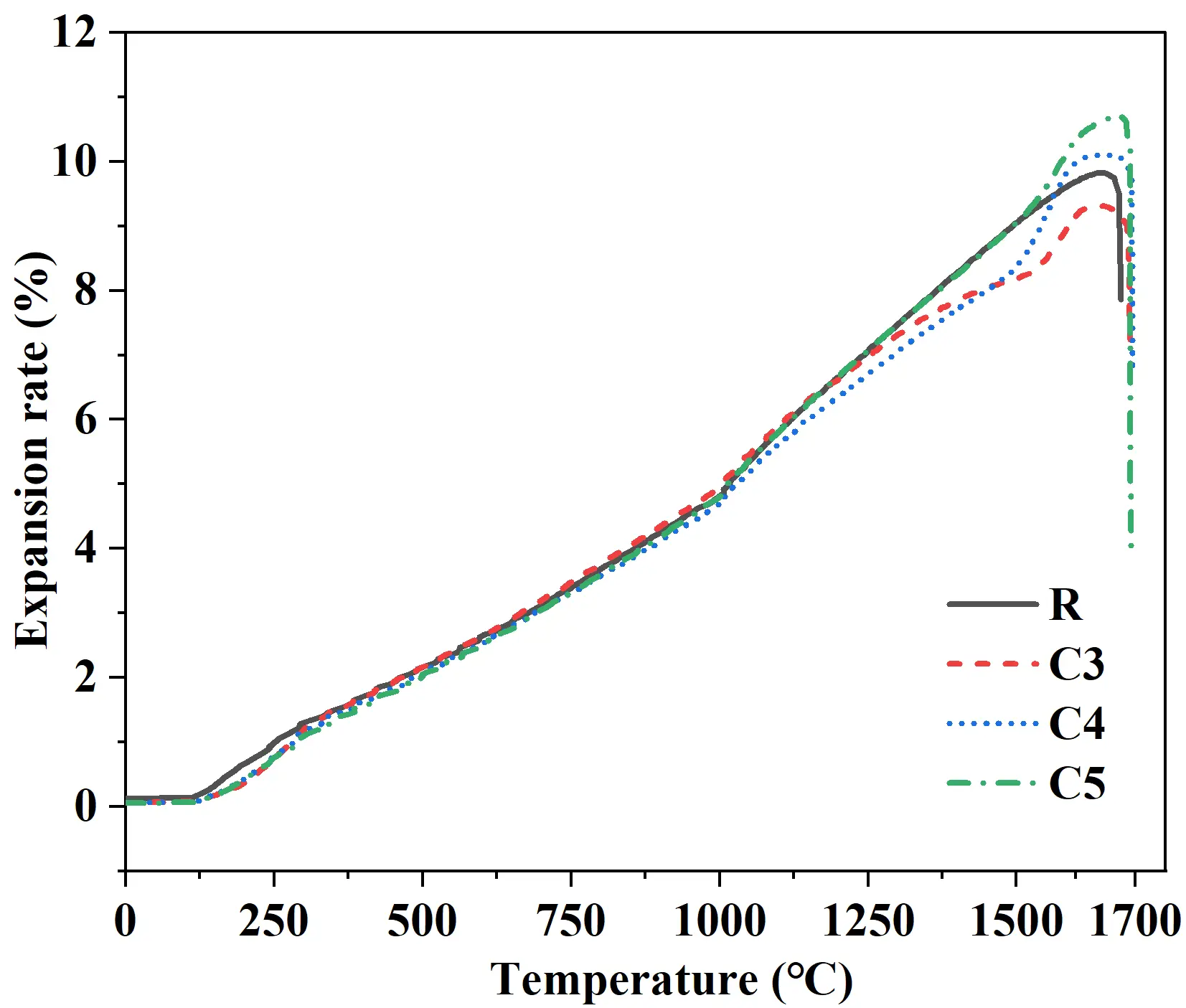
As shown in Figure 9, compared with the traditional silica brick (R), the quartz-based castables exhibited higher refractoriness under load. The C3 and C4 samples showed a pronounced “expansion peak” within the temperature range of 1500−1700 °C, whereas the R and C5 samples displayed relatively smooth and gradual expansion curves without distinct peaks. This difference was primarily attributed to the incomplete mineralization in C3 and C4, which resulted in a higher content of residual quartz. The concentrated phase transformation of this residual quartz at elevated temperatures led to the observed expansion phenomenon. In contrast, the R and C5 samples contained less residual quartz, thus exhibited similar and stable expansion behaviors. These results indicated that optimizing the cement content effectively promoted the pre-transformation of quartz, suppressed harmful expansion during service, and thereby enhanced the high-temperature dimensional stability.
