Detailed Analyses of Light Intensity Dependence to Uncover Multielectron Oxygen-Reduction Mechanism by Platinum-Loaded Tungsten(VI) Oxide

Detailed Analyses of Light Intensity Dependence to Uncover Multielectron Oxygen-Reduction Mechanism by Platinum-Loaded Tungsten(VI) Oxide
Mai Takashima 1,2,*,† Chiharu Yamada 2 Bunsho Ohtani 1,2,‡
Received: 07 November 2025 Revised: 17 December 2025 Accepted: 26 February 2026 Published: 09 March 2026

© 2026 The authors. This is an open access article under the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/).
1. Introduction
In the field of photocatalysis, one of the most important objectives is to develop a fundamental understanding of the parameter(s) that govern the activity of a photocatalyst, i.e., photocatalytic activity. Therefore, to the authors’ knowledge, all recent articles on photocatalysis discussed photocatalytic activity. The term “photocatalytic activity” is generally used to describe the ability of a photocatalyst to drive a specific photocatalytic reaction under defined conditions. In practice, however, such ability can only be evaluated in a relative manner by measuring the rate of photocatalytic reaction. Even when photocatalytic activity is expressed in terms of quantum efficiency, the estimation must be made based on the reaction rate [1].
From the viewpoint of physical chemistry, the observed reaction rate is influenced by the kinetic parameters, e.g., photon flux and the amount of adsorbed substrates, as long as thermodynamic parameters, e.g., conduction-band bottom/valence-band top energies of the photocatalyst and the standard electrode potential of a substrate, are satisfied. In other words, when the observed reaction rate is negligible or rather small, both kinetic and thermodynamic parameters need to be carefully examined. Consequently, in most cases, detailed kinetic analyses are imperative to fundamental understanding, although not all the studies on photocatalysis reported detailed kinetic analyses [2]. Late Professor David Ollis was a pioneer in the development of kinetic analysis on photocatalytic reactions, particularly for the practical applications such as environmental purification and decomposition of organic pollutants, from an engineering perspective [3,4,5].
Titanium(IV) oxide (titania) is currently the most widely used photocatalyst owing to its low cost, non-toxicity, and ease of availability [1]. However, the wide bandgap of ca. 3.2 eV of titania requires ultraviolet excitation, resulting in poor utilization of broad-spectrum light sources such as sunlight and indoor lighting. Alternatively, tungsten(VI) oxide (WO3) has a narrower bandgap of ca. 2.5 eV, enabling absorption up to ca, 470 nm, and has thus attracted attention as a visible light-responsive photocatalyst [6]. Nevertheless, pristine WO3 exhibits negligible activity for the degradation of organic compounds in the presence of oxygen, i.e., under aerobic conditions. This inactivity is attributed to the relatively low conduction band-bottom energy of WO3 (+0.50 V vs. SHE), which is a more cathodic potential than the one-electron reduction potential of oxygen, resulting in insufficient driving force for the reduction reaction of oxygen. In contrast, it has been reported that platinum-loaded WO3 (Pt/WO3) exhibits significantly enhanced photocatalytic activity for decomposition of organic compounds under visible-light irradiation [6]. This enhancement is assigned to multielectron oxygen reduction reactions, indicating that the photocatalytic activity is governed by the kinetics of the multielectron transfer process. However, the detailed role and quantitative contribution of multielectron transfer processes to govern the photocatalytic activity of Pt/WO3 remain unclear. On the other hand, for the reaction mechanism of oxygen reduction reactions, titania has been extensively studied as a model photocatalyst [7,8]. Photocatalytic oxygen reduction by pristine titania often proceeds via one-electron pathways, leading to the formation of superoxide (O2•−) and subsequent peroxide species [9,10]. For noble-metal loaded titania, for example, Pt nanoparticles serve as effective electron-accepting sites, promoting charge separation and facilitating oxygen reduction through one-electron or multistep pathways, including the formation of O2•− and peroxide species [11,12]. Those studies have primarily focused on activity enhancement and proposed reaction pathways based on intermediate detection or qualitative arguments. In contrast, we have demonstrated that light-intensity dependence (LID) analysis can serve as a powerful kinetic methodology to elucidate multielectron reaction mechanisms and cocatalyst functions, even in the absence of direct in situ detection of reaction intermediates [13,14,15]. In this study, we investigate the involvement of multielectron oxygen reduction and positive-hole oxidation and clarify the role of Pt as a cocatalyst by analyzing the LID of the reaction rate on Pt/WO3 photocatalysts.
