1. Introduction
CD8⁺ T cells are a specialized subset of lymphocytes that recognize and eliminate cells presenting foreign or abnormal peptides, including pathogen-derived antigens and tumor-associated neoantigens. These peptides are presented on MHC-I (Major Histocompatibility Complex class I) molecules on antigen presenting cells (APC) and recognized by the TCR (T Cell Receptors) on the T cells. Developmentally, immature T cells undergo positive and negative selection in the thymus, where the cells that can recognize MHC molecules (MHC-restricted) and have a moderate reactivity to self-antigens, are chosen to survive and mature [
1]. Such mature, but still naïve CD8 T cells recirculate between the blood stream and secondary lymphoid organs (SLO) such as spleen and lymph nodes to surveil the organism for infections and pathological conditions [
2]. Lymph nodes, in turn, are connected to various peripheral tissues via the lymphatic vessels, which transport metabolites, inflammatory mediators and cells.
Antigen presenting cells (APC), dendritic cells (DC) in particular, migrate in these vessels to carry information about the status of the tissue by integrating and relaying inflammatory cues and antigenic information [
3]. Once in the lymph node, DC can be efficiently accessed by naïve T cells that reach the lymph node via the blood stream [
2]. Thus, the lymph nodes provide a communication hub for the DC and CD8 T cells and are specialized organs for efficient CD8 T cell activation. Once activated by a specific antigen, CD8 T cells start to vigorously proliferate and differentiate into a spectrum of cellular states ranging from effector to stem-like memory cells. Effector T cells are generally short-lived and can be identified by expressing KLRG1 and CX3CR1 [
4]. Their major function is to quickly lyse infected or abnormal cells due to the presence of non-self-antigens on their surface MHC-I molecules. Accordingly, they can effectively and precisely identify and eliminate cancer and infected cells among healthy cells by releasing cytotoxic molecules such as perforin and granzymes. Additionally, they produce various effector cytokines such as interferon gamma (IFN-γ) or tumor necrosis factor (TNF) [
5]. Memory CD8 T cells, on the other hand are long-lived and reside either within lymph nodes where they form a central memory (Tcm) compartment or within tissues, where they reside as tissue resident memory (Trm). Central memory CD8 T cells can be identified by high expression of CCR7 and CD62L, which enables their homing and recirculation between lymph nodes. They serve as self-renewing reservoirs that can produce IL-2, quickly proliferate and differentiate into effector cells upon re-challenge. Tissue resident memory T cells can be identified by CD69 and CD103 expression. They are strategically localized within the sites of previous infections and hence can rapidly respond in case of secondary challenge [
6,
7,
8,
9]. During chronic infections or cancer, an additional CD8 T cell subset, namely exhausted T cells (T
ex), emerge. These again form a spectrum of cellular states ranging from long-lived cells with stem-like features to T
ex cells with a limited capacity to produce inflammatory cytokines. Stem-like cells express CD62L, PD-1 and the transcription factor Tox and are able to proliferate or produce effector cytokines, including IL-2. By contrast, T
ex cells display a markedly reduced ability to secrete cytokines, although their direct cytotoxic activity appears preserved [
10]. These T
ex cells are characterized by permanent expression of inhibitory molecules such as PD-1, TIM-3, and LAG-3 [
11,
12]. Hence, in cancer patients, stem-like CD8 T cells can be activated by checkpoint inhibitors, triggering their proliferation in lymphoid tissues. This process, in turn, has many parallels to the initial priming process of CD8 T cells that regulates the differentiation of both memory and effector T cells [
13,
14,
15]. In following sections, we will review our previous knowledge on the priming of CD8 T and discuss novel concepts and implications based on the recent advances in understanding this process.
2. CD8 T Cell Priming
2.1. Integration of Classical Signals, CD4 Help, DC Licensing and Conceptual Challenges
Optimal priming of CD8 T cells requires the integration of three signals (i) TCR engagement through recognition of peptide–MHC class I complexes, (ii) costimulatory signaling via CD28–CD80/86 interactions, and (iii) inflammatory cues
, such as IL-12 or type I interferons (IFN-I) [
14,
16]. Following this initial activation, CD8 T cells were believed to differentiate autonomously—a concept termed “CD8 T cells on autopilot” by Michael Bevan [
17]. However, evidence from tumor models challenged this view, revealing that the full expansion and differentiation of CD8 T cells into functional CTLs requires an additional component: CD4 helper T cell support [
13,
18,
19]. In vitro studies identified two critical helper signals provided by CD4 T cells: interleukin-2 (IL-2) and CD40 ligand (CD40L) [
13].
