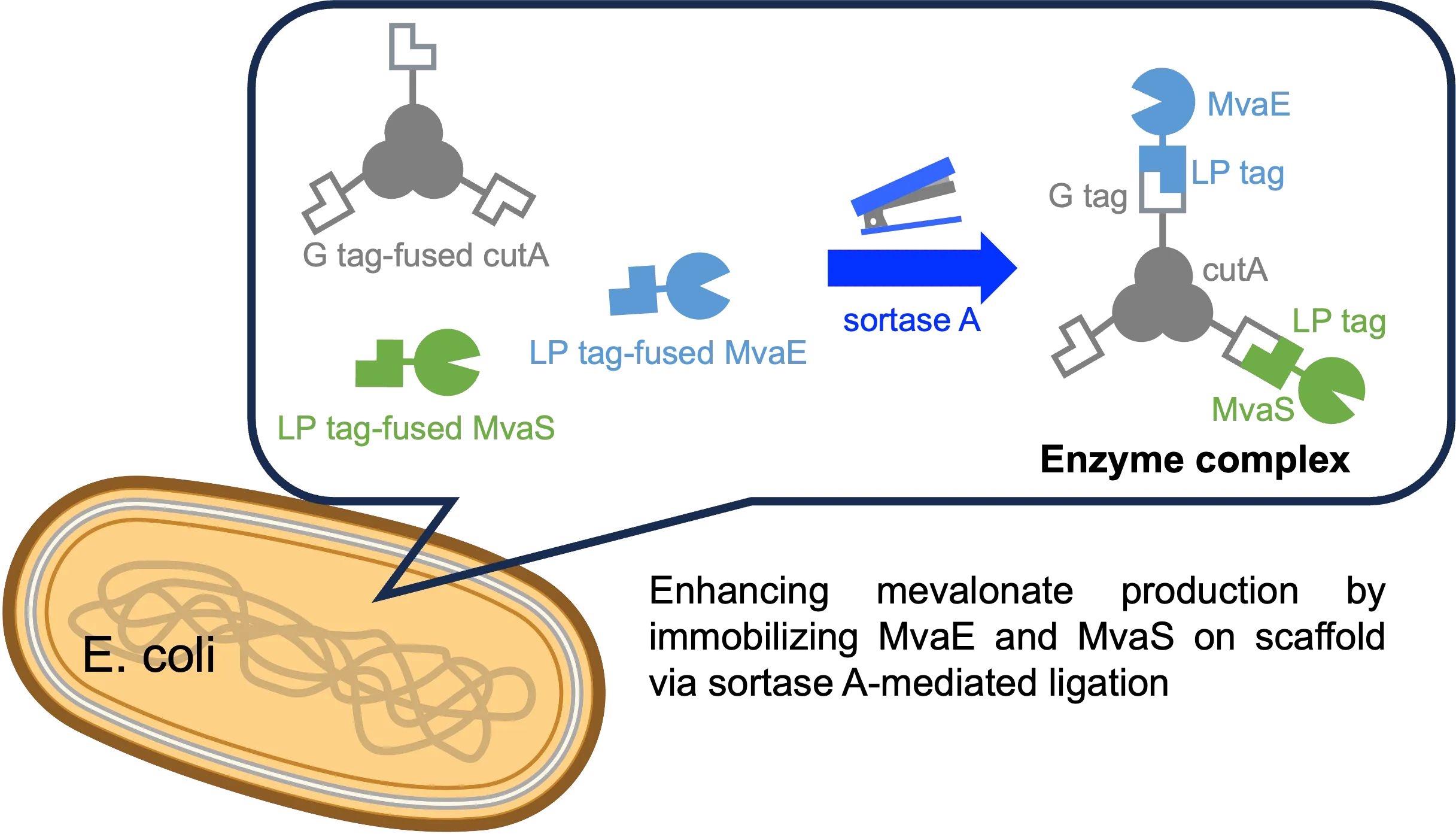
1. Introduction
In recent years, biorefineries, technologies, and industries that produce biofuels and chemicals from biomass have gained significant attention amid concerns regarding petroleum resource depletion and environmental issues. The use of renewable resources and energy-efficient bioprocesses in these technologies holds promise for reducing CO
2 emissions and promoting a circular economy [
1]. In particular, the production of valuable chemicals using microorganisms has been the subject of extensive research.
Escherichia coli is a well-studied microorganism with great potential for converting biomass into valuable compounds, such as fuels, basic chemicals, and pharmaceuticals. Over the past few decades, the primary focus has been on efficiently producing useful chemicals through strain engineering to achieve sufficient titer, yield, and productivity in
E. coli-based bioproduction [
2,
3].
Researchers have often employed metabolic and genetic engineering techniques to enhance production. Enhancing the metabolic flux from substrate to product through pathway engineering is a key strategy for improving bioproduction. Common approaches include the introduction of heterologous pathways [
4], the overexpression of endogenous genes [
5], and the elimination of competitive pathways [
6]. Although these methods have been successfully applied in designing desirable strains, conventional gene deletion techniques are limited by their inapplicability to essential genes involved in cell growth and maintenance. To address these limitations, sophisticated and systematic approaches have been developed in recent years. Some genetic tools offer advantages, such as portability, conditionality, and adjustability, surpassing standard utility methods [
7].
Metabolic channeling is another approach to improving strains for bioproduction [
8]. Generally, small metabolites are processed either by individual metabolic enzymes or through substrate channeling involving multi-enzyme complexes that perform a series of reactions in a stepwise manner. Substrate channeling facilitates the transfer of intermediates between two enzymes that catalyze sequential metabolic reactions in cells. This process prevents the diffusion of intermediates and enhances the efficiency of metabolic cascades [
9,
10]. These studies suggest that positioning metabolic enzymes in close proximity can facilitate substrate channeling of metabolic cascades and enhance the production of target chemicals by microorganisms.
