1. Introduction
Neuroinflammation is characterised by abnormal inflammation or an adaptive immune response against self-antigens in the central nervous system (CNS) that impairs neuronal activity [
1,
2,
3]. These conditions can manifest with a wide range of neurological and psychosis symptoms, varying greatly in presentation and severity. Among these, the autoimmune encephalitides have emerged as a prime example of how autoantibody-mediated processes are linked to severe psychosis manifestations, often mimicking primary psychotic disorders [
1].
The discovery of specific antigenic targets for autoimmune encephalitis has revolutionized the field of clinical neurology. With anti-N-methyl-D-aspartate (NMDA) receptor encephalitis being one of the most well characterized disorders, further research has identified and delineated a range of neuropsychiatric entities like LGI1, CASPR2, GABAR,
etc. [
4,
5,
6], and the association between autoantibodies and psychosis symptoms may expand our mechanistic understanding of mental disorders. Current treatment approaches for autoimmune encephalitis center around humoral mechanisms, such as plasma exchange (PLEX) and intravenous immunoglobulin (IVIG), to reduce the antibody titre from circulation. In contrast, other CNS autoimmune diseases and various animal models have shown that T cells, brain resident microglia, and astrocytes play a critical role in disease development.
In this review, we define psychosis symptom related diseases irrespective of neurological or psychosis diagnostic origin in the clinic. By synthesizing research on both humoral and cellular immune mechanisms underlying psychosis symptoms of suspected autoimmune origin, this review aims to contribute to the evolving conceptualization of these disorders. This integrated approach may help bridge the current gap between clinical observations and mechanistic understanding.
2. Autoimmunity and Psychosis Disorder
Firstly, Autoimmune encephalitis-related psychosis is not yet a formally established diagnostic category in the clinic, in contrast, Autoimmune neuropsychiatric disorders is widely accepted terminology. In this review, we are using the term ‘autoimmune encephalitis-related psychosis’ to describe conditions where the body’s immune system attacks the brain, leading to mental health issues. These psychosis symptoms might show up alongside typical neurological problems, or they might be the only visible sign that something’s wrong, but not clear neurological problems. While autoimmune encephalitis with psychosis manifestation represents a clear disease entity, we acknowledge that the concept of autoimmune encephalitis-related psychosis without neurological involvement requires further validation.
Autoimmunity involves the immune system attacking the body’s own tissues. The field of autoimmune research began in the 1940s with the discovery of antinuclear antibodies in patients with Systemic Lupus Erythematosus (SLE) [
7]. This led to a focus on B cells and autoantibody (autoreactive antibody) detection in clinical studies. In contrast, autoreactive T cell research in the clinic was initially less developed due to technical challenges in studying their antigen specificity. We are going to elaborate on the humoral and cellular autoimmunity in the later paragraphs.
Two major caveats challenge the initial hypothesis that neuronal autoimmunity induces psychosis. First, the CNS, including the brain, was traditionally considered immune-privileged, making it less likely to be targeted by the immune system [
8,
9]. However, increasing evidence has proven that the brain has its own lymphoid organs, like deep cervical lymph nodes and meninges, and maintains a tight communication with the peripheral immune system. Secondly, psychosis is often classified as a mental health disorder rather than being recognized as a manifestation of physical or neurological conditions [
10]. Patients with autoimmune encephalitis frequently present with psychosis, complicating the differentiation from schizophrenia and providing direct evidence linking autoimmunity to psychosis. The discovery of autoimmune encephalitis has significantly contributed to the notion of autoimmune psychosis. Notably, as early as 1938, a case of cerebellar degeneration was reported in a patient with ovarian cancer [
11]. Similarly, in 1968, a lung cancer patient exhibited distinct neurological symptoms, such as hallucinations and seizures, without evidence of brain metastases, leading to the identification of “limbic encephalitis” [
12]. Therefore, there is a long history in the clinic for autoimmunity and psychosis association.
A major advancement in understanding the antigen specificity in these patients occurred in 2007 with the identification of NMDAR (N-methyl-D-aspartate receptor) as a neuronal antigen associated with psychosis symptoms in ovarian cancer patients [
13]. Following this, anti-NMDAR autoreactive B cells and antibodies or oligoclonal bands were confirmed in patients’ CSF with autoimmune encephalitis, establishing anti-NMDAR autoimmune encephalitis as a recognized neurological disorder [
14,
15]. In a 1114 schizophrenia patient cohort study, 3–4% patients also tested positive for serum anti-NMDAR [
16]. It has been proposed that anti-NMDAR antibody may cause both encephalitis and psychosis as immunopsychiatric continuum factor [
17]. The scope of autoimmunity pathogenicity was later extended to other neuronal surface antigens, including CASPR [
18], AMPAR [
19], and others. This autoimmune encephalitis typically presents with both neurological and psychosis symptoms. However, there is increasing recognition of a subgroup of patients who test serum or cerebrospinal fluid positive for antibodies against neuronal protein but exhibit minimal or no neurological symptoms. Based on these findings, scientists have proposed the term “autoimmune psychosis” to define patients whose psychosis symptoms may be caused by autoimmunity [
1]. This review does not suggest that all primary psychosis disorders share an autoimmune etiology with conditions like NMDAR encephalitis. Rather, we examine how understanding immunopathogenic mechanisms across the spectrum of autoimmune neuropsychiatric conditions may inform our understanding of immune contributions to psychosis symptoms more broadly.
3. The Antineuronal Autoimmunity in Peripheral and Central Nerval System
In contrast to the well-documented presence of immune cells in the central nervous system (CNS) and the efficacy of immunotherapy, the mechanisms underlying the occurrence of autoimmunity in the brain remain intriguing. CNS autoimmunity may originate either peripherally or within the CNS itself. However, it is uncertain whether a distinct boundary exists between these origins.
The association between autoimmune encephalitis and peripheral conditions such as cancer or teratomas suggests that autoimmunity can traverse from the periphery to the CNS [
20,
21]. Notably, neuronal protein expression within teratoma tissues can induce antineuronal autoimmunity, with autoantibodies or autoreactive immune cells migrating to the CNS and causing damage. This is further supported by clinical observations where the resolution of teratomas correlates with the remission of psychosis symptoms in patients [
20], indicating a peripheral origin of autoimmunity that later impacts the CNS. In addition, clinical cases have also shown anti-neuronal antibody, neurofascin 186 antibodies, in the peripheral can be associated with CNS [
22], which also supports the immune cells peripheral entry to CNS. Autoreactive cells can infiltrate the CNS via several routes, including the blood-brain barrier, choroid plexus, meninges, or the olfactory pathway [
23]. The efficacy of anti-VLA-4 antibody treatment, specifically natalizumab, in multiple sclerosis (MS) highlights the critical role of α4β1 integrin (VLA-4) in the pathogenesis of EAE [
24]. Preclinical studies demonstrate that natalizumab significantly reduces disease severity when administered prior to EAE induction, but is less effective post-induction [
25]. Clinically, early natalizumab intervention in MS patients leads to better outcomes, lower relapse rates, and rapid symptomatic relief [
26,
27]. The natalizumab treatment for autoimmune encephalitis in clinical trials also showed preliminary, promising results [
28]. These findings underscore the difficulty of treating CNS autoimmunity once autoreactive cells have entered and begun self-perpetuation within the brain parenchyma. Nevertheless, questions remain regarding the specific sites within the brain where autoimmunity is sustained and amplified.
The recent discovery of meningeal lymphatic vessels (MLVs) has significantly advanced our understanding of the brain’s immune privilege. MLVs function not only as a drainage system for CNS components but also as an immune cell hub, akin to a lymphoid organ. For instance, B cells can complete their development and maturation cycle within the MLVs [
29]. Further research has identified dural-associated lymphoid tissue, particularly in the rostral-rhinal hub region of the meninges, as a key site for B cell maturation [
30]. In experimental autoimmune encephalomyelitis (EAE) animal models, meningeal inflammation is evident before the onset of inflammation in the brain parenchyma [
31,
32]. Post-mortem studies of CNS autoimmune diseases, such as multiple sclerosis (MS) and autoimmune encephalitis, consistently reveal meningeal inflammation [
33,
34,
35]. Additionally, peripheral antigens can accumulate in the meninges, leading to the activation of immune cells [
30]. This not only validates the meninges as a critical site for CNS autoimmune pathogenesis but also suggests that, in addition to the infiltration of peripheral autoreactive immune components into the brain, self-antigens from the periphery may induce autoimmunity within the CNS via the meninges.
