Refractory castables are widely used in high-temperature applications due to their excellent thermal resistance, mechanical strength, and faster, more cost-effective and more energy-efficient installation (than bricklaying). These materials are typically composed of a coarse and medium grain fraction (often up to 6 mm, but also coarser grains up to, e.g., 15 mm can be possible) and a fine matrix (particles < 45 μm). The distribution of raw materials in refractory castables follows multimodal packing models, where finer particles fill the pores between coarser grains, enhancing packing density and minimizing porosity [
1,
2,
3,
4]. This has led to continuous improvements in particle size reduction, with the finest materials now averaging around 0.5 μm in d
50 [
5]. Using highly sintered and (very) finely ground alumina raw materials increases the specific surface area of the ceramic matrix of refractory castables. Such refinements not only reduce porosity but also affect the castables processing, setting, and sintering behaviour. The influence on the initial stiffening and setting behaviour shall be systematically investigated in this study.
The use of dispersing agents is essential to counteract agglomeration of the particles [
6]. Dispersing agents, which are surface-active organic or inorganic molecules, play an essential role in tailoring the flow properties of refractory castables. Common dispersing agents like polycarboxylate ethers (PCE), acrylates or polyphosphates, adsorb onto particle surfaces. They cause electrostatic, steric, or electrosteric dispersion and thus prevent particle agglomeration [
7]. In the alkaline environment created by CA-cement, surfaces as of Al
2O
3- or SiO
2-particles carry an overall negative charge (thermodynamic equilibrium between negative and positive charges due to protolytic reactions with the pore water and its OH
− and H
3O
+, corresponding to a negative zeta potential in the distinct alkaline range) [
8,
9,
10,
11,
12,
13,
14]. Common dispersing agent systems exhibit a negative charge on their backbone. They, therefore, adsorb at the few positive partial charges on the surfaces (Ca
2+) and cause an increase in electrostatic repulsion due to their charge and/or steric (respectively electrosteric) repulsion due to their molecular structure [
15,
16,
17,
18]. The particles do not agglomerate, keeping the refractory castable is in a flowable state. Studies by Kasper et al. [
19] and Waldstädt et al. [
20] show that the measurement of particle surface charges using zeta potential is a suitable method for identifying dispersion mechanisms and the collapse of the rheological system, which correlates with the initial stiffening of refractory castables.
An important factor in the dispersion mechanism is the interaction between dispersing agents and Ca
2+-ions, which are released during the dissolution of CA-cement. These ions adsorb onto particle surfaces, influencing the adsorption behaviour of the dispersing agents and, consequently, the overall rheology of the castable. More precisely, the adsorption of dispersing agents with a negative charge on negative surface groups is mediated by Ca
2+. This results in increased adsorption of deflocculants and the dispersion is improved [
21].
Some dispersing agents, like polyphosphates or acrylates, precipitate with Ca
2+ at a critical concentration (supersaturation of Ca-dispersant-compounds), leading to a disruption in the castables flow properties and a stiffening effect [
18,
22]. Other dispersing agents, for example, PCE, with long side chains, do not precipitate together with Ca
2+ when the concentration of Ca
2+ increases due to the dissolution of the CA-cement in the pore water. This allows the rheological system to remain stable until the point at which hydration removes water from the system [
15,
16,
18,
23]. Furthermore, hydrate phases that form can be dispersed by a significant amount of free PCE molecules in the pore water. This leads to a longer, stable rheological system [
18].
Achieving the right balance between the concentration of the dispersing agent and the surface area is crucial. In practice, however, this is often determined empirically, which can lead to overdosing or underdosing without realising it. This affects both cost efficiency and performance.
The setting behaviour of CA-cement bonded refractory castables is essentially determined by the dissolution and crystallisation reaction of CA-cement to CAH phases. CA-cement consists mainly of CA, CA
2, and traces of C
12A
7 [
24]. Upon contact with water, Ca
2+-ions initially dissolve out of the surface of the CA-cement particles, thereby increasing the pH-value in the pore solution [
25]. The literature describes that following the initial dissolution of Ca
2+, an Al-O-H passivation layer is formed on the CA-cement particles, which inhibits further dissolution of the CA-cement (dormant period) [
26,
27]. It is essential that the passivation layer undergoes a period of ageing in order to facilitate further rapid dissolution of the CA-cement. Cracking or the pH-dependent dissolution of the Al-O-H passivation layer are among the discussed paths in the existing literature [
18,
26,
27,
28]. Once the passivation layer has undergone degradation, CAH
10, C
2AH
8 and C
3AH
6 (the latter accompanied by the simultaneous formation of AH
3) precipitate as cement hydrate phases, depending on the ions present in the pore solution of the refractory castable and the prevailing temperature [
29,
30]. However, there is evidence that at the same time a precipitation hampering takes place [
18].
Combined with the specific surface area of the particles in the refractory castable matrix, the surface of the particles can be interpreted as a Ca
2+ sink that removes Ca
2+ from the pore solution, thus inhibiting the formation of CAH phases and the degradation of the passivation layer. At the same time, Al
2O
3 is present as a raw material component in almost all refractory castable matrices. It is known from the state of the art that Al
2O
3 and AlO-OH are leached from raw materials with increasing pH-value and that the hydroxylated ions are present in the pore solution [
18,
31,
32,
33,
34,
35,
36,
37,
38]. As a result, during the reaction of CA-cement and dissolved water, an increased amount of Al(OH)
4- is present in the pore solution in addition to Ca
2+, which influences the reaction equilibrium of the hydration reactions.
In this study, a CA-cement based refractory model castable was modified to alter the particle size distribution and specific surface area of the matrix by using highly sintered, finely and very finely ground Al
2O
3 raw materials. Six refractory model castables were developed with varying specific surface areas due to differing ceramic matrix compositions. The objective was to examine how these changes affect the castables initial stiffening and setting behaviour. Three different dispersing agents were added to reduce water demand while maintaining processing characteristics. The dispersing agents and their quantities were systematically varied in order to study their impact on matrix surfaces.
2.1. Experimental Materials
2.1.1. Raw Materials and Their Characteristics
The particle size (-distributions) of the alumina matrix fractions were analysed using laser granulometry (Mastersizer 2000, Malvern Instruments, Malvern, UK) according to the Mie scattering measuring principle, while the specific surface areas were determined using the nitrogen adsorption method (BET) (SA-9600 Series Surface Area Analyzer, Horiba, Kyoto, Japan). The results are summarized in
.
.
Particle sizes and specific surface areas of the Al2O3 matrix components.
|
RG4000 |
CTC20 |
T60/64 -45 MY LI |
| BET in m2/g |
6.9 |
1.6 |
0.6 |
| d50 Cilas in µm |
0.54 |
1.80 |
16.00 |
2.1.2. Refractory Castable Compositions
Six self-flowing, high-alumina (Al
2O
3 raw materials supplied by Almatis GmbH, Ludwigshafen, Germany), CA-cement (Secar 71, Imerys S.A., Paris, France) containing refractory model castables with a maximum grain size of 6 mm were developed by varying the proportions of the individual ceramic Al
2O
3 components of the matrix, which differ in grain size and hence specific surface area, and/or by omitting individual components (
). For an appropriate interpretation of this contribution, it must be mentioned that highly sintered and (very) finely ground Al
2O
3 raw materials were used as a component of the ceramic matrix. Thus, the specific surface area of the particles only results from the (very) fine milling/grinding and not from the internal surface of weakly sintered porous alumina (such as calcined alumina).
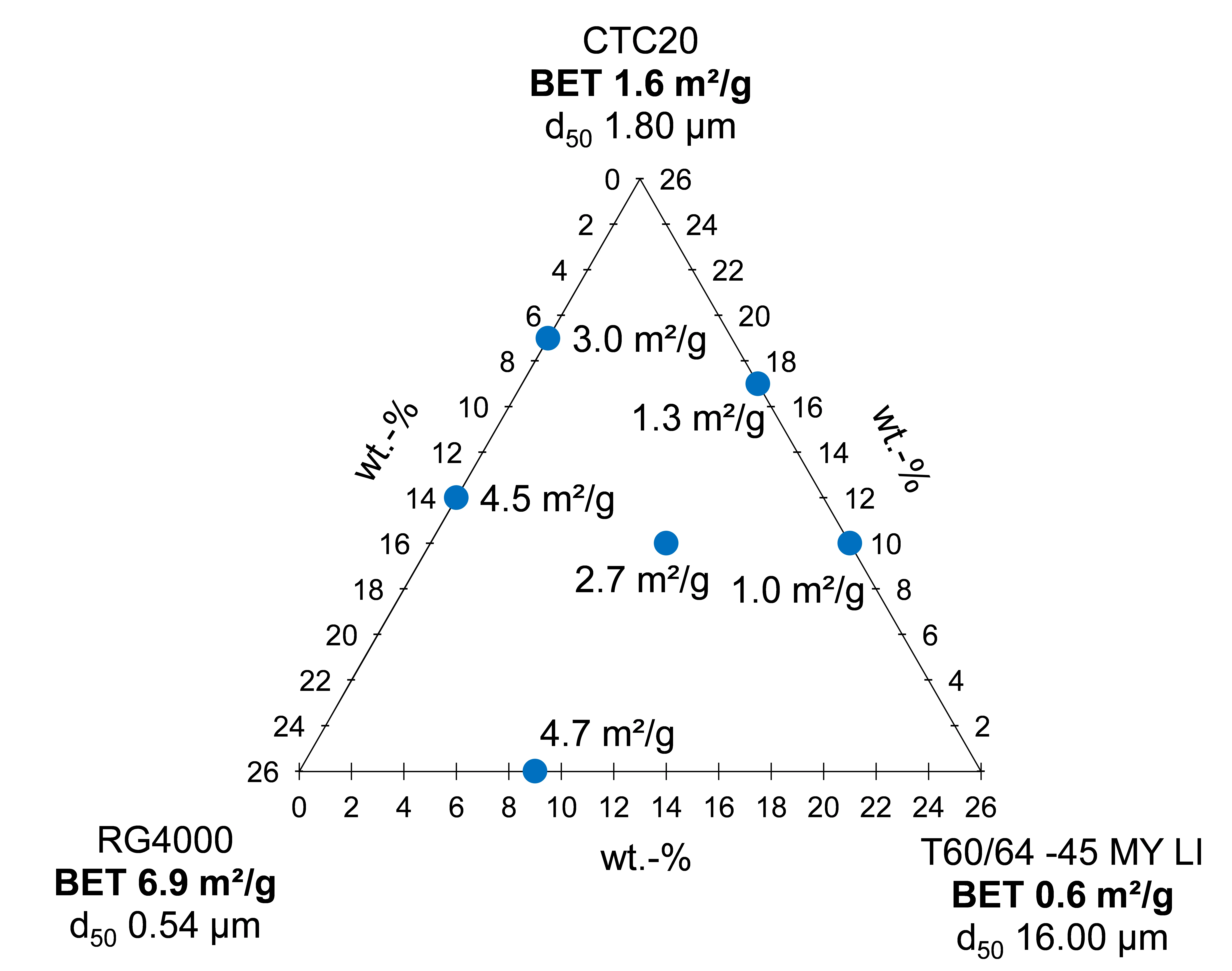
and
depict the corresponding compositions resulting from the variations in the individual matrix components (Al
2O
3 raw materials).
.
Compositions of refractory model castables with varied matrix components for different particle size distributions and specific surface areas (values for BET and d50 in the designation of the castables represent the matrix composition of the Al2O3 raw materials).
| BET in m2/g |
1.0 |
1.3 |
2.7 |
3.0 |
4.5 |
4.7 |
| d50 in µm |
7.1 |
3.2 |
3.0 |
1.8 |
1.4 |
2.0 |
| Component in wt.-% |
|
|
|
|
|
|
| Al2O3 |
|
|
|
|
|
|
| T60/64 3–6 mm |
16 |
16 |
16 |
16 |
16 |
16 |
| T60/64 1–3 mm |
21 |
21 |
21 |
21 |
21 |
21 |
| T60/64 0.5–1 mm |
11 |
11 |
11 |
11 |
11 |
11 |
| T60/64 0.2–0.6 mm |
10 |
10 |
10 |
10 |
10 |
10 |
| T60/64 0–0.2 mm |
11 |
11 |
11 |
11 |
11 |
11 |
| T60/64 -45 MY LI |
16 |
9 |
9 |
- |
- |
9 |
| Reactive alumina (CTC20) |
10 |
17 |
10 |
19 |
12 |
- |
| Reactive alumina (RG4000) |
- |
- |
7 |
7 |
14 |
17 |
| CA-cement (70 wt.-% Al2O3) |
5 |
5 |
5 |
5 |
5 |
5 |
| Sum |
100 |
100 |
100 |
100 |
100 |
100 |
| Water |
depending on dispersing agent () |
| Dispersing agent |
|
.