In vivo, effective help required CD4 and CD8 T cells to recognize the same antigen presented by the same dendritic cell (DC) [
20]. This presented a significant conceptual challenge: how can the immune system coordinate productive interactions among three distinct and rare cell types—antigen-specific CD4 T cells, CD8 T cells, and appropriate DCs—within the constrained timeframe of an immune response? Key insights into this process emerged with the advent of intravital microscopy, dramatically enhancing our understanding of T cell priming dynamics. This technique revealed how rapidly T cells migrate, how they navigate the lymph node microenvironment, and how many T cells a single DC can engage per hour. When antigen-specific T cells encounter cognate peptide-presenting DCs, they typically form stable interactions lasting up to 24 h, after which they disengage and begin to proliferate. However, considering the extremely low precursor frequencies of naïve CD4 and CD8 T cells (estimated at 1 in 10
5–10
6), it became evident that stable tri-cellular (CD4–CD8–DC) interactions are statistically unlikely. This led to hypotheses involving chemoattractant signaling to promote localized cell clustering. Yet, the concept of stable three-cell interactions was ultimately set aside when studies demonstrated that CD4 and CD8 T cells are typically primed by distinct DC subsets following viral infections [
20,
21]. Moreover, the timing of their activation is often asynchronous: for instance, CD8 T cells are activated earlier than CD4 T cells in vaccinia virus infections, whereas the reverse is observed in HSV infection models [
20,
21].
An explanation to this paradox came from the work of Matzinger’s group, who showed that CD4 T cells can “license” DCs through CD40L–CD40 interactions. Once licensed, these DCs are empowered to prime CD8 T cells subsequently, allowing for sequential rather than simultaneous interactions [
22]. This model also explained why CD4 depletion profoundly impairs anti-tumor CD8 T cell responses, yet has minimal effects on primary antiviral responses. In viral infections, the presence of PAMPs and high levels of inflammation can compensate for the absence of CD4 help by directly activating DCs. However, CD4 T cell help remains essential for effective memory formation even in viral settings. Multiple studies have since confirmed the critical role of CD40L-mediated DC licensing, which is carried out exclusively by the DC1 subset. The emerging model can be summarized as follows: CD4 and CD8 T cells are primed independently through prolonged interactions with antigen-bearing DCs. After this priming phase, both populations resume motility, allowing CD4 T cells to interact with DC1s, delivering CD40L-mediated licensing signals. Licensed DC1s subsequently engage CD8 T cells, delivering the necessary secondary signals for full CTL differentiation. But how does IL-2 fit into this picture? How can paracrine signals be transmitted effectively if the cells involved are actively travelling through the lymph node? Another unresolved issue emerged: depletion of DC1 during vaccinia virus infection significantly impaired the formation of memory CD8 T cell responses, yet had minimal impact on the acute effector phase. If DC licensing is indeed restricted to the DC1 subset, how can this be reconciled with the observation that CD4 T cells are critical for driving effector responses in tumors, but appear largely dispensable during the primary response to viral infections?
2.2. Solving the Conundrum—A Second Priming Phase in a Specialized Niche
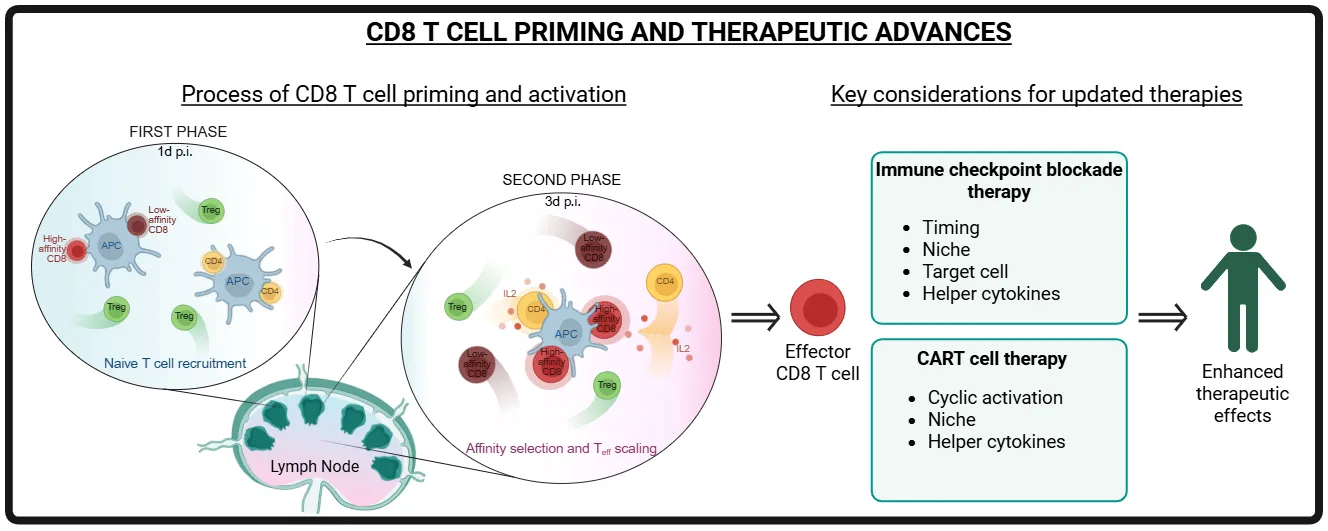
Our recent findings solved this conundrum by detailed investigation of later events during CD8 T cell priming. Using intravital microscopy on days 2 and 3 post vaccinia virus infection, we accessed deeper lymph node regions. We discovered that CD8 T cells regained their responsiveness and reengaged with DC for a second time for several hours. After the CD8 T cells were activated for the first time (first phase), they proliferated and upregulated CXCR3, allowing them to access the secondary subfollicular niche and form long-lasting interactions with DC [
23].