Moreover, since the meninges also serve as a drainage pathway for the CNS, the possibility of autoimmunity occurring independently within the CNS itself is plausible. Therefore, the role of the meninges in blurring the distinction between peripheral and CNS origins of autoimmunity highlights the complexity of CNS autoimmune pathogenesis. The mechanisms by which peripheral immune cells or autoantigens enter the meninges and contribute to autoimmunity warrant further investigation.
4. Autoantibody Pathogenicity for Neuropsychiatric Symptoms
Among all self-antigens associated with psychosis, the N-methyl-D-aspartate receptor (NMDAR) has been the focus of extensive research. The NMDAR is a neuron-specific ion channel composed of four subunits: two mandatory Grin1 subunits and two additional subunits selected from six different options. Serum or cerebrospinal fluid Autoantibodies targeting the NMDAR or other neuronal proteins are screened in some clinical settings for autoimmune encephalitis patients, and patients with schizophrenia or first-episode psychosis [
1,
36]. This practice is strongly supported by the NMDAR hypofunction theory induced by autoantibody binding []. Specifically, the NMDAR antagonist ketamine, when administered at relevant doses, blocks NMDAR ion channel function and induces hallucinations. Similarly, the binding of anti-NMDAR autoantibodies to the receptor leads to its internalization, resulting in hypofunction [
14]. This internalization is mediated by the cross-linking of NMDARs by the two Fab arms of intact antibodies, an effect not observed with the isolated Fab fragments [
37]. Additionally,
in vitro studies demonstrate that anti-NMDAR autoantibodies preferentially bind to extrasynaptic NMDARs, thereby reducing receptor density at the synapse [
38]. Although NMDAR hypofunction is a primary mechanism of interest, research on other self-antigens halve also heavily emphasized the role of autoantibodies in psychosis symptoms.
In addition to inducing receptor internalization, autoantibodies can interact with other immune cells. For instance, the anti-GluN1 antibody-NMDAR complex can be phagocytosed and removed from neurons by microglia in an
in vitro co-culture system [
39]. This microglial engulfment, triggered by the Fc portion of the anti-NMDAR antibody, involves two distinct pathways. The first pathway relies on direct engagement of the antibody’s Fc region with the microglial Fc receptor (CD64), while the second pathway involves the formation of an antibody-complement immune complex that binds to the microglial complement receptor (CD11b). These pathways appear to have compensatory roles in facilitating microglial phagocytosis [
39]. Notably, astrocytes and oligodendrocytes also express Fc and complement receptors; however, in vivo evidence for their interaction with antibodies to modulate neuronal activity remains lacking [
40,
41].
Microglial activation has also been observed in patients with anti-NMDAR encephalitis. Elevated levels of soluble-TREM2, CD44, and MMP9 in patient serum and cerebrospinal fluid (CSF) support this observation [
42]. Additionally, a recent study demonstrated microglial activation in the hippocampal region of mice immunized with NMDAR peptides. In these mice, IgG and NMDAR particles were found colocalized within microglia, further suggesting a role for microglia in the pathological process [
43]. Thus, autoantibodies not only induce microglial phagocytosis but also activate microglia, contributing to the establishment of a pro-inflammatory environment. However, the precise neuronal consequences of microglial activation in the CNS and the associated behavioural changes in patients remain unclear [].
. The antibody pathogenicity for autoimmune psychosis: ① The NMDAR antagonist Ketamine induces hypofunction of the NMDAR by blocking Ca<sup>2+</sup> influx through the receptor into the cell. Anti-NMDAR antibodies bind to the receptor and cause NMDAR hypofunction through two mechanisms: ② cross-linking receptors, leading to their internalization, and ③ binding to extra-synaptic receptors, preventing their migration and contributing to receptor scarcity at the synapse. ④ The antibody-NMDAR complex can be phagocytosed by microglia via the antibody’s Fc part, either through Fc receptors or complement receptors. Unknown aspects remain, ⑤ such as whether the same mechanism applies to other receptor and its antibody. Additionally, there is a significant need to understand the roles of ⑥ astrocytes and ⑦ oligodendrocytes in this process.
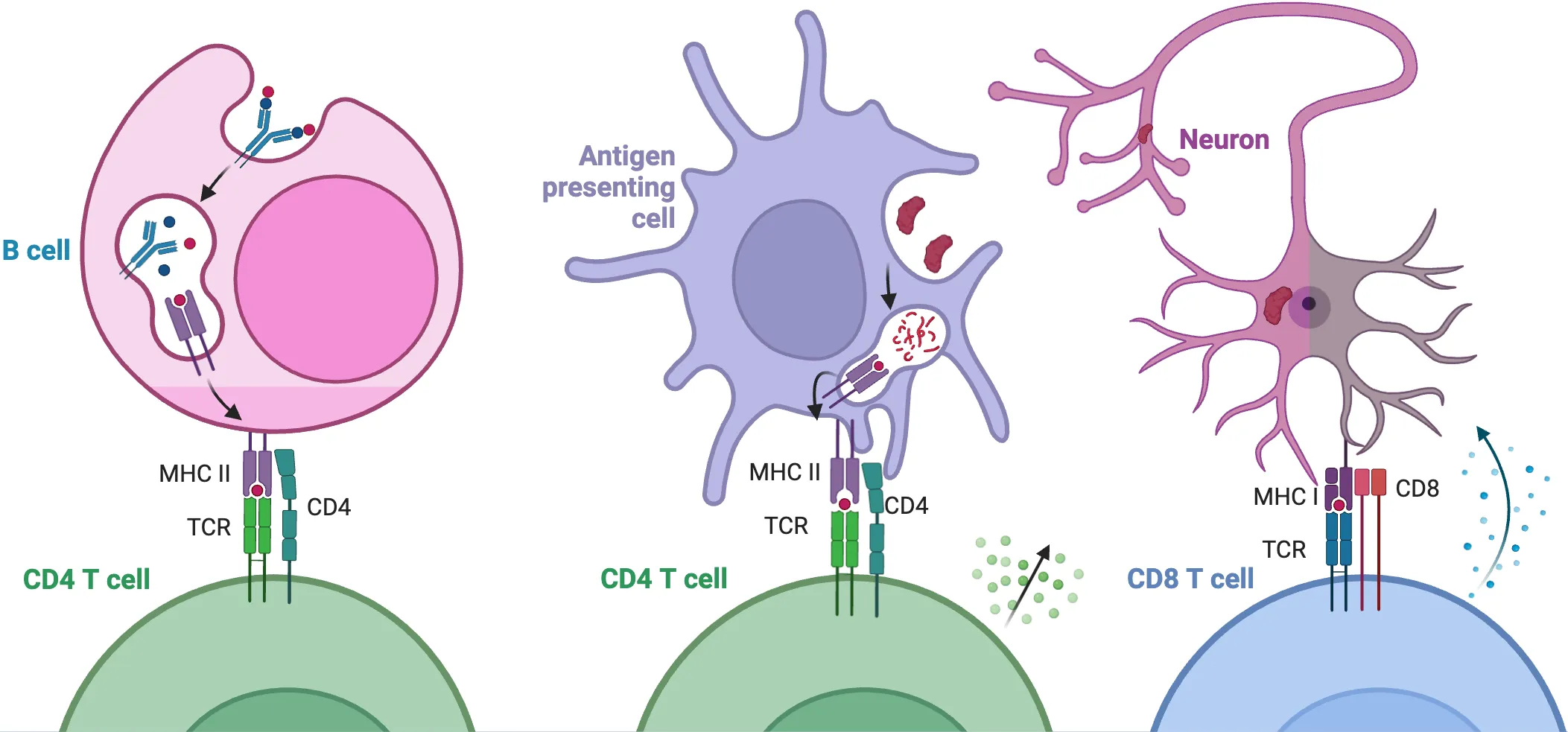
5. The Hidden Auto-Reactive T Cell
T cells, alongside B cells, constitute the two central pillars of adaptive immunity. Unlike B cells, which bind directly to antigens regardless of their size, T cells recognize segmented antigens presented by major histocompatibility complex (MHC) molecules on antigen-presenting cells (APCs). T cells contribute to autoimmune pathogenesis through two primary mechanisms: the secretion of pro-inflammatory cytokines or the direct killing of antigen-presenting cells []. Under healthy conditions, T cells actively survey the CNS and comprise approximately 80% of immune cells in the cerebrospinal fluid (CSF) that bathes the brain and spinal cord [
44]. Thus, it is plausible that T cells might play a part in the development of CNS autoimmune diseases.