Dispersing agents, quantities and water amount.
| Dispersing Agent System |
Dispersing Agent |
Amount in wt.-% |
Water in wt.-% |
Polycarboxylate ether
(PCE)
|
Castament FS60 |
0.075/0.10/0.15 |
4.8 |
| Polymethacrylate (PMA) |
Darvan 7S
+ Citric acid
|
0.07/0.10/0.13
0.01
|
6.3 |
Sodium tripolyphosphate
(S-TPP)
|
N25-15
+ Citric acid
|
0.05
0.010/0.015/0.020
|
6.7 |
Three different dispersing agent systems were selected for investigation of the interaction between matrix composition (particle size distribution and specific surface area), dispersing agent, and its concentration (
). The corresponding concentrations of the different dispersing agents were determined through experimental means and established in preliminary tests. As polycarboxylate ether (PCE), Castament FS60 (BASF Construction Polymers GmbH, Trostberg, Germany) was chosen. Darvan 7S (Vanderbilt Minerals, LLC, Norwalk, CT, USA) was selected as polymethacrylate (PMA) in combination with 0.01 wt.-% citric acid (food grade). Sodium tripolyphosphate (S-TPP) N25-15 (Chemische Fabrik Budenheim KG, Budenheim, Germany) was investigated. The S-TPP was furthermore combined with citric acid. However, the concentration of the polyphosphate was maintained at 0.05 wt.-%, while the proportion of citric acid was varied. The reason for that is that citric acid exhibits a stronger dispersion intensity.
The ideal water content of the model castables was determined separately for each of the three dispersing agents in preliminary flow behaviour tests. A spread-flow (according to DIN EN ISO 1927-4) of the multimodal distributed castables 2.7 m
2/g as a kind of reference was set at around 270 ± 20 mm. The water demand was kept constant for all model castables with varying specific surface areas to ensure comparability between differing matrix compositions (
and
). Considering all experiments of this study, spread-flow values between 114.8 and 355.5 mm were achieved. The effect of potential overdosing of dispersing agents was not part of this research and was not investigated.
2.1.3. Preparation of Refractory Castables
The model castables (6 kg mixtures) were mixed in an intensive mixer (type R02E, Eirich GmbH & Co. KG, Hardheim, Germany). A star agitator in co-current flow was used for all experiments. The castables were mixed 1 min dry and 5 min wet after water addition with a rotation speed of 79 rpm. In order to induce the least possible mixing energy and to facilitate the observation of the greatest number of differences resulting from the different matrix compositions, the lowest adjustable speed of the agitator in the mixer was utilised.
. Model castables, shown as points in a triangular diagram with axes in wt.-% to illustrate the compositions of the different matrix components (Al<sub>2</sub>O<sub>3</sub> raw materials) represented in the corners of the scheme.
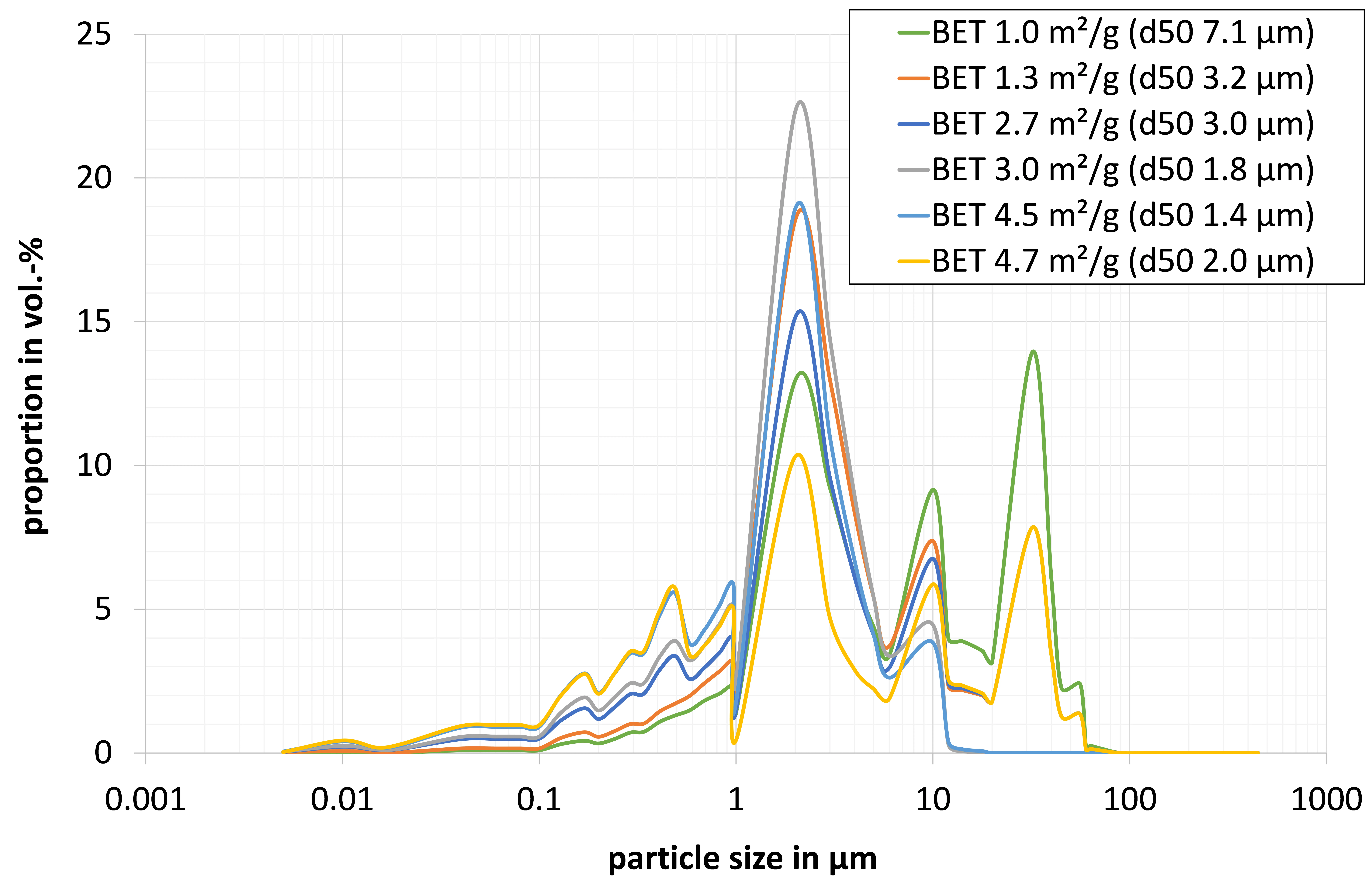
. Particle size distribution curves of the matrix components (Al<sub>2</sub>O<sub>3</sub> raw materials) from the developed refractory model castables with different matrix compositions from .
The sonic velocity and temperature development during the setting of the model castables were measured over a period of 48 h. All samples were stored in a climatic chamber at 20 °C and 95% relative humidity. Sonic velocity and temperature were recorded every 30 s using a test equipment Ultrasonic-Multiplex-Tester, IP 8 (UltraTest GmbH Dr. Steinkamp & Büssenschütt, Bremen, Germany). The measurements were performed directly after mixing the refractory castables. The castable temperature after mixing was recorded in each case.
The main focus was not set on the absolute heights of sonic velocity values but on the curve progression and characteristic points in time for a relative comparison of the castables. Characteristic points in time of the initial stiffening, main hydration, and temperature maximum during setting were determined visually by considering the curve progression of sonic velocity and temperature profile. The time values were rounded to 0.25 h to allow for inaccuracies in the determination.
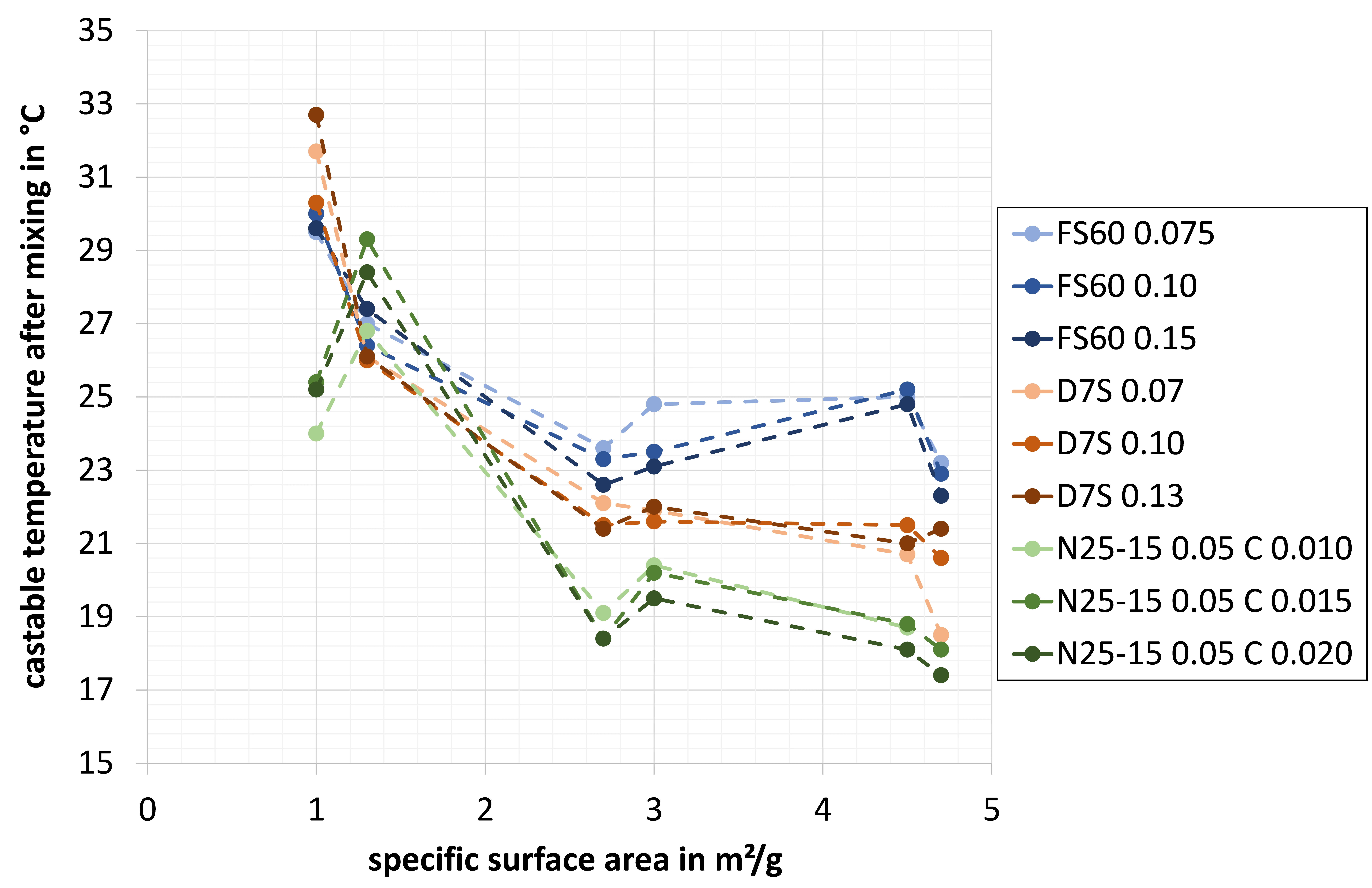
3.1. Temperature of the Castables after Mixing Process
The mass temperature influences the refractory castable properties, such as initial stiffening and setting. There are clear differences in the castable temperature after mixing, which are mainly due to the different specific surface areas of the matrix (and also the dispersing agent used, which influences the required water content). The warming of the castables is mainly caused by the mixing process (and also by the room, raw material and water temperature). The measured temperatures directly after mixing the refractory castables can be seen in
. The following trends can be recognised:
-
-
With a higher specific surface area, the refractory castables heat up less during mixing. This can be explained by the dilatant behaviour of the formulations with low specific surface areas. Castables with higher specific surface areas tend to exhibit rheological Newtonian or shear-thinning behaviour, requiring less mixing energy. As a result, they heat up less [39]. The temperature range extends from 22.9 to 17.4 °C for castables with high surface areas (4.7 m2/g) to 32.7 to 24.0 °C for castables with low surface areas (1.0 m2/g).
-
-
The influence of the dispersing agent system used is reflected indirectly via the temperature of the castables. Model castables dispersed with PCE (Castament FS60) show a higher temperature after mixing than with PMA (Darvan 7S in combination with citric acid). The lowest castable temperature is observed for formulations dispersed with S-TPP (N25-15) and citric acid. The greatest influence here is the different water demand, which increases in this order: 4.8 wt.-%, 6.3 wt.-% and 6.7 wt.-% (Table 2 and Table 3). The adjustments of the water content were necessary due to the reduced dispersion strength of PMA and S-TPP compared to PCE. A higher water content leads to a reduction in the interparticle forces at the high shear rates of mixing, which results in castables with a higher water content heating up less [39].
-
-
In most cases, higher concentrations of the respective dispersing agents result in lower castable temperatures. This can be explained by the lower mixing energy required when more dispersing agent is used [39]. The increased effectiveness of the dispersing agents when the concentrations are increased and thus weaker interparticle forces in the mixing process result in lower castable temperatures.
-
-
More monomodally distributed refractory castable matrix compositions (as is the case with 3.0 m2/g with a high proportion of a single particle fraction ((19 wt.-% of CTC20), and ) tend to lead to dilatancy (although weakly pronounced) [6], which requires higher mixing energy, resulting in (slightly) higher mass temperatures [39]. Multimodal particle size distributions of the matrix (2.7 m2/g) can have a positive influence on the mixing energy [5,6,40,41,42]. This leads to lower castable temperatures. However, these assumptions still need to be verified in further tests.
