Generative Artificial Intelligence for Function-Driven De Novo Enzyme Design

Generative Artificial Intelligence for Function-Driven De Novo Enzyme Design
Xuan Qi 1,2,3 Dehang Wang 1,2 Zhenkun Shi 1,2 Xiaoping Liao 1,2,* Hongwu Ma 1,2,*
Received: 28 August 2025 Revised: 10 September 2025 Accepted: 26 September 2025 Published: 29 September 2025

© 2025 The authors. This is an open access article under the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/).

Graphical Abstract
1. Introduction
Enzymes are the catalytic engines of biological systems, enabling precise and efficient transformations under mild conditions. Their vast potential in biomanufacturing, medicine, and environmental applications has driven increasing efforts to discover, engineer, and even design new enzymes beyond the natural repertoire [1,2,3,4]. Traditional approaches to enzyme engineering, such as directed evolution and rational design, have achieved remarkable success, especially in improving stability and catalytic ability based on the starting enzyme [5,6,7,8]. Rational enzyme design relies on structural knowledge to make targeted mutations for improved function, but its success is limited by the complexity of protein engineering and the risk of local fitness optimum. Directed evolution mimics natural selection by generating diverse mutant libraries and screening for improved variants. However, it is labor-intensive, costly, and often constrained by the difficulty of identifying rare high-performing mutants [9,10].
De Novo Enzyme Design aims to design artificial enzymes from scratch to catalyze chemical reactions that either do not exist in nature or are inefficiently catalyzed by natural enzymes [11,12]. Early efforts largely reused natural protein scaffolds—using tools such as RosettaMatch [13] to place theozyme-derived catalytic motifs (built from transition-state models) into unrelated backbones, followed by local sequence redesign. Owing to limitations in scoring/energy functions, incomplete active-site preorganization, and neglected conformational dynamics, the resulting catalysts typically displayed activities orders of magnitude below natural enzymes, precluding industrial use [14,15]. Subsequently, domain- and chimera-based strategies emerged, recombining fragments from related enzyme families and integrating evolutionary constraints with structural-compatibility filters [16,17]. By preserving the active center, these approaches better maintain proper folding; however, they remain inherently constrained by the structures and functions of existing protein backbones.
The advent of Generative Artificial Intelligence (GAI) no longer relies solely on pre-existing structural templates. Instead, it enables the generation of entirely novel architectures from first principles to meet predefined catalytic objectives. This shift expands the accessible design space beyond the limits of natural evolution, allowing the rational creation of artificial enzymes with bespoke catalytic functions that can overcome inherent limitations of natural enzymes [18,19,20]. Early demonstrations include the design of artificial luciferases with improved stability and broadened substrate tolerance, highlighting the feasibility of this approach [21]. Building on this foundation, a particularly notable advance was achieved by the David Baker laboratory, which recently applied GAI to design a fully de novo serine hydrolase with catalytic efficiencies (kcat/Km) up to 2.2 × 105 M−1·s−1 and folds distinct from natural hydrolases. Importantly, the artificial enzyme backbone is unprecedented in nature, highlighting the capacity of GAI to explore structural space inaccessible to evolutionary processes. This paradigm shift is driven by a new generation of AI-powered frameworks, including advanced backbone-generation and inverse-folding models (e.g., RFdiffusion [22], SCUBA-D [23], ProteinMPNN [24], and LigandMPNN [25]), that enable the de novo construction of protein scaffolds with tailored topological features, as well as mechanism-informed approaches that incorporate catalytic mechanisms directly into the design process.
Recent research in the field has exemplified a typical de novo enzyme design workflow. It begins with defining the catalytic requirements of the target reaction, followed by the identification of the active sites that establish the essential catalytic geometry. These active sites then serve as constraints for generating compatible protein backbones using generative models, followed by sequence design through inverse-folding frameworks to ensure structural integrity and chemical preorganization of the active site. The resulting candidates undergo iterative refinement through computational evaluation—including structural prediction and active-site geometry scoring—to enrich functional designs before experimental testing. Recent advances in GAI and structure-based modeling have enabled a more systematic and predictive design cycle, thereby supporting the realization of artificial enzymes with tailored catalytic functions. In the following sections, this review will systematically explore these computational methods and their applications across the enzyme design pipeline. It will provide a detailed discussion of the typical workflow for de novo enzyme design.
2. Identification of Active Sites
Although naturally occurring enzymes are extraordinarily efficient biocatalysts capable of mediating highly selective chemical transformations under mild conditions, natural enzymes still exhibit limitations in practical applications. Many important biosynthetic reactions lack corresponding natural enzymes, and the intrinsic properties of natural enzymes—such as stability and substrate specificity—often fall short of the stringent requirements of industrial applications. This challenge has driven an urgent demand for de novo design of novel enzymes, aiming to create entirely new catalytic proteins through rational and computational approaches in the absence of natural evolutionary templates. A central difficulty in this field lies in the fact that the catalytic efficiency of natural enzymes depends on the precise atomic-scale arrangement of residues within the active site relative to the substrate; for reactions that have not evolved in nature, no pre-existing structural templates are available for guidance. To address this challenge, two foundational design strategies have emerged in enzyme engineering: “data-driven design” centered on the Identification of consensus structures, and “rational design” exemplified by the Theozyme model. This section provides a detailed discussion of both strategies.
2.1. Consensus Structure Identification: A Data-Driven Approach
This data-driven approach extracts conserved geometrical features from families of natural enzymes using large structural databases such as the Protein Data Bank. This method aims to uncover highly conserved spatial relationships and hydrogen-bonding networks associated with catalytic function within a protein family [26,27]. The core concept is the identification of a “consensus shape”—a pseudo-protein that distills and summarizes the essential structural information of the protein family (Figure 1A). Through sequence and structural alignment [27,28], this approach can reveal conserved distances, angles, and dihedral distributions between catalytic residues.
A canonical example of consensus structure recognition is the catalytic triad (Ser-His-Asp) of serine hydrolases [26,29]. Despite their evolutionary divergence, many distinct serine protease families, including trypsin and subtilisin, have independently evolved this identical catalytic mechanism. Studies have shown that, beyond the triad itself, the adjacent oxyanion hole—a microenvironment formed by backbone amide hydrogen atoms—plays a critical role in stabilizing the tetrahedral intermediate of the reaction. Statistical analysis of these characteristic distances and angles provides reliable guidance for designing active sites for similar reactions.
The primary advantage of consensus structure identification lies in its low computational cost and ability to directly leverage evolutionary solutions honed over millions of years. It is particularly suitable for designing reactions for which natural templates exist or for catalytic chemistries similar to those found in nature. However, its main limitations are restricted transferability and coverage. The method cannot be applied to reactions that occupy “entirely novel chemical space,” where no natural template is available. Moreover, as a statistical abstraction [30], it does not explain why a particular geometry is optimal, nor does it provide insight at the level of first-principles theory [29].
In addition to structural alignment, recent advances in sequence-based models provide complementary means of identifying consensus features [31,32,33]. For example, protein language models such as ESM2 [34,35,36] and evolutionary approaches such as Evmutation [37] can highlight conserved residues or predict mutational tolerance through saturation mutagenesis scoring, thereby offering insight into positions critical for catalytic activity. Importantly, while sequence conservation and statistical scores can suggest putative active sites, because they reflect general evolutionary constraints rather than explicit catalytic geometries, their functional relevance often requires further validation—either through experimental mutagenesis or by integrating orthogonal computational strategies (e.g., molecular dynamics, energetic analysis). This ensures that residues inferred from sequence data are not solely artifacts of alignment but are mechanistically relevant to catalysis.
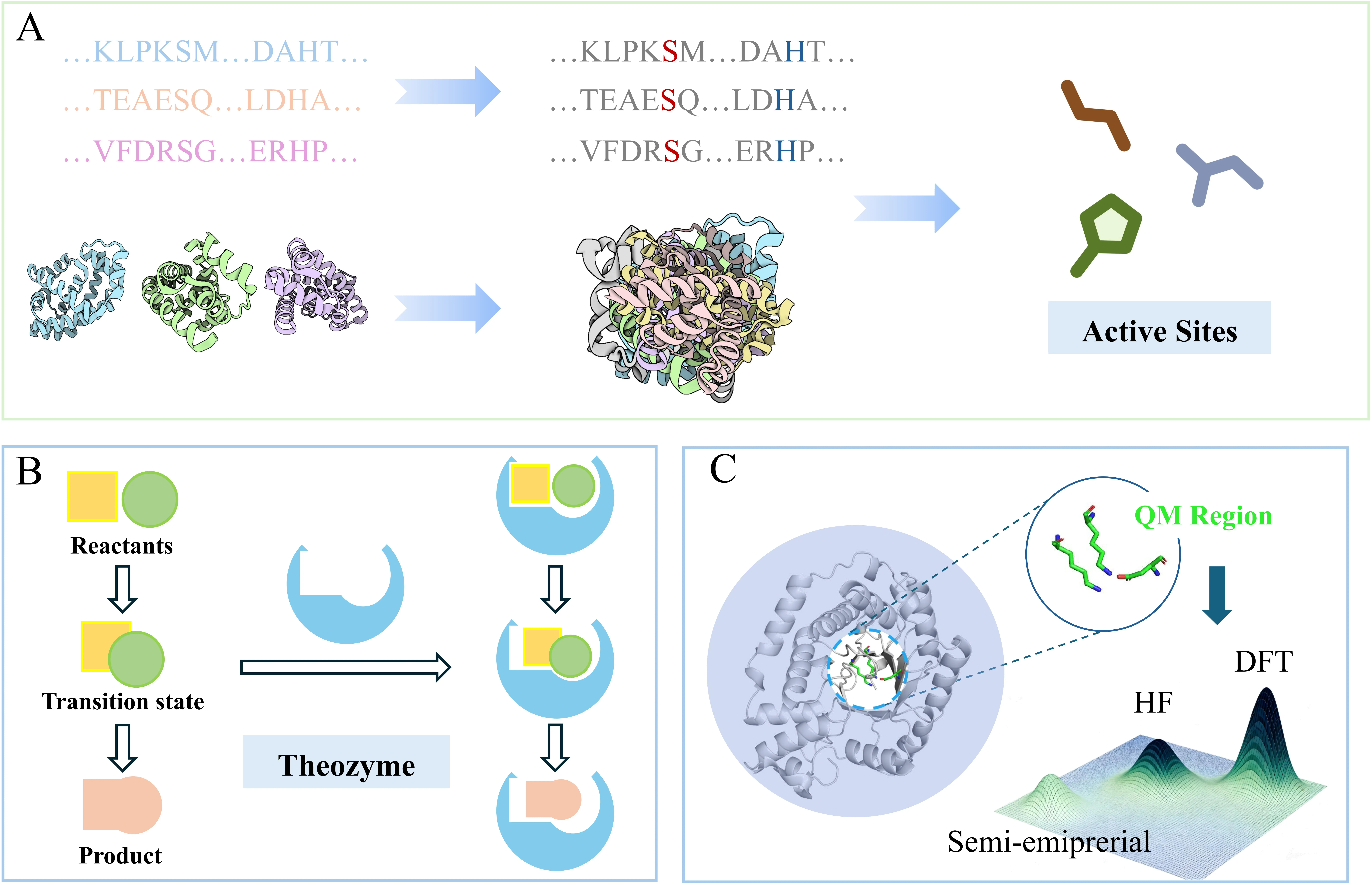
Figure 1. Computational strategies for catalytic center design in de novo enzymes. (A) Conserved residues are identified by sequence alignment (upper) or structural superposition (lower), providing consensus positions that define putative catalytic sites. (B)Theoretical enzyme model (theozyme). A minimal active-site representation is built by placing key catalytic residues around a transition-state analogue, thereby encoding the geometric and electrostatic requirements necessary to stabilize the reaction pathway. (C) QM-based theozyme construction. A localized active-site is optimized using quantum-chemical methods. The lower-right inset illustrates different electronic-structure approaches, including semi-empirical, Hartree–Fock, and density functional theory (DFT), with peak height representing accuracy and color shading indicating computational cost.