The adaptive transfer of antibodies to naïve animals is a well-established method for confirming the causality of autoantibodies. This approach has been extensively utilized in myasthenia gravis research, demonstrating that autoantibodies against the acetylcholine receptor (AChR) are causal for muscle weakness [
45,
46]. While antibody pathogenicity is a central focus in autoimmune psychosis diseases, the results from passive transfer experiments in these contexts have been less conclusive in establishing antibody causality. For instance, when anti-NMDAR (N-methyl-D-aspartate receptor) antibodies derived from patients are transferred into recipient mice, only mild behavioral changes are observed, both during development and in adulthood [
47,
48]. These changes are not as severe as the psychosis symptoms seen in patients with autoimmune encephalitis, suggesting that an additional factor, like T cells, could be a critical missing component contributing to the disease pathogenesis. Supporting this, studies have shown that T cells are essential for inducing behavioral phenotypes in mice immunized with the NMDAR holoreceptor [
49]. In the clinic, some autoimmune encephalitis patients’ CSF samples also shown with oligoclonal bands but without significant pleocytosis or protein, suggesting a potential causal role of cytotoxic T cell [
50]. This highlights the potential role of T cells in the development of autoimmune psychosis conditions and suggests that both antibodies and T cells may be required to fully replicate the disease phenotype in animal models.
Among autoimmune diseases, multiple sclerosis (MS) is widely recognized as an autoimmune disorder mediated by T cells that erroneously recognize and attack the body’s myelin, the protective sheath surrounding nerve fibers. Post-mortem analysis of brain tissue from MS patients reveals increased T cell infiltration, with a predominance of CD8
+ T cells over CD4
+ T cells [
51,
52,
53,
54,
55]. Intriguingly, there is a strong correlation between MS and Epstein-Barr virus (EBV) infection. In early-stage MS patients, approximately 13% of T cells in the CSF are specific for EBV-infected autologous B cells [
56]. Furthermore, in the experimental autoimmune encephalomyelitis (EAE) mouse model for MS, the adoptive transfer of T cells from diseased mice to healthy recipients induces disease, underscoring the pathogenic potential of autoreactive T cells.
While direct evidence of autoreactive T cells interacting with neurons to cause autoimmune psychosis symptoms is lacking, patients experiencing first-episode psychosis often exhibit elevated levels of T cell secreted pro-inflammatory cytokines such as IL-6, IFN-γ, and TNF-α [
57]. These cytokines may modulate neuronal function or recruit and activate other immune cells, thereby altering the immune environment in the CNS. There was an increased percentage of monocytes and T cell within the CSF in psychosis patients [
58]. This data suggested that the T cell promotes monocyte CNS infiltration via cytokines.
T cells also play a critical role in supporting B cell function, particularly in the process of B cell receptor (BCR) affinity maturation. This collaboration results in the production of high-affinity antibodies that effectively neutralize specific antigens. Through BCR or antibody sequencing, somatic hypermutation—a hallmark of B cell activation—has been observed in CSF derived B cells or plasma cells of autoimmune anti-NMDAR [
14] or anti-LGI [
59,
60,
61] 1 encephalitis patients. These findings highlight the role of T cells in facilitating autoimmune development by aiding B cells.
The potential role of autoreactive T cells in autoimmune psychosis diseases is further suggested by the involvement of intracellular antigens in autoimmune encephalitis. While membrane receptors such as NMDAR, AMPAR, and LGI1 are well-known targets, intracellular antigens such as Hu [
62], Ma2 [
21,
63,
64], CRMP5 [
65], and Amphiphysin [
66] have also been implicated. For instance, Hu is an intracellular RNA-binding protein that cannot be targeted by antibodies alone but becomes accessible to T cells when presented via MHC molecules. Although autoantibodies against these intracellular antigens are commonly detected, identifying autoreactive T cells specific to these antigens remains challenging. This difficulty stems from technical limitations in identifying pathogenic autoantigen epitopes and constructing MHC-antigen complexes to probe T cell specificity. Nevertheless, the presence of intracellular autoantibodies indirectly suggests the involvement of autoreactive T cells, as these autoantibodies often arise in response to intracellular antigens processed and presented to T cells.
In conclusion, the evidence strongly supports the pivotal role of autoreactive T cells in driving autoimmune diseases, including those affecting the CNS. Advancements in technology and methodology will be crucial to unravel the precise mechanisms by which these T cells contribute to disease pathogenesis, particularly in autoimmune psychosis conditions [].
. T cells play crucial roles in modulating immune responses. Firstly, CD4⁺ T follicular helper (Tfh) cells support B cells in undergoing somatic hypermutation of their receptors, leading to the production of high-affinity antibodies against antigens. Secondly, CD4⁺ T cells are presented with antigen peptides by antigen-presenting cells and secrete cytokines or chemokines, significantly modulating the immune environment. Thirdly, CD8⁺ T cells can directly interact with neurons via the MHC I-antigen complex and induce neuronal death.
6. The Inflammation and Glia Cells
Brain resident glia cell bridges the autoreactive T or B cells and the neuronal antigen. There is also growing evidence showing that the glia cell can also be activated by autoimmune processes, helping create a self-reinforcing inflammatory environment that alters neuronal activity.
Microglia, the CNS’s primary immune cells, rapidly respond to autoantibodies and inflammatory cytokines. In anti-NMDAR encephalitis, microglia phagocytose antibody-receptor complexes via FcγR (CD64) and complement receptors (CD11b), leading to synaptic stripping and NMDAR hypofunction [
67,
68]. Activated microglia also secrete IL-1β, TNF-α, and CCL2, which amplify neuroinflammation and recruit peripheral immune cells [
69,
70]. Notably, microglial TREM2 and MMP9 are elevated in patient CSF, correlating with psychosis severity [
67]. Preclinical models show that microglial depletion (via PLX5622) reverses inflammation-induced anhedonia and cognitive deficits, underscoring their central role in behavior [
71].
Astrocytes transition from homeostatic supporters to inflammatory mediators in autoimmune conditions. Pro-inflammatory A1 astrocytes, induced by microglial TNF-α and IL-1α, release reactive oxygen species and glutamate, exacerbating neuronal damage [
69,
72]. Conversely, A2 astrocytes promote tissue repair via BDNF and glial scar formation. For inflammation-induced depression-like behavior, astrocytic calcium dysregulation (e.g., Orai1-mediated signaling) disrupts tripartite synapse function, impairing glutamate clearance and amplifying excitotoxicity [
73].
Oligodendrocytes extend beyond myelination to actively modulate neuroinflammation. They express TLR3 and MHC-I, enabling direct interactions with autoreactive T cells [
74,
75]. In multiple sclerosis models, Th17 cells induce oligodendrocyte process retraction via glutamate release and CD29 integrin signaling, impairing remyelination [
74]. Oligodendrocyte precursor cells (OPCs) further exacerbate inflammation by secreting CCL2 and CXCL10, which recruit microglia and T cells [
76,
77]. Myelin breakdown releases DAMPs (e.g., MBP peptides), perpetuating autoimmune responses and disrupting axonal conduction—a potential substrate for psychosis [
76,
77].
Glia cell also form a network, which in turn regulates each other’s and immune cell’s activity. Microglia-astrocyte crosstalk amplifies neuroinflammation: microglial IL-1α and TNF-α drive A1 astrocyte polarization, while astrocytic CCL2 enhances microglial motility and phagocytosis [
69,
72]. This loop sustains cytokine storms that reduce synaptic density in prefrontal and hippocampal regions, regions critical for mood and cognition [
70,
71]. OPCs modulate microglial activation states via chemokine release (e.g., CXCL1). Depleting OPCs in Parkinson’s models increases dopaminergic neuron loss, highlighting their protective role [
77]. However, in acute inflammation, OPCs exacerbate macrophage and Th17 infiltration, creating a vicious cycle of demyelination and neurotoxicity [
74,
76].
7. The Treatment and Mechanism
The autoimmune psychosis disease mechanism strongly focuses on antibody-mediated pathogenicity. Consequently, treatments and clinical trials on autoimmune psychosis disease are primarily focused on dampening antibody-driven autoimmunity. For instance, first-line treatments such as plasma exchange (PLEX) and intravenous immunoglobulins (IVIg) aim to remove pathogenic autoantibodies.