2. Materials and Methods
2.1. Materials
Commercial WO3 powder was purchased from Kojundo Chemical Laboratory (Saitama, Japan) and fractionated according to a previously reported method [6], since the as-received WO3 contained a broad distribution of particle sizes. Briefly, a 10-g portion of WO3 powder was dispersed in 100 mL of ultrapure water using an ultrasonic homogenizer for 10 min. The resulting suspension was centrifuged at 1000 rpm for 10 min, and ca. 80% of the supernatant containing smaller particles was carefully collected. Ultrapure water was added to the remaining sediment containing larger particles to restore the total volume to 100 mL, followed by ultrasonic dispersion and centrifugation under the same conditions. This procedure was repeated five times to collect approximately 400 mL of combined supernatant enriched with small particles. The collected supernatant was subsequently centrifuged at 10,000 rpm for 15 min and dried at 393 K for 12 h. The obtained sample consisted of δ-phase WO3 with a specific surface area of ca. 9.7 m2·g−1.
2.2. Preparation of Pt/WO3 Photocatalysts and Determination of Pt Surface Area by CO Pulse Adsorption
Samples of Pt/WO3 were prepared via a photodeposition method. The fractionated WO3 (0.6 g) was dispersed in 25 mL of ultrapure water using an ultrasonic homogenizer for 10 min. An aqueous solution of chloroplatinic acid (H2PtCl6, Wako Pure Chemical, Osaka, Japan) was then added, followed by further ultrasonic dispersion for 10 min. A 300-W xenon lamp (Cermax LX-300F, Excelitas, Pittsburgh, PA, USA) equipped with a cutoff filter (L-42, Asahi Spectra, Tokyo, Japan) was used for light irradiation, excluding UV light below 400 nm to ensure visible-light irradiation only. After irradiation for 2 h, 2.9 mL of methanol (Wako Pure Chemical) was added as a sacrificial reagent, and the irradiation was continued for an additional 4 h. The resulting suspension was then centrifuged at 4500 rpm for 15 min, the supernatant was removed, and the obtained precipitate was dried at 343 K for 12 h.
The Pt surface area was measured by carbon monoxide (CO)-pulse adsorption using a catalyst analyzer (BEL-CAT, MicrotracBEL Corp., Osaka, Japan). Prior to the measurement, the samples were heated to 343 K at 7 K·min−1 under a helium atmosphere to remove adsorbed water and other volatile impurities, and the temperature was maintained at 343 K for 30 min. CO pulses were then introduced at 323 K under a continuous helium flow. Adsorption equilibrium was considered reached when the variation in the detected CO amount across three consecutive pulses was less than 2%.
2.3. Light-Intensity Dependence Analysis of Photocatalytic Activity
2.3.1. Measurement of Photon-Density Distribution and Calibration of Light Intensity and Reaction Rate
The spatial distribution of the LED’s photon density varied with light intensity. Therefore, the light intensity inhomogeneity was corrected by calculating the effective irradiation area (A95 cm2) [13], defined as the area covering 95% of the total photon density. The calibrated light intensity, IL (mW·cm−2), was then calculated as
where Pa (mW) is the total light power measured using a power meter (Hioki 3664, Hioki E.E., Tokyo, Japan) equipped with a 9742 optical sensor. Similarly, the reaction rate (r) was calibrated based on the CO2 evolution rate (Ra) obtained from photocatalytic activity tests as
2.3.2. Photocatalytic Oxidation of Acetic Acid with High-Intensity LED Irradiation
To investigate the multielectron oxygen reduction mechanisms, as well as positive-hole oxidation, over WO3 and Pt/WO3, the LID of the acetic acid-oxidation reaction, CH3COOH + 2O2 → 2CO2 + 2H2O, was evaluated. A photocatalyst powder (30 mg), a magnetic stirrer bar, and 3 mL of 5vol% aqueous acetic acid (Wako Pure Chemical; 99.7%) solution were placed in a quartz reaction cell. The cell was sealed with a rubber septum and Parafilm, and irradiated through a 10 mm × 10 mm square mask using a high-intensity UV-LED (NS Lighting, Osaka, Japan, ULEDN-101; 365 nm). The evolution of CO2 was monitored every 30 min using a gas chromatograph equipped with a thermal conductivity detector (Shimadzu GC-8A equipped with a Porapak-Q column, Shimadzu Corporation, Kyoto, Japan), and the reaction rates (Ra μmol·h−1) were calculated accordingly.