2.2. Theoretical Enzyme Models: Cornerstones of Rational Design
In contrast, the Theozyme (short for “theoretical enzyme”) represents an “inside-out” strategy (Figure 1B), introduced by the research group of Houk [38], in the late twentieth century. A theozyme is an idealized minimal active site model composed solely of the transition state of the target reaction together with catalytic groups capable of forming stabilizing interactions—typically simplified amino acid side chains or backbone fragments [39]. Through quantum mechanical (QM) calculations, this approach delineates the optimal spatial arrangement of catalytic groups required to maximally stabilize the transition state of a given chemical reaction.
The design philosophy of the theozyme is conceptually rooted in the transition-state theory proposed by Linus Pauling and Richard Wolfenden, which posits that efficient enzymes accelerate reactions primarily through tightly binding and stabilizing the transition state, thereby significantly lowering the activation barrier. The theozyme provides an atomically precise “blueprint” for this idealized state. Beyond insights into substrate orientation, bond dynamics, and molecular rearrangements [40,41,42,43,44,45], it enables quantitative assessment of how individual atoms or catalytic groups contribute to the overall reaction rate.
The Theozyme construction process follows a QM-based workflow (Figure 1C). Initially, the transition-state structure of the target reaction is precisely located under idealized conditions, in the absence of any external catalytic groups, using QM methods such as Hartree–Fock theory (HF), density functional theory (DFT), or semi-empirical approaches [38]. Catalytic residue models—typically side-chain fragments or backbone functional groups—are systematically positioned around this transition state. To reduce complexity, these residues are truncated and capped with hydrogens [46]. With the transition state fixed as a constraint, the geometry of the entire supramolecular system is optimized, yielding an arrangement that maximally stabilizes the transition state and minimizes the reaction barrier. This process distills key geometric parameters—distances, angles, and dihedrals—that guide subsequent enzyme design algorithms.
In practice, the hybrid functional B3LYP/6-31+G* remains one of the most widely applied methods for theozyme calculations [46]. B3LYP combines a portion of exact Hartree–Fock exchange with DFT-based exchange–correlation, balanced by three empirical parameters [47,48]. This hybridization provides a favorable compromise between accuracy and efficiency in describing organic thermochemistry and geometries, typically predicting activation energies with an approximate error of ~1 kcal·mol−1. The 6-31+G* basis set defines atomic orbitals with sufficient flexibility at a reasonable cost. While not flawless in reproducing relative transition-state energies, the B3LYP/6-31+G* level of theory generally provides geometrical precision sufficient to guide theozyme-based enzyme design.
In summary, the Theozyme model and consensus-structure identification thus serve as complementary tools for active-site design: the former provides a rigorous atomistic blueprint rooted in transition-state stabilization, while the latter reduces arbitrariness by leveraging structural motifs conserved through evolution. Integrating these strategies holds promise for overcoming current bottlenecks in de novo enzyme design.
3. GAI Is Reshaping Enzyme Backbone Design
A central objective in de novo enzyme design is to construct a backbone capable of precisely accommodating catalytically essential residues [15]. Traditional approaches, such as RosettaMatch [49], rely on identifying compatible sites within a predefined scaffold library to position the active-site geometry. While successfully generating a range of artificial enzymes, these methods are inherently constrained by the geometric limitations of the available scaffold libraries [50]. As a result, achieving a balance between global scaffold stability and the precise spatial requirement of the catalytic center often proves elusive, limiting the realization of ideal active-site geometries within physically plausible protein structures.
To overcome these limitations, recent efforts have shifted toward active-site-constrained scaffold generation. Unlike conventional template-based strategies, this approach does not rely on natural topologies or predefined backbones. Instead, it seeks to generate novel scaffolds that are intrinsically compatible with the intended catalytic geometry via sampling, recombination, or de novo construction. Early machine learning-based methods in this area were often restricted by the diversity and size of existing scaffold libraries, limiting their ability to explore novel geometries. However, advances in generative modeling have opened new avenues. In particular, diffusion-based models (such as SMCDiff [51] and RFdiffusion [22]) and flow-matching models (such as RFdiffusion2 [20]) have marked a paradigm shift in protein scaffold design.
This section outlines the principles and emerging applications of these generative models, examining their advantages and limitations in the context of enzyme design.
3.1. Diffusion Model Opens New Era for Enzyme Backbone Design
Diffusion model [52] is a class of probabilistic generative models that operate by progressively corrupting input structures with noise during a forward process, and then learning to reverse this process to recover clean samples [52,53]. The core idea is to interpolate between two distributions: a simple, tractable noise distribution, typically Gaussian, and the complex, desired data distribution. A neural network is trained to iteratively denoise samples, thereby transforming random noise into structured outputs that reflect the target distribution (Figure 2A).
A notable advancement in protein backbone generation was introduced by Trippe and colleagues in 2022, who leveraged E(3)-equivariant graph neural networks [54,55] to develop SMCDiff [51] (Sequential Monte Carlo Diffusion), a two-stage generative framework. The process begins with ProtDiff, an unconditional generative model trained to capture the distribution of protein backbone geometries. Conditional sampling is then applied to embed user-defined catalytic motifs within the generated scaffolds. This strategy enables the construction of diverse backbone architectures of up to 80 residues while preserving structural plausibility at scale. The two-step approach—unconditional modeling followed by conditional refinement-offers a practical balance between geometric diversity and functional fidelity. However, a critical limitation arises from the use of E(3)-equivariant networks, which inherently cannot distinguish between left- and right-handed helices. As a result, approximately 45% of generated scaffolds contain erroneous left-handed helices, rendering them incompatible with downstream design and ultimately non-functional.
To address these stereochemical limitations and enhance generative scalability, the Baker laboratory introduced RFdiffusion in 2023 [22,56], a pioneering framework that applies diffusion models directly to the generation of three-dimensional protein coordinates, including the backbones of enzymes such as serine hydrolases and carbonic anhydrases. (These applications are discussed in detail in the case study section.) This approach marked a breakthrough in navigating high-dimensional conformational spaces. By precisely defining both the sequence positions and spatial orientations of catalytic residues, RFdiffusion integrates RoseTTAFold [57] with an SE(3)-equivariant diffusion model and employs guided sampling to enforce strict geometric constraints. The model can generate protein structures with complex topologies exceeding 600 residues and demonstrates a 10–20% improvement in design success rate over prior methods. Furthermore, it significantly accelerates the generative process-structures with 100 residues can be produced in approximately 11 seconds. Nonetheless, the imposition of fine-grained, residue-level geometric constraints increases sampling complexity and occasionally yields physically implausible motifs, particularly under stringent design specifications. This may be because the model is difficult to efficiently explore the high-dimensional conformational space and satisfy fine local constraints simultaneously during the iterative denoising process. In the absence of external potential energy guidance, it is easy to generate geometrically reasonable but stereochemically or energetically unstable structures.
Complementing these efforts, Liu et al. [23] introduced SCUBA-D (Side Chain-Unknown Backbone Arrangement-Diffusion), a novel generative framework that operates independently of external structure prediction tools such as RoseTTAFold [57]. During diffusion, SCUBA-D conditions generation on the backbone coordinates of predefined catalytic residues and incorporates protein sequence representations as geometric constraints. SCUBA-D introduces an adversarial loss function during training to improve the model’s robustness and generalizability. This mechanism effectively reduces the risk of generating unrealistic structures when encountering out-of-distribution samples. Remarkably, SCUBA-D is highly efficient, capable of generating a 100-residue backbone in approximately 30 seconds on an RTX 3090 GPU. The framework also demonstrated excellent control over the placement of functionally critical residues, achieving an average all-atom RMSD of 0.1 Å, comparable to that of RFdiffusion.
3.2. Flow Matching Unlocks Next-Gen Enzyme Backbone Design
Advances in diffusion models have significantly accelerated the progress of de novo enzyme design, enabling the generation of highly diverse and geometrically constrained protein backbones. However, one key limitation of diffusion-based approaches is their computational inefficiency during inference. Generating high-quality samples often requires thousands of forward passes, rendering large-scale sampling computationally prohibitive, particularly for models with substantial parameter counts. To address this challenge, flow matching has emerged as an efficient alternative [58]. Flow matching [59] models are based on continuous-time ordinary differential equations (ODEs) or stochastic differential equations (SDEs) that define smooth transformations (flows) from a noise distribution to the target data distribution (Figure 2B). Compared to diffusion models, flow-based approaches offer higher sampling efficiency, enable continuous trajectory generation, and provide greater flexibility for incorporating conditional guidance, thereby improving control over structural and functional attributes.