Tippmann et al. advanced the concept of metabolic channeling by applying affibody-based scaffolds. Their study demonstrated the assembly of two metabolic enzymes on anti-idiotypic affibody pairs in
Saccharomyces cerevisiae to enhance farnesene production and the assembly of three metabolic enzymes on affibody scaffolds in
E. coli to enhance polyhydroxybutyrate production. The implementation of these two- and three-component scaffolds resulted in enhanced production of farnesene and polyhydroxybutyrate, respectively [
11]. Notably, this approach to metabolic channeling offers a significant advantage in directing metabolic flux towards the desired pathway without necessitating genetic disruption or manipulation.
Although conventional gene fusion protocols offer usability and versatility, they frequently suffer from inactivation issues [
12,
13]. This is particularly problematic for metabolic enzymes with complex structures, where gene fusions are susceptible to protein misfolding. Consequently, there is a pressing need for posttranslational connection methodologies [
14]. An alternative approach that has gained attention is the colocalization of metabolic enzymes. A notable example is a study that focused on geraniol production [
15]. We employed a protein scaffold to colocalize the three principal enzymes involved in the geraniol biosynthetic pathway. This colocalization approach enhanced both the rate and yield of geraniol synthesis, primarily by mitigating the detrimental effects of pathway intermediates on cell growth.
Metabolic enzyme linkages via sortase A (SrtA) have been reported to connect enzymes within
E. coli cells. SrtA, produced by
Staphylococcus aureus, is the most extensively studied sortase. It recognizes the leucine-proline-X-threonine-glycine sequence in substrate proteins (LP tag), cleaves the peptide bond between threonine and glycine residues, and subsequently forms a native peptide bond between the threonine carboxyl group and the amino group of the N-terminal glycine oligomers (G tag). Our group previously demonstrated metabolic enzyme ligation using a transpeptidase (staphylococcal SrtA) in
E. coli to redirect metabolic flux through metabolic channeling and successfully achieved metabolic channeling. The expression of SrtA enhances the binding of PFL-LP and G-PTA in
E. coli, resulting in enhanced acetate production [
16]. Although SrtA-mediated ligation offers an effective approach for posttranslational enzyme coupling, it is limited by its restriction of linking only two enzymes. Moreover, N-terminal modifications, which are required for SrtA-mediated ligation, can sometimes have unfavorable effects on protein expression.
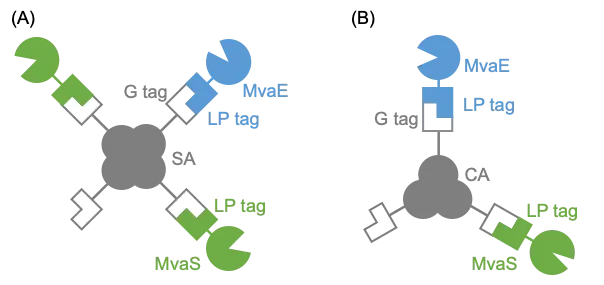
To address these limitations, we explored a method for enzyme immobilization on a scaffold using SrtA. Metabolic enzyme complexes, formed through the colocalization of multiple enzymes on scaffold proteins, are anticipated to enhance production by accelerating the consumption of toxic intermediates. The selection of an appropriate scaffold is crucial for this approach. In the present study, we investigated two potential scaffolds: streptavidin (SA), a heterologous tetrameric protein, and CutA (CA), an endogenous trimeric protein. illustrates the concept of metabolic enzyme complex formation using scaffold proteins. There are also reports of successfully expressing streptavidin and cutA [
17,
18,
19]. They should be improved in future studies [
20,
21], so we attempted to form these complexes using SA and CA as scaffolds. To validate this concept, the production of mevalonate was used as a model system. Mevalonate is a crucial raw material in cosmetics and pharmaceuticals and an important intermediate in terpenoid synthesis [
22,
23]. The biosynthetic production of mevalonate offers a distinct advantage in that it enables the selective synthesis of the desired enantiomer, (R)-mevalonate, without removing the (S)-mevalonate byproduct. By introducing a specific set of enzyme-encoding genes into
E. coli, selective production of (R)-mevalonate can be achieved [
24]. The introduction of
atoB,
mvaE, and
mvaS, which encode key enzymes for mevalonate biosynthesis, into
E. coli enables mevalonate production (Supplementary Figure S1) [
24]. However, in the engineered mevalonate biosynthesis pathway in
E. coli, the reaction that consumes the intermediate 3-hydroxy-3-methylglutaryl CoA (HMG-CoA) represents a rate-limiting step. Consequently, HMG-CoA, which is toxic to
E. coli, tends to accumulate. Accelerating the consumption of HMG-CoA would not only alleviate the metabolic burden on
E. coli but also enhance the overall efficiency of the mevalonate biosynthesis pathway. In this study, we investigated the formation of metabolic enzyme complexes by immobilizing MvaE and MvaS on scaffold proteins via SrtA-mediated ligation to enhance mevalonate production in
E. coli. Our approach utilizes multimeric scaffolds that provide multiple attachment sites, enabling the assembly of more than two enzymes. Although we demonstrated this concept with two enzymes in this study, our method is designed to be scalable for systems requiring three or more enzymes, making it applicable to more complex metabolic pathways in future studies.
. Schematic illustration of metabolic enzyme complex formation on (<b>A</b>) SA or (<b>B</b>) CA scaffold.
2. Materials and Methods
2.1. Bacterial Strains and Growth Conditions
E. coli DH5α cells were used for plasmid generation. Cells were transformed with the plasmid for cloning via heat shock and electroporation. Cells were cultured overnight at 37 °C in Luria–Bertani medium (20 g/L LB broth, Lenox, Nacalai Tesque, Kyoto, Japan) containing 100 μg/mL ampicillin and 20 mg/L kanamycin.