PLEX involves withdrawing blood from one vein, separating the plasma from blood cells via membrane filtration or centrifugation, and reinfusing the blood with a plasma substitute into another vein. This process removes antibodies and other factors, like cytokines, that promote autoimmunity [
78]. PLEX was demonstrated to produce positive outcomes in 1976 for treating myasthenia gravis, an autoimmune neurological disorder caused by autoantibodies targeting acetylcholine receptors [
79].
IVIg is a treatment that uses pooled immunoglobulin G from thousands of healthy donors to induce anti-inflammatory and immunomodulatory effects. It was first applied to immunedeficient patients to provide temporary passive immunity, and its ability to mitigate autoimmune processes may be through multiple mechanisms [
80]. Firstly, it can compete with autoantibodies for binding to antigens and complement system factors. In addition, it accelerates autoantibody catabolism by saturating the neonatal Fc receptor (FcRn), thereby enhancing autoantibody clearance. Efgartigimod, a novel FcRn antagonist, has shown promising results in clinical trials for various autoimmune diseases. A case report has demonstrated its efficiency in eliminating psychosis symptoms of autoimmune encephalitis patients [
81]. Therefore, both PLEX and IVIg have the potential to be used to treat autoimmune disease, like autoimmune encephalitis [
82,
83,
84]. Corticosteroids, another first-line therapy, are potent immunosuppressive agents that reduce inflammation globally.
Antibody depletion is particularly relevant if the antigen is extracellular. Regarding the intracellular antigen, the autoimmunity may be primarily driven by the T cells [
85]. In this scenario, the antibody is a by-product of the autoimmunity, and removing it may not contribute to the autoimmunity attenuation. The T cell depletion therapy is not there yet in the clinic. But we know B cells play an important role in antigen-specific T cell’s activation, and there are different treatment choices to remove B cell in the clinic.
Once the first-line therapies fail, second-line treatments such as rituximab are employed. Rituximab, a monoclonal antibody targeting CD20, depletes B cells and is considered a more aggressive intervention. This antibody binds to B cells and induces their apoptosis either directly or by engaging effector cells such as natural killer (NK) cells, macrophages, or other immune cells. This interaction triggers antibody-dependent cellular cytotoxicity (ADCC) or complement-dependent cytotoxicity (CDC), leading to B cell depletion [
86]. It was initially approved by the FDA for treating B-cell non-Hodgkin lymphomas resistant to other chemotherapy regimens. Rituximab effectively depletes B cells with minimal impact on plasma cells, as plasma cells exhibit limited CD20 expression. Since the early 2000s, rituximab has been repurposed for autoimmune diseases, demonstrating efficacy in conditions such as rheumatoid arthritis [
87], systemic lupus erythematosus (SLE) [
88], and autoimmune encephalitis [
89,
90]. Inspired by these successes, rituximab and other B-cell depletion strategies are being actively explored for treating psychosis and schizophrenia in clinical trials to treat antineuronal antibody positive patients [
91,
92].
Chimeric antigen receptor T (CAR-T) cell therapy, which can be employed to eliminate pathogenic B cells, is a rapidly evolving treatment [
93]. Unlike B cell-targeting antibodies, CAR-T cells directly deplete B cells without the need for other immune cells, achieving more extensive and profound B cell depletion within the affected tissue. The development of allogeneic CAR-T cells has also paved the way for large-scale industrial manufacturing [
94]. Additionally, CAR-T cells may cross the blood-brain barrier more efficiently than antibodies, potentially offering superior B cell depletion for CNS autoimmune diseases [
95]. Two case reports have confirmed the safety and efficacy of CAR-T therapy in treating neurological autoimmune diseases [
96,
97]. Furthermore, nine registered clinical trials are currently investigating its potential for treating CNS autoimmune diseases [
93]. Over the coming years, we anticipate gaining a deeper understanding of CAR-T therapy’s efficacy in addressing autoimmune psychosis diseases.
8. The B Cells on CNS Autoimmunity
B cells, as a whole, exhibit a dual role in autoimmune disorders, acting as both promoters and suppressors of neuroinflammation [
98,
99,
100]. While their role as antibody-producing cells after further development is well-established, emerging evidence highlights their capacity to modulate immune responses through antigen presentation and cytokine secretion—functions critical to understanding the variable efficacy of B cell-targeted therapies in autoimmune psychosis [
101,
102].
B cells process and present antigens via MHC class II with 10,000-fold greater efficiency than nonspecific mechanisms, enabling them to dominate immune responses even against low-abundance neuronal antigens [
103]. In the multiple sclerosis (MS) disease animal model, meningeal B cells form ectopic lymphoid structures that present myelin antigens to autoreactive T cells, sustaining neuroinflammation through several mechanisms [
98,
99]. These B cell-T cell interactions amplify the immune response as B cells sustain T cell reactivation through CD86/CD80 stimulation, produce GM-CSF to recruit pro-inflammatory myeloid cells, and secrete IL-6 to promote Th17 differentiation [
98,
99]. Clinically, anti-CD20 therapies like rituximab achieve 79–89% relapse-free rates in MS by depleting pathogenic meningeal B cell clusters while sparing regulatory subsets [
98,
99]. Paradoxically, these therapies leave plasma cells intact, demonstrating that antibody production alone cannot explain B cells’ contribution to autoimmunity [
100].
Experimental autoimmune encephalomyelitis (EAE) reveals context-dependent B cell functions [
98,
99,
100]. The pathogenic role partially relies on B cells MHC II dependent antigen presentation function [
104]. Depleting these cells during established EAE reduces disease severity [
105]. Conversely, CD1dhiCD5+B10 cells (IL10 expressing B cells) demonstrate a protective role by suppressing EAE initiation via IL-10 [
106]. Early B cell depletion removes this population, increasing CNS monocyte infiltration [
99,
107]. Notably, adoptive transfer of B10 cells normalizes EAE in B cell-depleted mice, confirming their regulatory function [
106].
In addition to depleting B cell directly via targeting the protein on its cell surface, like CD19, other principles have also been applied for B cell depletion. Like Atacicept, it is a recombinant protein drug that combines with the extracellular part of TACI and Fc part of IgG1. The Atacicept application absorbs B cell survival cytokines: BAFF (B-cell activating factor) and APRIL (a proliferation-inducing ligand), causing B cell death. However, the phase II trial of Atacicept on Multiple Sclerosis [
108]. Despite reducing peripheral B cells and immunoglobulins, atacicept increased annualized relapse rates by 126–158% in MS patients [
109]. Mouse study reveals Atacicept depletes IL-10
+ B10 cells while sparing meningeal pathogenic B cells, disrupting immune homeostasis [
99]. This contrasts with SLE, where BAFF/APRIL inhibition benefits patients by targeting antibody-driven pathology [
110]. In EAE, preventive B cell depletion exacerbates disease via B10 cell loss, whereas depletion during established disease suppresses symptoms by removing antigen-presenting B cells [
106]. This temporal duality underscores the need for precision timing in B cell-targeted therapies for patients.
The role of B cells in autoimmune psychosis remains relatively elusive due to the lack of robust animal models and clinical evidence. In an anti-NMDAR autoimmune encephalitis model, behavioral abnormalities, including shortened lifespan and hyperactivity following active immunization, were ameliorated in B cell-deficient mouse strains [
49]. This finding demonstrates the pathogenic role of B cells, independent of their functions in antigen presentation or antibody production. Recent clinical trials investigating B cell depletion therapies (primarily rituximab) in psychosis-related disorders have shown promising but preliminary results, with variable success rates [
36,
92,
111,
112]. These variable outcomes may be explained by the dichotomous functions of B cells in CNS autoimmunity, which underscore the critical need for additional animal models and expanded clinical studies.
9. Conclusions and Future Research
This review traces the history and evolving understanding of autoimmune mechanisms underlying psychosis disorders. Neuroinflammatory conditions, such as autoimmune encephalitis and related autoimmune encephalitis-related psychosis, exemplify the complex interaction between the immune system and the CNS. The identification of specific autoantibodies linked to psychosis symptoms has advanced the concept of autoimmune encephalitis-related psychosis in both basic research and clinical practice. However, the role of autoantibodies in treatment is not definitive, as these antibodies may sometimes be disease-irrelevant. Therefore, despite acknowledging that autoimmunity can cause psychosis disorders, the clinical practice of diagnosing and treating autoimmune-induced psychosis patients remains limited. This knowledge and practice gap is largely due to our insufficient mechanistic understanding of these diseases.