3. Results and Discussion
Figure 1 shows the LID of the reaction rates plotted on a double logarithmic scale. In the case of bare WO3, the reaction order shifted from first order to 0.5th order with increasing light intensity in the intermediate IL region. This behavior suggests a two-electron-transfer process, consistent with a previous report [16], and can be interpreted using a kinetic model based on a radical-chain mechanism involving peroxy radicals as chain carriers.

Figure 1. Double-logarithmic plots of reaction rate (r) of photocatalytic CO2 evolution (blue: bare, red: Pt loaded) from aqueous acetic acid under air (closed circles) or oxygen (open circles) as a function of light intensity (IL).
In contrast, the Pt/WO3 photocatalyst exhibited enhanced reaction rates across the entire IL region, with the reaction order remaining approximately 0.5th except in the intermediate IL region. These fundamental features will be discussed in detail later.
A characteristic feature of the Pt/WO3 case is that the reaction order changed from 0.5th order to zero in the intermediate IL region and increased again in higher IL region. When the reaction was conducted under an oxygen atmosphere, the zero-order region disappeared. Therefore, this zero-order behavior is attributed to the diffusion-limited supply of oxygen as one of the reactants.
To further investigate this hypothesis, a series of Pt/WO3 photocatalysts with different Pt loading amounts was prepared, and their Pt surface areas were estimated by CO adsorption, as shown in Figure 2. The Pt surface area per unit mass of photocatalyst (30 mg) increased with increasing Pt loading amount. Moreover, a good correlation was observed between the Pt surface area and the reaction rate in the zero-order region at lower Pt loadings (0.5–2wt%) (Figure 2 left). These results indicate that the primary active sites for photoexcited-electron transfer in the Pt/WO3 photocatalyst are located on the Pt surface, and that increasing the available Pt surface area enhances the amount of adsorbed oxygen, thereby leading to an increased reaction rate.
In contrast, a significant decrease in the reaction rate was observed for the 4wt% Pt/WO3 sample. The average number of Pt particles per one WO3 particle, estimated from the respective surface area of Pt and WO3, is shown in Figure 2 (table). It was found that more than two Pt particles were loaded on a single WO3 particle on average in the 4wt% Pt/WO3 sample. This suggests that photoexcited electrons generated within a single WO3 particle may migrate to different Pt particles, thereby suppressing the multielectron oxygen reduction reaction and consequently decreasing the overall reaction rate. The relatively low activity of the 1wt% Pt/WO3 sample in Figure 2 left can also be interpreted by a slightly higher number of Pt particles per one WO3 particle (1.5). Similar behavior has been reported for the cocatalyst-loaded titania in oxygen evolution from water [13].
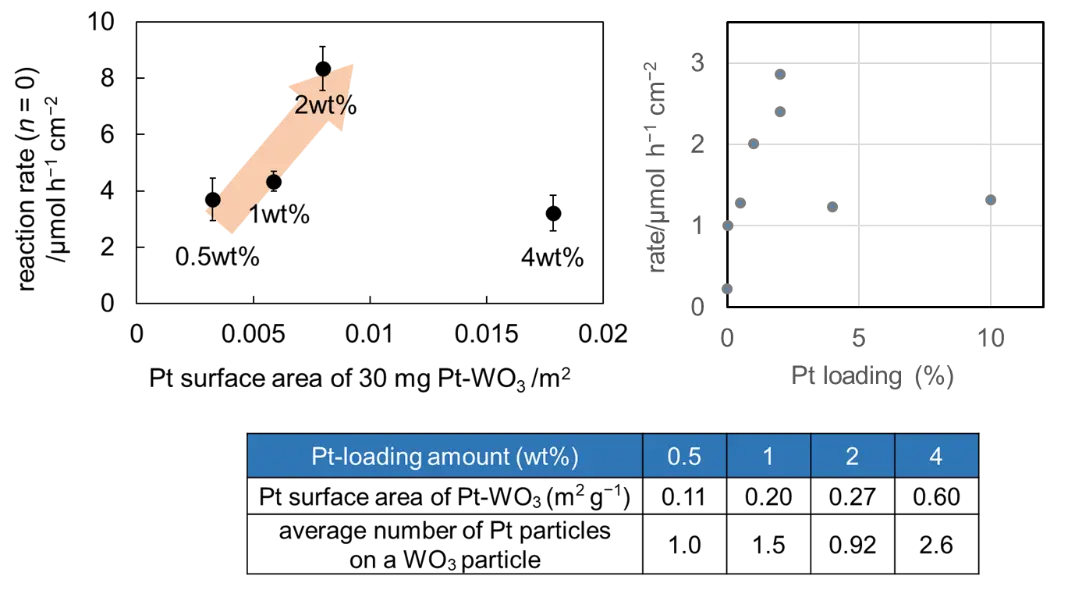
Figure 2. (Left) Photocatalytic reaction rate (r) as a function of Pt surface area, (bottom) average number of Pt particles per one WO3 particle, (right) reaction rate at 2.72 mW·cm−2 (0.5th-order region for all the samples) as a function of Pt-loading amount.