E. coli strain BW25113 was used for mevalonate production. Each plasmid was introduced into cells by heat shock and electroporation methods and cultured overnight at 37 °C in LB medium containing 100 μg/mL ampicillin and 20 mg/L kanamycin. Transformants were incubated overnight at 37 °C, 180 rpm in test tubes containing 4 mL of LB liquid medium containing 100 μg/mL ampicillin and 20 mg/L kanamycin. Precultures were inoculated into 16φ test tubes containing 4 mL of M9Y liquid medium having 100 μg/mL ampicillin sodium, 50 μg/mL kanamycin sulfate, and 20 g/L glucose and incubated at 37 °C, 200 rpm until optical density at 600 nm (OD
600) reached 0.5 to 0.8. Protein expression was then induced by adding 0.5 mM isopropyl-β-D-thiogalactopyranoside (IPTG) at a final concentration of 0.1 mM and incubated at 37 °C, 200 rpm for 48 h.
2.2. Plasmid Construction
The plasmids used in this study are shown in Table S1, and the primers used are listed in Table S2. Polymerase chain reactions were performed using KOD Plus or KOD One Master Mix (Toyobo Co., Ltd., Osaka, Japan). The vectors and inserts were ligated using NEBuilder (New England Biolabs Inc., Ipswich, MA, USA) following the manufacturer’s protocol. Using pMev plasmids as templates [
25], primers 1 and 2 were used to amplify the
mvaE gene with a C-terminal LPETG tag, primers 3 and 4 were used to amplify the
mvaS gene with a C-terminal LPETG tag, and primers 5 and 6 were used to amplify the gene fragment of the vector containing
atoB. Each gene fragment was subcloned into the pTrcHisB vector, and the resulting plasmid was named pMevlp. The SA gene with a N-terminal G tag was amplified using primers 7 and 8 with the pColdI-SA-LPETG vector as the template [
26]. The amplified fragment was inserted into the
SmaI site of pZAsrtA (supplemental information), and the resulting plasmid was named pZAsrtA_s. Primers 9 and 10 were used to amplify the CA gene with N-terminal G tag using
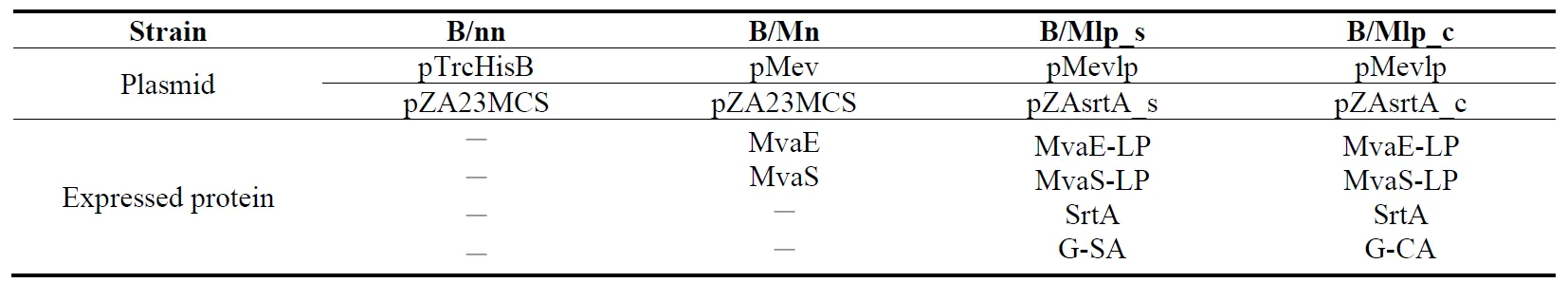
E. coli DH5α as a template. The gene fragments of the vector were amplified using primers 11 and 12 and pZAsrtA as a template. These two gene fragments were ligated, and the resulting plasmid was named pZAsrtA_c. In summary, five strains were constructed as follows: B/nn, which is BW25113 transformed with pTrcHisB and pZA23MCS; B/Mn, which is BW25113 transformed with pMev and pZA23MCS; B/Mlp, which is BW25113 transformed with pMevlp and pZAsrtA; B/Mlp_s, which is BW25113 transformed with pMevlp and pZAsrtA_s; and B/Mlp_c, which is BW25113 transformed with pMevlp and pZAsrtA_c ().
. The constructed strains and their components.
The cells were resuspended in 50 mM Tris-HCl buffer (pH 7.5) and adjusted to an OD
600 of 5.0. The cell suspension was collected in microtubes and sonicated using a BIORUPTOR UCD-250 instrument (Tosho Electric Co., Ltd., Tokyo, Japan). SDS treatment was performed by mixing equal amounts of SDS sample buffer with the protein solution after sonication and heating at 95 °C for 5 min. Proteins were separated using SDS-PAGE (4.5% or 15% [
w/
v] acrylamide) and stained with Coomassie Brilliant Blue.
2.4. Metabolite Analysis
After 48 h of cultivation, 500 μL of the culture medium was collected, centrifuged (13,000 rpm, 2 min, 4 °C), and 300 μL of the supernatant was collected. To the supernatant solution, 10 µL of 5 M HCl was added and mixed at 50 °C and 1000 rpm for 40 min. Subsequently, the organic phase was mixed with ethyl acetate in a 1:1 ratio, and 150 µL of each sample was collected in a sample tube. Gas chromatography (Nexis GC-2030, Shimadzu Co., Kyoto, Japan) equipped with a flame ionization detector was performed using SH-Stabilwax (30 m × 0.25 mm × 0.25 µm). The oven was set to 120 °C and held for 1 minute. Then, the temperature was increased from 30 °C/min to 250 °C. The temperature was raised to 250 °C at 30 °C/min and held for 4 min. The temperature of the detector was maintained at 250 °C.
Glucose concentrations were measured using the Glucose CII Test kit (Wako Pure Chemical, Osaka, Japan) and the Multiskan GO microplate reader (Thermo Scientific, Rochester, NY, USA).