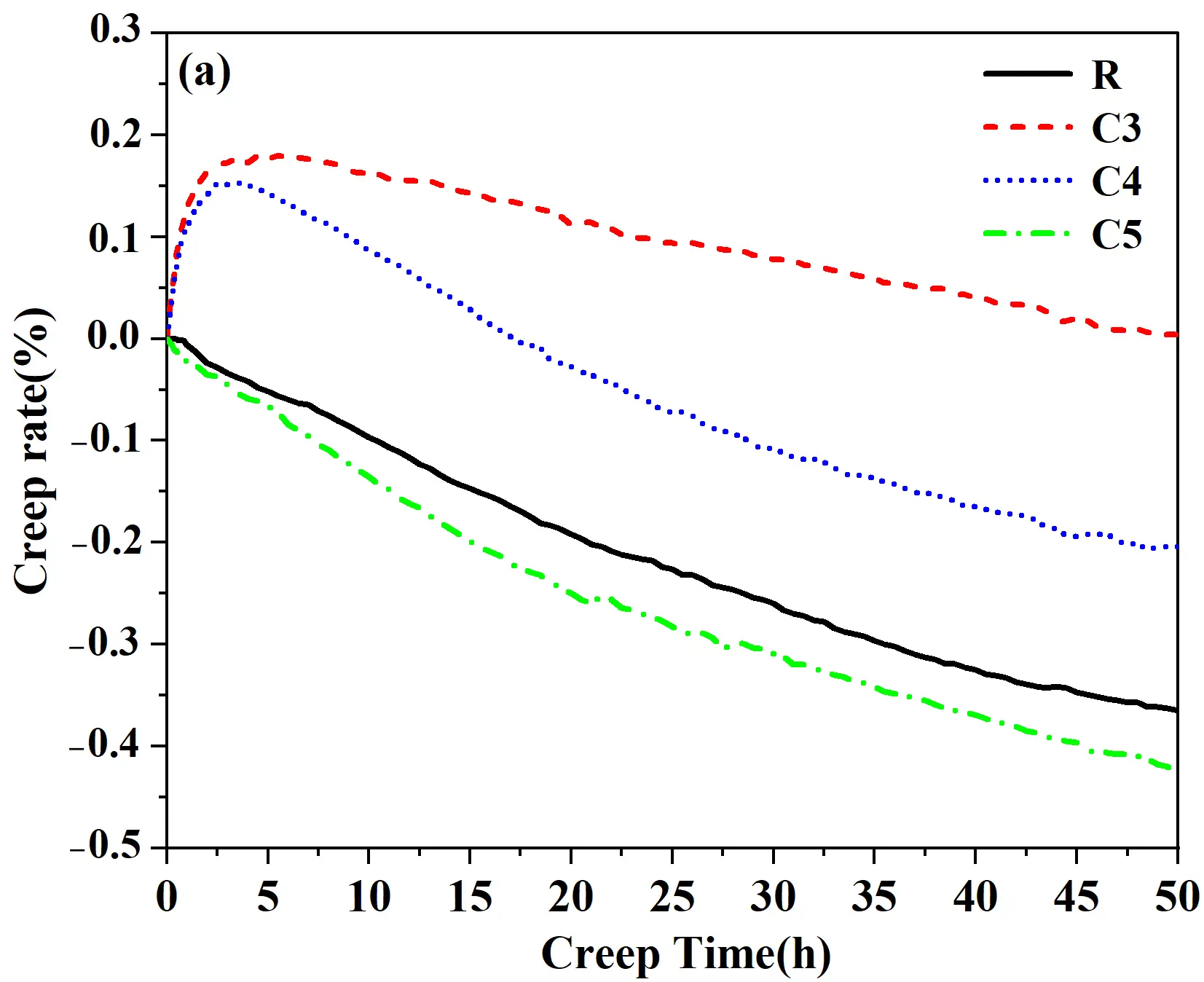
Additionally, to evaluate the high-temperature creep behavior of the quartz-based castables, the specimens calcined at 1430 °C with improved mineralization were subjected to creep testing at 1500 °C for 50 h (Figure 10a). At the initial stage (0−5 h), quartz progressively transformed into cristobalite and tridymite with associated volumetric expansion. Due to its higher residual quartz content, sample C3 exhibited a more pronounced positive expansion than sample C4. As the transformation proceeded to completion, the expansion effect diminished, and sintering densification together with structural stabilization became dominant, resulting in a gradual decrease in creep rate. For sample C4, the higher mineralizer content facilitated greater liquid phase formation at elevated temperature, leading to enhanced densification and overall shrinkage. In contrast, sample C5, with negligible residual quartz, showed continuous shrinkage throughout the test and no apparent expansion associated with phase transformation; the creep rate was about 0.06% higher than that of the reference silica brick (R sample). This behavior was attributed to sintering densification and the formation of calcium silicate [32]. In summary, increasing cement addition reduced the residual quartz content and shifted the creep behavior of the castables from expansion-dominated to shrinkage-dominated.
The XRD patterns and relative crystalline contents of samples after the creep test are shown in Figure 10b. Compared to Figure 5b, the diffraction peaks of residual quartz in samples C3 and C4 decreased significantly, whereas no obvious changes were observed in sample C5. A comparison with Table 3 revealed that after the creep test, the tridymite content decreased and the cristobalite content increased in the conventional silica brick (R sample). In sample C3, both cristobalite and tridymite contents increased, with a more pronounced rise in cristobalite (∼8%). By contrast, samples C4 and C5 exhibited an increase in tridymite content but a decrease in cristobalite and residual quartz. These trends could be explained as follows: In the conventional brick (R sample), which had a low initial quartz content, tridymite transformed into cristobalite at high temperature. In sample C3, the higher residual quartz content allowed direct transformation to cristobalite or partial dissolution-recrystallization into tridymite at 1500 °C; however, the direct phase transformation dominated owing to insufficient mineralization. In samples C4 and C5, the higher cement enhanced mineralization and promoted the formation of a low-eutectic liquid phase, which facilitated the conversion of residual quartz into tridymite via dissolution-recrystallization. For sample C5, owing to its very low initial quartz content, the XRD patterns showed negligible change after creep, though a slight increase in tridymite was inferred.