Once CD8 T cells re-cluster with DC, they obtain additional TCR stimulation and receive IL-2 from CD4 helper T cells that transiently stop at this niche. By developing a novel mouse model that allowed for selective depletion of CD4 helper T cells while sparing regulatory T cells, we found that CD4 help is essential for CD8 T cell effector generation during viral infections. Thus, the delivery of IL-2 by CD4 T helper cells requires their concomitant, yet transient, presence at the site of DC-CD8 T cell interaction. Notably, the presence of DC1 is dispensable for the relay of IL-2 and thus CD8 T cell effector generation [
23]. Importantly, CD4 T cells at the second phase of priming delivered their help predominantly to high affinity T cells, as CD8 T cells with low affinity could not access the secondary niche. As a consequence, they did not efficiently expand to form effector cells one week post infection [
23].
In summary, while the first priming phase activates a broad range of antigen-specific CD8 T cells, the second priming phase selectively expands high affinity CD8 T cells but also orchestrates DC-licensing and thus optimal memory formation. To promote these events, activated CD8 T cells re-cluster with DC to receive an additional round of TCR signaling as well as IL-2 from CD4 T helper cells. Subsequently, these high affinity T cells efficiently differentiate into effector cells with cytotoxic properties.
These results raise an important question: If optimal effector T cell differentiation and selection during the priming process requires specialized niches, does a similar requirement exist during chronic activation, such as in cancer or unresolved infections? Available data suggest that optimal CD8 T cell effector differentiation during chronic infection requires continuous CD4 T cell help, not only during the initial priming phase but also throughout the response [
24,
25,
26,
27]. We hypothesize that the fundamental principles during chronic conditions do resemble those of priming—such as CD8 T cell arrest and transient interactions with CD4 T cells to enable paracrine signaling—yet important details may differ, including the participating cell types and their phenotype as well as the local cytokine milieu. Indeed, CD4 T cells have been shown to produce IL-21, rather than IL-2, to drive the differentiation of the cytolytic CX3CR1 effector population during chronic viral infection. Currently, the spatiotemporal dynamics of IL-21 delivery to CD8 T cells in this setting and the precise composition of the local niche remain to be elucidated but could provide important new angles for therapeutic intervention.
The situation in chronic infection differs from that in the tumor context, where the extent and nature of CD4 T cell help vary considerably between tumor entities. An especially intriguing aspect is that, in tumors, CD4 T cells seem to provide help not only in the lymph node but also directly within the tumor microenvironment—the effector site itself [
28]. The precise role of CD4 T cell help in the tumor setting remains incompletely understood, but it seems to contribute to the complete differentiation of CD8 effector T cells and support their survival [
29,
30]. However, the quality of such help could be limited due to suppressive signals in the tumor microenvironment. Thus, there is likely a complex interplay between dendritic cells, metabolic factors, local suppressive cues and stromal cells that critically shape the CD8 T cell differentiation within the tumor niche. Following the initial priming of CD8 T cell, if a subsequent second signal occurs at the effector site, it confines these cells within the tumor tissue, leading them towards a systematic loss of function resulting in exhaustion [
31]. In line with this, recent spatial data have highlighted spatially coordinated interactions between DC1-CD8 T cell subpopulations within tumor and how they might shape the further differentiation of CD8 T cells [
32]. Another lung carcinoma model study shows the importance of efficient TCR engagement with DCs to form and maintain the stem-like population to provide anti-tumor immunity [
33]. Additionally, the presence of metabolites like methionine could shape the differentiation of T cells following TCR engagement, highlighting the role of microenvironment [
34]. Multiple factors influencing CD8 T cell differentiation in the tumor and its local microenvironment have been identified, but their precise mechanism and coordination remain fully understood.