3. Results and Discussion
3.1. Evaluation of Mevalonate Production by Using SA as a Scaffold
Our previous research demonstrated successful acetic acid production through SrtA-mediated linkage of metabolic enzymes [
16]. Building on this approach, we first applied a similar method for mevalonate production in the present study. However, tagging MvaE and MvaS with a G-tag at their N-termini resulted in a significant reduction in mevalonate production (Supplementary Figure S2). When the LP tag was applied to both MvaE and MvaS, mevalonate production levels remained similar to those observed in the absence of tags. Mevalonate production was significantly decreased by the addition of the G-tag to MvaS. These results suggest that simple ligation of MvaE and MvaS by SrtA does not enhance mevalonate production because of the N-terminal modification of the G tag.
To address this issue, we explored using scaffolds in this study. We hypothesized that a metabolic enzyme complex could be formed by linking the G-tag-appended scaffold and LP-tag-appended MvaE (MvaE-LP) and MvaS (MvaS-LP) via SrtA. We first evaluated the effect of G-SA as a scaffold for mevalonate production (A).
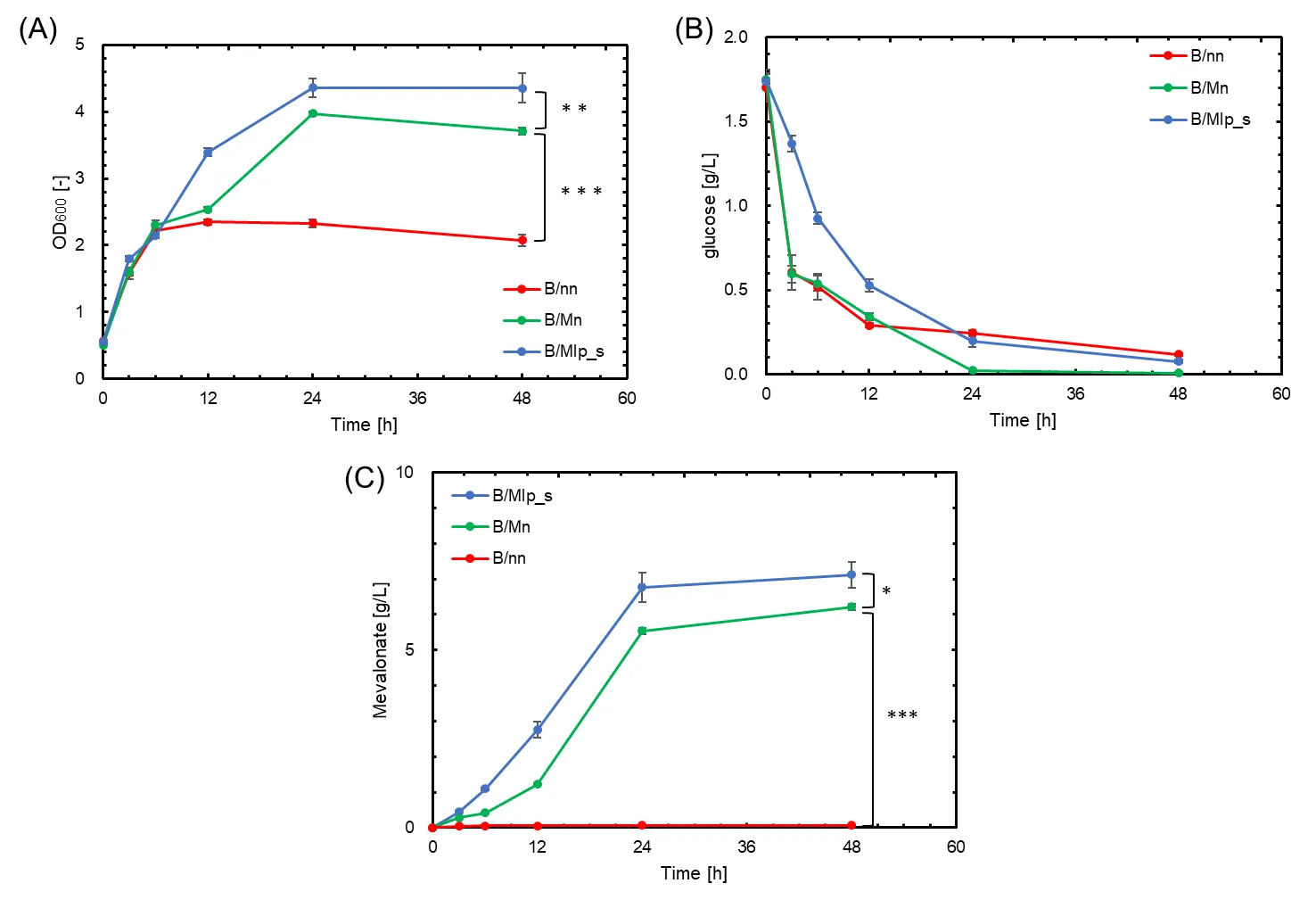
The formation of metabolic enzyme complexes using G-SA as a scaffold was confirmed using SDS-PAGE (). The expected molecular weight of the metabolic enzyme complex comprising MvaE-LP, MvaS-LP, and G-SA is approximately 200 kDa. A band observed at this position in suggests the formation of this complex. shows the culture results and experimental outcomes for B/nn, B/Mn, and B/Mlp_s strains, comparing conditions with and without G-SA and SrtA. B/Mlp_s, which harbors pMevlp and pZAsrtA_s (containing the G-SA scaffold), produced 7.21 g/L of mevalonate after 48 h, representing a 1.11-fold improvement over the B/Mn strain, which harbors pMevlp and pZA23MCS (C). The B/Mlp_s strain initially consumed less glucose than the other strains immediately after expression induction but eventually reached similar glucose consumption levels over time (A). We postulate that this initial lag in glucose consumption may be due to the incomplete formation of the metabolic enzyme complex immediately after expression induction, with glucose being primarily metabolized by unbound enzymes.