In addition to the established autoantibody causality, this review explores the potential roles of T cells and the origins of autoimmunity—whether peripheral or CNS-based—as alternative perspectives on autoimmune encephalitis-related psychosis. Extended from the T cell, this review also illustrated that the brain resident glia cells play an important role in sensing inflammation and modulating neuronal activity. Based on the current clinical treatment test, the B cell function was also further exploited. It is clear that the need for animal models to understand T cell causality, the role of B cell, and also different glia cells. Though, there are animal models that have demonstrated that anti-NMDAR autoimmunity can cause behavioural symptoms. However, these models have not been consistently replicated across laboratories, potentially due to technical challenges. We believe that a more accessible animal model would greatly facilitate mechanistic research. Furthermore, animal studies provide critical insights into the efficacy and safety of new treatment approaches, accelerating the translation of findings into clinical applications.
A deeper exploration of CNS immunity, especially the role of meningeal functions during disease progression, represents a crucial frontier in this field. The meninges, serving as the interface between the peripheral immune system and the CNS, may hold the key to understanding how autoimmunity transitions from the periphery to the CNS. If autoimmunity requires propagation within the meninges rather than the periphery, targeted treatment at the meningeal level might be more effective than global approaches. Investigating the immunological dynamics within the meninges during autoimmune psychosis diseases promises to reveal how autoimmunity develops and persists in the CNS.
Future research should integrate clinical findings, animal models, and immunological insights to establish a comprehensive framework for understanding autoimmune-mediated psychosis disorders. Leveraging advanced tools like single-cell sequencing, spatial transcriptomics, and high-resolution imaging can uncover the complex mechanisms underlying autoimmune psychosis diseases. This knowledge will enhance our diagnostic and treatment capabilities and pave the way for innovative strategies to prevent the onset and progression of these disorders.
Author Contributions
Writing–Original Draft Preparation, L.H.; Writing–Review & Editing, V.P., T.N.
Ethics Statement
Not applicable.
Informed Consent Statement
Not applicable.
Funding
This work was supported by the Wellcome Trust (226779/Z/22/Z) and by the Francis Crick Institute, which receives its core funding from Cancer Research UK (CC2222), the UK Medical Research Council (CC2222), and the Wellcome Trust (CC2222).
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
-
1.
Pollak TA, Lennox BR, Müller S, Benros ME, Prüss H, Tebartz van Elst L, et al. Autoimmune psychosis: an international consensus on an approach to the diagnosis and management of psychosis of suspected autoimmune origin.
Lancet Psychiatry 2020,
7, 93–108.
[Google Scholar]
-
2.
Shastri A, Bonifati DM, Kishore U. Innate immunity and neuroinflammation.
Mediat. Inflamm. 2013,
2013, 342931.
[Google Scholar]
-
3.
Rathod SS, Agrawal YO, Nakhate KT, Meeran MFN, Ojha S, Goyal SN. Neuroinflammation in the Central Nervous System: Exploring the Evolving Influence of Endocannabinoid System.
Biomedicines 2023,
11, 2642.
[Google Scholar]
-
4.
Chen HJ, Appelman B, Willemen H, Bos A, Prado J, Geyer CE, et al. Transfer of IgG from Long COVID patients induces symptomology in mice.
BioRxiv 2024. doi:10.1101/2024.05.30.596590.
[Google Scholar]
-
5.
Nersesjan V, Amiri M, Nilsson AC, Wamberg C, Jensen VV, Petersen CB, et al. SARS-CoV-2 and autoantibodies in the cerebrospinal fluid of COVID-19 patients: Prospective multicentre cohort study.
Brain Commun. 2023,
5, fcad274.
[Google Scholar]
-
6.
Hansen N. Psychiatric Symptoms in Acute and Persisting Forms of COVID-19 Associated with Neural Autoantibodies.
Antibodies 2023,
12, 49.
[Google Scholar]
-
7.
Mackay IR. Travels and travails of autoimmunity: A historical journey from discovery to rediscovery.
Autoimmun. Rev. 2010,
9, A251–A258.
[Google Scholar]
-
8.
Ransohoff RM, Brown MA. Innate immunity in the central nervous system.
J. Clin. Investig. 2012,
122, 1164–1171.
[Google Scholar]
-
9.
Carson MJ, Doose JM, Melchior B, Schmid CD, Ploix CC. CNS immune privilege: hiding in plain sight.
Immunol. Rev. 2006,
213, 48–65.
[Google Scholar]
-
10.
Malla A, Joober R, Garcia A. “Mental illness is like any other medical illness”: a critical examination of the statement and its impact on patient care and society.
J. Psychiatry Neurosci. 2015,
40, 147–150.
[Google Scholar]
-
11.
Brouwer B, Biemond A. Les affections parenchymateuses du cervelet et leur signification au point de vue de l’anatomie et de la physiologie de cet organe.
J. Belg. de Neurol. et de Psychiat. 1938,
38, 691–757.
[Google Scholar]
-
12.
Corsellis JA, Goldberg GJ, Norton AR. “limbic encephalitis” and its association with carcinoma.
Brain 1968,
91, 481–496.
[Google Scholar]
-
13.
Dalmau J, Tüzün E, Wu HY, Masjuan J, Rossi JE, Voloschin A, Baehring JM, et al. Paraneoplastic Anti-N-methyl-D-aspartate Receptor Encephalitis Associated with Ovarian Teratoma.
Ann. Neurol. 2007,
61, 25–36.
[Google Scholar]
-
14.
Kreye J, Wenke NK, Chayka M, Leubner J, Murugan R, Maier N, et al. Human cerebrospinal fluid monoclonal
N. -methyl-D-aspartate receptor autoantibodies are sufficient for encephalitis pathogenesis.
Brain 2016,
139, 2641–2652.
[Google Scholar]
-
15.
Rozenberg A, Shelly S, Vaknin-Dembinsky A, Friedman-Korn T, Benoliel-Berman T, Spector P, et al. Cognitive impairments in autoimmune encephalitis: the role of autoimmune antibodies and oligoclonal bands.
Front. Immunol. 2024,
15, 1405337.
[Google Scholar]
-
16.
Luykx JJ, Visscher R, Winter-van Rossum I, Waters P, de Witte LD, Fleischhacker WW, et al. Clinical symptoms and psychosocial functioning in patients with schizophrenia spectrum disorders testing seropositive for anti-NMDAR antibodies: a case–control comparison with patients testing negative.
Lancet Psychiatry 2024,
11, 828–838.
[Google Scholar]
-
17.
Hansen N. NMDAR autoantibodies in psychiatric disease—An immunopsychiatric continuum and potential predisposition for disease pathogenesis.
J. Transl. Autoimmun. 2022,
5, 100165.
[Google Scholar]
-
18.
Joubert B, Saint-Martin M, Noraz N, Picard G, Rogemond V, Ducray F, et al. Characterization of a Subtype of Autoimmune Encephalitis with Anti–Contactin-Associated Protein-like 2 Antibodies in the Cerebrospinal Fluid, Prominent Limbic Symptoms, and Seizures.
JAMA Neurol. 2016,
73, 1115.
[Google Scholar]
-
19.
Lai M, Hughes EG, Peng X, Zhou L, Gleichman AJ, Shu H, et al. AMPA receptor antibodies in limbic encephalitis alter synaptic receptor location.
Ann. Neurol. 2009,
65, 424–434.
[Google Scholar]
-
20.
Dalmau J, Gleichman AJ, Hughes EG, Rossi JE, Peng X, Lai M, et al. Anti-NMDA-receptor encephalitis: case series and analysis of the effects of antibodies.
Lancet Neurol. 2008,
7, 1091–1098.
[Google Scholar]
-
21.
Alamowitch S. Limbic encephalitis and small cell lung cancer. Clinical and immunological features.
Brain 1997,
120, 923–928.
[Google Scholar]
-
22.
Hansen N, Sagebiel A, Rentzsch K, Hirschel S, Wiltfang J, Schott BH, et al. Case report: Amnestic mild cognitive impairment in multiple domains associated with neurofascin 186 autoantibodies: Case series with follow-up and review.
Front. Psychiatry 2023,
13, 1054461.
[Google Scholar]
-
23.
Wilson EH, Weninger W, Hunter CA. Trafficking of immune cells in the central nervous system.
J. Clin. Investig. 2010,
120, 1368–1379.
[Google Scholar]
-
24.
Parker Harp CR, Archambault AS, Cheung M, Williams JW, Czepielewski RS, Duncker PC, et al. Neutrophils promote VLA-4–dependent B cell antigen presentation and accumulation within the meninges during neuroinflammation.