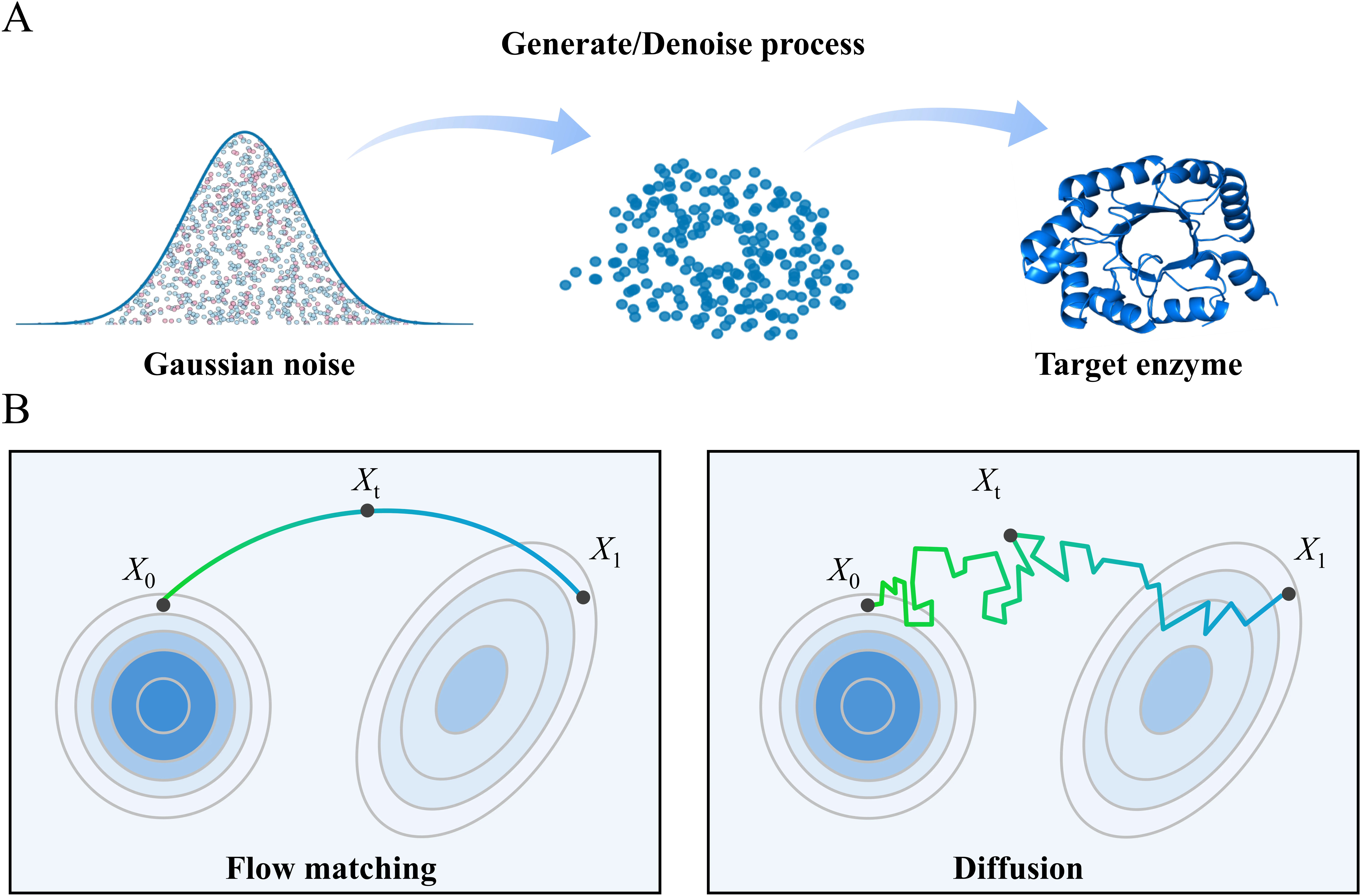
Figure 2. Application of GAI in enzyme backbone design. (A) Protein diffusion models are trained to recover protein structures corrupted by noise, progressively denoising random Gaussian inputs to generate coarse geometric point-cloud representations that are subsequently refined into complete enzyme backbones with well-defined secondary structures. (B) Two continuous-time processes (Xt, 0 ≤ t ≤ 1) transforming a source sample X0 to a target sample X1. (Left) A flow matching process, where the sample evolves deterministically along a smooth trajectory in continuous state space via an ODE-defined velocity field. (Right) A diffusion process, where the sample undergoes stochastic evolution with noisy intermediate states, converging to the target distribution through an SDE-driven reverse denoising process.
Expanding beyond diffusion-based sampling, FrameFlow [60] was introduced in 2023 as a protein backbone generation framework based on SE(3) [61] flow matching, representing a substantial improvement over the previous diffusion-based model FrameDiff [62]. As the first model to apply flow matching in SE(3) space, FrameFlow learns continuous vector fields that efficiently transform Gaussian noise into structured protein conformations. Its architecture incorporates the Invariant Point Attention (IPA) mechanism originally developed in AlphaFold and employs SE(3)-equivariant graph neural networks to capture geometric relationships and structural constraints between residues. Compared to the diffusion-based sampling of FrameDiff, FrameFlow adopts an ODE-based generation strategy that reduces the number of sampling steps by approximately fivefold and doubles the design efficiency. These features make it particularly well-suited for constructing large and diverse backbone libraries, capable of generating a 100-residue backbone in just 5.7 s (on an NVIDIA A100 GPU). Despite its promising computational performance, however, FrameFlow has not yet undergone in vitro experimental validation. The foldability and functional viability of its generated structures remain to be systematically assessed.
Pushing the field further toward function-directed design, the Baker laboratory introduced RFdiffusion2 in 2025 [19,20], a next-generation deep learning framework that enables direct protein backbone generation based solely on the spatial positions of functional groups—without requiring predefined sequences or exhaustive rotamer sampling. By incorporating a flow matching strategy, RFdiffusion2 learns the distribution of protein structures that support designated catalytic geometries, thereby avoiding the combinatorial explosion that limits traditional design workflows. Experimental validation involved three distinct catalytic motifs—an aldolase, a cysteine protease, and a zinc hydrolase. Fewer than 96 designed sequences were tested for each case, yet active catalysts were successfully identified for all three functional sites. In benchmarking studies, RFdiffusion2 generated 41 distinct catalytic site configurations, significantly outperforming the original RFdiffusion model, which succeeded in only 16 cases. These results highlight the model’s superior design capacity, geometric generalization, and practical utility in scaffold construction for functionally critical active sites.
Recent advances (Table 1) in structure-driven protein design highlight a transition from residue-level modeling to atomic-level precision, shifting the focus from structure generation to function-oriented molecular engineering. Innovations in generative strategies-particularly the adoption of flow matching and ODE-based sampling-have substantially accelerated backbone construction. Despite these advances, most current approaches still rely on post hoc side-chain packing and sequence optimization, which can limit overall design fidelity and throughput. The development of end-to-end, all-atom generative models capable of co-designing both sequence and structure holds immense potential for increasing design accuracy, efficiency, and scalability, ultimately paving the way for fully integrated pipelines in enzyme design.
Table 1. Summary of recent studies in backbone design.
|
Category |
Model |
Release Date |
Key Innovation |
Constraint and Performance |
|---|---|---|---|---|
|
Diffusion Model |
ProtDiff/SMCDiff [51] |
2022 |
F irst diffusion model for backbone design |
Length < 80 residues; cannot distinguish left- and right-handed helices . |
|
FrameDiff [62] |
2023 |
SE(3)-equivariant local frame diffusion for backbone design |
Length < 500 residues 100-residue backbone in 4.4 s (NVIDIA A100) |
|
|
RFdiffusion [22] |
2023 |
Designs backbone based on provided geometric data of possible active site configurations |
Length < 6 00 residues 100- residues in 11 s (NVIDIA A4000) |
|
|
SCUBA-D [23] |
2024 |
A backbone design model trained independently to overcome the limitations of pretrained models |
100- residues in 30 s ( RTX 3090 ) |
|
|
Flow Matching |
FrameFlow [60] |
2023 |
Uses flow matching instead of a diffusion model to directly learn the structural transformation trajectory |
100- residues in 5.7 s (NVIDIA V100) |
|
RFdiffusion2 [20] |
2025 |
Requires only a defined theozyme active site, without pre-indexed atomic positions or preset rotamers |
150- residues in 5 min (NVIDIA A40, including backbone design, sequence design, and evaluation) |
4. Sequence Design on Fixed Backbones
Once the backbone has been defined, identifying a suitable amino acid sequence to fully describe the protein involves a level of complexity that goes far beyond first impressions [63]. This is because it requires inferring an amino acid chain that can reliably fold into a given three-dimensional backbone structure that is not only energetically optimal but also structurally stable. This “reverse” process(Figure 3A), wherein the goal is to design a sequence compatible with a predefined structure, is known as the inverse protein folding problem [24].
A direct approach to addressing these issues is to use deep learning to learn the mapping between the structure and sequence. (Figure 3B). Early progress was marked by the introduction of ESM-IF1 [64] and ProteinMPNN [24] in 2022, which became foundational models in AI-driven protein sequence design. ESM-IF defines inverse folding as a structure-to-sequence problem, focusing primarily on the protein backbone structure while disregarding the complexity of side chains. By leveraging 12 million protein structures predicted by AlphaFold2 and using the coordinates of the N, Cα, and C atoms as backbone inputs, ESM-IF employs an autoregressive framework to predict the natural amino acid sequence from the three-dimensional positions of the backbone atoms. In contrast, ProteinMPNN is built upon the general message-passing neural network (MPNN) framework, has been applied to de novo sequence design for carbonic anhydrases and artificial luciferases. (These applications are discussed in detail in the case study section.) It utilizes experimentally determined protein crystal structures and incorporates interatomic distance features. Importantly, instead of using a fixed decoding order from the N to C terminus, ProteinMPNN adopts an order-agnostic autoregressive approach for sequence generation, thereby overcoming a key limitation of conventional autoregressive models, which generate amino acids sequentially and can only condition on past residues but not future ones. By inverting the structure prediction process, ESM-IF1 designs sequences that fit given backbone traces, achieving a sequence recovery of 51%. ProteinMPNN attains a recovery rate of 52.4%.