. SDS-PAGE analysis of whole-cell fractions using G-SA scaffold. Lane 1: B/Mn, containing MvaE and MvaS; Lane 2: B/Mlp_s, containing MvaE-LP, MvaS-LP and G-SA.
. Growth, glucose consumption, and mevalonate production in strains with and without G-SA and SrtA. Red, green, and blue symbols indicate results for B/nn, B/Mn: containing MvaE and MvaS, and B/Mlp_s: containing MvaE-LP, MvaS-LP and G-SA, respectively. (<b>A</b>) Cell growth (OD<sub>600</sub>); (<b>B</b>) Glucose concentration (g/L); (<b>C</b>) Mevalonate production (g/L). Data are shown as the mean of five independent experiments, each starting from separately isolated colonies, and error bars represent standard deviations. Statistical significance was determined by Student’s <i>t</i>-test (<sup>∗</sup> <i>p</i> < 0.10; <sup>∗∗</sup> <i>p</i> < 0.05; <sup>∗∗∗</sup> <i>p</i> < 0.01).
Although using SA as a scaffold led to a modest increase in mevalonate production, this improvement was not as substantial as anticipated. We hypothesized that this limited enhancement may be due to issues with SA complex formation. SA can form inclusion bodies, which can be mitigated by biotin addition [
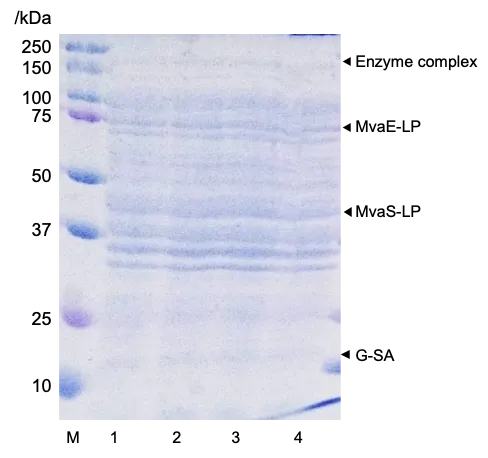
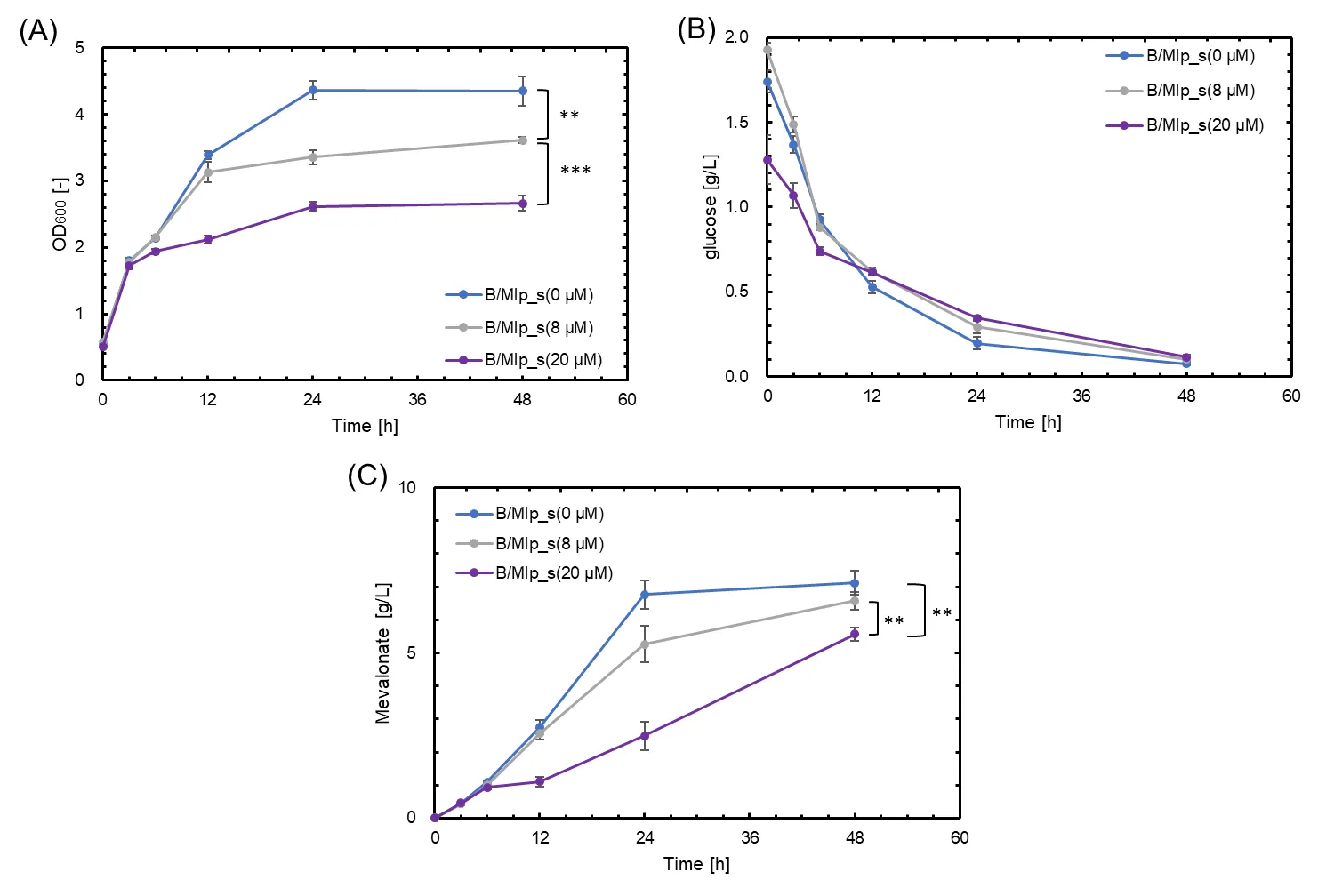
27]. To address this issue and further enhance our system, we investigated the effects of biotin addition. During expression induction, biotin was added at final concentrations of 0, 8, and 20 µM to promote proper intracellular folding. The strains were cultured under various conditions, and the results of SDS-PAGE analysis are shown in . Similar to , a band was observed at approximately 200 kDa, confirming the formation of metabolic complexes in the presence of biotin. However, there were no noticeable differences in band intensity or other characteristics, indicating no significant improvement in metabolic enzyme complex formation with biotin addition. The corresponding culture experiment results are presented in . Contrary to expectations, mevalonate production decreased by adding biotin (C). Moreover, OD
600 decreased with biotin supplementation (A). Glucose consumption rates were low in biotin-supplemented cultures after 12 h of incubation (B). These observations suggest that biotin addition may have inhibited cell growth and consequently did not effectively stimulate metabolic complex formation in