. Temperatures of all model castables with different specific surface areas and dispersing agents in different concentrations directly after mixing ( and and and ).
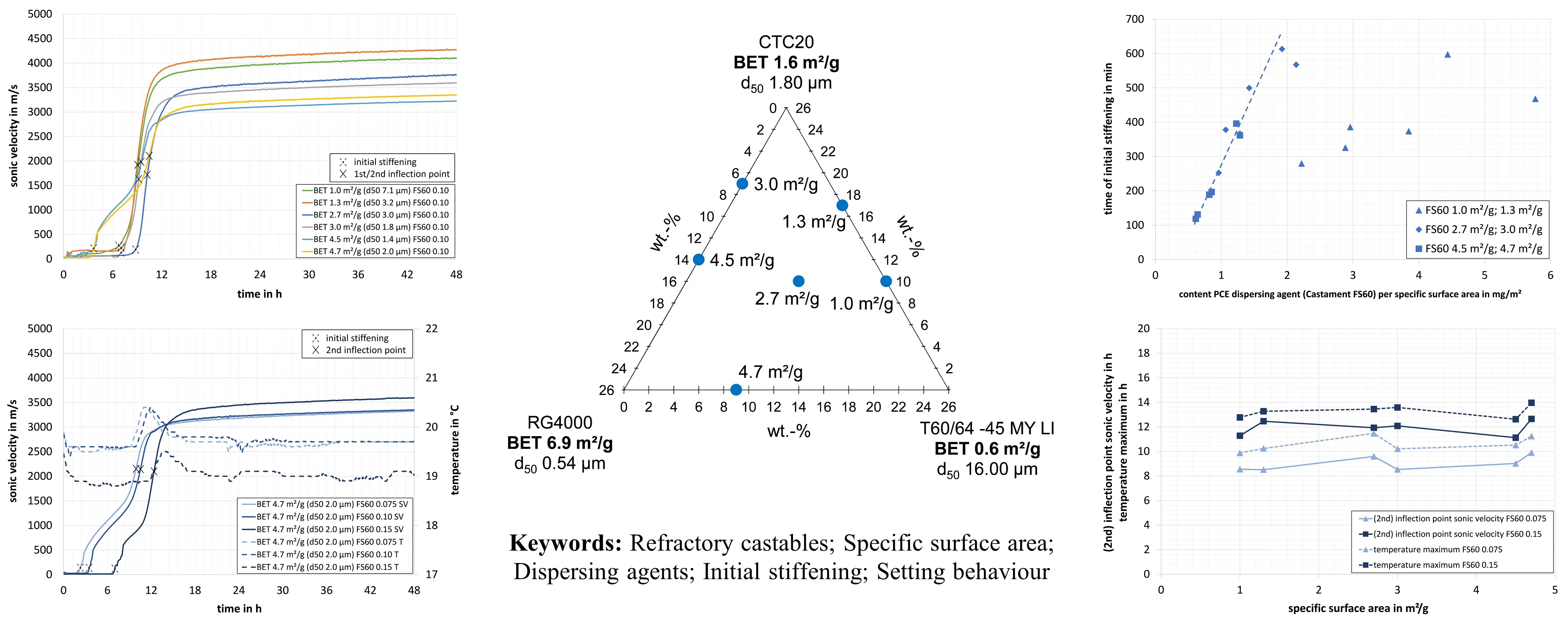
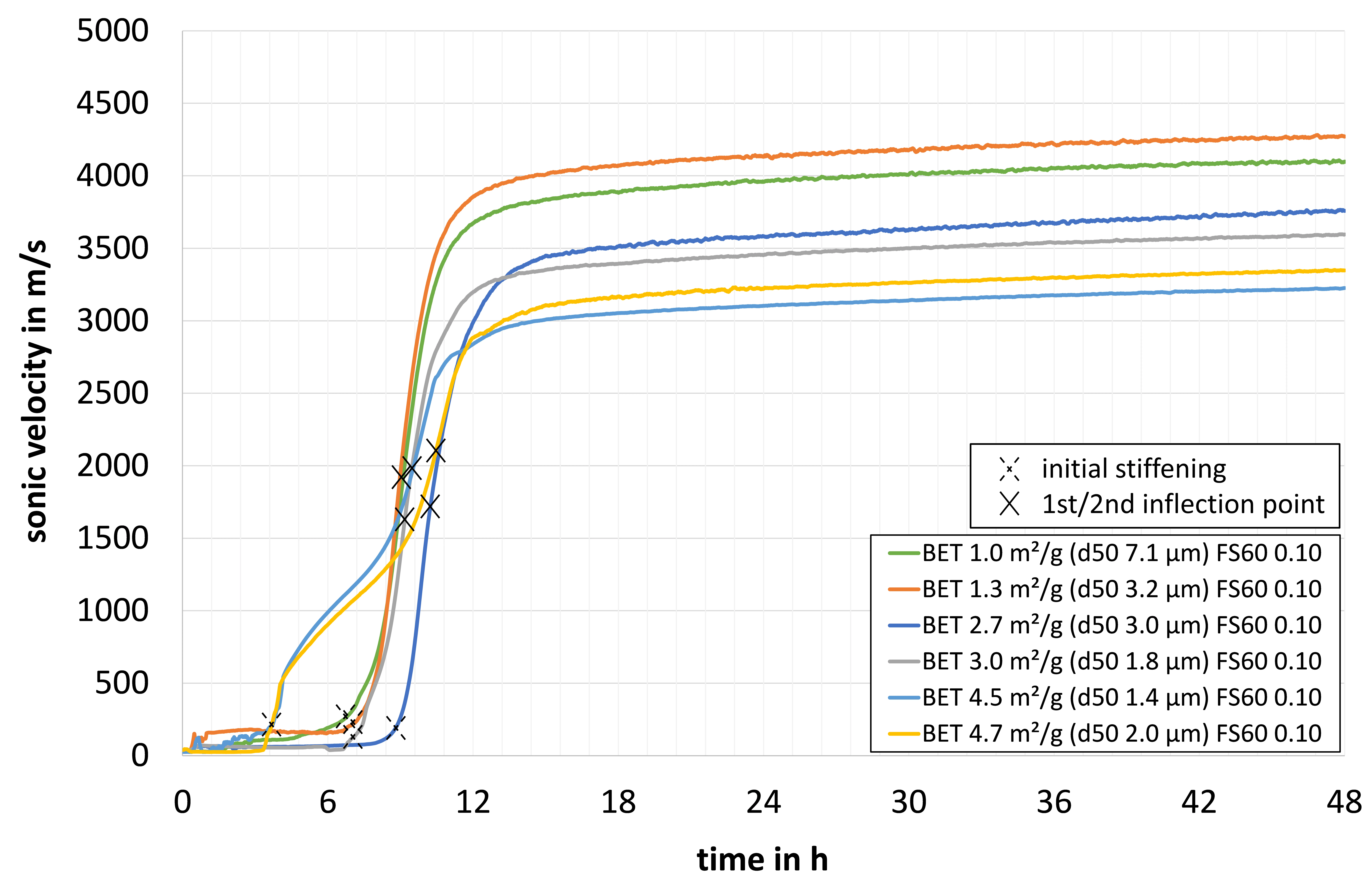
The results, shown in
, indicate that the first significant increase in sonic velocity (enormous increase in the rate of change of sonic signal) of the PCE dispersed refractory castables (0.10 wt.-% of Castament FS60), attributed to the initial stiffening depends on the specific surface area of the matrix composition. All castables in this series exhibit a relatively extended processing time period after mixing and moulding, during which the sonic velocity does not increase. This phenomenon can be attributed to the mode of action of this PCE, which enables the dispersion of newly formed hydrate phases [
18].
For the high specific surface area mixtures, a first (significant) increase in sonic velocity can be observed at first after the start of the test. The refractory castable with a surface area of 4.7 m
2/g reaches initial stiffening after 3.00 h, followed by the mixture with a surface area of 4.5 m
2/g at 3.25 h. Castable 3.0 m
2/g stiffens after 6.00 h and 2.7 m
2/g after 8.50 h. In these cases, the initial stiffening of the refractory castables appears to depend very systematically on the specific surface area of the matrix. Formulations with higher specific surface areas stiffen earlier. This can be attributed to more rapid consumption of the dispersing agent on the high specific surface areas of the matrix particles than in castables with lower specific surface areas. At lower specific surface areas of the matrix, there is more free dispersing agent (PCE) in the pore water available so that hydrates that form can still be dispersed over a longer period of time. This is why mixtures with low specific surface areas stiffen at a later point in time. Here, the different specific surface areas (4.7, 4.5, 3.0, and 2.7 m
2/g) appear to be the dominant effect, leading to different stiffening times (earlier points in time with higher specific surface areas). Zeta potential measurements on matrix suspensions correlated with this established trend. Matrix suspensions with higher specific surface areas dispersed with the same PCE stiffened earlier than those with lower specific surface areas [
20].
. Sonic velocity of model castables with different specific surface areas using the PCE dispersing agent Castament FS60 in a concentration of 0.10 wt.-% ( and ).
Castables 1.0 and 1.3 m
2/g exhibited stiffening after 6.25 h and 6.50 h, respectively. This is not in line with the expected trend that these castables should stiffen at later points in time (due to their low specific surface area). One explanation could be that the first stiffening is sensitive to temperature. Higher castable temperatures from 1.0 and 1.3 m
2/g (30.0 and 26.4 °C) towards 2.7, 3.0, 4.5 and 4.7 m
2/g (23.3, 23.5, 25.2 and 22.9 °C) were found (
, see explanation for the differences in castable temperatures under Section 3.1). For formulations with a low specific surface area (1.0 and 1.3 m
2/g), the effect of the castable temperature seems to have an increased influence, which is why these castables stiffen earlier (than in line with the expected trend of a later stiffening).
In mathematical terms, different inflection points can be determined in the curves of the sonic velocity. Mixtures 4.5 and 4.7 m
2/g show two inflection points (one earlier, close to the first stiffening, and one later). The other compositions each show only one inflection point. For the following evaluations, only the 2nd inflection point of the sonic velocity curves of the mixtures 4.5 and 4.7 m
2/g and the only inflection point of the other refractory castables are considered. This two-stage mechanism (two inflection points) in the early time range of castables 4.5 and 4.7 m
2/g could possibly be cautiously explained as follows: Some research indicates the early formation of amorphous calcium aluminum hydrate phases immediately after mixing. While these early hydrates might not be the primary cause of the initial stiffening defined by rheological changes, their formation on the high surface area of the matrix particles could contribute to changes in the suspension stability and influence the effectiveness of the PCE, indirectly affecting the stiffening behaviour. The absence of a measurable temperature increase during their formation supports an amorphous nature (
). In the discussion of tabular alumina-based refractory castables from Kasper [
18], the data showed that hydrate water was already present immediately after mixing and increased significantly in the first hours. This early binding of water could be attributed to the formation of poorly crystallized or gel-like hydrates (which might not be detected by X-ray diffraction) [
18]. However, the nature of these early water-binding phases (e.g., amorphous hydrates or gels) requires careful consideration and potentially complementary techniques to fully understand.
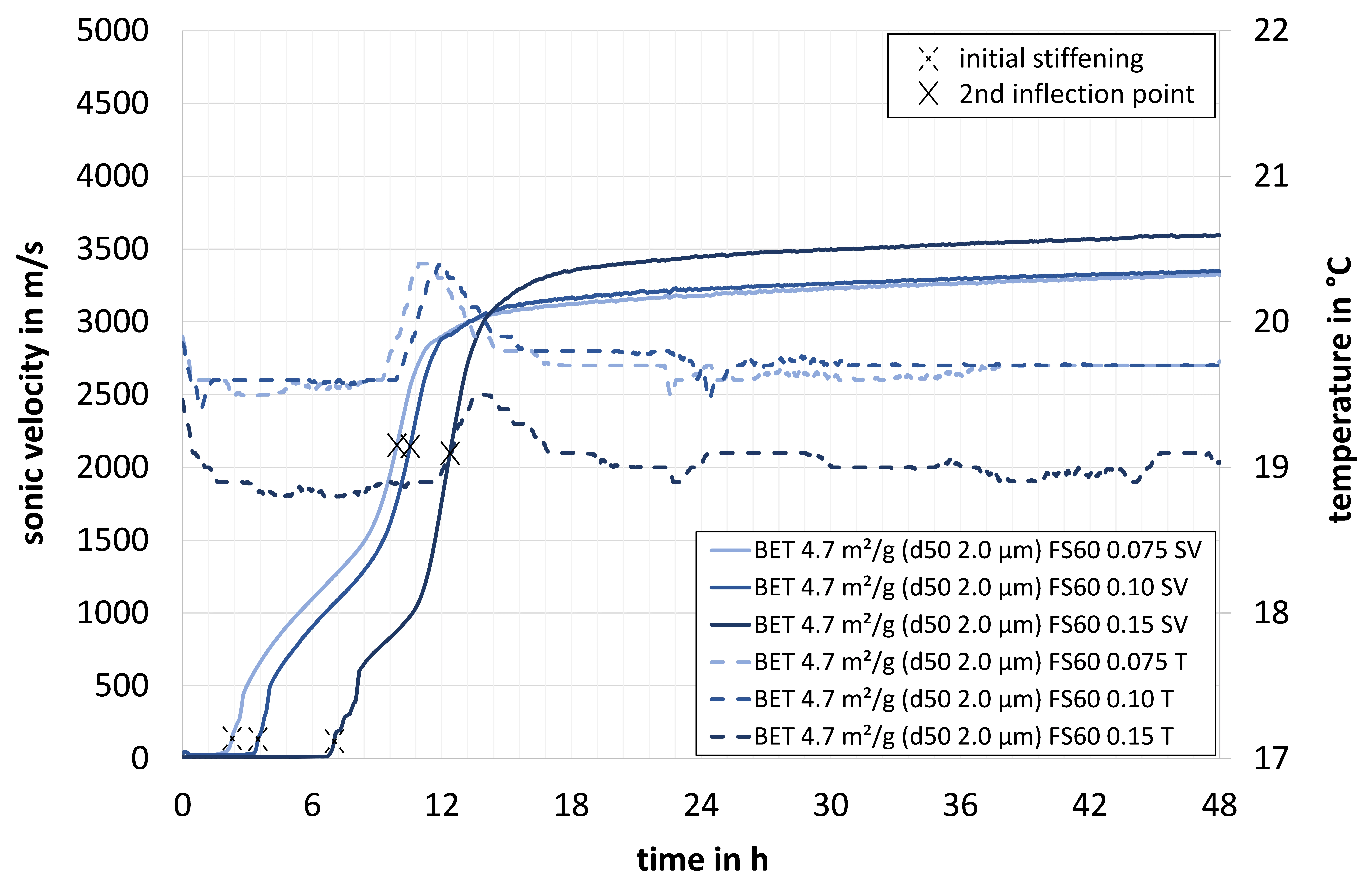
. Sonic velocity and temperature development of the castables with a specific surface area of 4.7 m<sup>2</sup>/g with concentrations of the PCE dispersing agent Castament FS60 of 0.075, 0.10, and 0.15 wt. % ( and ).