2.3. Cyclical Priming of CD8⁺ T Cells and the Path to Optimal Function or Dysfunction
Hitherto, it was believed that in acute infections, CD8 T cells cluster with antigen presenting cells just once to get activated, after which they proliferate without the need of further stimulation [
17,
35,
36]. However, our recent study about the later events in T cell priming showed that a second activation step is crucial for optimal CD8 T cell response. It is thus conceivable that CD8 T cells go through multiple cycles of TCR de- and re-sensitization and subsequent activation, including migrational arrest and retention within specific niches. The duration of these cycles could differ depending on TCR affinity, where high-affinity CD8 T cells undergoing shorter cycling times. This raises important questions with potential therapeutic implications: If CD8 T cell priming occurs in cycles, at what point does this process shift into exhaustion? Is this transition driven purely by quantitative factors (e.g., number of cycles) or also by qualitative changes in the activation environment? If so, what are these factors, and can they be therapeutically targeted? During persistent infections, the continuous activation of CD8 T cells favors a hierarchical loss of function, leading towards functionally exhausted populations rather than robust effectors. This shift is marked by high expression of inhibitory markers like PD-1, Lag3, Tim-3 and lower cytokine and chemokine polyfunctionality [
11,
37]. In addition to continuous antigen stimulation, the lack of CD4 help and suppressive signals from inhibitory molecules contribute to the dysfunction [
11]. These data support a quantitative model in which CD8 T cell dysfunction progressively increases with successive activation cycles and ultimately becomes fixed. However, the process of T cell exhaustion appears to differ markedly between chronic infection and cancer. In the tumor context, the induction of exhaustion and T cell dysfunction has been observed to be initiated during the initial contact between T cells and DC presenting tumor antigens [
38]. Inflammatory cues that activate DC and T cells are an important factor here. It will therefore be crucial to distinguish tumors according to their inflammatory environments, both within the tumor itself and in associated lymphoid structures, to enable the development of tailored therapeutic strategies.
In summary, T cell activation proceeds in cycles during both acute infection and chronic stimulation. The surrounding microenvironment plays a decisive role in determining whether CD8 T cells undergo effective differentiation or progress toward dysfunction. Defining the precise localization, cellular composition, and molecular principles of these environments will be essential for optimizing CD8 T cell activation and advancing immunotherapy.
3. Implications for Therapy
3.1. Mechanisms of Immune Checkpoint Blockade and Strategies for Optimization
As CD8 T cells get activated to become cytotoxic, numerous mechanisms are employed by the immune system to avert collateral tissue damage by an uncontrolled response of these cells. Among such mechanisms are immune checkpoint molecules, which primarily interfere with TCR signaling and costimulation and thereby increase the activation threshold and limit the reactivity of T cells. Molecules already targeted in the clinics include PD-L1/2 interactions with PD-1, CD80/CD86 with CTLA4, GAL9 with Tim3, MHCII with LAG3 and others [
39]. It was initially thought that the efficacy of immune checkpoint blockade (ICB) stemmed from the reinvigoration of T
ex cells, leading to enhanced effector cytokine production within the tumor microenvironment [
40]. Preclinical studies, however, demonstrate that the critical mode of action occurs at the interface between stem-like T cells and DC within lymphoid tissues, where these interactions drive T cell proliferation and expand the pool of T
ex cells [
41,
42,
43]. Accordingly, also patients with PD-L1 negative tumors benefited from PD-1/PD-L1 blockade [
44,
45,
46,
47,
48]. Nevertheless, the level of PD-L1 expression in patient tumors may carry significance, since it mirrors the local inflammatory state and correlates with immune cell infiltration.
As outlined above it is very likely that during the reinvigoration of stem-like T cells in the context of cancer and chronic infections similar rules apply as during the second priming phase that we recently discovered. In particular, there is a requirement for cytokines like IL-2 or IL-21. Consistently, it was shown that IL-2 synergizes with PD-1 therapy and promotes the generation of highly effective cytotoxic effector cells [
49,
50]. An important future challenge is how to deliver cytokines such as IL-2, IL-21, or other common γ-chain cytokines locally into the niche between dendritic cells and stem-like CD8 T cells in combination with checkpoint blockade. One potential strategy would be to fuse cytokines to antibodies to achieve targeted release, as recently shown [
51]. We see considerable potential in optimizing the timing of checkpoint therapy. As noted above, CD8 T cell reactivity is cyclical, suggesting that checkpoint-mediated activation should be aligned with these cycles. Our preclinical data indicate that, during the priming phase of high-affinity T cells, the cycle lasts approximately 2–3 days. Defining the cycle length of polyclonal, tumor-reactive CD8 T cell populations in humans will be critical for modeling the optimal timing of antibody administration. The use of short-lived antibodies may further enhance control over the duration of checkpoint activity.
3.2. CAR T Cell Therapy—Mechanism of Action and Limitations
Chimeric antigen receptor (CAR) T cell therapy was developed to overcome the problem of MHC class I restriction. This was achieved by fusing the antigen-recognition domain of an antibody, typically a single-chain variable fragment (scFv), with the intracellular signaling machinery of a T cell. In addition to the CD3ζ signaling domain, CAR constructs often incorporate costimulatory domains, most commonly derived from CD28 or 4-1BB, to enhance T cell activation, proliferation, and persistence [
52,
53]. As a result, CAR T cells can directly recognize and bind tumor-associated antigens expressed on the surface of malignant cells, thereby eliminating target cells in an MHC-independent manner. CAR T cells face several challenges despite their clinical success in hematological malignancies. Antigen escape, on-target/off-tumor toxicity, limited efficacy in solid tumors, and issues related to persistence and exhaustion remain major obstacles. Ongoing efforts therefore focus on improving CAR design, introducing logic-gated or dual-targeting receptors to reduce antigen escape, engineering safety switches to enhance controllability, and modifying T cells to better overcome the immunosuppressive tumor microenvironment. These innovations aim to extend the transformative potential of CAR T therapy beyond lymphomas to a broader range of malignancies.