E. coli to enhance mevalonate production.
. Effect of biotin on protein complex formation: SDS-PAGE analysis of whole-cell fractions. Lane 1: B/Mn, containing MvaE and MvaS; Lane 2: B/Mlp_s, containing MvaE-LP, MvaS-LP and G-SA without biotin; Lane 3: B/Mlp_s with 8 μM biotin; Lane 4: B/Mlp_s with 20 μM biotin.
. The effect of biotin concentration on growth, glucose consumption, and mevalonate production in B/Mlp_s, containing MvaE-LP, MvaS-LP and G-CA. Blue, gray, and purple symbols indicate results with 0, 8, and 20 µM biotin, respectively. (<b>A</b>) Cell growth (OD<sub>600</sub>); (<b>B</b>) Glucose concentration (g/L); (<b>C</b>) Mevalonate production (g/L). Data are shown as the mean of five independent experiments, each starting from separately isolated colonies, and error bars represent standard deviations. Statistical significance was determined by Student’s <i>t</i>-test (<sup>∗∗</sup> <i>p</i> < 0.05; <sup>∗∗∗</sup> <i>p</i> < 0.01).
Given the limited success of SA as a scaffold, alternative protein scaffolds were sought. CA, a 12.3 kDa
E. coli-derived protein that forms homotrimers, has been used as a scaffold in various studies [
28,
29]. Therefore, we investigated the use of CA as an alternative to SA (B).
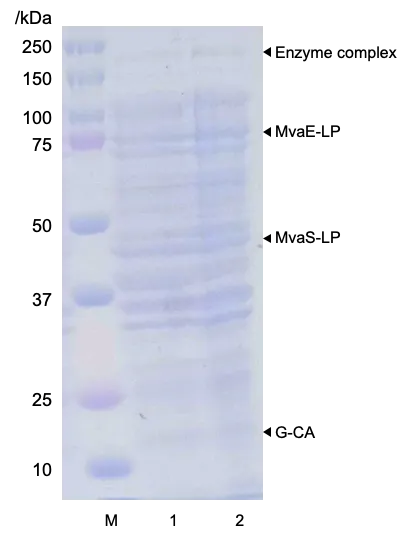
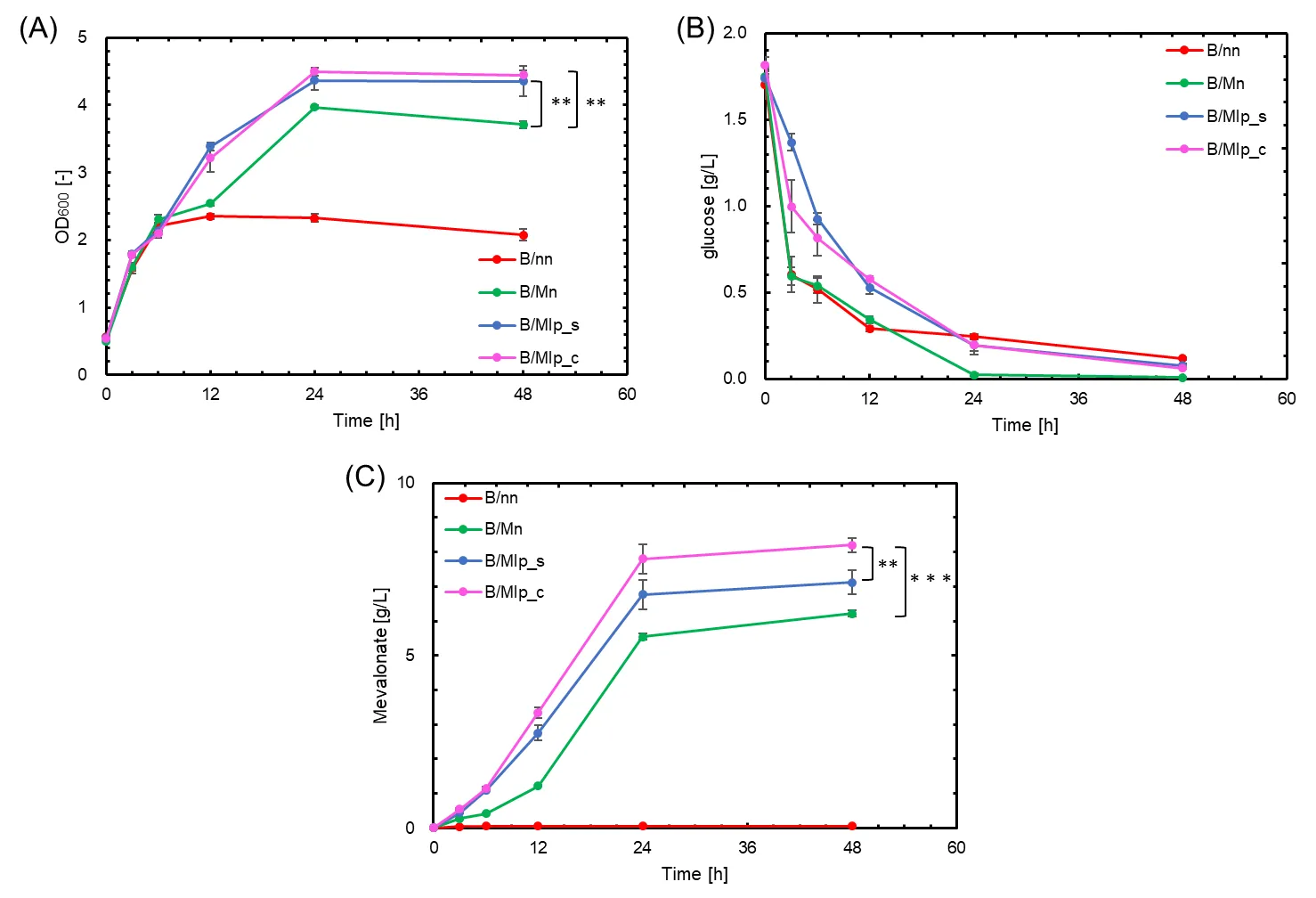
SDS-PAGE confirmed the formation of a metabolic enzyme complex using G-CA as a scaffold, as bands corresponding to the metabolic enzyme complexes of MvaE-LP, MvaS-LP, and G-CA were observed at approximately 200 kDa (). The results of the culture experiments are also shown in . Notably, mevalonate production by B/Mlp_c, which harbored pMevlp and pZAsrtA_c (containing the G-CA scaffold), reached 8.20 g/L. This is 1.32 times higher than that of B/Mn and 1.15 times higher than that of B/Mlp_s (C). B/Mlp_c exhibited a similar behavior to B/Mlp_s in terms of glucose consumption (B). The OD
600 results indicated that the growth of B/Mlp_c was consistently lower than that of B/Mlp_s throughout the cultivation period (A). These results demonstrated that G-CA serves as a more effective scaffold than G-SA for enhancing mevalonate production in
E. coli. While our current study demonstrates the influence of G-tagged scaffolds on mevalonate production, further characterization of these scaffold assemblies and their impact on enzyme activity remains an important area for future investigation. Such studies could potentially lead to improved efficiency and control of our metabolic enzyme complexes.
. SDS-PAGE analysis of whole-cell fractions using G-CA scaffold. Lane 1: B/Mn strain, containing MvaE and MvaS-LP; Lane 2: B/Mlp_c, containing MvaE-LP, MvaS-LP and G-CA.
. Comparison of growth, glucose consumption, and mevalonate production using G-CA and G-SA scaffolds. Red, green, blue, and pink symbols indicate results for B/nn, B/Mn: containing MvaE and MvaS, B/Mlp_s: containing MvaE-LP, MvaS-LP and G-SA, and B/Mlp_c: containing MvaE-LP, MvaS-LP and G-CA, respectively. (<b>A</b>) Cell growth (OD<sub>600</sub>); (<b>B</b>) Glucose concentration (g/L); (<b>C</b>) Mevalonate production (g/L). Data are shown as the mean of five independent experiments, each starting from separately isolated colonies, and error bars represent standard deviations. Statistical significance was determined by Student’s <i>t</i>-test (<sup>∗∗</sup> <i>p</i> < 0.05; <sup>∗∗∗</sup> <i>p</i> < 0.01).