 |
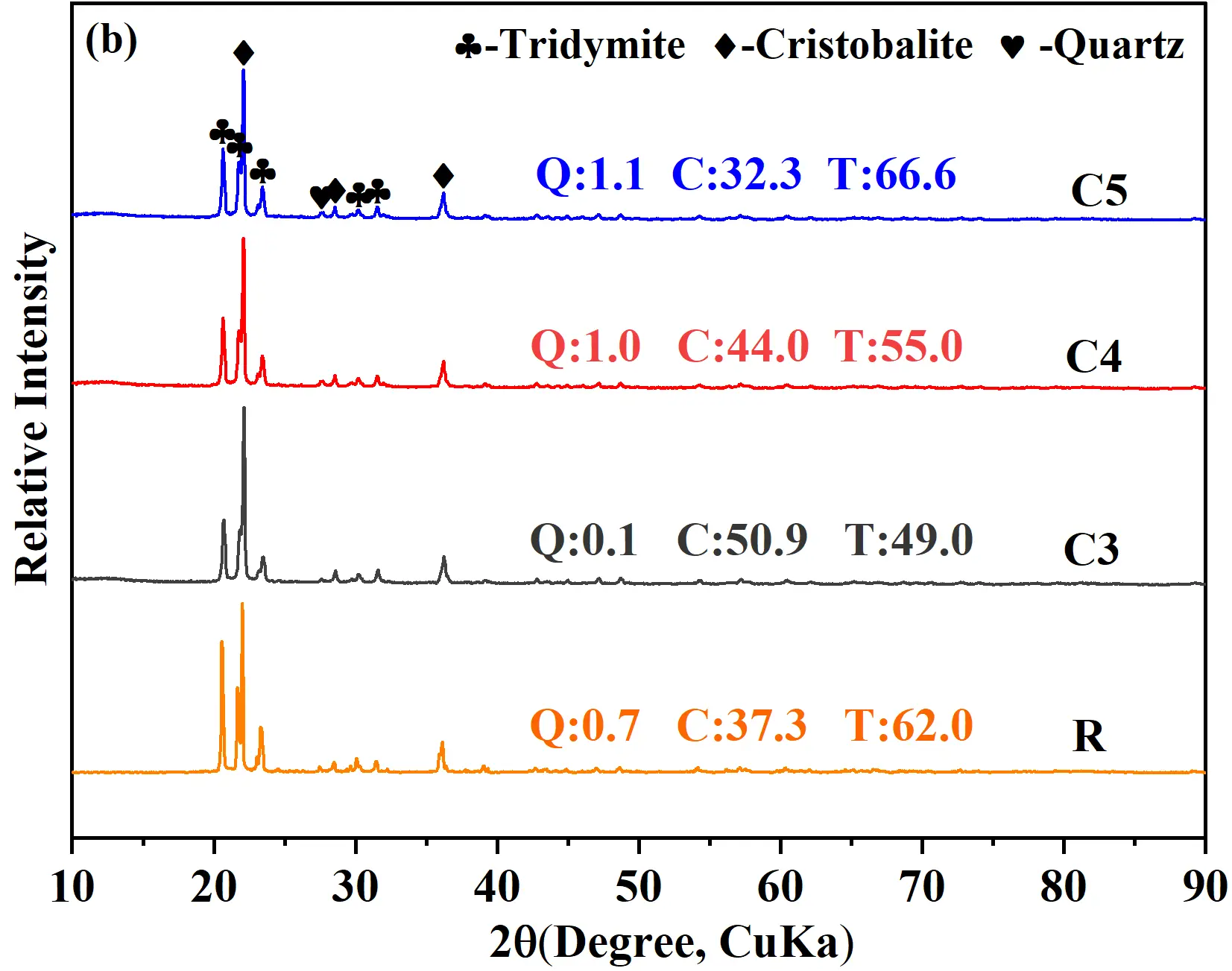 |
Figure 10. (a) Creep curves of the quartz-based castable fired at 1430 °C, tested at 1500 °C for 50 h. (b) XRD patterns of samples after high-temperature creep test (Q: Quartz; C: Cristobalite; T: Tridymite, wt%).
The SEM micrographs of the specimen after creep are shown in Figure 11. Compared with Figure 7, the dark regions corresponded to epoxy-filled pores, the gray regions represented aggregates and matrices, and the bright regions were the glass phase formed by the reaction between CaO and SiO2. It could be observed that more numerous and finer “roof-tile” cracks had developed inside the aggregates of both conventional silica bricks and quartz-based castables, and the mineralizer had penetrated the interior of the aggregates along these cracks. This indicated that cubic high-temperature cristobalite underwent a phase transformation to tetragonal low-temperature cristobalite at high temperatures [28]; however, this cubic-to-tetragonal transformation process did not appear to be directly correlated with the content of cristobalite.