The reaction order and the effect of Pt loading on the quantum efficiency were evaluated based on a kinetic analysis described as follows. Following a previous study on acetic-acid oxidation over titania [16] and studies on radical-chain oxidation mechanisms involving peroxy radicals [3,4,5], a kinetic model was constructed under the following assumptions: (1) a radical chain reaction occurs, in which peroxy radicals act as chain carriers, and (2) only a two-electron oxygen reduction pathway proceeds over WO3 because of its low conduction band potential [6]. The reaction steps and the corresponding reaction rates are summarized as follows:
where IL represents the calibrated light intensity, ψ0 and ψ1 are the photoabsorption efficiencies of a photocatalyst particle <PC> and <PC>(1e−), respectively, φ is the quantum efficiency of electron–hole pair generation, and the k’s denote the corresponding rate constants. The working hypothesis of this mechanism is that: (i) <PC>(1e−) is deactivated via reaction (5) without driving a photocatalytic reaction unless further accumulation of electrons and holes occurs through reaction (4) and (ii) once <PC>(2e−) is formed via photoabsorption of <PC>(1e−), the two electrons are immediately transferred to oxygen, while oxidation of the organic substrate (RH) proceeds simultaneously without deactivation. It should be emphasized that Equation (6), Equation (7), Equation (8) and Equation (9) describe radical-chain oxidation reactions rather than reduction reactions. In the present system, the reductive process is limited to multielectron oxygen reduction, whereas the oxidation of organic substrates proceeds via radical intermediates generated in subsequent chain reactions. The termination step, as expressed by Equation (9), is introduced as a formal kinetic description of radical recombination processes, as conventionally assumed for autooxidation reactions. It does not imply the accumulation of any stable tetroxide species (RO4R) on the photocatalyst surface, but decomposes spontaneously. As a result, the overall reaction rate is governed by the kinetics of multielectron oxygen reduction and positive-hole oxidation, which control the initiation and sustainability of the subsequent radical-chain oxidation of organic substrates. For simplicity, it is assumed that no back reaction from <PC>(2e−) to <PC>(1e−) occurs. By applying the steady-state approximation to the concentrations of R•, RO2•, <PC>(1e−), and <PC>(2e−), the overall rate r was derived as follows (see SI for details of the derivation):
The first term represents the direct oxidation of acetic acid by photogenerated holes, while the second term corresponds to the radical-chain reaction consuming acetic acid. The former can be neglected by assuming a sufficiently high chain length. Moreover, the rate equation can be simplified in the lower and higher light-intensity limits as follows:
These rate equations are consistent with the experimentally observed change in reaction order for WO3; first order at lower IL and 0.5th order at higher IL regions. The threshold intensity, IL,thr, between these two regions can be described as follows:
On the assumption that photoabsorption coefficient (ψ1: presumably depends only on the photocatalyst particle size [9]) and intrinsic quantum efficiency (φ) of electron-hole generation remain constant irrespective of the Pt loading amount, the disappearance of the first-order region for Pt/WO3 can be attributed to a decrease in the deactivation rate constant (kd) due to the presence of Pt. The loaded Pt may serve as an electron reservoir, prolonging the lifetime of <PC>(1e−). Based on the estimated IL,thr values of 2.8 and <0.3 mW·cm−2 for WO3 and Pt/WO3, respectively, the kd was reduced to approximately one-tenth upon Pt loading.