Analysis of the (2nd) inflection points of the sonic velocity curves reveals that, in contrast to the initial phase of stiffening, they tend to occur within a narrow time frame for all refractory castables. At this concentration of the PCE (Castament FS60, 0.10 wt.-%), the inflection points are observed to occur within a time period of 9.00 to 10.50 h. For the same dispersing agent concentration, there appears to be no dependency on the specific surface area of the matrix. The previously described characteristics of the sonic velocity progression were also observed for the other two concentrations of PCE (Castament FS60, 0.075 and 0.15 wt.-%).
depicts that as the dispersing agent content increases, the first (significant) increase in sonic velocity, attributed to the first stiffening and the (2nd) inflection point of the sonic velocity curves, are both shifted to a later point in time. At the lowest concentration of 0.075 wt.-% the refractory castable 4.7 m²/g exhibited stiffening after 2.00 h. The castable with 0.10 wt.-% started stiffening after 3.25 h, while dispersion with 0.15 wt.-% required 6.50 h. This can be explained by the fact that the ratio of the dispersing agent to a specific surface area increases with a higher dispersing agent concentration. As a result, there is proportionally more free dispersing agent (PCE) present in the pore water. Therefore, more hydrates that form can be dispersed before the rheological system collapses. This results in a later initial stiffening. During measurements of the zeta potential matrix, suspensions with the same specific surface area but a higher content of the same PCE (Castament FS60) stiffened distinctly later than those with lower concentrations [
20]. The (2nd) inflection points of the curves are at 10.00 h for 0.075 wt.-%, 10.50 h for 0.10 wt.-% and 12.50 h for 0.15 wt.‑%. At higher dispersing agent contents, the (2nd) inflection points are shifted to a later point in time approximately linearly proportional to the increase in the concentration of the dispersing agent (as with the initial stiffening).
Another value measured and depicted in
is the temperature profile. The maximum temperature in each castable corresponds to an exothermic reaction, which can be attributed to the main formation of hydrate phases. These exothermic peaks (temperature maximum) are observed at 11.25 h for a concentration of 0.075 wt.-%, 12.25 h at 0.10 wt.-%, and 14.00 h at 0.15 wt.-%. All temperature peaks coincide with the respective (2nd) inflection points of the sonic velocity, which can, therefore, be correlated with the time of main hydration. This correlation has been documented in previous literature [
18,
43].
The phenomenon of delayed initial stiffening of castables with increased PCE content and the subsequent delay (linearly proportional) in the (2nd) inflection point of the sonic velocity curve was also observed in all other refractory castables with different matrix compositions dispersed with the PCE.
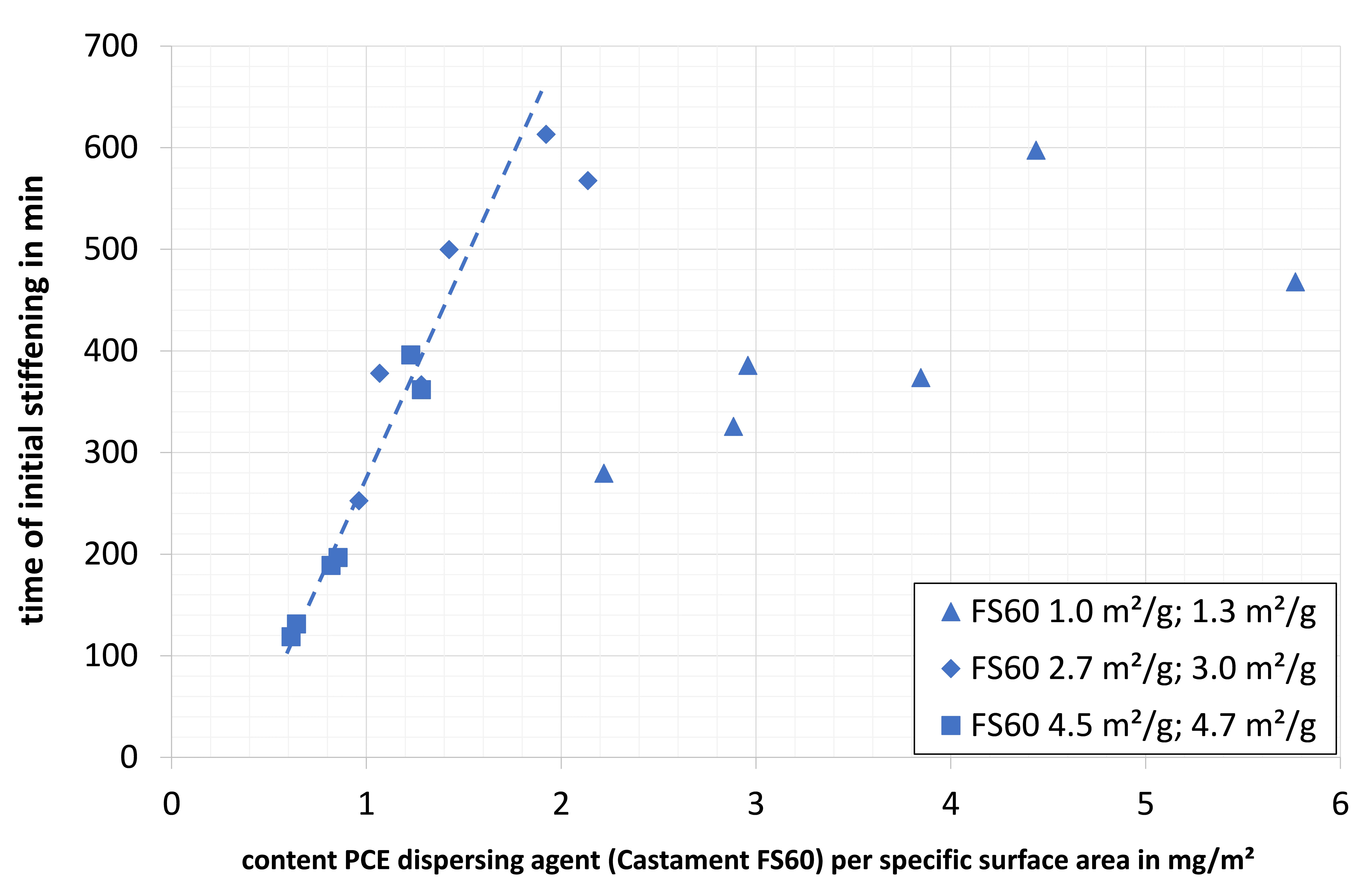
The described correlations regarding the initial stiffening and hydration are highly systematic for all refractory castables with different specific surface areas dispersed with PCE (Castament FS60) as becomes evident in
and
. The first stiffening shows a clear dependency on the specific surface area of the matrix (
). Considering the content of the dispersing agent per specific surface area (a dashed line shows an approximate trend), a linearly proportional dependency can be recognised that the time of the first stiffening is shifted to a later time with a lower specific surface area and an increasing content of dispersing agent (for castables 2.7, 3.0, 4.5 and 4.7 m
2/g). Times of an initial stiffening of 120 to 400 min (4.5; 4.7 m
2/g) and 250 to 600 min (2.7; 3.0 m
2/g) were established. This trend is not apparent for mixtures 1.0 and 1.3 m
2/g (first stiffening after 275 to 600 min). One explanation for the earlier initial stiffening of these formulations (than in line with the expected trend) may be the significantly higher castable temperature after the mixing process (
, see explanation for the differences in castable temperatures under Section 3.1).
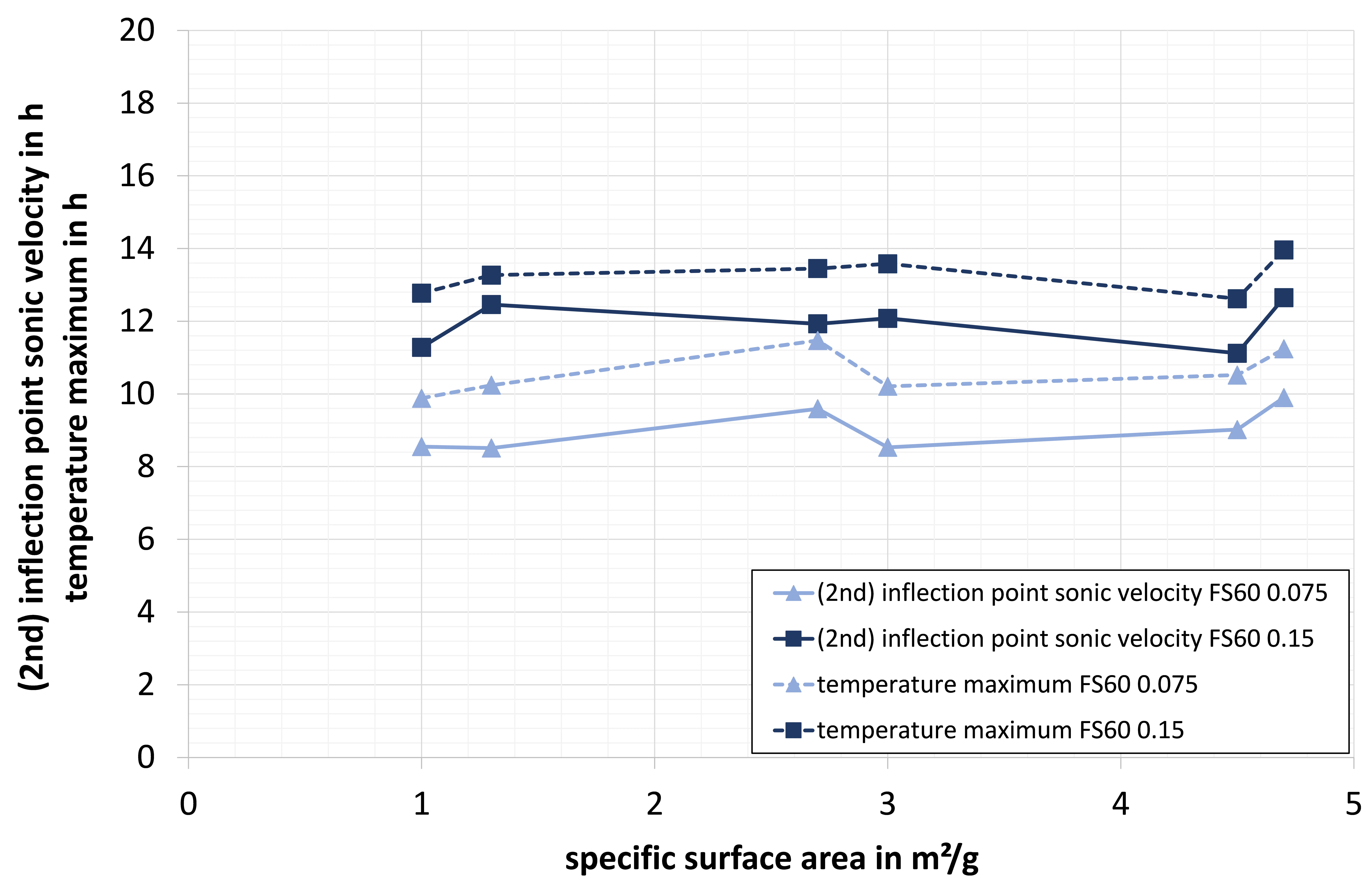
The inflection points (2nd inflection point of the sonic velocity curves of the mixtures 4.7 and 4.5 m
2/g and the only inflection points of 1.0, 1.3, 2.7 and 3.0 m
2/g) of the sonic velocity curve seem with times of 8.50 to 10.00 h (0.075 wt.-%) and 11.25 to 12.50 h (0.15 wt.-%) to be independent of the matrix compositions (
). A clear dependence on the concentration of the PCE can be recognised. A higher concentration leads to a later hydration range (irrespective of the specific surface areas). The temperature maximum during setting is equally delayed and matches with the (2nd) inflection point of the sonic velocity in each case, respectively and can therefore be related to the time of main hydration.