4. Summary and Conclusions
In this study, calcium silicate cement is successfully synthesized, and this work confirms the dual functionality (binding and mineralizing) of calcium silicate cement in quartz-based castables. It presents a simplified and efficient technical pathway for manufacturing large-sized silica precast components, eliminating the need for separate additions of binder and mineralizer, with potential benefits for intelligent masonry in high-temperature industrial applications. Based on the results obtained, the following conclusions can be drawn:
- (1)
-
The optimal formulation for achieving a balance between workability and early-age strength is determined to be 0.015 wt% citric acid (retarder) and 5 wt% calcium silicate cement. This composition provides adequate setting control and a demolding strength sufficient for handling large-sized precast components.
- (2)
-
The fine CaO and SiO2 derived from the decomposition of cement hydrates during firing act as a highly effective mineralizer. It significantly promotes quartz transformation into tridymite. After firing at 1430 °C for 20 h, the sample with 5 wt% cement addition achieves a remarkably low residual quartz content of 1%, comparable to conventional silica bricks, and requires lower mineralization temperature and time.
- (3)
-
The incorporation of calcium silicate cement influences the microstructure by facilitating liquid-phase sintering at high temperatures, which enhances the bonding between aggregates and the matrices. Consequently, the room-temperature mechanical strengths of the castables improve with increasing cement content, and the CCS (above 100 MPa) is 2.5 times higher than the conventional silica bricks.
- (4)
-
The high-temperature creep mechanism is fundamentally influenced by the cement content. At low cement additions, the behavior is “expansion-dominant”, driven by the phase transformation of residual quartz. With increasing cement content (up to 5 wt%), the creep mechanism shifts to “shrinkage-dominant”, characterized by densification and the formation of a eutectic liquid phase, resulting in a creep rate approaching that of conventional silica bricks.
- (5)
-
For the industrial application of large-sized quartz-based precast components, pre-firing is an essential and highly recommended process; the industrial pre-firing regime of 1430 °C for 20 h is proposed, which enables complete SiO2 polymorph transformation (residual quartz < 1%) and effectively eliminates harmful volume expansion during the service of components under high temperatures.
Statement of the Use of Generative AI and AI-Assisted Technologies in the Writing Process
During the preparation of this manuscript, the authors used AI language assistance tools in order to improve language expression and academic writing. After using this tool/service, the authors reviewed and edited the content as needed and take full responsibility for the content of the published article.
Author Contributions
Conceptualization, Z.C., Y.L. and N.L.; Methodology, Z.C.; Software, Z.C.; Validation, Z.C., Y.L., N.L. and W.L.; Formal Analysis, Z.C. and W.L.; Investigation, Z.C.; Resources, Y.L., N.L., W.B., D.X., W.Y. and B.T.; Data Curation, Z.C.; Writing—Original Draft Preparation, Z.C. and W.L.; Writing—Review & Editing, Y.L., N.L. and W.L.; Visualization, Z.C.; Supervision, Z.C.; Project Administration, Y.L. and N.L.; Funding Acquisition, Y.L. and S.J. All authors have read and agreed to the published version of the manuscript.