Proc. Natl. Acad. Sci. USA 2019,
116, 24221–24230.
[Google Scholar]
-
25.
Mindur JE, Ito N, Dhib-Jalbut S, Ito K. Early Treatment with Anti-VLA-4 mAb Can Prevent the Infiltration and/or Development of Pathogenic CD11b+CD4+ T Cells in the CNS during Progressive EAE.
PLoS ONE 2014,
9, e99068.
[Google Scholar]
-
26.
Graf J, Leussink VI, Soncin G, Lepka K, Meinl I, Kümpfel T, et al. Relapse-independent multiple sclerosis progression under natalizumab.
Brain Commun. 2021,
3, fcab229.
[Google Scholar]
-
27.
Kappos L, O'Connor PW, Polman CH, Vermersch P, Wiendl H, Pace A, et al. Clinical effects of natalizumab on multiple sclerosis appear early in treatment course.
J. Neurol. 2013,
260, 1388–1395.
[Google Scholar]
-
28.
Bastiaansen AEM, de Jongste AHC, de Bruijn MAAM, Crijnen YS, Schreurs MWJ, Verbeek MM, et al. Phase II trial of natalizumab for the treatment of anti-Hu associated paraneoplastic neurological syndromes.
Neurooncol. Adv. 2021,
3, vdab145.
[Google Scholar]
-
29.
Brioschi S, Wang WL, Peng V, Wang M, Shchukina I, Greenberg ZJ, et al. Heterogeneity of meningeal B cells reveals a lymphopoietic niche at the CNS borders.
Science 2021,
373, eabf9277.
[Google Scholar]
-
30.
Fitzpatrick Z, Ghabdan Zanluqui N, Rosenblum JS, Tuong ZK, Lee CYC, Chandrashekhar V, et al. Venous-plexus-associated lymphoid hubs support meningeal humoral immunity.
Nature 2024,
628, 612–619.
[Google Scholar]
-
31.
Christy AL, Walker ME, Hessner MJ, Brown MA. Mast cell activation and neutrophil recruitment promotes early and robust inflammation in the meninges in EAE.
J. Autoimmun. 2013,
42, 50–61.
[Google Scholar]
-
32.
Bhargava P, Kim S, Reyes AA, Grenningloh R, Boschert U, Absinta M, et al. Imaging meningeal inflammation in CNS autoimmunity identifies a therapeutic role for BTK inhibition.
Brain 2021,
144, 1396–1408.
[Google Scholar]
-
33.
Pikor NB, Prat A, Bar-Or A, Gommerman JL. Meningeal Tertiary Lymphoid Tissues and Multiple Sclerosis: A Gathering Place for Diverse Types of Immune Cells during CNS Autoimmunity.
Front. Immunol. 2016,
6, 165433.
[Google Scholar]
-
34.
Zhang PP, He ZC, Yao XH, Tang R, Ma J, Luo T, et al. COVID-19-associated monocytic encephalitis (CAME): histological and proteomic evidence from autopsy.
Signal Transduct. Target. Ther. 2023,
8, 1–11.
[Google Scholar]
-
35.
Yamakawa M, Hogan KO, Leever J, Jassam YN. Autopsy Case of Meningoencephalomyelitis Associated with Glial Fibrillary Acidic Protein Antibody.
Neurol. Neuroimmunol. Neuroinflamm. 2021,
8, e1081.
[Google Scholar]
-
36.
Lennox BR, Tomei G, Vincent SA, Yeeles K, Pollard R, Palmer-Cooper E, et al. Study of immunotherapy in antibody positive psychosis: feasibility and acceptability (SINAPPS1).
J. Neurol. Neurosurg. Psychiatry 2019,
90, 365–367.
[Google Scholar]
-
37.
Hughes EG, Peng X, Gleichman AJ, Lai M, Zhou L, Tsou R, et al. Cellular and Synaptic Mechanisms of Anti-NMDA Receptor Encephalitis.
J. Neurosci. 2010,
30, 5866–5875.
[Google Scholar]
-
38.
Jézéquel J, Johansson EM, Dupuis JP, Rogemond V, Gréa H, Kellermayer B, et al. Dynamic disorganization of synaptic NMDA receptors triggered by autoantibodies from psychotic patients.
Nat. Commun. 2017,
8, 1791.
[Google Scholar]
-
39.
Rahman KA, Orlando M, Boulos A, Andrzejak E, Schmitz D, Ziv NE, et al. Microglia actively remove NR1 autoantibody-bound NMDA receptors and associated post-synaptic proteins in neuron microglia co-cultures.
Glia 2023,
71, 1804–1829.
[Google Scholar]
-
40.
Nakahara J, Tan-Takeuchi K, Seiwa C, Gotoh M, Kaifu T, Ujike A, et al. Signaling via Immunoglobulin Fc Receptors Induces Oligodendrocyte Precursor Cell Differentiation.
Dev. Cell 2003,
4, 841–852.
[Google Scholar]
-
41.
Pekna M, Pekny M. The Complement System: A Powerful Modulator and Effector of Astrocyte Function in the Healthy and Diseased Central Nervous System.
Cells 2021,
10, 1812.
[Google Scholar]
-
42.
Chang H, Ma J, Feng K, Feng N, Wang X, Sun J, et al. Elevated blood and cerebrospinal fluid biomarkers of microglial activation and blood‒brain barrier disruption in anti-NMDA receptor encephalitis.
J. Neuroinflamm. 2023,
20, 172.
[Google Scholar]
-
43.
Maudes E, Planagumà J, Marmolejo L, Radosevic M, Serafim AB, Aguilar E, et al. Neuro-immunobiology and treatment assessment in a mouse model of anti-NMDAR encephalitis.
Brain 2025,
148, 2023–2037.
[Google Scholar]
-
44.
Svenningsson A, Andersen O, Edsbagge M, Stemme S. Lymphocyte phenotype and subset distribution in normal cerebrospinal fluid.
J. Neuroimmunol. 1995,
63, 39–46.
[Google Scholar]
-
45.
Patrick J, Lindstrom J. Autoimmune Response to Acetylcholine Receptor.
Science 1973,
180, 871–872.
[Google Scholar]
-
46.
Lindstrom JM, Seybold ME, Lennon VA, Whittingham S, Duane DD. Antibody to acetylcholine receptor in myasthenia gravis: Prevalence, clinical correlates, and diagnostic value.
Neurology 1976,
26, 1054–1059.
[Google Scholar]
-
47.
Kuchling J, Jurek B, Kents M, Kreye J, Geis C, Wickel J, et al. Impaired functional connectivity of the hippocampus in translational murine models of NMDA-receptor antibody associated neuropsychiatric pathology.
Mol. Psychiatry 2023,
29, 85–96.
[Google Scholar]
-
48.
García-Serra A, Radosevic M, Pupak A, Brito V, Ríos J, Aguilar E, et al. Placental transfer of NMDAR antibodies causes reversible alterations in mice.
Neurol. (R) Neuroimmunol. Neuroinflamm. 2021,
8, e915.
[Google Scholar]
-
49.
Jones BE, Tovar KR, Goehring A, Jalali-Yazdi F, Okada NJ, Gouaux E, et al. Autoimmune receptor encephalitis in mice induced by active immunization with conformationally stabilized holoreceptors.
Sci. Transl. Med. 2019,
11, 1–13.
[Google Scholar]
-
50.
Blinder T, Lewerenz J. Cerebrospinal Fluid Findings in Patients With Autoimmune Encephalitis—A Systematic Analysis.
Front. Neurol. 2019,
10, 804.
[Google Scholar]
-
51.
Peeters LM, Vanheusden M, Somers V, Van Wijmeersch B, Stinissen P, Broux B, et al. Cytotoxic CD4+ T Cells Drive Multiple Sclerosis Progression.
Front. Immunol. 2017,
8, 1160.
[Google Scholar]
-
52.
Salou M, Nicol B, Garcia A, Laplaud DA. Involvement of CD8+ T Cells in Multiple Sclerosis.
Front. Immunol. 2015,
6, 604.
[Google Scholar]
-
53.
Booss J, Esiri MM, Tourtellotte WW, et al. Immunohistological analysis of T lymphocyte subsets in the central nervous system in chronic progressive multiple sclerosis.
J. Neurol. Sci. 1983,
62, 219–232.
[Google Scholar]
-
54.
Hauser SL, Bhan AK, Gilles F, Kemp M, Kerr C, Weiner HL. Immunohistochemical analysis of the cellular infiltrate in multiple sclerosis lesions.