An often overlooked aspect of CAR T cell therapy is that CAR T cells not only act as immediate effectors but also need to persist as a stem-like population within lymphoid tissues. This stem-like compartment is essential for continuous activation and the generation of new effector cells, thereby enabling a sustained anti-tumor response. For this process, CAR T cells must encounter their cognate antigen within lymphoid tissues and receive the optimal signals, including cytokines such as IL-2—again a process likely similar to the second priming phase. This requirement is often fulfilled in B cell malignancies since the malignant cells reside in lymphoid organs. In contrast, in solid tumors, the relevant antigen is typically absent from lymphoid sites, unless lymph node metastases are present. However, these metastatic lymph nodes are frequently functionally compromised, limiting their capacity to support the maintenance of stem-like CAR T cells [
54]. The absence of detectable antigen for CAR T cells in the tumor-draining lymph nodes of solid tumors has been addressed by Irvine’s team, who combined CAR T cell therapy with a vaccination strategy to reactivate CARs. In this approach, synthetic nanoparticles presenting the CAR target antigen were used as a vaccine, enabling antigen recognition in lymphoid tissues, boosting CAR T cell expansion, and enhancing their persistence and anti-tumor activity [
55]. To further supply IL-2 within a proliferation-supporting niche for CAR T cells, others have developed an elegant synNotch-based approach. In this strategy, CAR engagement triggers autocrine IL-2 production, promoting optimal proliferation and enhancing tumor control [
56]. Other approaches attempted to create a structured immune environment mimicking lymphoid tissues: CAR T cells that secrete IL-7 and CCL19 were shown to decrease tumor burden significantly [
57].
Another missing, or rather insufficient signal, comes from the receptor itself, as CARs have lower antigen sensitivity than native TCRs, and the signal duration is less sustained [
58]. Recent studies on Synthetic TCR and Antigen Receptors (STAR) use receptors that resemble native TCRs more closely. Consequently, STAR T cells appear to be more effective than CAR T cells [
52]. Despite these advances, we consider the induction of CAR T cell proliferation and differentiation within lymphoid organs to be a central challenge for achieving success against solid tumors.
As highlighted above, cyclic antigen exposure accompanied by appropriate costimulatory signals promotes efficient effector function, and we assume that such periodic activation is critical for preserving the capacity of stem-like T cells. In contrast, CAR T cells frequently encounter tonic signaling through their receptors, which leads to functional exhaustion and impaired persistence. Hence, periodic cessation of tonic CAR signaling through a pharmacological off-switch, such as dasatinib, promotes the acquisition of a memory-like phenotype and prevents exhaustion [
52]. Such pharmacological ON/OFF switches provide temporal control over CAR signaling and may help to better mimic physiological patterns of antigen encounter.
Whether in immune checkpoint blockade or CAR T cell therapy, the optimal activation, proliferation, and differentiation of T cells clearly depend on dedicated niches that provide not only the necessary signals but also critical nutrients. Equally important, successful strategies must align the timing of these signals with the cyclic nature of physiological T cell activation to maximize therapeutic benefit. Unraveling the nature of these niches and the rules that govern them will provide a foundation for the rational design of improved cancer immunotherapies. Pursuing this line of investigation holds promise for advancing cellular therapies across a broad spectrum of diseases, including cancer, autoimmunity, and viral infections.
Author Contributions
Writing—Review & Editing: K.J., D.S. and W.K.
Ethics Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Funding
W.K. is supported by the Max Planck Society (Max Planck Research Groups), the German Research Foundation (DFG) (CRC/TRR 338 Nr. 452881907 and CRC1583 Nr. 492620490) and the European Research Council (ERC) W.K. (819329-STEP2).
Declaration of Competing Interest
The authors declare that they have no competing interests.
References
-
1.
Ashby KM, Hogquist KA. A guide to thymic selection of T cells.
Nat. Rev. Immunol. 2023,
23, 697–697. doi:10.1038/s41577-023-00911-8.
[Google Scholar]
-
2.
Miller MJ, Wei SH, Cahalan MD, Parker I. Autonomous T cell trafficking examined in vivo with intravital two-photon microscopy.
Proc. Natl. Acad. Sci. USA 2003,
100, 2604–2609.
[Google Scholar]
-
3.
Martín-Fontecha A, Sebastiani S, Höpken UE, Uguccioni M, Lipp M, Lanzavecchia A, et al. Regulation of dendritic cell migration to the draining lymph node: impact on T lymphocyte traffic and priming.
J. Exp. Med. 2003,
198, 615–621.