. Times of initial stiffening of the model castables with different specific surface areas of the matrix with all considered concentrations of the PCE dispersing agent Castament FS60 of 0.075, 0.010, and 0.15 wt.-% with regard to the content of dispersing agent per specific surface area ( and ).
. (2nd) inflection point (2nd inflection point of the sonic velocity curves of the mixtures 4.5 and 4.7 m<sup>2</sup>/g and the only inflection point of 1.0, 1.3, 2.7, 3.0 m<sup>2</sup>/g) of the sonic velocity curve and temperature maximum of the model castables with different specific surface areas of the matrix with concentrations of the PCE dispersing agent Castament FS60 of 0.075 and 0.15 wt.-% ( and )
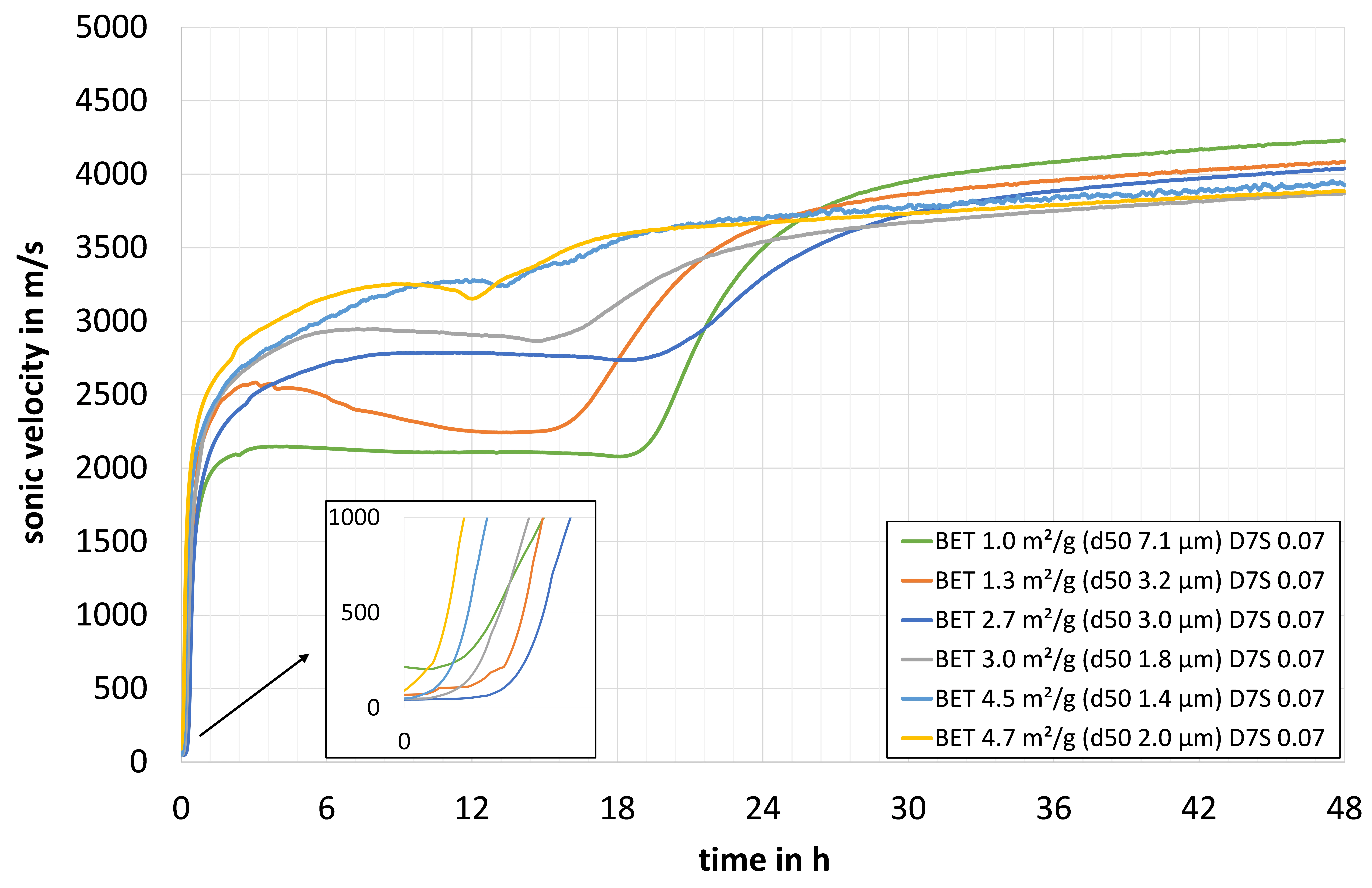
Using PMA (0.07 wt.-% of Darvan 7S) with citric acid instead of PCE (Castament FS60) leads to notable differences in the sonic velocity curve progression, although a clear dependence on the specific surface area can still be observed (
). A first significant increase in sonic velocity (enormous increase in the rate of change of sonic signal) can be determined within a couple of minutes, which is attributed to the initial stiffening of the refractory castables. This comparatively rapid initial stiffening (in contrast to PCE) can be explained by the mode of action of the PMA, as it precipitates at a critical concentration with Ca
2+ (supersaturation of Ca-dispersant-compounds) [
18,
22,
44]. The zoomed-in section clearly shows that formulations with a higher specific surface area of the matrix demonstrate an earlier onset of initial stiffening. Castables 4.7 and 4.5 m
2/g stiffened after ca. 5 min, 3.0 m
2/g within ca. 10 min and 2.7 m
2/g after ca. 20 min. This is because more dispersing agent adsorbs on higher specific surface areas. This means that there is less free dispersing agent present in the pore water. When Ca
2+ is dissolved from the cement, the free PMA (but also citric acid) is consumed first. Subsequently, adsorbed PMA (and citric acid) are removed from the surfaces, which gradually leads to coagulation by precipitation of sparingly soluble Ca-PMA-compounds (and Ca-citrate-compounds). Therefore, at high specific surface areas, initial stiffening occurs earlier, as adsorbed PMA (and citric acid) are removed from the surfaces faster (which previously maintained the dispersion). As is also the case with PCE, the different specific surface areas (4.7, 4.5, 3.0, and 2.7 m
2/g) appear to be the dominant effect, which leads to different times of stiffening (earlier points in time with higher specific surface areas). However, due to the completely different dispersion mechanism at a much earlier point in time than with PCE. Zeta potential measurements on matrix suspensions correlated with this trend. Matrix suspensions with higher specific surface areas dispersed with the same PMA (combined with the same citric acid) stiffened at earlier points in time than those with lower specific surface areas [
20].
. Sonic velocity of the model castables with different specific surface areas using the PMA dispersing agent Darvan 7S in a concentration of 0.07 wt.-% and citric acid ( and ).
Castables 1.0 and 1.3 m
2/g exhibited initial stiffening after ca. 10 and ca. 15 min, respectively, and do not follow the expected trend that these castables should stiffen at later points in time (due to their low specific surface area). As the first stiffening is sensitive to temperature, higher castable temperatures from 1.0 and 1.3 m
2/g with 31.7 and 26.1 °C towards 2.7, 3.0, 4.5 and 4.7 m
2/g with 22.1, 21.9, 20.7 and 18.5 °C seem to have an increased influence here (as with PCE), which is why the castables with a low specific surface area of the matrix stiffen at an earlier point in time (than expected (
, explanation for the differences in castable temperatures under Section 3.1)).
Similar to the initial stiffening, the 2nd inflection points of sonic velocity curves demonstrate a dependency on the specific surface areas of the matrix. In the model castables with a lower surface area, the 2nd inflection point, which also indicates the hydration range, is shifted to a later point in time. At a concentration of PMA dispersing agent (Darvan 7S) of 0.07 wt.-%, the 2nd inflection points can be detected after 12.00 and 13.25 h for refractory castables with surface areas of 4.7 and 4.5 m
2/g, respectively. Castables 3.0 m
2/g exhibits a 2nd inflection point at 15.00 h and 2.7 m
2/g at 19.00 h. As already observed during the initial stiffening, the 2nd inflection points of the castables 1.0 m
2/g at 18.25 h and 1.3 m
2/g at 15.25 h do not correspond to the trend of later hydration of formulations with lower specific surface areas. This could again be explained by the higher refractory castable temperature of 1.0 and 1.3 m
2/g compared to 2.7, 3.0, 4.5, and 4.7 m
2/g (
). The temporal shift of the 2nd inflection point of the sonic velocity curve occurs over a much longer period of time than the relatively short temporal shift associated with the initial stiffening. The described trends in relation to the sequence of the 2nd inflection points (and also the times of the initial stiffening) could also be observed for all other model castables dispersed with PMA (Darvan 7S) in different concentrations.
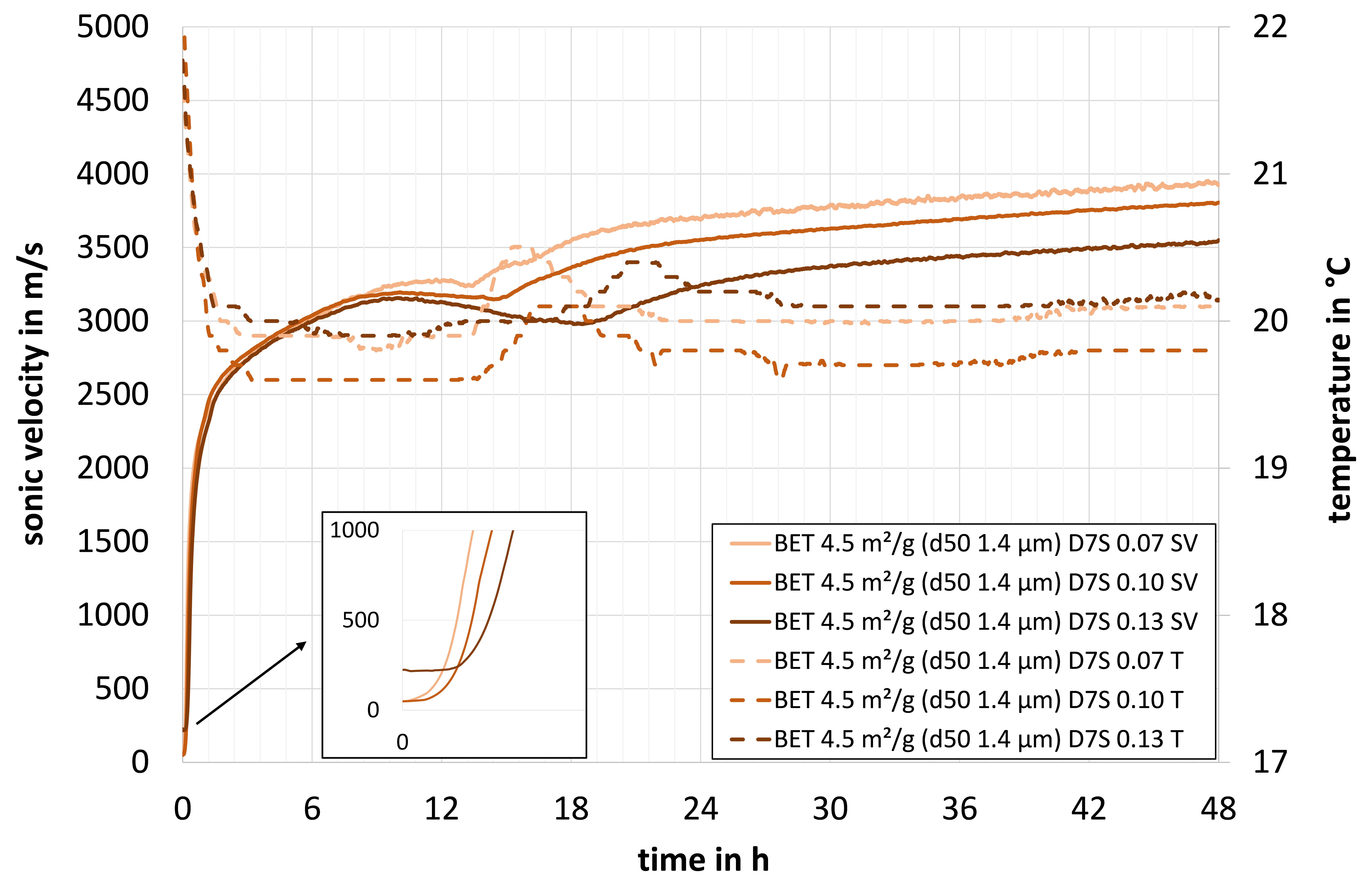
An extremely systematic pattern within the sonic velocity curves was observed as the dispersing agent content of PMA was increased (
). There is a recognisable trend that a first significant increase in sonic velocity (enormous increase in the rate of change of sonic signal), indicating initial stiffening and also the 2nd inflection point of the sonic velocity progression to be shifted to a later point in time. As depicted in the zoomed-in view, the refractory castable 4.5 m²/g exhibited earlier stiffening with a concentration of 0.07 wt.-% Darvan 7S (at ca. 5 min) than with 0.10 wt.-% (at ca. 10 min) and 0.13 wt.-% (at ca. 15 min) with an approximately linearly proportional dependency. Due to a higher concentration, there is more free dispersing agent present in the pore water. As Ca
2+ is dissolved from the cement, free PMA (but also citric acid) is consumed first. Afterwards, adsorbed PMA (and citric acid) is removed from the surfaces, gradually leading to coagulation by precipitation of sparingly soluble Ca-PMA-compounds (and Ca-citrate-compounds). Therefore, at higher concentrations, initial stiffening occurs later, as more PMA (and citric acid) is in the pore water available and simultaneously adsorbed PMA (and citric acid) is removed from the surfaces more slowly (whereby the dispersion mechanism can be maintained over a longer period of time). During zeta potential measurements, matrix suspensions with the same specific surface area but a higher content of the same PMA (Darvan 7S) and the same citric acid showed a later stiffening than those with lower concentrations [
20].