4. Conclusions
In this study, we explored using SrtA-mediated ligation to form metabolic enzyme complexes on scaffold proteins to enhance mevalonate production in E. coli. Although the G-SA scaffold showed limited improvement in mevalonate production, using G-CA as an alternative scaffold led to a modest yet notable increase in mevalonate production, achieving a 1.32-fold improvement compared to the control strain.
SDS-PAGE analysis confirmed the formation of metabolic enzyme complexes in both scaffolds, and SA, CA were found to be capable of further assembly. However, only the G-CA scaffold led to appreciable improvement in mevalonate production. These results suggest that G-CA is a more suitable scaffold for mevalonate-producing strains in E. coli when SrtA-mediated enzyme ligation is used. This approach shows promise for improving the metabolic pathway efficiency, although further optimization may be required to achieve substantial production enhancements.
Supplementary Materials
The following supporting information can be found at https://www.sciepublish.com/article/pii/334, Table S1. Strains and plasmids used in this study; Table S2. Primers used in this study; Table S3. The gene sequence of synthetic gene flagment; Figure S1. Overview of the mevalonate biosynthetic pathway; Figure S2. Comparison of mevalonate production using LP- or G-tagged mvaE and mvaS. Data are shown as the mean of three independent experiments, each starting from separately isolated colonies, and error bars represent standard deviations.
Acknowledgments
We would like to thank Editage (www.editage.com) for English language editing.
Author Contributions
Conceptualization, M.H., M.F. and T.M.; Methodology, M.H., M.F. and T.M.; Investigation, M.H., M.F.; Resources, T.M. and R.Y.; Data Curation, M.H. and T.M.; Writing—Original Draft Preparation, M.H.; Writing—Review & Editing, T.M.; Supervision, R.Y. and H.O.; Project Administration, T.M.; Funding Acquisition, T.M.
Ethics Statement
Not applicable.
Informed Consent Statement
Not applicable.
Funding
This work was supported by JSPS KAKENHI Grant Number 24K08687.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
-
1.
Keasling J, Garcia Martin H, Lee TS, Mukhopadhyay A, Singer SW, Sundstrom E. Microbial production of advanced biofuels.
Nat. Rev. Microbiol. 2021,
19, 701–715.
[Google Scholar]
-
2.
Becker J, Wittmann C. Advanced Biotechnology: Metabolically Engineered Cells for the Bio-Based Production of Chemicals and Fuels, Materials, and Health-Care Products.
Angew. Chem. Int. Ed. 2015,
54, 3328–3350.
[Google Scholar]
-
3.
Chung H, Yang JE, Ha JY, Chae TU, Shin JH, Gustavsson M, et al. Bio-based production of monomers and polymers by metabolically engineered microorganisms.