[Google Scholar]
-
4.
Gerlach C, Moseman EA, Loughhead SM, Alvarez D, Zwijnenburg AJ, Waanders L, et al. The Chemokine Receptor CX3CR1 Defines Three Antigen-Experienced CD8 T Cell Subsets with Distinct Roles in Immune Surveillance and Homeostasis.
Immunity 2016,
45, 1270–1284.
[Google Scholar]
-
5.
Rausch L, Kallies A. Molecular Mechanisms Governing CD8 T Cell Differentiation and Checkpoint Inhibitor Response in Cancer.
Annu. Rev. Immunol. 2025,
43, 515–543.
[Google Scholar]
-
6.
Sallusto F, Lenig D, Förster R, Lipp M, Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions.
Nature 1999,
401, 708–712.
[Google Scholar]
-
7.
Bachmann MF, Wolint P, Schwarz K, Jäger P, Oxenius A. Functional Properties and Lineage Relationship of CD8
+ T Cell Subsets Identified by Expression of IL-7 Receptor α and CD62L1.
J. Immunol. 2005,
175, 4686–4696.
[Google Scholar]
-
8.
Martin MD, Badovinac VP. Defining Memory CD8 T Cell.
Front. Immunol. 2018,
9, 2692.
[Google Scholar]
-
9.
Crowl JT, Heeg M, Ferry A, Milner JJ, Omilusik KD, Toma C, et al. Tissue-resident memory CD8
+ T cells possess unique transcriptional, epigenetic and functional adaptations to different tissue environments.
Nat. Immunol. 2022,
23, 1121–1131.
[Google Scholar]
-
10.
Sandu I, Cerletti D, Claassen M, Oxenius A. Exhausted CD8
+ T cells exhibit low and strongly inhibited TCR signaling during chronic LCMV infection.
Nat. Commun. 2020,
11, 4454.
[Google Scholar]
-
11.
Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion.
Nat. Rev. Immunol. 2015,
15, 486–499.
[Google Scholar]
-
12.
Giles JR, Globig AM, Kaech SM, Wherry EJ. CD8
+ T cells in the cancer-immunity cycle.
Immunity 2023,
56, 2231–2253.
[Google Scholar]
-
13.
Busselaar J, Tian S, van Eenennaam H, Borst J. Helpless Priming Sends CD8
+ T Cells on the Road to Exhaustion.
Front. Immunol. 2020,
11, 592569.
[Google Scholar]
-
14.
Cui W, Joshi NS, Jiang A, Kaech SM. Effects of Signal 3 during CD8 T cell priming: Bystander production of IL-12 enhances effector T cell expansion but promotes terminal differentiation.
Vaccine 2009,
27, 2177–2187.
[Google Scholar]
-
15.
Snell LM, MacLeod BL, Law JC, Osokine I, Elsaesser HJ, Hezaveh K, et al. CD8
+ T cell priming in established chronic viral infection preferentially directs differentiation of memory-like cells for sustained immunity.
Immunity 2018,
49, 678–694.e5.
[Google Scholar]
-
16.
Welten SP, Melief CJ, Arens R. The distinct role of T cell costimulation in antiviral immunity.
Curr. Opin. Virol. 2013,
3, 475–482.
[Google Scholar]
-
17.
Bevan MJ, Fink PJ. The CD8 response on autopilot.
Nat. Immunol. 2001,
2, 381–382.
[Google Scholar]
-
18.
Borst J, Ahrends T, Babala N, Melief CJM, Kastenmuller W. CD4(+) T cell help in cancer immunology and immunotherapy.
Nat. Rev. Immunol. 2018,
18, 635–647.
[Google Scholar]
-
19.
Ossendorp F, Mengedé E, Camps M, Filius R, Melief CJ. Specific T helper cell requirement for optimal induction of cytotoxic T lymphocytes against major histocompatibility complex class II negative tumors.
J. Exp. Med. 1998,
187, 693–702.
[Google Scholar]
-
20.
Eickhoff S, Brewitz A, Gerner MY, Klauschen F, Komander K, Hemmi H, et al. Robust Anti-viral Immunity Requires Multiple Distinct T Cell-Dendritic Cell Interactions.
Cell 2015,
162, 1322–1337.
[Google Scholar]
-
21.
Hor JL, Whitney PG, Zaid A, Brooks AG, Heath WR, Mueller SN. Spatiotemporally distinct interactions with DC subsets facilitates CD4 and CD8 T cell activation to localized viral infection.
Eur. J. Immunol. 2016,
46, 56.
[Google Scholar]
-
22.
Ridge JP, Di Rosa F, Matzinger P. A conditioned dendritic cell can be a temporal bridge between a CD4+ T-helper and a T-killer cell.
Nature 1998,
393, 474–478.
[Google Scholar]
-
23.
Jobin K, Seetharama D, Rüttger L, Fenton C, Kharybina E, Wirsching A, et al. A distinct priming phase regulates CD8 T cell immunity by orchestrating paracrine IL-2 signals.