In 2023, PiFold was proposed by Li et al. [65], introducing expressive structural features and a novel PiGNN module. Based on the local coordinate system of each residue, the model constructs distance, angle, and orientation features on both nodes and edges to ensure rotational and translational invariance. At the same time, learnable virtual atoms are incorporated to capture complementary information from real atoms. Furthermore, unlike previous autoregressive or iterative models (Table 1) [66,67,68], PiFold completely removes the autoregressive decoder by stacking additional PiGNN layers, enabling one-shot protein sequence generation without sacrificing accuracy. Experiments show that PiFold could achieve 51.66% recovery on CATH 4.2, while the inference speed is 70 times faster than the autoregressive competitors.
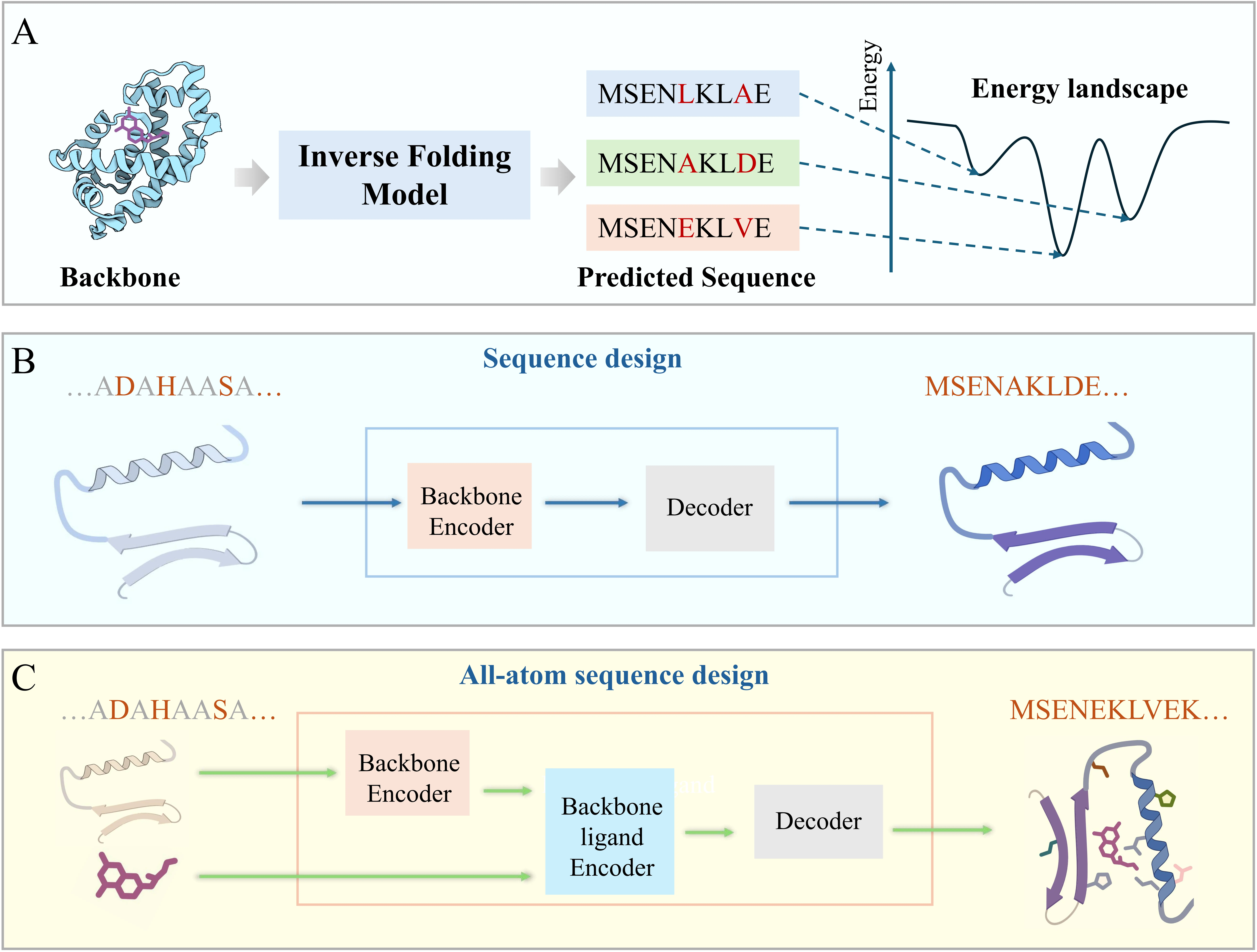
Figure 3. Sequence design on fixed backbones. (A) Overview of Inverse folding: a predefined protein backbone is provided as input to predict amino acid sequences compatible with the target structure. Candidate sequences are evaluated on the energy landscape to identify low-energy solutions with favorable folding propensities. (B) A backbone-based sequence design framework is where structural information of the backbone is encoded and decoded into a compatible sequence, enabling recovery of secondary structure elements and overall fold. (C) All-atom sequence design framework that augments backbone features with explicit side-chain and ligand representations through an additional backbone–ligand encoder, thereby enabling the decoder to generate sequences optimized not only for backbone compatibility but also for atomic-level interactions and binding requirements.
Subsequently, numerous inverse folding frameworks were developed (Table 2) [69,70,71,72,73], aiming to improve sequence recovery accuracy and generative efficiency. However, like earlier inverse folding models, these approaches remained focused on backbone-based design and lacked the capacity to tailor proteins for specific biochemical functions. This limitation is particularly critical in enzyme design, where atomic-level control over active sites and ligand interactions is essential. To bridge this gap, the introduction of LigandMPNN [25] in 2025 represented a breakthrough by enabling protein sequence generation in the context of explicit ligand environments, which has been demonstrated in the de novo sequence design of serine hydrolases and metal hydrolases, with these applications discussed in detail in the case study section, particularly for serine hydrolases. The model constructs distinct graph representations for the protein backbone, ligand atoms, and their mutual interactions, allowing it to capture fine-grained geometric, chemical, and physical properties of the binding site. LigandMPNN automatically optimizes residue types and side-chain conformations by incorporating ligand-aware features, enhancing binding interface complementarity. Beyond sequence generation, it also predicts side-chain rotamer angles (chi angles), ensuring the designed structures closely resemble experimentally validated conformations. This development marks a critical step toward functionally aware sequence design. LigandMPNN exhibits excellent sequence recovery accuracy in protein design, achieving 63.3% for residues near small molecules, 50.5% near nucleotides, and 77.5% near metals, significantly higher than ProteinMPNN. (50.4%, 34.0%, 40.6% respectively). Furthermore, the model excels in side-chain packing accuracy, with a chi1 score of 86.1% near small molecules, and high generation efficiency, designing 100 residues in just 0.9 s on a single CPU.
In summary, ESM-IF1 and ProteinMPNN laid the groundwork for backbone-based sequence design. Building on this foundation, the advent of LigandMPNN marks a pivotal step toward atomistic, ligand-aware sequence design by explicitly incorporating ligand information into the generative process.
Table 2. Summary of recent studies in Inverse folding.
|
Inverse Folding Models |
Method |
Main Architecture |
|---|---|---|
|
ESM-IF1 [64] |
Backbone |
Transformer [74] ; Autoregressive |
|
ProteinMPNN [24] |
Backbone |
Message Passing Neural Network [75] ; Autoregressive |
|
PiFold [65] |
Backbone |
Graph Neural Network [76] |
|
GCA [66] |
Backbone |
Graph Neural Network; Autoregressive |
|
StructGNN [67] |
Backbone |
Graph Neural Network; Autoregressive |
|
GVP [68] |
Backbone |
Graph Neural Network; Autoregressive |
|
GRADE-IF [69] |
Backbone |
Diffusion Model |
|
LaGDif [70] |
Backbone |
Diffusion Model |
|
Bridge-IF [71] |
Backbone |
Diffusion Model |
|
CarbonDesign [72] |
Backbone |
Transformer; Markov random fields model |
|
MapDiff [73] |
Backbone |
Diffusion Model |
|
LigandMPNN [25] |
Backbone + Ligand |
Message Passing Neural Network; Autoregressive |
5. Virtual Screening to Ensure Success in De Novo Enzyme Design
Artificial enzymes with potential catalytic functions can be designed from scratch by precisely constructing active catalytic sites using quantum chemical methods, subsequently tailoring backbones with generative models, and completing amino acid sequence design via inverse folding models [22,77]. However, in practice, many of these candidate enzymes often exhibit deviations in catalytic geometries, misfolding, low activity, or even complete activity loss, which largely limit their experimental feasibility and functional reliability.
To improve the success rate of enzyme design and reduce the number of non-functional candidates, filtering steps are typically introduced after sequence design. A common strategy is to use Rosetta to filter candidate designs. FastRelax [78] is applied to the designs to relieve local strain and optimize overall stability. The resulting structures are evaluated for their ability to recapitulate the catalytic motif geometry, confirming that the spatial arrangement of catalytic residues is maintained to preserve chemical functionality. The shape complementarity between the substrate-binding pocket and the substrate is assessed to ensure that the binding site can accommodate the substrate snugly and support effective catalysis (Figure 4A). On this basis, structure prediction tools such as AlphaFold2 (Figure 4B) [79] were applied to evaluate folding constraints and eliminate designs unlikely to adopt correct folds. Designs were considered acceptable if they achieved RMSD < 2.0 Å, with TM-score > 0.5 reported as a supplemental criterion [80].