Ethics Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
Funding
This research was funded by the Hubei Provincial Special Fund for Central-Guided Local S&T Development, grant number 2025CSA017.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Arahori T, Suzuki T. Transformation of tridymite to cristobalite below 1470 °C in silica refractories. J. Mater. Sci. 1987, 22, 2248–2252. DOI:10.1007/BF01132967 [Google Scholar]
- Fan J, Li Y, Zhang X, Sun Y, Ma C, Gao Y. Performance of silica bricks with ferrosilicon nitride as the mineralizer. Ceram. Int. 2022, 48, 26791–26799. DOI:10.1016/j.ceramint.2022.05.377 [Google Scholar]
- Nevřivová L, Kotouček M, Lang K. Possibilities of Reducing the Apparent Porosity of Silica Bricks for the Coke Batteries. Adv. Mater. Res. 2014, 897, 121–124. DOI:10.4028/www.scientific.net/AMR.897.121 [Google Scholar]
- Li S, Zhang H, Zou Y, Gu H, Huang A, Fu L, et al. Microstructural evolution during H2 corrosion of Al2O3–SiO2 based refractory aggregates. Ceram. Int. 2023, 49, 27788–27795. DOI:10.1016/j.ceramint.2023.05.265 [Google Scholar]
- Heimann RB. Classic and Advanced Ceramics: From Fundamentals to Applications; John Wiley & Sons: Hoboken, NJ, USA, 2010. [Google Scholar]
- Fan J, Li Y, Gao Y, Zhang X, Jiang P. Properties of both Chinese silica brick and silica raw material. Ironmak. Steelmak. 2022, 49, 495–505. DOI:10.1080/03019233.2021.2022870 [Google Scholar]
- Zhang XH, Li Y, Cui YY, Tian ZH, Sun LJ, Ma CH, et al. Corrosion mechanism of silica bricks containing high amorphous for hot stoves. Ceram. Int. 2023, 49, 40746–40753. DOI:10.1016/j.ceramint.2023.10.058 [Google Scholar]
- Manivasakan P, Rajendran V, Rauta PR, Sahu BB, Sahu P, Panda BK, et al. Effect of TiO2 Nanoparticles on Properties of Silica Refractory. J. Am. Ceram. 2010, 93, 2236–2243. DOI:10.1111/j.1551-2916.2010.03727.x [Google Scholar]
- Zhang X, Ma C, Qian Y, Yang J, Sun Y, Li Y. Reaction Mechanism of Mineralizers Ferrosilicon Nitride and Calcium Carbonate in Silica Bricks. J. Chin. Ceram. Soc. 2023, 51, 594–601. DOI:10.14062/j.issn.0454-5648.20220859 [Google Scholar]
- Wang J, Dong B, Yu C, Deng C, Ding J, Zhu H. Effect of Quaternary Mineralizer on Microstructure and Physical Properties of Silica Brick. Bull. Chin. Ceram. Soc. 2023, 42, 1115–1121. Available online: https://www.researching.cn/ArticlePdf/m00115/2023/42/3/2023-03-1115.pdf (accessed on 11 March 2026).
- Allen AJ, Thomas JJ, Jennings HM. Composition and density of nanoscale calcium–silicate–hydrate in cement. Nat. Mater. 2007, 6, 311–316. DOI:10.1038/nmat1871 [Google Scholar]
- Peng G, Chan S, Anson M. Chemical kinetics of CSH decomposition in hardened cement paste subjected to elevated temperatures up to 800 °C. Adv. Cem. Res. 2001, 13, 47–52. DOI:10.1680/adcr.2001.13.2.47 [Google Scholar]
- GB/T 5072-2023; Refractory Products—Determination of Cold Compressive Strength. Standardization Administration of China (SAC): Beijing, China, 2023. [Google Scholar]
- GB/T 3001-2017; Refractory Materials—Determination of Cold Modulus of Rupture. Standardization Administration of China (SAC): Beijing, China, 2017. [Google Scholar]
- GB/T 2997-2015; Test Method for Bulk Density, Apparent Porosity and True Porosity of Dense Shaped Refractory Products. China National Technical Committee for Standardization of Refractory Materials: Beijing, China, 2015. [Google Scholar]
- GB/T 5073-2005; Refractory Products—Test Method of Creep in Compression. Standardization Administration of China (SAC): Beijing, China, 2006. [Google Scholar]