Science 2025,
388, eadq1405.
[Google Scholar]
-
24.
Tian Y, Zajac AJ. IL-21 and T cell differentiation: consider the context.
Trends Immunol. 2016,
37, 557–568.
[Google Scholar]
-
25.
Zander R, Kasmani MY, Chen Y, Topchyan P, Shen J, Zheng S, et al. Tfh cell-derived interleukin 21 sustains Effector CD8+ T cell responses during chronic viral infection.
Immunity 2022,
55, 475–493.e5.
[Google Scholar]
-
26.
Isvoranu G, Chiritoiu-Butnaru M. Therapeutic potential of interleukin-21 in cancer.
Front. Immunol. 2024,
15, 1369743.
[Google Scholar]
-
27.
Asao H. Interleukin-21 in Viral Infections.
Int. J. Mol. Sci. 2021,
22, 9521.
[Google Scholar]
-
28.
Prokhnevska N, Cardenas MA, Valanparambil RM, Sobierajska E, Barwick BG, Jansen G, et al. CD8+T cell activation in cancer comprises an initial activation phase in lymph nodes followed by effector differentiation within the tumor.
Immunity 2023,
56, 107–124.
[Google Scholar]
-
29.
Di Pilato M, Kfuri-Rubens R, Pruessmann JN, Ozga AJ, Messemaker M, Cadilha BL, et al. CXCR6 positions cytotoxic T cells to receive critical survival signals in the tumor microenvironment.
Cell 2021,
184, 4512–4530.
[Google Scholar]
-
30.
Schietinger A, Philip M, Liu RB, Schreiber K, Schreiber H. Bystander killing of cancer requires the cooperation of CD4 and CD8 T cells during the effector phase.
J. Exp. Med. 2010,
207, 2469–2477.
[Google Scholar]
-
31.
Takahashi M, So TY, Chamberlain-Evans V, Hughes R, Yam-Puc JC, Kania K, et al. Intratumoral antigen signaling traps CD8(+) T cells to confine exhaustion to the tumor site.
Sci. Immunol. 2024,
9, eade2094.
[Google Scholar]
-
32.
Piot C, Pereira da Costa M, Biram A, Minutti CM, Lim KHJ, Green M, et al. Spatial Organisation of Tumour cDC1 States Correlates with Effector and Stem-Like CD8(+) T Cells Location.
Eur. J. Immunol. 2025,
55, e70011.
[Google Scholar]
-
33.
Lan X, Mi T, Alli S, Guy C, Djekidel MN, Liu X, et al. Antitumor progenitor exhausted CD8(+) T cells are sustained by TCR engagement.
Nat. Immunol. 2024,
25, 1046–1058.
[Google Scholar]
-
34.
Sharma P, Guo A, Poudel S, Boada-Romero E, Verbist KC, Palacios G, et al. Early methionine availability attenuates T cell exhaustion.
Nat. Immunol. 2025,
26, 1384–1396.
[Google Scholar]
-
35.
van Stipdonk MJ, Lemmens EE, Schoenberger SP. Naïve CTLs require a single brief period of antigenic stimulation for clonal expansion and differentiation.
Nat. Immunol. 2001,
2, 423–429.
[Google Scholar]
-
36.
Kaech SM, Ahmed R. Memory CD8 T cell differentiation: initial antigen encounter triggers a developmental program in naive cells.
Nat. Immunol. 2001,
2, 415–422.
[Google Scholar]
-
37.
Wherry EJ. T cell exhaustion.
Nat. Immunol. 2011,
12, 492–499.
[Google Scholar]
-
38.
Rudloff MW, Zumbo P, Favret NR, Roetman JJ, Detres Roman CR, Erwin MM, et al. Hallmarks of CD8(+) T cell dysfunction are established within hours of tumor antigen encounter before cell division.
Nat. Immunol. 2023,
24, 1527–1539.
[Google Scholar]
-
39.
Guo Z, Zhang R, Yang AG, Zheng G. Diversity of immune checkpoints in cancer immunotherapy.
Front. Immunol. 2023,
14, 1121285.
[Google Scholar]
-
40.
Immune Checkpoint Inhibitors—NCI. 2019. Available online: https://www.cancer.gov/about-cancer/treatment/types/immunotherapy/checkpoint-inhibitors (accessed on 9 April 2025).
-
41.
Peng Q, Qiu X, Zhang Z, Zhang S, Zhang Y, Liang Y, et al. PD-L1 on dendritic cells attenuates T cell activation and regulates response to immune checkpoint blockade.
Nat. Commun. 2020,
11, 4835.
[Google Scholar]
-
42.
Ruiz-Torres DA, Wise JF, Zhao BY, Oliveira-Costa JP, Cavallaro S, Sadow PM, et al. Dendritic cell effector mechanisms and tumor immune microenvironment infiltration define TLR8 modulation and PD-1 blockade.