Figure 3 illustrates the proposed reaction mechanism. Under lower IL conditions, accumulation of four electrons within a single WO3 particle is difficult; therefore, the reaction proceeds via a two-electron transfer process coupled with a radical-chain mechanism. The reaction exhibits first-order dependence, which presumably results from the combined effects of a second-order dependence for two-electron accumulation and a 0.5th-order dependence from the radical-chain process (Figure 3a). As the IL increases and two-photon absorption within a single WO3 particle becomes more probable, the reaction efficiency reaches saturation, resulting in a transition from first order to 0.5th order behavior (Figure 3a).
In the case of Pt/WO3, the photoexcited electrons are rapidly transferred to the Pt particles, thereby suppressing electron–hole recombination. As a result, two-electron accumulation is consistently maintained on the Pt surface, and the first-order dependence is no longer observed (Figure 3c). Furthermore, two key experimental observations, (i) the disappearance of the zero-order region under an oxygen atmosphere, and (ii) the linear correlation between the reaction rate and the Pt surface area, support the hypothesis that the two-electron oxygen reduction predominantly proceeds on the Pt surface (Figure 3d).
However, the increase in reaction order observed under extremely high IL conditions cannot be explained solely by the two-electron reduction pathway. One possible interpretation is that a four-electron transfer process becomes operative. Moreover, under these high-intensity conditions, the oxygen supply to the Pt surface appears to be diffusion limited. Therefore, the observed increase in reaction order suggests that the oxygen reduction also occurs on the WO3 surface in addition to the Pt surface (Figure 3e). In other words, the behavior observed under extremely high IL regions cannot be explained solely by reactions on the Pt surface, as overcoming the diffusion limitation of oxygen requires additional reduction sites. Consequently, a four-electron oxygen reduction pathway is likely to proceed on both bare WO3 and Pt/WO3 photocatalysts under such conditions.
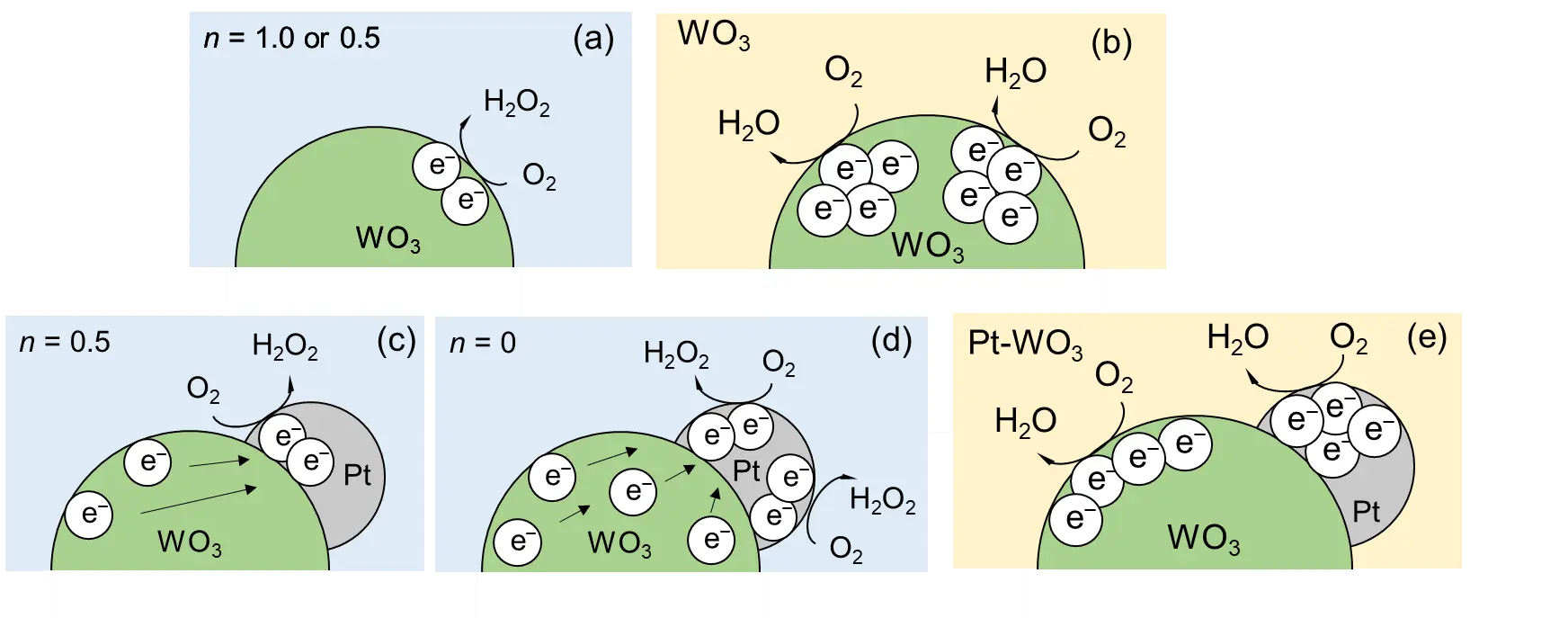
Figure 3. Schematic illustration of photocatalytic reaction mechanisms for (a,b) bare WO3 and (c–e) Pt-loaded WO3. Light blue and yellow background colors indicate relatively low IL and high IL regions, respectively.