The 2nd inflection points of the sonic velocity curves occur at 13.25, 14.50, and 18.25 h with concentrations of Darvan 7S of 0.07 wt.-%, 0.10 wt.-% and 0.15 wt.-%, respectively, indicating a linearly proportional correlation. The temporal shift is significantly more pronounced in the hydration range than in the relatively brief temporal shift observed during the initial stiffening phase. A similar systematic can be identified when considering the temperature development. The temperature maximum values for this test series are 15.50 h at a concentration of 0.07 wt.-%, 17.25 h at 0.10 wt.-%, and 21.25 h at 0.13 wt.-%. As also observed with PCE, the time of the temperature maximum is found to correlate with the time of main hydration in each case.
. Sonic velocity and temperature development of the castables with a specific surface area of 4.5 m<sup>2</sup>/g with concentrations of the PMA dispersing agent Darvan 7S of 0.07, 0.10, and 0.13 wt.-% and citric acid ( and ).
These described correlations regarding the initial stiffening and hydration are highly systematic for all refractory castables with different specific surface areas dispersed with PMA (Darvan 7S) and citric acid, as depicted in
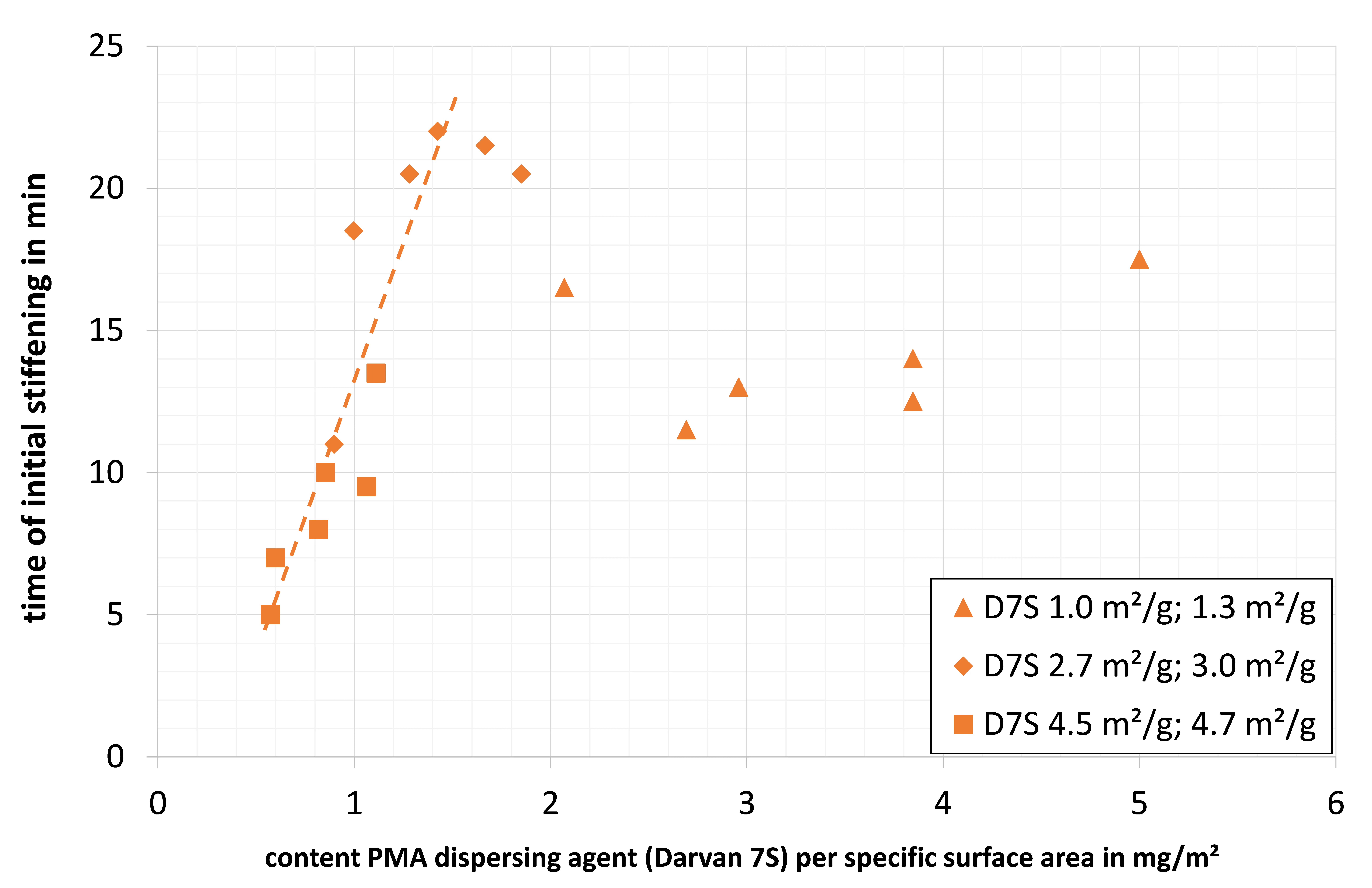
and
. The first stiffening shows a clear dependency on the specific surface area of the matrix (
). As also found with PCE, when considering the content of dispersing agent per specific surface area (a dashed line shows an approximate trend), a linearly proportional dependency can be recognised that the time of the initial stiffening is shifted to a later point in time with a lower specific surface area and increasing content of PMA dispersing agent (for castables 2.7, 3.0, 4.5 and 4.7 m
2/g). Times of the initial stiffening of 5 to 15 min (4.5; 4.7 m
2/g) and 10 to 25 min (2.7; 3.0 m
2/g) were measured. This trend is not apparent for mixtures 1.0 and 1.3 m
2/g (first stiffening after 10 to 20 min). An explanation for the earlier stiffening of these formulations (than in line with the expected trend) can be the significantly higher castable temperature after the mixing process (
, see an explanation for the differences in castable temperatures under Section 3.1). This behaviour was also observed and described with PCE.
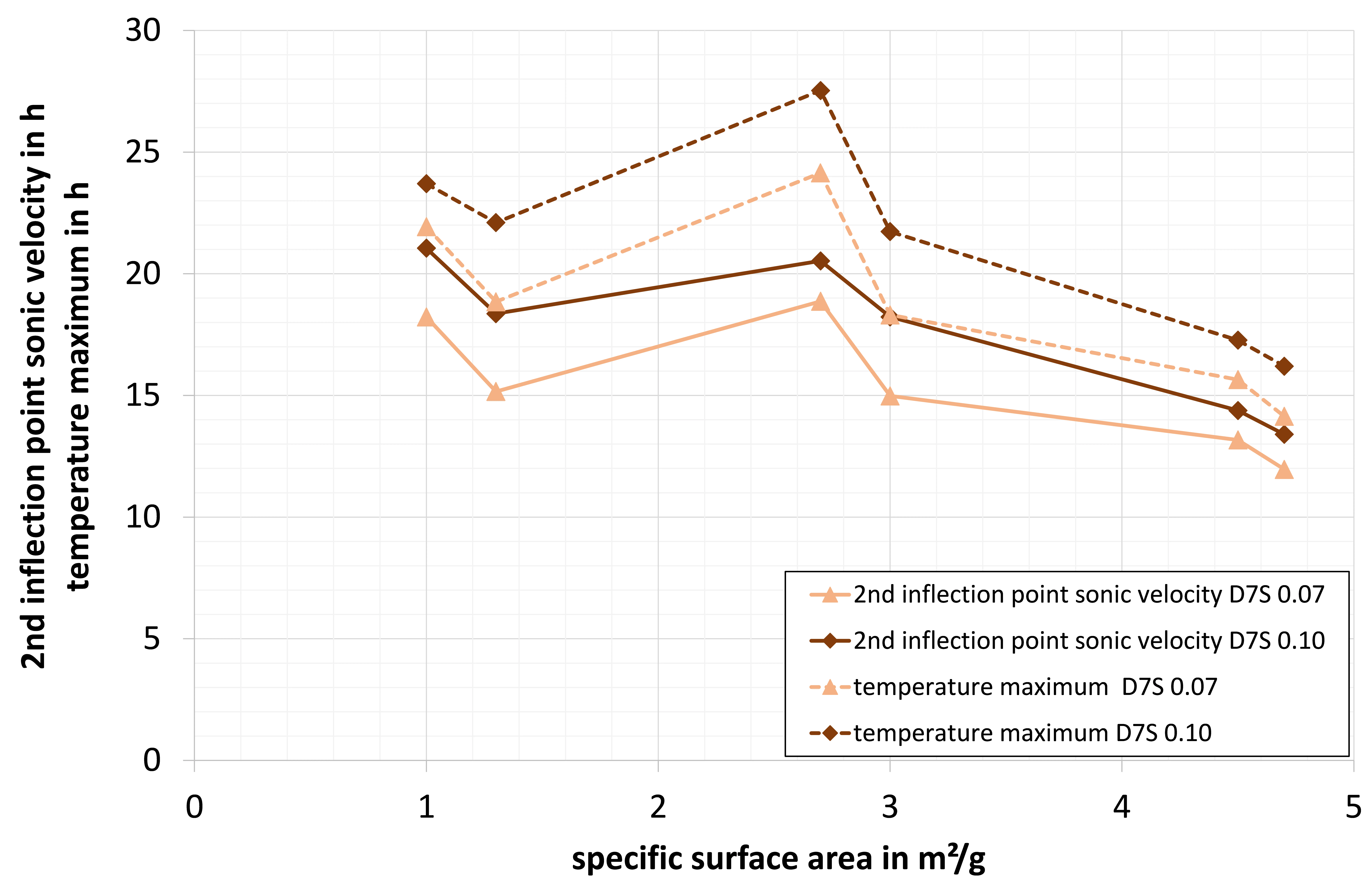
As visualised in
, the 2nd inflection points of the sonic velocity curves (marking main hydration) appear to be clearly dependent on the matrix compositions (in contrast to PCE, where independence was established). Refractory castables dispersed with PMA (Darvan 7S) with a higher specific surface area of the matrix show an earlier point of hydration compared to those with lower specific surface areas. The use of a higher concentration of PMA has been observed to result in a delayed hydration range. Hydration times of 12.00 to 19.00 h (0.07 wt.-%) and 13.50 to 21.00 h (0.15 wt.-%) were observed. The temperature maximum during the setting exhibits a highly systematic correlation with the time of main hydration (2nd inflection point of the sonic velocity) in each case. The temperature maxima also established at later points in time with lower specific surface areas and with higher PMA contents.
. Times of initial stiffening of the model castables with different specific surface areas of the matrix with all considered concentrations of the PMA dispersing agent Darvan 7S of 0.07, 0.10, and 0.13 wt.-% and citric acid with regard to the content of dispersing agent per specific surface area ( and ).
. 2nd inflection point of the sonic velocity curve and temperature maximum of the model castables with different specific surface areas of the matrix with concentrations of the PMA dispersing agent Darvan 7S of 0.07 and 0.10 wt.-% and citric acid ( and ).
displays that the sonic velocity of the refractory model castables using the S-TPP dispersing agent N25-15 (0.05 wt.-%) and citric acid (0.015 wt.-%) shows a high systematicity and demonstrates a clear correlation with the specific surface area of the matrix. The sonic velocity progression exhibits a comparable profile to those observed when using PMA (Darvan 7S) in combination with citric acid as a dispersing agent. Within a few minutes at the beginning, a first significant increase in sonic velocity (enormous increase in the rate of change of sonic signal) is observed due to the initial stiffening of the refractory castables. As with PMA, this comparatively rapid initial stiffening can be explained by the mode of action of the S-TPP, which precipitates at a critical concentration with Ca
2+ (supersaturation of Ca-dispersant-compounds) [
18,
45]. The zoomed-in view clarifies that castables with a higher specific surface area of the matrix exhibit an earlier onset of initial stiffening. The model castable with a specific surface area of 4.7 m
2/g stiffened after ca. 5 min, 4.5 m
2/g stiffened within ca. 15 min, 3.0 m
2/g after ca. 20 min, and 2.7 m
2/g within ca. 25 min. For the first stiffening, the same effect occurs as for PMA: More dispersing agent adsorbs on higher specific surface areas. Thus, less free dispersing agent is available in the pore water. When Ca
2+ is dissolved from the cement, the free S-TPP but also citric acid are consumed first. Subsequently, adsorbed S-TPP and citric acid are removed from the surfaces, which gradually leads to coagulation by precipitation of sparingly soluble Ca-TPP-compounds and Ca-citrate-compounds. Therefore, initial stiffening occurs earlier at high specific surface areas, as adsorbed S-TPP and citric acid are removed from the surfaces faster (which previously maintained the dispersion). As with PCE and PMA, the different specific surface areas (4.7, 4.5, 3.0, and 2.7 m
2/g) appear to be the dominant effect, leading to different stiffening times (earlier points in time with higher specific surface areas). Mixtures 1.3 and 1.0 m
2/g stiffen after ca. 15 and ca. 20 min, respectively, and do not follow the expected trend that castables with lower specific surface areas stiffen at later points in time. As the first stiffening appears to be sensitive to temperature, higher castable temperatures from 1.0 and 1.3 m
2/g with 25.4 and 29.3 °C towards 2.7, 3.0, 4.5, and 4.7 m
2/g with 18.4, 20.2, 18.8 and 18.1 °C most probably have an increased influence here (as with PCE and PMA at 1.0 and 1.3 m
2/g). This could be why the castables with a low specific surface area of the matrix stiffen at an earlier point in time (than expected (
, explanation for the differences in castable temperatures under Section 3.1)).