- GB/T 5989-2023, Refractory Products—Determination of Refractoriness under Load (Differential Method with Rising Temperature). Standardization Administration of China (SAC): Beijing, China, 2023. [Google Scholar]
- GB/T 5988-2022; Test Method for Permanent Linear Change of Refractories Upon Heating. Standardization Administration of China (SAC): Beijing, China, 2022. [Google Scholar]
- Simedru AF, Becze A, Cadar O, Scurtu DA, Simedru D, Ardelean I. Structural characterization of several cement-based materials containing chemical additives with potential application in additive manufacturing. J. Mol. Sci. 2023, 24, 7688. DOI:10.3390/ijms24097688 [Google Scholar]
- Wang S, Zhao P, Tian Y, Liu J. Effects of CSH Seed Prepared by Wet Grinding on the Properties of Cement Containing Large Amounts of Silica Fume. Polymers 2024, 16, 2769. DOI:10.3390/polym16192769 [Google Scholar]
- Zhu Z, Wang Z, Zhou Y, Chen Y, Wu K. Identification of chemical bonds and microstructure of hydrated tricalcium silicate (C3S) by a coupled micro-Raman/BSE-EDS evaluation. Materials 2021, 14, 5144. DOI:10.3390/ma14185144 [Google Scholar]
- Schleiting M, Wetzel A, Göbel D, Krooß P, Frenck JM, Niendorf T, et al. Growth of C–S–H phases on different metallic surfaces. J. Microsc. 2022, 286, 148–153. DOI:10.1111/jmi.13089 [Google Scholar]
- Xia X, Yu Y, Qi F, Li P, Qu C, Xu P, et al. Experimental Investigation of a CSH Nanocrystalline Nucleus Modified with PCE Dispersant on the Early-Age Mechanical Behavior of Oil Well Cement Paste. Materials 2025, 18, 326. DOI:10.3390/ma18020326 [Google Scholar]
- Chen W, Shui Z, Li Y. Early age hydration of cement paste monitored with ultrasonic velocity and numerical simulation. J. Wuhan Univ. Technol. Sci. Ed. 2010, 25, 704–707. DOI:10.1007/s11595-010-0075-2 [Google Scholar]
- Ning H, Yan C, Zhang J, Li J, Nie W, Lv Z, et al. Study of micro-expansion deformation of cement-based materials enhanced by soybean-wax-coated carbide slag composites. Mater. Chem. Phys. 2025, 332, 130165. DOI:10.1016/j.matchemphys.2024.130165 [Google Scholar]
- Ma Y, Jin M, Li W, Zhang J, Huang J, Shen X, et al. Effect of drying method on calcium silicate hydrate (CSH): Experiments and molecular dynamics simulations study. Constr. Build. Mater. 2024, 411, 134367. DOI:10.1016/j.conbuildmat.2023.134367 [Google Scholar]
- Tantawy M. Effect of high temperatures on the microstructure of cement paste. J. Mater. Sci. Chem. Eng. 2017, 5, 33–48. DOI:10.4236/msce.2017.511004 [Google Scholar]
- Dai Y, Yin Y, Xu X, Jin S, Li Y, Harmuth H. Effect of the phase transformation on fracture behaviour of fused silica refractories. J. Eur. Ceram. 2018, 38, 5601–5609. DOI:10.1016/j.jeurceramsoc.2018.08.040 [Google Scholar]
- Schneider H. High-temperature transformation of tridymite single crystals to cristobalite. Z. für Krist. Cryst. Mater. 1986, 175, 165–176. DOI:10.1524/zkri.1986.175.14.165 [Google Scholar]
- Jusnes KF, Tangstad M, Ringdalen E. Phase transformations from quartz to cristobalite. In Extraction 2018: Proceedings of the First Global Conference on Extractive Metallurgy; Springer: Cham, Switzerland, 2018; pp. 717–727. [Google Scholar]
- Peng C, Li N, Han B. Effect of zircon on sintering, composition and microstructure of magnesia powders. Sci. Sinter. 2009, 41, 11–17. DOI:10.2298/SOS0901011P [Google Scholar]
- Fan J, Li Y, Gao Y, Zhang X, Jiang P. Evaluation of the morphology and pore characteristics of silica refractory using X-ray computed tomography. Ceram. Int. 2021, 47, 18084–18093. DOI:10.1016/j.ceramint.2021.03.124 [Google Scholar]