4. Conclusions
In this study, light-intensity dependences (LIDs) of the reaction rates for photocatalytic oxygen reduction, as well as positive-hole oxidation, were systematically investigated to elucidate the multielectron transfer mechanism and the role of a cocatalyst, based on a kinetic model, using Pt/WO3 under high-intensity LED irradiation. Bare WO3 exhibited first, 0.5th, and first-order behaviors under low (<2 mW), intermediate (2–100 mW), and high (>100 mW) IL conditions, respectively, whereas Pt/WO3 showed 0.5th, zero, and higher-than-first-order behaviors under the corresponding low (<20 mW), intermediate (20–100 mW), and high (>100 mW) regions.
From a thermodynamic perspective, WO3 facilitates a two-electron reduction reaction of oxygen because its conduction band edge lies at a more cathodic potential than the standard electrode potential of the one-electron oxygen reduction. The observed LID behavior in the lower IL region can be reasonably interpreted using a kinetic model based on a radical-chain mechanism initiated by peroxy radicals, under the assumption that oxygen reduction proceeds only when two electrons are accumulated on a single photocatalyst particle.
In the case of Pt/WO3, the absence of first-order behavior indicates that the accumulation of two electrons is consistently maintained due to suppressed electron-hole recombination (i.e., a reduced kd) brought about by Pt loading. Furthermore, the reaction rate in the zero-order region increased proportionally with Pt surface area (up to 2wt%), suggesting that the reaction predominantly occurs on the Pt surface. The zero-order dependence is attributed to the oxygen diffusion limitation, as evidenced by the enhanced reaction rate under an oxygen-rich atmosphere.
Finally, the pronounced increase in reaction rate observed under extremely high IL region implies that the oxygen reduction also proceeds on the WO3 surface. This finding indicates that the overall reaction rate can be further improved by spatially concentrating the incident light without changing the total energy of light beam.
Supplementary Materials
The following supporting information can be found at: https://www.sciepublish.com/article/pii/898, Details on derivation of rate equations were described in Supplementary Materials.
Acknowledgment
As described in the Special Issue Information for A Commemorative Issue in Honor of Professor David Ollis: A Pioneer and Mentor, David had significantly advanced photocatalysis research by publishing papers, particularly in the field of kinetic analysis. The authors’ works, including the present paper, on the kinetic analysis of photocatalysis by clarifying the light-intensity dependence would not have been possible without the fruitful discussion with David at the scientific conferences. Here, we would like to express our deepest gratitude to the late David Ollis.
Author Contributions
Conceptualization, B.O. and M.T.; Operation of photocatalytic reaction and data acquisition, C.Y.; Writing—Original Draft Preparation, C.Y. and M.T.; Writing—Review and Editing, all authors.
Ethics Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Raw data were generated at Institute for Catalysis, Hokkaido University (North 21, West 10, Sapporo 001-0021, Japan). Derived data supporting the findings of this study are available from the corresponding author M.T. on request.