. Sonic velocity of the model castables with different specific surface areas using the S TPP dispersing agent N25-15 in a concentration of 0.05 wt.-% in combination with citric acid in a concentration of 0.015 wt.-% ( and ).
The 2nd inflection points of the sonic velocity also correlate with the specific surface area of the refractory castables’ matrices (as observed with PMA). In the mixtures with a lower specific surface area, the 2nd inflection point, marking the hydration of CA-cement, is shifted highly systematically to a later point in time. The hydration times achieved in descending order of specific surface area from 4.7 to 1.0 m
2/g are 15.25, 17.25, 21.00, 22.25, 23.00 and 25.50 h. The temporal shift in hydration occurs over a much longer period of time than the relatively short temporal shift associated with the initial stiffening. The aforementioned trends (initial stiffening and hydration) were also observed in all other model castables with S-TPP dispersing agent N25-15 and citric acid at different concentrations.
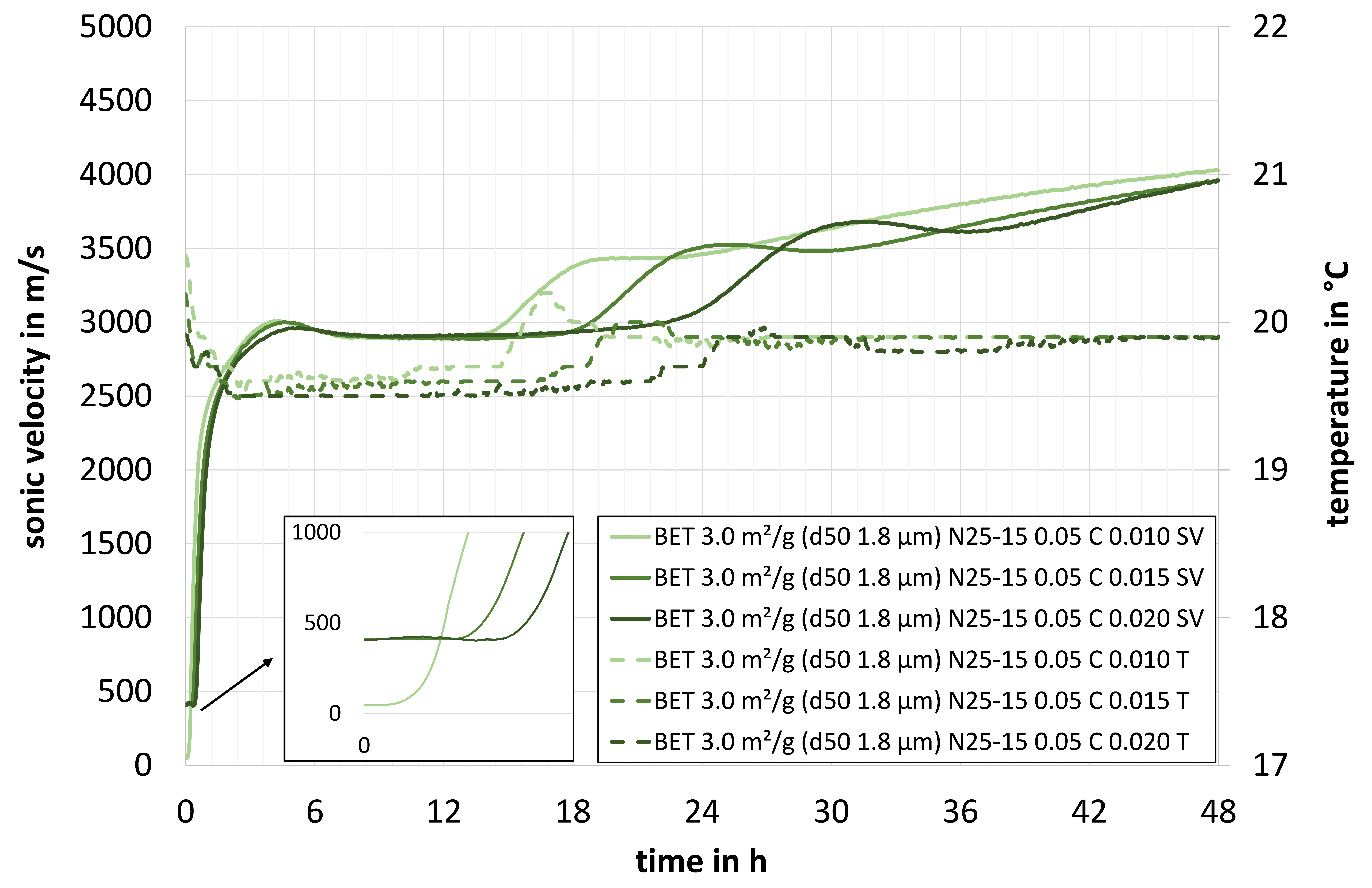
depicts that the sonic velocity and the temperature evolution of the castable 3.0 m²/g with S-TPP dispersing agent N25-15 (0.05 wt.-%) in combination with citric acid at concentrations of 0.010, 0.015, and 0.020 wt.-% exhibit a high degree of systematic order. The first significant increase in sonic velocity (enormous increase in the rate of change of sonic signal), indicating a first stiffening, and also the 2nd inflection point of the sonic velocity curves are shifted to a later point in time with increasing dispersing agent content (in this case citric acid). In the zoomed-in section, it can be observed that the refractory castable with a concentration of 0.010 wt.-% citric acid exhibits earlier stiffening (after ca. 10 min) than that with 0.015 wt.-% (after ca. 20 min) and 0.020 wt.-% (after ca. 30 min) with a linearly proportional dependency. The same reasoning as for PMA can be used to explain this: Due to a higher concentration, there is more free dispersing agent present in the pore water. As Ca
2+ is dissolved from the cement, free S-TPP and citric acid are consumed first. Afterwards, adsorbed S-TPP and citric acid are removed from the surfaces. This gradually leads to coagulation by precipitation of sparingly soluble Ca-TPP-compounds and Ca-citrate-compounds. At higher concentrations (of citric acid), initial stiffening occurs later, as more S-TPP and citric acid are in the pore water available and simultaneously adsorbed S-TPP and citric acid are removed from the surfaces more slowly. Thus, the dispersion mechanism can be maintained over a longer period of time.
The main hydration ranges of the castables are 16.75 h (0.010 wt.-%), 21.00 h (0.015 wt.-%), and 26.50 h (0.020 wt.-%). With an increased citric acid concentration at a constant specific surface area, the times of hydration are markedly delayed. A similar trend can be identified when examining the temperature maxima. The values of the temperature maxima, indicating the predominant time of formation of hydrate phases, are determined at fairly identical points in time. It is noticeable that the height of the temperature maximum peaks decreases with increasing citric acid content. This could indicate that the exothermic reaction is less intense, and consequently, fewer hydrate phases are formed (not systematically analysed). The temporal shift of the hydration range is considerably more pronounced than that observed for the relatively brief temporal shift in the first stiffening.
. Sonic velocity and temperature development of the castables with a specific surface area of 3.0 m<sup>2</sup>/g with the S TPP dispersing agent N25-15 in a concentration of 0.05 wt.-% in combination with citric acid in concentrations of 0.010, 0.015 and 0.020 wt.-% ( and ).
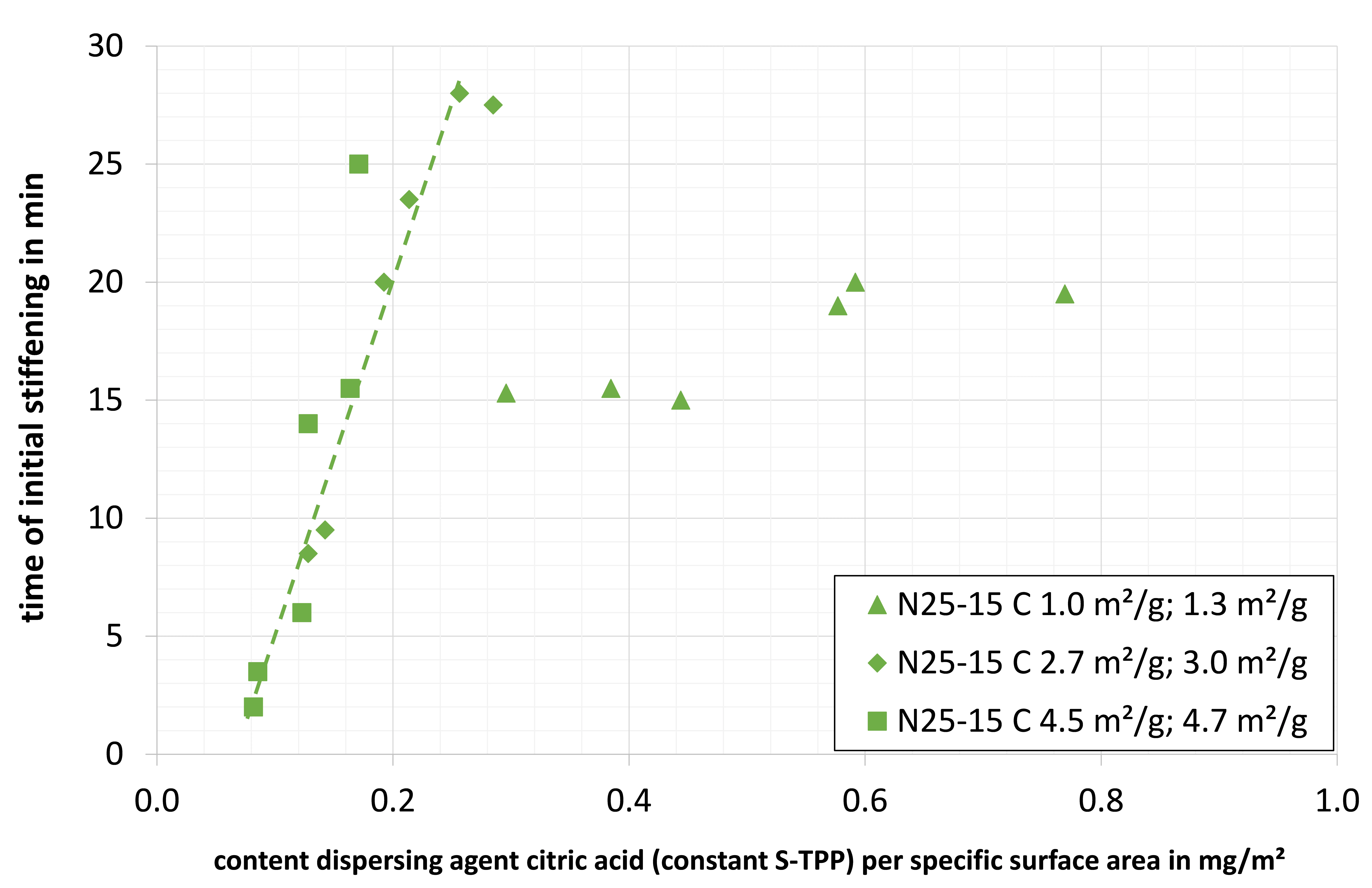
and
underline the described highly systematic correlations regarding the initial stiffening and hydration for all refractory castables with different specific surface areas using S-TPP (N25-15) and citric acid. The first stiffening is clearly dependent on the specific surface area of the matrix (
). As with PCE and PMA, examining the content of the dispersing agent per specific surface area (a dashed line shows an approximate trend) reveals a linearly proportional dependency. The time of the first stiffening is shifted to a later time with a lower specific surface area and an increasing content of (S-TPP and) citric acid dispersing agent (for castables 2.7, 3.0, 4.5 and 4.7 m
2/g). Times of the initial stiffening of < 5 to 25 min (4.5; 4.7 m
2/g) and 10 to 30 min (2.7; 3.0 m
2/g) were measured. This trend is not apparent for mixtures 1.0 and 1.3 m
2/g (first stiffening after 15 to 20 min). The earlier initial stiffening of these formulations (than in line with the expected trend) can be explained by the significantly higher castable temperature after the mixing process (
, see explanation for the differences in castable temperatures under Section 3.1). This behaviour was also observed and described with PCE and PMA.