In addition, molecular dynamics (MD) simulations are frequently employed at this stage to probe the conformational stability of designed scaffolds and to assess whether catalytic residues and substrate-binding pockets remain geometrically compatible under dynamic conditions (Figure 4C). By monitoring side-chain rearrangements, hydrogen-bonding persistence, and local flexibility across nanosecond-to-microsecond trajectories [81], MD provides a virtual screening layer that helps eliminate candidates prone to unfolding or incapable of maintaining catalytically relevant interactions [82,83,84].
Current computational approaches struggle to accurately capture enzymatic preorganization, and although MD simulations provide dynamic insights, their high cost, limited sampling, and difficulty in modeling multi-state catalytic cycles mean that critical side-chain–transition state interactions are often missed, resulting in many designed enzymes with low catalytic efficiency [18,85]. To address this, the Baker laboratory developed PLACER (Protein-Ligand Atomistic Conformational Ensemble Resolver), an all-atom modeling network for protein–ligand interactions (Figure 4D) [85]. PLACER takes as input the coordinates of the protein backbone surrounding a binding or catalytic pocket, together with atomic-level descriptions of the bonded geometry of small molecules and side chains. Using a graph neural network, it generates predicted protein–ligand binding conformations through a denoising process and iteratively refines them to form a conformational ensemble. Simulating multiple key states along the catalytic cycle, including the apo, substrate-bound, and tetrahedral intermediate states, PLACER enables assessment of structural compatibility across dynamic conformations, guiding the rational optimization of enzyme design.
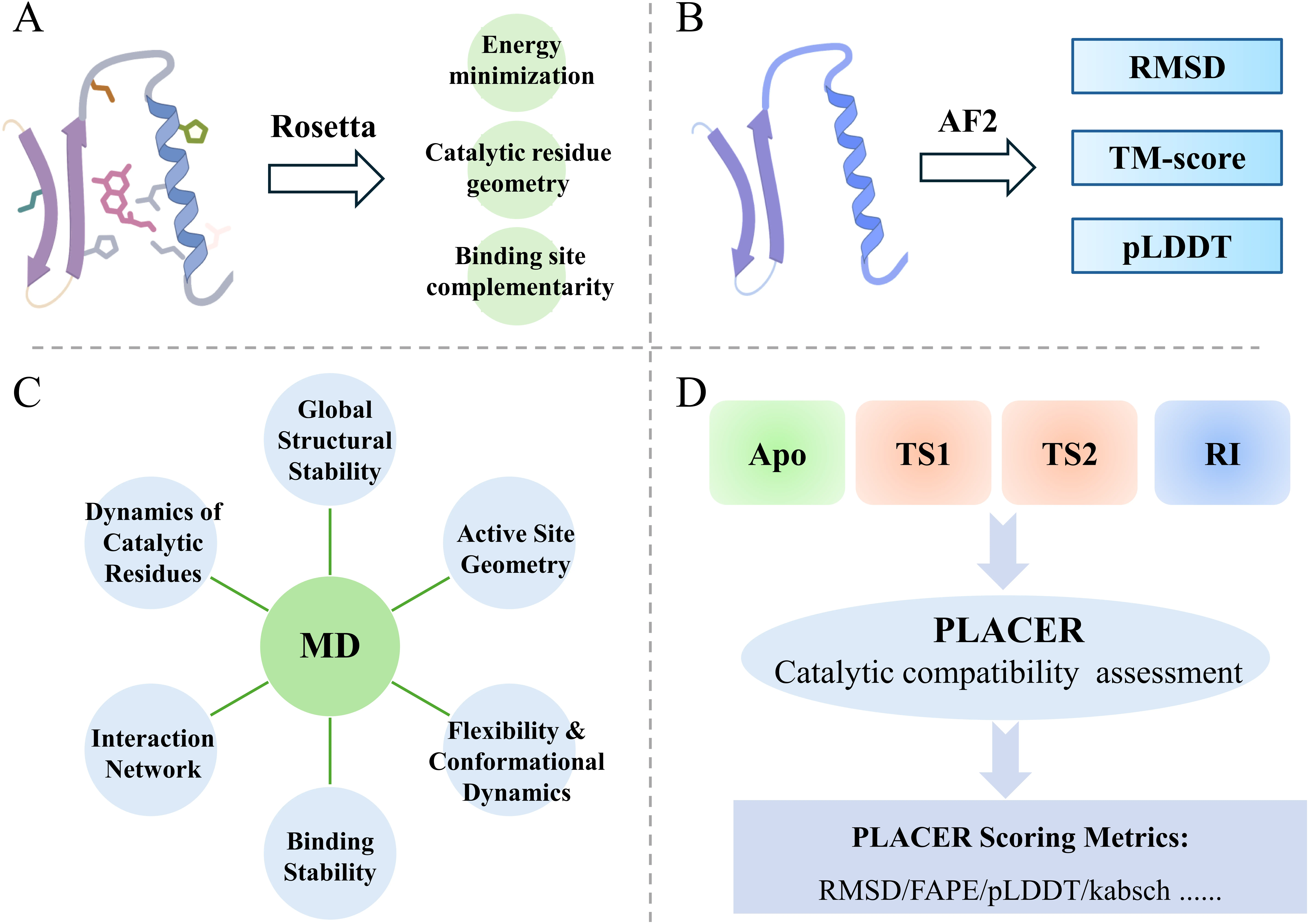
Figure 4. Computational strategies for structural and functional evaluation in de novo enzyme design. (A) Rosetta-based relaxation and filtering. Candidate enzyme models undergo energy minimization (e.g., FastRelax) to relieve local strain and optimize backbone/side-chain packing. The resulting structures are assessed for catalytic residue geometry (satisfaction of catalytic distance/angle/dihedral constraints) and binding-site complementarity (shape and interaction compatibility with the substrate), ensuring that the active site is chemically plausible. (B) Designs are evaluated with AlphaFold2, using RMSD, TM-score, and pLDDT to filter out structures unlikely to fold correctly. (C) Molecular dynamics simulations probe whether catalytic residues, active-site geometry, and substrate-binding pocket remain stable under thermal fluctuations, examining global structural stability, side-chain dynamics, interaction networks, flexibility, and binding stability over time. (D) PLACER generates atomistic ensembles across key catalytic states (including, but not limited to, apoenzyme: apo, transition states: TS1, TS2, and reactive intermediate: RI) to assess catalytic preorganization. Scoring combines geometric and confidence metrics (RMSD, FAPE, pLDDT, alignment scores) to prioritize designs with both static and dynamic fidelity.
In summary, de novo enzyme design combines multiple computational strategies to generate functional catalysts. Post-design filtering, such as structure relaxation, catalytic motif validation, substrate-pocket assessment, and folding evaluation-helps remove unstable or misfolded candidates. Finally, atomistic modeling tools like PLACER allow simulation of dynamic conformational states to assess catalytic compatibility. Together, these methods form an integrated, structure-and function-informed workflow that guides the rational design and optimization of artificial enzymes. Overall, PLACER integrates deep learning with enzymology knowledge to conformational ensembles for protein–ligand complexes, enabling the evaluation of conformational changes during dynamic catalysis, and thereby guiding the rational optimization of enzyme design.
6. Case Study
In the long-term vision of computational protein design, the de novo creation of enzymes capable of catalyzing arbitrary chemical reactions has remained a central objective [18,81]. Traditional approaches typically rely on embedding a small theoretical model of catalytic residues (theozyme) into natural protein scaffolds, as exemplified by Retro-Aldol enzymes [15] and Kemp elimination catalysts [14]. However, this strategy is constrained by the fixed scaffold, and as a result, the designed enzymes typically exhibit activities several orders of magnitude lower than those of natural enzymes.
With the advancement of deep learning, artificial luciferases were designed in 2023 using a “family-wide hallucination” approach. These de novo enzymes overcome limitations of natural enzymes in stability and substrate specificity, with the most notable variant being small (13.9 kDa), thermostable (>95 °C), and exhibiting superior specificity with catalytic activity comparable to natural luciferases [21]. Yet their initial activity still required improvement through mutagenesis, highlighting the need for new methods. In 2024, Hu et al. developed the Generative Redesign in Artificial Computational Enzymology (GRACE) workflow [86], integrating RFdiffusion for backbone generation, ProteinMPNN for sequence optimization, and molecular dynamics for screening. Applied to carbonic anhydrase, GRACE designed two functional sequences from a pool of 10,000 candidates, achieving activities up to 400 WAU/mL. Despite these advances, the overall success rate and catalytic performance of de novo enzymes remain low, largely because designs have focused on simplified active sites optimized for a single state.
In 2025, the David Baker laboratory achieved the de novo design of a serine hydrolase [18], integrating GAI and multistate constraints, marking a shift from static to dynamic scaffold design. In the early stages, the first two design rounds built simple Ser-His dyad active sites with a single oxyanion hole contact, using RFdiffusion to generate the backbones and LigandMPNN to design the corresponding sequences. Round 1 designs were filtered using AlphaFold2, yielding 139 functional sequences from 214 K candidates, while Round 2 designs were further screened with PLACER ensembles of the apo state to ensure key Ser-His hydrogen bonding, resulting in 261 sequences. These steps improved the fraction of activated serines and detectable esterase activity, though the designs remained limited to the initial nucleophilic attack. To enhance success, the third round added a histidine-stabilizing catalytic acid and a second oxyanion hole donor, expanding PLACER screening to the acyl-enzyme intermediate (AEI), yielding the first de novo enzyme capable of multiple turnovers. Multistate design with LigandMPNN and FastRelax further optimized reaction coordinates and active-site geometry. Integration with RFdiffusion produced complete enzymes with novel oxyanion hole networks, generating “momi” (kcat/Km = 1240 M−1·s−1), optimized to “momi120” (4300 M−1s−1), and extended to PET hydrolase mimics (“momi120_103”), achieving up to 2.2 × 105 M−1·s−1. Crystallography confirmed entirely non-natural folds surpassing traditional directed evolution.