Funding
This research received no external funding.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Ohtani B. Photocatalysis A to Z—What we know and what we do not know in a scientific sense. J. Photochem. Photobiol. C 2010, 11, 157–178. DOI:10.1016/j.jphotochemrev.2011.02.001 [Google Scholar]
- Fleischauer PD, Alen Kan HK, Schepherd JR. Quantum yields of silver ion reduction on titanium dioxide and zinc oxide single crystals. J. Am. Chem. Soc. 1972, 94, 283–285. DOI:10.1021/ja00756a055 [Google Scholar]
- Pruden AL, Ollis DF. Photoassisted heterogeneous catalysis: The degradation of trichloroethylene in water. J. Catal. 1983, 82, 404–417. DOI:10.1016/0021-9517(83)90207-5 [Google Scholar]
- Ollis DF, Pelizzetti E, Serpone N. Photocatalyzed destruction of water contaminants. Environ. Sci. Technol. 1991, 25, 1522–1529. DOI:10.1021/es00021a001 [Google Scholar]
- Ollis DF. Kinetics of liquid phase photocatalyzed reactions: An illuminating approach. J. Phys. Chem. B 2005, 109, 2439–2444. DOI:10.1021/jp040236f [Google Scholar]
- Abe R, Takami H, Murakami N, Ohtani B. Pristine simple oxides as visible light driven photocatalysts: Highly efficient decomposition of organic compounds over platinum-loaded tungsten oxide. J. Am. Chem. Soc. 2008, 130, 7780–7781. DOI:10.1021/ja800835q [Google Scholar]
- Chen H, Nanayakkara CE, Grassian VH. Titanium dioxide photocatalysis in atmospheric chemistry. Chem. Rev. 2012, 112, 5919–5948. DOI:10.1021/cr3002092 [Google Scholar]
- Henderson MA, Lyubinetsky I. Molecular-level insights into photocatalysis from scanning probe microscopy studies on TiO2(110). Chem. Rev. 2013, 113, 4428–4455. DOI:10.1021/cr300315m [Google Scholar]
- Burek OB, Bahnemann WD, Bloh ZJ. Modeling and Optimization of the Photocatalytic Reduction of Molecular Oxygen to Hydrogen Peroxide over Titanium Dioxide. ACS Catal. 2019, 9, 25–37. DOI:10.1021/acscatal.8b03638 [Google Scholar]
- Zuo G, Li B, Guo Z, Wang L, Yang F, Hou W, et al. Efficient photocatalytic hydrogen peroxide production over TiO2 passivated by SnO2. Catalysts 2019, 9, 623. DOI:10.3390/catal9070623 [Google Scholar]
- Kobayashi H, Teranishi M, Negishi R, Naya S, Tada H. Reaction mechanism of the multiple-electron oxygen reduction reaction on the surfaces of gold and platinum nanoparticles loaded on titanium (IV) oxide. J. Phys. Chem. Lett. 2016, 7, 5002–5007. DOI:10.1021/acs.jpclett.6b02026 [Google Scholar]
- Zhang Z, Wang X, Long J, Ding Z, Fu X, Fu X. H2–O2 promoting effect on photocatalytic degradation of organic pollutants in an aqueous solution without an external H2 supply. Appl. Catal. A Gen. 2010, 380, 178–184. DOI:10.1016/j.apcata.2010.03.057 [Google Scholar]
- Takeuchi S, Takashima M, Takase M, Ohtani B. Digitally controlled kinetics of titania-photocatalyzed oxygen evolution. Chem. Lett. 2018, 47, 373–376. DOI:10.1246/cl.171093 [Google Scholar]
- Ketwong P, Yoshihara S, Takeuchi S, Takashima M, Ohtani B. Light intensity-dependence studies on the role of surface deposits for titania-photocatalyzed oxygen evolution: Are they really cocatalysts? J. Chem. Phys. 2020, 153, 124709. DOI:10.1063/5.0014913 [Google Scholar]
- Hori H, Takashima M, Takase M, Ohtani B. Kinetic analysis supporting multielectron reduction of oxygen in bismuth tungstate-photocatalyzed oxidation of organic compounds. Catal. Today 2018, 313, 218–223. DOI:10.1016/j.cattod.2018.01.001 [Google Scholar]
- Torimoto T, Aburakawa Y, Kawahara Y, Ikeda S, Ohtani B. Light intensity dependence of the action spectra of photocatalytic reactions with anatase titanium(IV) oxide. Chem. Phys. Lett. 2004, 392, 220–224. DOI:10.1016/j.cplett.2004.05.077 [Google Scholar]