Front. Immunol. 2024,
15, 1440530.
[Google Scholar]
-
43.
Gardner A, de Mingo Pulido Á, Ruffell B. Dendritic Cells and Their Role in Immunotherapy.
Front. Immunol. 2020,
11, 924.
[Google Scholar]
-
44.
Zhao B, Zhao H, Zhao J. Efficacy of PD-1/PD-L1 blockade monotherapy in clinical trials.
Ther. Adv. Med. Oncol. 2020,
12, 1758835920937612.
[Google Scholar]
-
45.
Brahmer J, Reckamp KL, Baas P, Crinò L, Eberhardt WEE, Poddubskaya E, et al. Nivolumab versus Docetaxel in Advanced Squamous-Cell Non-Small-Cell Lung Cancer.
N. Engl. J. Med. 2015,
373, 123–135.
[Google Scholar]
-
46.
Carlino MS, Long GV, Schadendorf D, Robert C, Ribas A, Richtig E, et al. Outcomes by line of therapy and programmed death ligand 1 expression in patients with advanced melanoma treated with pembrolizumab or ipilimumab in KEYNOTE-006: A randomised clinical trial.
Eur. J. Cancer 2018,
101, 236–243.
[Google Scholar]
-
47.
Fuchs CS, Doi T, Jang RW. Safety and efficacy of pembrolizumab monotherapy in patients with previously treated advanced gastric and gastroesophageal junction cancer: Phase 2 clinical KEYNOTE-059 trial.
JAMA Oncol. 2018,
4, e180013.
[Google Scholar]
-
48.
Mehra R, Seiwert TY, Gupta S, Weiss J, Gluck I, Eder JP, et al. Efficacy and safety of pembrolizumab in recurrent/metastatic head and neck squamous cell carcinoma: Pooled analyses after long-term follow-up in KEYNOTE-012.
Br. J. Cancer 2018,
119, 153–159.
[Google Scholar]
-
49.
Hashimoto M, Araki K, Cardenas MA, Li P, Jadhav RR, Kissick HT, et al. PD-1 combination therapy with IL-2 modifies CD8 T cell exhaustion program.
Nature 2022,
610, 173–181.
[Google Scholar]
-
50.
West EE, Jin HT, Rasheed AU, Penaloza-MacMaster P, Ha SJ, Tan WG, et al. PD-L1 blockade synergizes with IL-2 therapy in reinvigorating exhausted T cells.
J. Clin. Investig. 2013,
123, 2604–2615.
[Google Scholar]
-
51.
Tichet M, Wullschleger S, Chryplewicz A, Fournier N, Marcone R, Kauzlaric A, et al. Bispecific PD1-IL2v and anti-PD-L1 break tumor immunity resistance by enhancing stem-like tumor- reactive CD8+T cells and reprogramming macrophages.
Immunity 2023,
56, 162–179.
[Google Scholar]
-
52.
Yu L, Liu Y, Lin X. Transitioning from native to synthetic receptors: broadening T-cell engineering and beyond.
Cell. Mol. Immunol. 2025,
22, 712–729.
[Google Scholar]
-
53.
Zhu L, Liu J, Li J, Wang N, Zhao Y, Su H. Research progress on HER2-specific chimeric antigen receptor T cells for immunotherapy of solid tumors.
Front. Immunol. 2025,
16, 1514994.
[Google Scholar]
-
54.
Rahim MK, Okholm TLH, Jones KB, McCarthy EE, Liu CC, Yee JL, et al. Dynamic CD8+T cell responses to cancer immunotherapy in human regional lymph nodes are disrupted in metastatic lymph nodes.
Cell 2023,
186, 1127–1143.
[Google Scholar]
-
55.
Ma LY, Hostetler A, Morgan DM, Maiorino L, Sulkaj I, Whittaker CA, et al. Vaccine-boosted CAR T crosstalk with host immunity to reject tumors with antigen heterogeneity.
Cell 2023,
186, 3148–3165.
[Google Scholar]
-
56.
Allen GM, Frankel NW, Reddy NR, Bhargava HK, Yoshida MA, Stark SR, et al. Synthetic cytokine circuits that drive T cells into immune-excluded tumors.
Science 2022,
378, eaba1624.
[Google Scholar]
-
57.
Duan DM, Wang KK, Wei C, Feng DD, Liu YH, He QY, et al. The BCMA-Targeted Fourth-Generation CAR-T Cells Secreting IL-7 and CCL19 for Therapy of Refractory/Recurrent Multiple Myeloma.
Front. Immunol. 2021,
12, 609421.
[Google Scholar]
-
58.
Salter AI, Rajan A, Kennedy JJ, Ivey RG, Shelby SA, Leung I, et al. Comparative analysis of TCR and CAR signaling informs CAR designs with superior antigen sensitivity and
in vivo function.
Sci. Signal. 2021,
14, eabe2606.
[Google Scholar]
 Deeksha Seetharama
1,†
Deeksha Seetharama
1,†