Similarly, David Baker’s laboratory applied the GAI method RFdiffusion2 to metal hydrolases [19], starting from DFT-modeled active-site geometries to generate zinc enzyme backbones and optimize sequences, achieving catalytic efficiencies up to 23,000 M−1·s−1. In addition,in 2025, Sarel J. Fleishman’s team [87] employed a non-generative de novo design approach, combining TIM-barrel fragment assembly with PROSS [88] and FuncLib [89] active-site optimization to produce the stable Kemp elimination enzyme Des27.7, achieving a catalytic efficiency of 123,000 M−1·s−1, close to the median of natural enzymes.
Recent advances in de novo enzyme design demonstrate that effective catalyst development relies on the coordinated integration of complementary computational strategies. Following the theoretical enzyme model, the precise specification of catalytic active sites establishes the geometric foundation for reactivity. Functional realization of these sites is enabled by scaffold generation methods that provide structurally compatible backbones and inverse folding models that optimize sequence–structure compatibility. Explicit consideration of conformational dynamics and multi-state reaction pathways has emerged as a key determinant for achieving turnover and overall catalytic efficiency. The successful de novo design of a serine hydrolase exemplifies the importance of this integrated workflow: active-site engineering, de novo backbone generation, sequence optimization, and post-design dynamic evaluation collectively underpin the creation of entirely new enzymes with high catalytic activity. These insights underscore that systematic, multi-layered design strategies are essential for realizing predictable and efficient artificial enzymes.
7. Conclusions and Perspectives
De novo enzyme design represents a core frontier technology in synthetic biology and biomanufacturing, offering the potential to overcome the functional limitations of natural enzymes. Evolutionary adaptation often limits the catalytic activity, substrate specificity, and stability of natural enzymes, making it difficult to meet the tailored demands of industrial production.
Recent advances in GAI are enabling true de novo enzyme design by integrating complementary strategies: QM calculations provide atomistic blueprints for catalytic site construction; backbone-generating and inverse folding models enable stable incorporation of active sites into protein frameworks; and tools such as PLACER assess structural compatibility across reaction coordinates. Together, these approaches substantially enhance the feasibility and success of de novo enzyme design, as exemplified by GAI-designed serine hydrolases that achieve high catalytic efficiency with folds entirely distinct from any natural homologs.
Future developments in GAI-driven enzyme design are expected to focus on fully integrated, mechanism-informed frameworks that enable rational and efficient engineering of catalytic proteins. Key directions include multiscale modeling that combines quantum mechanics, molecular mechanics, and AI to capture and optimize entire catalytic cycles; dynamic design approaches that incorporate protein motions and conformational changes directly into the design process; and closed-loop “design–build–test–learn” pipelines, leveraging laboratory automation to iteratively refine AI models. By uniting structural prediction, sequence optimization, and dynamic evaluation within a computational-experimental workflow, these advances will make on-demand, custom enzyme design increasingly feasible, opening new opportunities for synthetic biology and sustainable biomanufacturing.
Despite recent advances in de novo enzyme design, which now incorporate geometric constraints, folding prediction, and assessments of expressibility, designed enzymes often display only limited activity when tested experimentally. This discrepancy largely reflects the gap between the “static idealization” of computational models and the “dynamic optimization” achieved through natural evolution. Current approaches typically rely on a small number of idealized conformations, whereas real proteins in solution populate diverse ensembles, making the occupancy of catalytically competent geometries under dynamic equilibrium much lower than anticipated, thereby limiting transition-state stabilization [90]. Moreover, effective stabilization of the transition state depends not only on the spatial arrangement of first-shell residues, but also on subtle contributions from local electric fields, pKa tuning, and polarization effects—factors often insufficiently captured by rapid computational models [91,92]. Efficient catalysis in natural enzymes further relies on extended residue networks that cooperate in maintaining conformational stability, regulating solvent access, and suppressing side reactions, yet such higher-order interactions are frequently neglected in automated design. In addition, mismatches between design assumptions and experimental conditions can further diminish or even mask latent activity [93].
Future strategies must integrate generative modeling, screening, and experimental validation more tightly to improve hit rates while retaining throughput and speed. A promising approach is the adoption of hierarchical screening frameworks: rapid geometric and folding filters can be applied at the early stage to down-select large candidate pools; intermediate tiers may incorporate evaluations of electrostatic preorganization, pKa prediction, placement of key water molecules, and short-timescale enhanced-sampling MD to assess catalytic robustness; and only a small subset of candidates would then proceed to high-accuracy QM/MM calculations and experimental testing, thereby balancing computational cost with predictive precision. At the same time, generative models themselves should embed physical constraints—for example, optimizing local electrostatics or ensemble occupancy of transition-state geometries—to reduce the frequency of designs that are “statistically correct but dynamically ineffective”. Equally important is closed-loop experimental learning: small-scale but information-rich assays can provide real measurements of activity, stability, and expressibility, which are then fed back into the generative and scoring functions to refine predictions iteratively.
In the long term, truly effective enzyme design will require a framework that is mechanism-driven, dynamically informed, and experimentally closed-loop. Such an approach would consider the entire catalytic cycle during generation, incorporate multiscale physical modeling during screening, and leverage automated experimental platforms for rapid feedback. Together, these elements could enable simultaneous optimization of catalytic activity, stability, and manufacturability, gradually transforming the design of high-performance enzymes from theoretical possibility into reproducible and scalable reality.
Author Contributions
Conceptualization, X.Q. and H.M.; Writing—Original Draft Preparation, X.Q. and D.W.; Writing—Review & Editing, X.Q., Z.S., X.L. and H.M.; Supervision, X.L. and H.M.; Funding Acquisition, X.L. and H.M.
Ethics Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No data was used for the research described in the article.
Funding
This research was financially supported by “The Strategic Priority Research Program of the Chinese Academy of Sciences [XDC0110200] and National Natural Science Foundation of China [12326611]” and “The APC was funded by The Strategic Priority Research Program of the Chinese Academy of Sciences [XDC0110200]”.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
-
Bornscheuer UT, Huisman GW, Kazlauskas RJ, Lutz S, Moore JC, Robins K. Engineering the third wave of biocatalysis. Nature 2012, 485, 185–194. doi:10.1038/nature11117.[Google Scholar]
-
Miller DC, Athavale SV, Arnold FH. Combining chemistry and protein engineering for new-to-nature biocatalysis. Nat. Synth. 2022, 1, 18–23. doi:10.1038/s44160-021-00008-x.[Google Scholar]
-
Reetz MT, Qu G, Sun Z. Engineered enzymes for the synthesis of pharmaceuticals and other high-value products. Nature Synthesis 2024, 3, 19–32. doi:10.1038/s44160-023-00417-0.[Google Scholar]
-
Martinusen SG, Nelson SE, Slaton EW, Long LF, Pho R, Ajayebi S, et al. Protease engineering: Approaches, tools, and emerging trends. Biotechnol. Adv. 2025, 82, 108602. doi:10.1016/j.biotechadv.2025.108602.[Google Scholar]
-
Li A, Qu G, Sun Z, Reetz MT. Statistical Analysis of the Benefits of Focused Saturation Mutagenesis in Directed Evolution Based on Reduced Amino Acid Alphabets. ACS Catal. 2019, 9, 7769–7778. doi:10.1021/acscatal.9b02548.[Google Scholar]
-
Shi L, Liu P, Tan Z, Zhao W, Gao J, Gu Q, et al. Complete Depolymerization of PET Wastes by an Evolved PET Hydrolase from Directed Evolution. Angew. Chem. Int. Ed. Engl. 2023, 62, e202218390. doi:10.1002/anie.202218390.[Google Scholar]
-