Ann. Neurol. 1986,
19, 578–587.
[Google Scholar]
-
55.
Salou M, Garcia A, Michel L, Gainche-Salmon A, Loussouarn D, Nicol B, et al. Expanded CD 8 T-cell sharing between periphery and CNS in multiple sclerosis.
Ann. Clin. Transl. Neurol. 2015,
2, 609–622.
[Google Scholar]
-
56.
Gottlieb A, Pham HPT, Saltarrelli JG, Lindsey JW. Expanded T lymphocytes in the cerebrospinal fluid of multiple sclerosis patients are specific for Epstein-Barr-virus-infected B cells.
Proc. Natl. Acad. Sci. USA 2024,
121, e2315857121.
[Google Scholar]
-
57.
Kim H, Baek SH, Kim JW, Ryu S, Lee JY, Kim JM, Chung YC, Kim SW. Inflammatory markers of symptomatic remission at 6 months in patients with first-episode schizophrenia.
Schizophrenia 2023,
9, 1–7.
[Google Scholar]
-
58.
Räuber S, Heming M, Repple J, Ruland T, Kuelby R, Schulte-Mecklenbeck A, et al. Cerebrospinal fluid flow cytometry distinguishes psychosis spectrum disorders from differential diagnoses.
Mol. Psychiatry 2021,
26, 7661–7670.
[Google Scholar]
-
59.
Feng J, Fan S, Sun Y, Ren H, Guan H, Wang J. Comprehensive B-Cell Immune Repertoire Analysis of Anti-NMDAR Encephalitis and Anti-LGI1 Encephalitis.
Front. Immunol. 2021,
12, 717598.
[Google Scholar]
-
60.
Lehmann-Horn K, Irani SR, Wang S, Palanichamy A, Jahn S, Greenfield AL, et al. Intrathecal B-cell activation in LGI1 antibody encephalitis.
Neurol. Neuroimmunol. Neuroinflamm. 2020,
7, e669.
[Google Scholar]
-
61.
Irani SR, Lehmann-Horn K, Geschwind M, Wang S, Vincent A, von Büdingen HC. The active intrathecal B-cell response in LGI1-antibody encephalitis.
Lancet 2015,
385, S46.
[Google Scholar]
-
62.
Dalmau J, Graus F, Rosenblum MK, Posner JB. Anti-Hu-Associated Paraneoplastic Encephalomyelitis/Sensory Neuronopathy A Clinical Study of 71 Patients.
Medicine 1992,
71, 59–72.
[Google Scholar]
-
63.
Hoffmann LA, Jarius S, Pellkofer HL, Schueller M, Krumbholz M, Koenig F, et al. Anti-Ma and anti-Ta associated paraneoplastic neurological syndromes: 22 newly diagnosed patients and review of previous cases.
J. Neurol. Neurosurg. Psychiatry 2008,
79, 767–773.
[Google Scholar]
-
64.
Overeem S, Dalmau J, Bataller L, Nishino S, Mignot E, Verschuuren J, Lammers GJ. Hypocretin-1 CSF levels in anti-Ma2 associated encephalitis.
Neurology 2004,
62, 138–140.
[Google Scholar]
-
65.
Yu Z, Kryzer TJ, Griesmann GE, Kim K, Benarroch EE, Lennon VA. CRMP-5 neuronal autoantibody: marker of lung cancer and thymoma-related autoimmunity.
Ann. Neurol. 2001,
49, 146–154.
[Google Scholar]
-
66.
Antoine JC, Absi L, Honnorat J, Boulesteix JM, de Brouker T, Vial C, et al. Antiamphiphysin Antibodies Are Associated With Various Paraneoplastic Neurological Syndromes and Tumors.
Arch. Neurol. 1999,
56, 172.
[Google Scholar]
-
67.
Gao C, Jiang J, Tan Y, Chen SD. Microglia in neurodegenerative diseases: mechanism and potential therapeutic targets.
Signal Transduct. Target. Ther. 2023,
8, 359.
[Google Scholar]
-
68.
Jha MK, Jeon S, Suk K. Glia as a Link between Neuroinflammation and Neuropathic Pain.
Immune Netw. 2012,
12, 41–47.
[Google Scholar]
-
69.
Yang Q, Zhou J. Neuroinflammation in the central nervous system: Symphony of glial cells.
Glia 2019,
67, 1017–1035.
[Google Scholar]
-
70.
Zou J, Shang W, Yang L, Liu T, Wang L, Li X, et al. Microglia activation in the mPFC mediates anxiety-like behaviors caused by
Staphylococcus aureus strain USA300.
Brain Behav. 2022,
12, e2715.
[Google Scholar]
-
71.
Chen R, Routh BN, Straetker JE, Gibson CR, Weitzner AS, Bell KS, et al. Microglia depletion ameliorates neuroinflammation, anxiety-like behavior, and cognitive deficits in a sex-specific manner in Rev-erbα knockout mice.
Brain Behav. Immun. 2023,
114, 287–298.
[Google Scholar]
-
72.
Zhao Y, Huang Y, Cao Y, Yang J. Astrocyte-Mediated Neuroinflammation in Neurological Conditions.
Biomolecules 2024,
14, 1204.
[Google Scholar]
-
73.
Novakovic MM, Korshunov KS, Grant RA, Martin ME, Valencia HA, Scott Budinger GR, et al. Astrocyte reactivity and inflammation-induced depression-like behaviors are regulated by Orai1 calcium channels.
Nat. Commun. 2023,
14, 5500.
[Google Scholar]
-
74.
Larochelle C, Wasser B, Jamann H, Löffel JT, Cui QL, Tastet O, et al. Pro-inflammatory T helper 17 directly harms oligodendrocytes in neuroinflammation.
Proc. Natl. Acad. Sci. USA 2021,
118, e2025813118.
[Google Scholar]
-
75.
Kirby L, Jin J, Cardona JG, Smith MD, Martin KA, Wang J, et al. Oligodendrocyte precursor cells present antigen and are cytotoxic targets in inflammatory demyelination.
Nat. Commun. 2019,
10, 3887.
[Google Scholar]
-
76.
Ohashi K, Uemura N, Nagayasu K, Kaneko S, Maki T, Shirakawa H. Oligodendrocyte precursor cells exacerbate acute CNS inflammation via macrophage and T cell activation in a mouse model of multiple sclerosis.
bioRxiv 2024. doi:10.1101/2024.05.28.596190.
[Google Scholar]
-
77.
Boccazzi M, Raffaele S, Fumagalli M. Not only myelination: the immune-inflammatory functions of oligodendrocytes.
Neural Regen. Res. 2022,
17, 2661–2663.
[Google Scholar]
-
78.
Shumak KH, Rock GA. Therapeutic Plasma Exchange.
N. Engl. J. Med. 1984,
310, 762–771.
[Google Scholar]
-
79.
Pinching A. Remission of myasthenia gravis following plasma-exchange.
Lancet 1976,
308, 1373–1376.
[Google Scholar]
-
80.
Velikova T, Sekulovski M, Bogdanova S, Vasilev G, Peshevska-Sekulovska M, Miteva D, Georgiev T. Intravenous Immunoglobulins as Immunomodulators in Autoimmune Diseases and Reproductive Medicine.
Antibodies 2023,
12, 20.
[Google Scholar]
-
81.
Zhang Q, Yang W, Qian Y, Zhang Y, Zhao H, Shu M, et al. Case report: Rapid symptom relief in autoimmune encephalitis with efgartigimod: A three-patient case series.
Front. Immunol. 2024,
15, 1444288.
[Google Scholar]
-
82.
Lee ST, Lee HS, Lee WJ, Cha HA, Kim SH, Shin SY, et al. The safety and efficacy of intravenous immunoglobulin in autoimmune encephalitis.
Ann. Clin. Transl. Neurol. 2022,
9, 610–621.
[Google Scholar]
-
83.
Zhang Y, Huang HJ, Chen WB, Liu G, Liu F, Su YY. Clinical efficacy of plasma exchange in patients with autoimmune encephalitis.
Ann. Clin. Transl. Neurol. 2021,
8, 763–773.
[Google Scholar]
-
84.
Krouse A, Li H, Krenzer JA, Rose WN. Plasmapheresis, Rituximab, and Ceftriaxone Provided Lasting Improvement for a 27-Year-Old Adult Male with Pediatric Autoimmune Neuropsychiatric Disorders Associated with Streptococcal Infections (PANDAS).