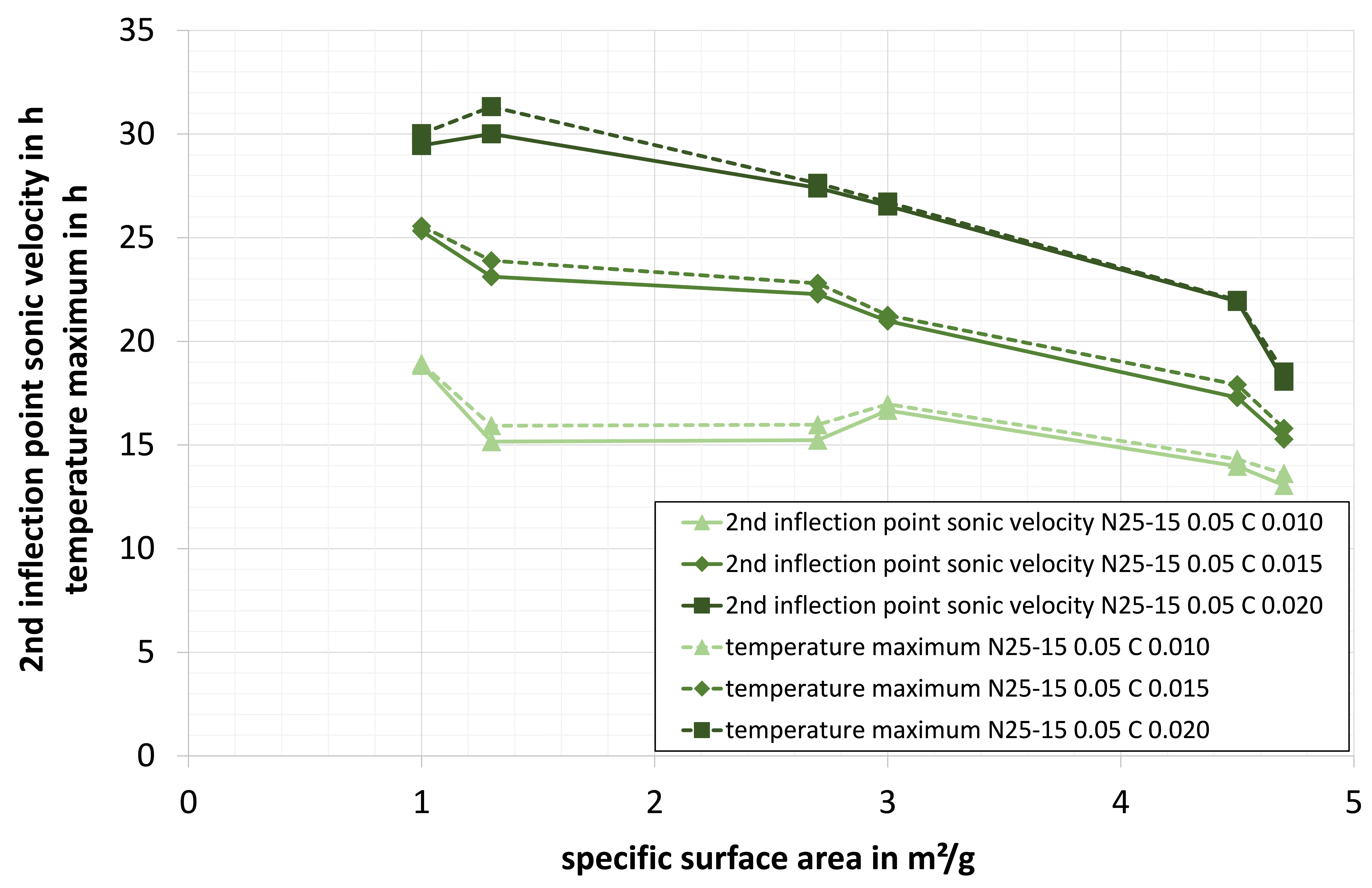
shows that the 2nd inflection points of the sonic velocity curve (marking main hydration) appear to be clearly dependent on the matrix compositions (as with PMA, but in contrast to PCE, where independence was established). Castables dispersed with S-TPP (N25-15) and citric acid with a higher specific surface area of the matrix show an earlier point of hydration compared to those with lower specific surface areas. A higher concentration of citric acid results in a delayed time of hydration. Hydration times of 13.00 to 19.00 h (0.010 wt.-%), 15.25 to 25.50 h (0.015 wt.-%) and 18.00 to 30.00 h (0.020 wt.-%) were observed. A highly systematic correlation of the temperature maximum during the setting and the time of main hydration (2nd inflection point of the sonic velocity) in each case can be observed. The temperature maxima were also established at later points in time with a lower specific surface area and with higher (S-TPP and) citric acid contents. The variation of citric acid in the system N25-15 is an effective means of systematically and finely controlling both the stiffening and hydration times. Another correlation that can be recognised is that at a low concentration of citric acid, there is only a small dependency on the specific surface area, but at higher concentrations, there appears to be an increased dependency on the specific surface area. One possible explanation could be that the specific surface area has less influence at low citric acid concentrations, as S-TPP (that is kept constant) primarily influences the hydration process. In this case, S-TPP plays the primary role in delaying hydration. At higher concentrations of citric acid, its surface-mediated effect becomes more pronounced (especially for high specific surface areas), making differences in the specific surface area of the refractory castables’ matrix more significant.
. Times of initial stiffening of the model castables with different specific surface areas of the matrix with all considered concentrations of the S-TPP dispersing agent N25-15 at a (constant) concentration of 0.05 wt.-% in combination with citric acid in concentrations of 0.010, 0.015 and 0.020 wt.-% with regard to the content of dispersing agent per specific surface area ( and ).
. 2nd inflection point of the sonic velocity curve and temperature maximum of the model castables with different specific surface areas of the matrix with a concentration of the S TPP dispersing agent N25-15 of 0.05 wt.-% in combination with citric acid in concentrations of 0.010, 0.015 and 0.020 wt.-% ( and ).
This study systematically investigates six developed self-flowing, high-alumina, CA-cement containing refractory castables with a maximum particle size of 6 mm. The matrix (particles ≤ 45 µm) was modified through targeted alteration of the particle size distribution and, therefore, the specific surface area (by using highly sintered and (very) finely ground alumina raw materials with high specific surface areas) to investigate the impact of the specific surface area of the matrix on initial stiffening and setting. Three different dispersing agents, polycarboxylate ether (Castament FS60), polymethacrylate (Darvan 7S, in combination with citric acid), sodium tripolyphosphate (N25-15), and citric acid and their concentrations were systematically varied for this purpose. The potential impact of overdosing dispersing agents was not investigated as part of this research.
The differences observed in the initial stiffening process concerning the specific surface areas can be explained by various effects (with different weighting). Overall, it was observed that the initial stiffening of the refractory model castables is linearly proportional to the increase of the specific surface area of the matrix, irrespective of the dispersing agent used. A higher specific surface area leads to an earlier onset of the first stiffening. With regard to the time of main hydration, clear differences were found depending on which dispersing agent was used. A systematic influence of the concentration was found for all three dispersing agents used. A higher concentration leads to a linearly proportional delayed first stiffening and time of main hydration but under the process of completely different mechanisms. However, the sonic velocity curves of the castables differ significantly in their curve progression in relation to the dispersing agent used.
Castables dispersed with PCE (Castament FS60) show a wide processing time period. A first significant increase in sonic velocity (enormous increase in the rate of change of sonic signal) indicates the initial stiffening. It occurs after 2 to 10 h. This is due to the mode of action of PCE: Forming cement hydrate phases can be dispersed, and a rheological stable matrix suspension can be maintained over a relatively long period of time. Formulations dispersed with PCE and with higher specific surface areas stiffen earlier than those with lower specific surface areas. This can be attributed to more rapid consumption of the dispersing agent on the high specific surface areas of the matrix particles. At lower specific surface areas of the matrix, there is more free PCE in the pore water available, so that hydrates that form can still be dispersed over a longer period of time, leading to a later stiffening. At a higher concentration of PCE, more free dispersing agent is present in the pore water, resulting in a later initial stiffening because the dispersion mechanism of hydrate phases can be maintained for a longer period of time.
When using PMA (Darvan 7S) in combination with citric acid and S-TPP (N25-15) with citric acid, the model castables establish an initial stiffening within < 5 to 30 min (in contrast to PCE, where this time span is several hours). This can be explained by the completely different dispersing mechanism of PMA and S-TPP compared to PCE. Again, castables with higher specific surface areas stiffen more quickly than mixtures with lower specific surface areas. This can be attributed to more rapid consumption of the dispersing agent on the high specific surface areas, whereby there is less free dispersing agent in the pore water available. When Ca
2+ is dissolved from the cement, the free PMA/S-TPP (but also citric acid) is consumed first. Subsequently, adsorbed PMA/S-TPP and citric acid are removed from the surfaces, which gradually leads to coagulation of the matrix suspension. Therefore, initial stiffening occurs earlier at high specific surface areas, as adsorbed PMA or S-TPP and citric acid are removed from the surfaces faster (which previously maintained the dispersion mechanism). At higher concentrations, there is even more free PMA/S-TPP and citric acid present in the pore water and simultaneously, adsorbed PMA/S-TPP and citric acid are removed from the surfaces more slowly. The dispersion mechanism can be maintained over a longer period of time, resulting in a later initial stiffening.
The time of initial stiffening is followed by an inflection point. In most cases, there is only one inflection point in the PCE sonic velocity curve. However, for castables with a high specific surface area, a 2nd inflection point may occur. These inflection points correlate with the respective temperature maxima (exothermic reaction) and mark the main hydration. For PCE, these inflection points are very close to the first stiffening (which occurs after several hours). For the same concentration of PCE, hydration takes place in a narrow time period and appears to be independent of the specific surface area of the matrix. As the PCE concentration is increased, the time of main hydration is systematically delayed (linearly proportional) with more PCE.
For PMA/S-TPP and citric acid, the 2nd inflection points mark the main hydration range and correlate with the respective temperature maxima (as for PCE). With PMA/S-TPP and citric acid, hydration occurs at times greater than 10 hours after the first stiffening. For the same concentrations of PMA/S-TPP and citric acid, hydration depends on the specific surface area of the matrix (in contrast to PCE) and occurs later at lower specific surface areas. An increase in PMA and (S-TPP with) citric acid leads to a linearly proportional delayed hydration (as with PCE).
At higher castable temperatures (due to low specific surface areas, which can lead to dilatancy and correspondingly high mixing energy input during the mixing process), regardless of the use of PCE, PMA, or S-TPP, the process of initial stiffening and hydration range (only with PMA and citric acid) can take place earlier, as this mechanism is influenced by the castable temperature.
It is already documented in the literature that PCE acts by steric repulsion and delays the initial stiffening [
15,
16,
18,
20,
23], while PMA and S-TPP (in combination with citric acid) act electrosterically and precipitate together with Ca
2⁺, which leads to earlier coagulation [
18,
20,
22,
44,
45]. It is also described for matrix suspensions that higher specific surface areas adsorb more dispersing agents [
20]. For PCE in particular, but also PMA, a relationship between concentration and processing time period (initial stiffening) is stated [
18,
20,
46]. Furthermore, it is reported that measurements of the sonic velocity (of all dispersing agent systems considered) show that the inflection point of the curve progression correlates with main hydration. A temperature maximum marks the exothermic reaction [
18,
43].
A new contribution to this research’s state of the art is the systematic proof of the linearly proportional dependency between the specific surface area of the castables’ matrix and the first stiffening, regardless of the dispersing agent system used. However, the stiffening times differ significantly between PCE/PMA/S-TPP/citric acid due to their completely different mechanisms of action. The results also showed an extension of the state of the art with regard to the time of main hydration. At constant PCE content, the specific surface area was independent. With a constant PMA/S-TPP/citric acid content, a very systematic dependency of the time of main hydration range on the specific surface area (which has not yet been shown in literature) could be demonstrated. Another effect that has only been empirically determined so far, and not systematically quantified, is the accelerating effect of mixing energy-induced temperature increase on the initial stiffening. In the present investigations, the higher temperature of the refractory castables can be identified as a disruptive influence that leads to deviations in the results that do not correspond to an expected trend. Methodologically innovative is the correlation of the required amount of dispersing agent per specific surface area with the time of the initial stiffening, allowing a targeted, predictable refractory castable formulation, which was previously only possible empirically.
These results can provide valuable information for developing or adapting refractory castables. The initial stiffening and main hydration can be precisely controlled or modified over time by selecting suitable dispersing agent systems and dosages. The influence of the castable matrix composition can also be considered in this context. This enables specific adjustments to be made to individual process parameters.
Conceptualization, F.H., T.W., J.K., C.D., O.K.; Methodology, F.H.; Validation, F.H., T.W., J.K.; Investigation, F.H.; Data Curation, F.H.; Writing—Original Draft Preparation, F.H.; Writing—Review & Editing, F.H., J.K., T.W.; Supervision, J.K., C.D., O.K.
Not applicable.
Not applicable.
The datasets generated and analyzed during the current study are available from the corresponding author on reasonable request.
This research project (IGF-No. 22499N) is being funded by the German Ministry of Economic Affairs and Climate Action (BMWK) according to a decision of the German Bundestag.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.