Tan Z, Tang Z, Wei H, Zhang R, Sun L, Liu W, et al. Helix Zipper Regulating Formolase Activity. ACS Catal. 2025, 15, 1586–1595. doi:10.1021/acscatal.4c07452.[Google Scholar]
-
Otten R, Pádua RAP, Bunzel HA, Nguyen V, Pitsawong W, Patterson M, et al. How directed evolution reshapes the energy landscape in an enzyme to boost catalysis. Science 2020, 370, 1442–1446. doi:10.1126/science.abd3623.[Google Scholar]
-
Bell EL, Hutton AE, Burke AJ, O’Connell A, Barry A, O’Reilly E, et al. Strategies for designing biocatalysts with new functions. Chem. Soc. Rev. 2024, 53, 2851–2862. doi:10.1039/d3cs00972f.[Google Scholar]
-
Albayati SH, Nezhad NG, Taki AG, Rahman R. Efficient and easible biocatalysts: Strategies for enzyme improvement. A review. Int. J. Biol. Macromol. 2024, 276, 133978. doi:10.1016/j.ijbiomac.2024.133978.[Google Scholar]
-
Bolon DN, Mayo SL. Enzyme-like proteins by computational design. Proc. Natl. Acad. Sci. USA 2001, 98, 14274–14279. doi:10.1073/pnas.251555398.[Google Scholar]
-
Kaplan J, DeGrado WF. De novo design of catalytic proteins. Proc. Natl. Acad. Sci. USA 2004, 101, 11566–11570. doi:10.1073/pnas.0404387101.[Google Scholar]
-
Zanghellini A, Jiang L, Wollacott AM, Cheng G, Meiler J, Althoff EA, et al. New algorithms and an in silico benchmark for computational enzyme design. Protein Sci. 2006, 15, 2785–2794. doi:10.1110/ps.062353106.[Google Scholar]
-
Rothlisberger D, Khersonsky O, Wollacott AM, Jiang L, DeChancie J, Betker J, et al. Kemp elimination catalysts by computational enzyme design. Nature 2008, 453, 190–195. doi:10.1038/nature06879.[Google Scholar]
-
Jiang L, Althoff EA, Clemente FR, Doyle L, Röthlisberger D, Zanghellini A, et al. De. Novo Computational Design of Retro-Aldol Enzymes. Science 2008, 319, 1387–1391. doi:10.1126/science.1152692.[Google Scholar]
-
Lipsh-Sokolik R, Khersonsky O, Schröder SP, De Boer C, Hoch S-Y, Davies GJ, et al. Combinatorial assembly and design of enzymes. Science 2023, 379, 195–201. doi:10.1126/science.ade9434.[Google Scholar]
-
Basanta B, Bick MJ, Bera AK, Norn C, Chow CM, Carter LP, et al. An enumerative algorithm for de novo design of proteins with diverse pocket structures. Proc. Natl. Acad. Sci. USA 2020, 117, 22135–22145. doi:10.1073/pnas.2005412117.[Google Scholar]
-
Lauko A, Pellock SJ, Sumida KH, Anishchenko I, Juergens D, Ahern W, et al. Computational design of serine hydrolases. Science 2025, 388, eadu2454. doi:10.1126/science.adu2454.[Google Scholar]
-
Kim D, Woodbury SM, Ahern W, Kalvet I, Hanikel N, Salike S, et al. Computational Design of Metallohydrolases. bioRxiv 2024. doi:10.1101/2024.11.13.623507.[Google Scholar]
-
Ahern W, Yim J, Tischer D, Salike S, Woodbury SM, Kim D, et al. Atom level enzyme active site scaffolding using RFdiffusion2. bioRxiv 2025. doi:10.1101/2025.04.09.648075.[Google Scholar]
-
Yeh AH-W, Norn C, Kipnis Y, Tischer D, Pellock SJ, Evans D, et al. De novo design of luciferases using deep learning. Nature 2023, 614, 774–780. doi:10.1038/s41586-023-05696-3.[Google Scholar]
-
Watson JL, Juergens D, Bennett NR, Trippe BL, Yim J, Eisenach HE, et al. De novo design of protein structure and function with RFdiffusion. Nature 2023, 620, 1089–1100. doi:10.1038/s41586-023-06415-8.[Google Scholar]
-
Liu Y, Wang S, Dong J, Chen L, Wang X, Wang L, et al. De novo protein design with a denoising diffusion network independent of pretrained structure prediction models. Nat. Methods 2024, 21, 2107–2116. doi:10.1038/s41592-024-02437-w.[Google Scholar]
-
Dauparas J, Anishchenko I, Bennett N, Bai H, Ragotte RJ, Milles LF, et al. Robust deep learning–based protein sequence design using ProteinMPNN. Science 2022, 378, 49–56. doi:10.1126/science.add2187.[Google Scholar]
-
Dauparas J, Lee GR, Pecoraro R, An L, Anishchenko I, Glasscock C, et al. Atomic context-conditioned protein sequence design using LigandMPNN. Nat. Methods 2025, 22, 717–723. doi:10.1038/s41592-025-02626-1.[Google Scholar]
-
Smith AJT, Müller R, Toscano MD, Kast P, Hellinga HW, Hilvert D, et al. Structural Reorganization and Preorganization in Enzyme Active Sites: Comparisons of Experimental and Theoretically Ideal Active Site Geometries in the Multistep Serine Esterase Reaction Cycle. J. Am. Chem. Soc. 2008, 130, 15361–15373. doi:10.1021/ja803213p.[Google Scholar]
-
Ilinkin I, Ye J, Janardan R. Multiple structure alignment and consensus identification for proteins. BMC Bioinform. 2010, 11, 71. doi:10.1186/1471-2105-11-71.[Google Scholar]
-
Jain A, Terashi G, Kagaya Y, Maddhuri Venkata Subramaniya SR, Christoffer C, Kihara D. Analyzing effect of quadruple multiple sequence alignments on deep learning based protein inter-residue distance prediction. Sci. Rep. 2021, 11, 7574. doi:10.1038/s41598-021-87204-z.[Google Scholar]
-
Buller AR, Townsend CA. Intrinsic evolutionary constraints on protease structure, enzyme acylation, and the identity of the catalytic triad. Proc. Natl. Acad. Sci. USA 2013, 110, E653–E661. doi:10.1073/pnas.1221050110.[Google Scholar]
-
Chew LP, Kedem K. Finding the Consensus Shape for a Protein Family. Algorithmica 2004, 38, 115–129. doi:10.1007/s00453-003-1045-2.[Google Scholar]
-
Carpentier M, Chomilier J. Protein multiple alignments: sequence-based versus structure-based programs. Bioinformatics 2019, 35, 3970–3980. doi:10.1093/bioinformatics/btz236.[Google Scholar]
-
Sternke M, Tripp KW, Barrick D. Consensus sequence design as a general strategy to create hyperstable, biologically active proteins. Proc. Natl. Acad. Sci. USA 2019, 116, 11275–11284. doi:10.1073/pnas.1816707116.[Google Scholar]
-
Modi V, Dunbrack RL. A Structurally-Validated Multiple Sequence Alignment of 497 Human Protein Kinase Domains. Sci. Rep. 2019, 9, 19790. doi:10.1038/s41598-019-56499-4.[Google Scholar]
-
Zhang Y, Zheng J, Zhang B. Protein Language Model Identifies Disordered, Conserved Motifs Driving Phase Separation. eLife 2025. doi:10.7554/elife.105309.1.[Google Scholar]
-
Zhang Z, Wayment-Steele HK, Brixi G, Wang H, Kern D, Ovchinnikov S. Protein language models learn evolutionary statistics of interacting sequence motifs. Proc. Natl. Acad. Sci. USA 2024, 121, e2406285121. doi:10.1073/pnas.2406285121.[Google Scholar]
-
Saadat A, Fellay J. Fine-tuning protein language models to understand the functional impact of missense variants. Comput. Struct. Biotechnol. J. 2025, 27, 2199–2207. doi:10.1016/j.csbj.2025.05.022.[Google Scholar]
-
Hopf TA, Ingraham JB, Poelwijk FJ, Schärfe CP, Springer M, Sander C, et al. Mutation effects predicted from sequence co-variation. Nat. Biotechnol. 2017, 35, 128–135. doi:10.1038/nbt.3769.[Google Scholar]
-
Tantillo DJ, Jiangang C, Houk KN. Theozymes and compuzymes: Theoretical models for biological catalysis. Curr. Opin. Chem. Biol. 1998, 2, 743–750. doi:10.1016/S1367-5931(98)80112-9.[Google Scholar]
-
Kiss G, Çelebi-Ölçüm N, Moretti R, Baker D, Houk KN. Computational Enzyme Design. Angew. Chem. Int. Ed. 2013, 52, 5700–5725. doi:10.1002/anie.201204077.[Google Scholar]
-
Noey EL, Tibrewal N, Jiménez-Osés G, Osuna S, Park J, Bond CM, et al. Origins of stereoselectivity in evolved ketoreductases. Proc. Natl. Acad. Sci. USA 2015, 112, E7065–E7072. doi:10.1073/pnas.1507910112.[Google Scholar]
-
Siegel JB, Zanghellini A, Lovick HM, Kiss G, Lambert AR, St. Clair JL, et al. Computational Design of an Enzyme Catalyst for a Stereoselective Bimolecular Diels-Alder Reaction. Science 2010, 329, 309–313. doi:10.1126/science.1190239.[Google Scholar]
-
Nakashima Y, Mitsuhashi T, Matsuda Y, Senda M, Sato H, Yamazaki M, et al. Structural and Computational Bases for Dramatic Skeletal Rearrangement in Anditomin Biosynthesis. J. Am. Chem. Soc. 2018, 140, 9743–9750. doi:10.1021/jacs.8b06084.[Google Scholar]
-
Li B, Guan X, Yang S, Zou Y, Liu W, Houk KN. Mechanism of the Stereoselective Catalysis of Diels–Alderase PyrE3 Involved in Pyrroindomycin Biosynthesis. J. Am. Chem. Soc. 2022, 144, 5099–5107. doi:10.1021/jacs.2c00015.[Google Scholar]
-
Lovelock SL, Crawshaw R, Basler S, Levy C, Baker D, Hilvert D, et al. The road to fully programmable protein catalysis. Nature 2022, 606, 49–58. doi:10.1038/s41586-022-04456-z.[Google Scholar]
-
Peccati F, Noey EL, Houk KN, Osuna S, Jiménez-Osés G. Interplay Between Substrate Polarity and Protein Dynamics in Evolved Kemp Eliminases. ChemCatChem 2024, 16, e202400444. doi:10.1002/cctc.202400444.[Google Scholar]
-
Dechancie J, Clemente FR, Smith AJ, Gunaydin H, Zhao YL, Zhang X, et al. How similar are enzyme active site geometries derived from quantum mechanical theozymes to crystal structures of enzyme-inhibitor complexes? Implications for enzyme design. Protein Sci. 2007, 16, 1851–1866. doi:10.1110/ps.072963707.[Google Scholar]
-
Hou Y, Chen J, Liu W, Zhu G, Yang Q, Wang X. Using the Theozyme Model to Study the Dynamical Mechanism of the Post-Transition State Bifurcation Reaction by NgnD Enzyme. Molecules 2024, 29, 5518. doi:10.3390/molecules29235518.[Google Scholar]
-
Becke AD. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. doi:10.1063/1.464913.[Google Scholar]