Case Rep. Psychiatry 2021,
2021, 1–4.
[Google Scholar]
-
85.
Burbelo PD, Iadarola MJ, Keller JM, Warner BM. Autoantibodies Targeting Intracellular and Extracellular Proteins in Autoimmunity.
Front. Immunol. 2021,
12, 548469.
[Google Scholar]
-
86.
de Sèze J, Maillart E, Gueguen A, Laplaud DA, Michel L, Thouvenot E, et al. Anti-CD20 therapies in multiple sclerosis: From pathology to the clinic.
Front. Immunol. 2023,
14, 1004795.
[Google Scholar]
-
87.
Edwards JC, Szczepański L, Szechiński J, Filipowicz-Sosnowska A, Emery P, Close DR, et al. Efficacy of B-Cell–Targeted Therapy with Rituximab in Patients with Rheumatoid Arthritis.
N. Engl. J. Med. 2004,
350, 2572–2581.
[Google Scholar]
-
88.
Leandro MJ, Edwards JC, Cambridge G, Ehrenstein MR, Isenberg DA. An open study of B lymphocyte depletion in systemic lupus erythematosus.
Arthritis Rheum. 2002,
46, 2673–2677.
[Google Scholar]
-
89.
Shang H, Shen X, Yu X, Zhang J, Jia Y, Gao F. B-cell targeted therapies in autoimmune encephalitis: mechanisms, clinical applications, and therapeutic potential.
Front. Immunol. 2024,
15, 1368275.
[Google Scholar]
-
90.
Nepal G, Shing YK, Yadav JK, Rehrig JH, Ojha R, Huang DY, et al. Efficacy and safety of rituximab in autoimmune encephalitis: A meta-analysis.
Acta Neurol. Scand. 2020,
142, 449–459.
[Google Scholar]
-
91.
Bejerot S, Eklund D, Hesser H, Hietala MA, Kariis T, Lange N, et al. Study protocol for a randomized controlled trial with rituximab for psychotic disorder in adults (RCT-Rits).
BMC Psychiatry 2023,
23, 1–17.
[Google Scholar]
-
92.
Lennox B, Yeeles K, Jones PB, Zandi M, Joyce E, Yu LM, et al. Intravenous immunoglobulin and rituximab versus placebo treatment of antibody-associated psychosis: Study protocol of a randomised phase IIa double-blinded placebo-controlled trial (SINAPPS2).
Trials 2019,
20, 1–12.
[Google Scholar]
-
93.
Konitsioti AM, Prüss H, Laurent S, Fink GR, Heesen C, Warnke C. Chimeric antigen receptor T-cell therapy for autoimmune diseases of the central nervous system: a systematic literature review.
J. Neurol. 2024,
271, 6526–6542.
[Google Scholar]
-
94.
Wang X, Wu X, Tan B, Zhu L, Zhang Y, Lin L, et al. Allogeneic CD19-targeted CAR-T therapy in patients with severe myositis and systemic sclerosis.
Cell 2024,
187, 4890–4904.e9.
[Google Scholar]
-
95.
Liu Y, Dong M, Chu Y, Zhou L, You Y, Pang X, et al. Dawn of CAR-T cell therapy in autoimmune diseases.
Chin. Med. J. 2024,
137, 1140–1150.
[Google Scholar]
-
96.
Fischbach F, Richter J, Pfeffer LK, Fehse B, Berger SC, Reinhardt S, et al. CD19-targeted chimeric antigen receptor T cell therapy in two patients with multiple sclerosis.
Med 2024,
5, 550–558.e2.
[Google Scholar]
-
97.
Faissner S, Motte J, Sgodzai M, Geis C, Haghikia A, Mougiakakos D, et al. Successful use of anti-CD19 CAR T cells in severe treatment-refractory stiff-person syndrome.
Proc. Natl. Acad. Sci. USA 2024,
121, e2403227121.
[Google Scholar]
-
98.
Gupta S, Simic M, Sagan SA, Shepherd C, Duecker J, Sobel RA, et al. CAR-T Cell–Mediated B-Cell Depletion in Central Nervous System Autoimmunity.
Neurol. Neuroimmunol. Neuroinflamm. 2023,
10, e200080.
[Google Scholar]
-
99.
Kumar G, Maria Z, Kohli U, Agasing A, Quinn JL, Ko RM, et al. CNS Autoimmune Responses in BCMA-Deficient Mice Provide Insight for the Failure of Atacicept in MS.
Neurol. Neuroimmunol. Neuroinflamm. 2021,
8, e973.
[Google Scholar]
-
100.
Lee DSW, Rojas OL, Gommerman JL. B cell depletion therapies in autoimmune disease: advances and mechanistic insights.
Nat. Rev. Drug Discov. 2021,
20, 179–199.
[Google Scholar]
-
101.
Al‐Diwani AA, Pollak TA, Irani SR, Lennox BR. Psychosis: an autoimmune disease?
Immunology 2017,
152, 388–401.
[Google Scholar]
-
102.
Engh JA, Ueland T, Agartz I, Andreou D, Aukrust P, Boye B, et al. Plasma Levels of the Cytokines B Cell-Activating Factor (BAFF) and A Proliferation-Inducing Ligand (APRIL) in Schizophrenia, Bipolar, and Major Depressive Disorder: A Cross Sectional, Multisite Study.
Schizophr. Bull. 2022,
48, 37–46.
[Google Scholar]
-
103.
Avalos AM, Ploegh HL. Early BCR Events and Antigen Capture, Processing, and Loading on MHC Class II on B Cells.
Front. Immunol. 2014,
5, 92.
[Google Scholar]
-
104.
Fazazi MR, Doss PM, Pereira R, Fudge N, Regmi A, Joly-Beauparlant C, et al. Myelin-reactive B cells exacerbate CD4+ T cell-driven CNS autoimmunity in an IL-23-dependent manner.
Nat. Commun. 2024,
15, 5404.
[Google Scholar]
-
105.
Molnarfi N, Schulze-Topphoff U, Weber MS, Patarroyo JC, Prod’homme T, Varrin-Doyer M, et al. MHC class II–dependent B cell APC function is required for induction of CNS autoimmunity independent of myelin-specific antibodies.
J. Exp. Med. 2013,
210, 2921–2937.
[Google Scholar]
-
106.
Matsushita T, Horikawa M, Iwata Y, Tedder TF. Regulatory B Cells (B10 Cells) and Regulatory T Cells Have Independent Roles in Controlling Experimental Autoimmune Encephalomyelitis Initiation and Late-Phase Immunopathogenesis.
J. Immunol. 2010,
185, 2240–2252.
[Google Scholar]
-
107.
Bennett JL, O'Connor KC, Bar-Or A, Zamvil SS, Hemmer B, Tedder TF, et al. B lymphocytes in neuromyelitis optica.
Neurol. Neuroimmunol. Neuroinflamm 2015,
2, e104.
[Google Scholar]
-
108.
Kaegi C, Steiner UC, Wuest B, Crowley C, Boyman O. Systematic Review of Safety and Efficacy of Atacicept in Treating Immune-Mediated Disorders.
Front. Immunol. 2020,
11, 433.
[Google Scholar]
-
109.
Kappos L, Hartung HP, Freedman MS, Boyko A, Radü EW, Mikol DD, et al. Atacicept in multiple sclerosis (ATAMS): a randomised, placebo-controlled, double-blind, phase 2 trial.
Lancet Neurol. 2014,
13, 353–363.
[Google Scholar]
-
110.
Asleh R, Vucicevic D, Petterson TM, Kremers WK, Pereira NL, Daly RC, et al. Sirolimus-Based Immunosuppression Is Associated with Decreased Incidence of Post-Transplant Lymphoproliferative Disorder after Heart Transplantation: A Double-Center Study.
J. Clin. Med. 2022,
11, 322.
[Google Scholar]
-
111.
Bejerot S, Stein SS, Welin E, Eklund D, Hylén U, Humble MB. Rituximab as an adjunctive treatment for schizophrenia spectrum disorder or obsessive-compulsive disorder: Two open-label pilot studies on treatment-resistant patients.
J. Psychiatr. Res. 2023,
158, 319–329.
[Google Scholar]
-
112.
Reda M, Jabbour R, Haydar A, Jaafar F, El Ayoubi N, Nawfal O, et al. Case report: Rapid recovery after intrathecal rituximab administration in refractory anti-NMDA receptor encephalitis: Report of two cases.
Front. Immunol. 2024,
15, 1369587.
[Google Scholar]