-
Richter F, Leaver-Fay A, Khare SD, Bjelic S, Baker D. De novo enzyme design using Rosetta3. PLoS ONE 2011, 6, e19230. doi:10.1371/journal.pone.0019230.[Google Scholar]
-
Song Y, Sohl-Dickstein JN, Kingma DP, Kumar A, Ermon S, Poole B. Score-Based Generative Modeling through Stochastic Differential Equations. arXiv 2020, arXiv:2011.13456.[Google Scholar]
-
Trippe BL, Yim J, Tischer DK, Broderick T, Baker D, Barzilay R, et al. Diffusion probabilistic modeling of protein backbones in 3D for the motif-scaffolding problem. arXiv 2022, arXiv:2206.04119.[Google Scholar]
-
Ho J, Jain A, Abbeel P. Denoising Diffusion Probabilistic Models. arXiv 2020, arXiv:2006.11239.[Google Scholar]
-
Yang L, Zhang Z, Song Y, Hong S, Xu R, Zhao Y, et al. Diffusion models: A comprehensive survey of methods and applications. ACM Comput. Surv. 2023, 56, 1–39. doi:10.48550/arXiv.2209.00796.[Google Scholar]
-
Batzner S, Musaelian A, Sun L, Geiger M, Mailoa JP, Kornbluth M, et al. E (3)-equivariant graph neural networks for data-efficient and accurate interatomic potentials. Nat. Commun. 2022, 13, 2453. doi:10.1038/s41467-022-29939-5.[Google Scholar]
-
Hoogeboom E, Garcia Satorras V, Vignac C, Welling M. Equivariant Diffusion for Molecule Generation in 3D. arXiv 2022, arXiv:2203.17003.[Google Scholar]
-
Watson JL, Juergens D, Bennett NR, Trippe BL, Yim J, Eisenach HE, et al. Broadly applicable and accurate protein design by integrating structure prediction networks and diffusion generative models. bioRxiv 2022. doi:10.1101/2022.12.09.519842.[Google Scholar]
-
Baek M, DiMaio F, Anishchenko I, Dauparas J, Ovchinnikov S, Lee GR, et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 2021, 373, 871–876. doi:10.1126/science.abj8754.[Google Scholar]
-
Cao H, Tan C, Gao Z, Xu Y, Chen G, Heng P-A, et al. A survey on generative diffusion models. IEEE Trans. Knowl. Data Eng. 2024, 36, 2814–2830. doi:10.48550/arXiv.2209.02646.[Google Scholar]
-
Lipman Y, Chen RTQ, Ben-Hamu H, Nickel M, Le M. Flow Matching for Generative Modeling. arXiv 2022, arXiv:2210.02747.[Google Scholar]
-
Yim J, Campbell A, Foong AYK, Gastegger M, Jiménez-Luna J, Lewis S, et al. Fast protein backbone generation with SE(3) flow matching. arXiv 2023, arXiv:2310.05297.[Google Scholar]
-
Bose AJ, Akhound-Sadegh T, Huguet G, Fatras K, Rector-Brooks J, Liu C-H, et al. SE(3)-Stochastic Flow Matching for Protein Backbone Generation. arXiv 2023, arXiv:2310.02391.[Google Scholar]
-
Yim J, Trippe BL, De Bortoli V, Mathieu E, Doucet A, Barzilay R, et al. SE (3) diffusion model with application to protein backbone generation. arXiv 2023, arXiv:2302.02277.[Google Scholar]
-
Yue K, Dill KA. Inverse protein folding problem: designing polymer sequences. Proc. Natl. Acad. Sci. USA 1992, 89, 4163–4167. doi:10.1073/pnas.89.9.4163.[Google Scholar]
-
Hsu C, Verkuil R, Liu J, Lin Z, Hie B, Sercu T, et al. Learning inverse folding from millions of predicted structures. bioRxiv 2022, 8946-8970. doi:10.1101/2022.04.10.487779.[Google Scholar]
-
Gao Z, Tan C, Li SZ. PiFold: Toward effective and efficient protein inverse folding. arXiv 2022, arXiv:2209.12643.[Google Scholar]
-
Tan C, Gao Z, Xia J, Hu B, Li SZ. Global-Context Aware Generative Protein Design. In Proceedings of the ICASSP 2023—2023 IEEE International Conference on Acoustics, Speech and Signal Processing (ICASSP), Rhodes Island, Greece, 4–10 June 2023; pp. 1–5. doi: 10.1109/ICASSP49357.2023.10095229.[Google Scholar]
-
Chou Y-T, Chang W-T, Jean JG, Chang K-H, Huang Y-N, Chen C-S. StructGNN: An efficient graph neural network framework for static structural analysis. Comput. Struct. 2024, 299, 107385. doi:10.1016/j.compstruc.2024.107385.[Google Scholar]
-
Jing B, Eismann S, Suriana P, Townshend RJ, Dror R. Learning from protein structure with geometric vector perceptrons. arXiv 2020, arXiv:2009.01411.[Google Scholar]
-
Yi K, Zhou B, Shen Y, Lio’ P, Wang YG. Graph Denoising Diffusion for Inverse Protein Folding. arXiv 2023, arXiv:2306.16819.[Google Scholar]
-
Wu T, Wang Y, Shen Y. LaGDif: Latent Graph Diffusion Model for Efficient Protein Inverse Folding with Self-Ensemble. In Proceedings of the 2024 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), Lisboa, Portugal, 3–6 December 2024; pp. 3850–3855. doi:10.48550/arXiv.2411.01737.[Google Scholar]
-
Zhu Y, Wu J, Li Q, Yan J, Yin M, Wu W, et al. Bridge-IF: Learning Inverse Protein Folding with Markov Bridges. arXiv 2024, arXiv:2411.02120.[Google Scholar]
-
Ren M, Yu C, Bu D, Zhang H. Accurate and robust protein sequence design with CarbonDesign. Nat. Mach. Intell. 2024, 6, 536–547. doi:10.1038/s42256-024-00838-2.[Google Scholar]
-
Bai P, Miljković F, Liu X, De Maria L, Croasdale-Wood R, Rackham O, et al. Mask-prior-guided denoising diffusion improves inverse protein folding. Nat. Mach. Intell. 2025, 7, 876–888. doi:10.1038/s42256-025-01042-6.[Google Scholar]
-
Vaswani A, Shazeer N, Parmar N, Uszkoreit J, Jones L, Gomez AN, et al. Attention is all you need. Adv. Neural Inf. Process. Syst. 2017, 30. doi:10.48550/arXiv.1706.03762.[Google Scholar]
-
Gilmer J, Schoenholz SS, Riley PF, Vinyals O, Dahl GE. Neural message passing for quantum chemistry. Int. Conf. Mach. Learn. 2017, 1263–1272. doi:10.48550/arXiv.1704.01212.[Google Scholar]
-
Scarselli F, Gori M, Tsoi AC, Hagenbuchner M, Monfardini G. The graph neural network model. IEEE Trans. Neural Netw. 2008, 20, 61–80. doi:10.1109/TNN.2008.2005605.[Google Scholar]
-
Braun M, Tripp A, Chakatok M, Kaltenbrunner S, Totaro M, Stoll D, et al. Computational design of highly active de novo enzymes. bioRxiv 2024. doi:10.1101/2024.08.02.606416.[Google Scholar]
-
Das R, Baker D. Macromolecular Modeling with Rosetta. Annu. Rev. Biochem. 2008, 77, 363–382. doi:10.1146/annurev.biochem.77.062906.171838.[Google Scholar]
-
Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. doi:10.1038/s41586-021-03819-2.[Google Scholar]
-
Yim J, Stärk H, Corso G, Jing B, Barzilay R, Jaakkola TS. Diffusion models in protein structure and docking. WIREs Comput. Mol. Sci. 2024, 14, e1711. doi:10.1002/wcms.1711.[Google Scholar]
-
Nam K, Shao Y, Major DT, Wolf-Watz M. Perspectives on Computational Enzyme Modeling: From Mechanisms to Design and Drug Development. ACS Omega 2024, 9, 7393–7412. doi:10.1021/acsomega.3c09084.[Google Scholar]
-
Childers MC, Daggett V. Insights from molecular dynamics simulations for computational protein design. Mol. Syst. Des. Eng. 2017, 2, 9–33. doi:10.1039/c6me00083e.[Google Scholar]
-
Kiss G, Pande VS, Houk KN. Molecular Dynamics Simulations for the Ranking, Evaluation, and Refinement of Computationally Designed Proteins. Methods Enzymol. 2013, 523, 145–170. doi:10.1016/B978-0-12-394292-0.00007-2.[Google Scholar]
-
Mak WS, Siegel JB. Computational enzyme design: Transitioning from catalytic proteins to enzymes. Curr. Opin. Struct. Biol. 2014, 27, 87–94. doi:10.1016/j.sbi.2014.05.010.[Google Scholar]
-
Anishchenko I, Kipnis Y, Kalvet I, Zhou G, Krishna R, Pellock SJ, et al. Modeling protein-small molecule conformational ensembles with ChemNet. bioRxiv 2024. doi:10.1101/2024.09.25.614868.[Google Scholar]
-
Hu R-E, Yu C-H, Ng IS. GRACE: Generative Redesign in Artificial Computational Enzymology. ACS Synth. Biol. 2024, 13, 4154–4164. doi:10.1021/acssynbio.4c00624.[Google Scholar]
-
Listov D, Vos E, Hoffka G, Hoch SY, Berg A, Hamer-Rogotner S, et al. Complete computational design of high-efficiency Kemp elimination enzymes. Nature 2025, 643, 1421–1427. doi:10.1038/s41586-025-09136-2.[Google Scholar]
-
Goldenzweig A, Goldsmith M, Hill Shannon E, Gertman O, Laurino P, Ashani Y, et al. Automated Structure- and Sequence-Based Design of Proteins for High Bacterial Expression and Stability. Mol. Cell 2016, 63, 337–346. doi:10.1016/j.molcel.2016.06.012.[Google Scholar]
-
Khersonsky O, Lipsh R, Avizemer Z, Ashani Y, Goldsmith M, Leader H, et al. Automated Design of Efficient and Functionally Diverse Enzyme Repertoires. Mol. Cell 2018, 72, 178–186.e175. doi:10.1016/j.molcel.2018.08.033.[Google Scholar]
-
Rakotoharisoa RV, Seifinoferest B, Zarifi N, Miller JDM, Rodriguez JM, Thompson MC, et al. Design of Efficient Artificial Enzymes Using Crystallographically Enhanced Conformational Sampling. J. Am. Chem. Soc. 2024, 146, 10001–10013. doi:10.1021/jacs.4c00677.[Google Scholar]
-
Pan X, Kortemme T. Recent advances in de novo protein design: Principles, methods, and applications. J. Biol. Chem. 2021, 296, 100558. doi:10.1016/j.jbc.2021.100558.[Google Scholar]
-
Ruiz-Pernía JJ, Świderek K, Bertran J, Moliner V, Tuñón I. Electrostatics as a Guiding Principle in Understanding and Designing Enzymes. J. Chem. Theory Comput. 2024, 20, 1783–1795. doi:10.1021/acs.jctc.3c01395.[Google Scholar]
-
Johnson SR, Fu X, Viknander S, Goldin C, Monaco S, Zelezniak A, et al. Computational scoring and experimental evaluation of enzymes generated by neural networks. Nat. Biotechnol. 2025, 43, 396–405. doi:10.1038/s41587-024-02214-2.[Google Scholar]