Curr. Opin. Biotechnol. 2015,
36, 73–84.
[Google Scholar]
-
4.
Markina NM, Kotlobay AA, Tsarkova AS. Heterologous Metabolic Pathways: Strategies for Optimal Expression in Eukaryotic Hosts.
Acta Naturae 2020,
12, 28–39.
[Google Scholar]
-
5.
Zhu F, Peña M, Bennett GN. Metabolic engineering of Escherichia coli for quinolinic acid production by assembling L-aspartate oxidase and quinolinate synthase as an enzyme complex.
Metab. Eng. 2021,
67, 164–172.
[Google Scholar]
-
6.
Song F, Qin Z, Qiu K, Huang Z, Wang L, Zhang H, et al. Development of a vitamin B5 hyperproducer in Escherichia coli by multiple metabolic engineering.
Metab. Eng. 2024,
84, 158–168.
[Google Scholar]
-
7.
McNerney MP, Watstein DM, Styczynski MP. Precision metabolic engineering: The design of responsive, selective, and controllable metabolic systems.
Metab. Eng. 2015,
31, 123–131.
[Google Scholar]
-
8.
Welch GR, Easterby JS. Metabolic channeling versus free diffusion: Transition-time analysis.
Trends Biochem. Sci. 1994,
19, 193–197.
[Google Scholar]
-
9.
Bauler P, Huber G, Leyh T, McCammon JA. Channeling by proximity: The catalytic advantages of active site colocalization using Brownian dynamics.
J. Phys. Chem. Lett. 2010,
1, 1332–1335.
[Google Scholar]
-
10.
Wheeldon I, Minteer SD, Banta S, Barton SC, Atanassov P, Sigman M. Substrate channelling as an approach to cascade reactions.
Nat. Chem. 2016,
8, 299–309.
[Google Scholar]
-
11.
Tippmann S, Anfelt J, David F, Rand JM, Siewers V, Uhlén M, et al. Affibody Scaffolds Improve Sesquiterpene Production in Saccharomyces cerevisiae.
ACS Synth. Biol. 2016,
6, 19–28.
[Google Scholar]
-
12.
Sørensen HP, Mortensen KK. Advanced
genetic
strategies
for
recombinant
protein
expression
in
Escherichia coli.
J. Biotechnol. 2005,
115, 113–128.
[Google Scholar]
-
13.
Kaur J, Kumar A, Kaur J. Strategies
for
Optimization
of
Heterologous
Protein
Expression
in
E. coli:
Roadblocks
and
Reinforcements.
Int. J. Biol. Macromol. 2018,
106, 803–822.
[Google Scholar]
-
14.
Rosano GL, Ceccarelli EA.
Recombinant
Protein
Expression
in
Escherichia coli:
Advances
and
Challenges.
Front. Microbiol. 2014,
5, 172.
[Google Scholar]
-
15.
Xiao L, Wang X, Zhang J, Zhou Y, Wang F, Zhang Y, et al. Co-localizing
key
pathway
enzymes
by
protein
scaffold
to
enhance
geraniol
production
in
Escherichia coli.
Ind. Crops Prod. 2023,
203, 117144.
[Google Scholar]
-
16.
Matsumoto T, Furuta K, Tanaka T, Kondo A. Sortase
A-Mediated
Metabolic
Enzyme
Ligation
in
Escherichia coli.
ACS Synth. Biol. 2016,
5, 1284–1289.
[Google Scholar]
-
17.
Veĭko VP, Gul’ko LB, Okorokova NA, D’iakov NA, Debabov VG. Klonirovanie
gena
streptavidina
iz
Streptomyces
avidinii
i
ego
ékspressiia
v
Escherichia coli.
Sekretsiia
streptavidina
kletkami
E. coli [Cloning
of
streptavidin
gene
from
Streptomyces
avidinii
and
its
expression
in
Escherichia coli.
Secretion
of
streptavidin
by
E. coli cells].
Bioorg. Khim. 1999,
3, 184–188.
[Google Scholar]
-
18.
Gallizia A, de Lalla C, Nardone E, Santambrogio P, Brandazza A, Sidoli A, et al. Production
of
a
soluble
and
functional
recombinant
streptavidin
in
Escherichia coli.
Protein Expr. Purif. 1998,
2, 192–196.
[Google Scholar]
-
19.
Matsuura Y, Ota M, Tanaka T, Takehira M, Ogasahara K, Bagautdinov B, et al.
Remarkable
improvement
in
the
heat
stability
of
CutA1
from
Escherichia coli by
rational
protein
design.
J. Biochem. 2010,
4, 449–458.
[Google Scholar]
-
20.
Matsumoto T, Isogawa Y, Minamihata K, Tanaka T, Kondo A. Twigged streptavidin polymer as a scaffold for protein assembly.
J. Biotechnol. 2016,
225, 61–66.
[Google Scholar]
-
21.
Castellana M, Wilson MZ, Xu Y, Joshi P, Cristea IM, Rabinowitz JD, et al.
Enzyme
clustering
accelerates
processing
of
intermediates
through
metabolic
channeling.
Nat. Biotechnol. 2014,
10, 1011–1018.
[Google Scholar]
-
22.
Wada K, Toya Y, Banno S, Yoshikawa K, Matsuda F, Shimizu H.
13C-metabolic
flux
analysis
for
mevalonate-producing
strain
of
Escherichia coli.
J. Biosci. Bioeng. 2017,
123, 177–182.
[Google Scholar]
-
23.
Liao P, Hemmerlin A, Bach TJ, Chye ML.
The
potential
of
the
mevalonate
pathway
for
enhanced
isoprenoid
production.
Biotechnol. Adv. 2016,
34, 697–713.
[Google Scholar]
-
24.
Shin J, South EJ, Dunlop MJ.
Transcriptional
Tuning
of
Mevalonate
Pathway
Enzymes
to
Identify
the
Impact
on
Limonene
Production
in
Escherichia coli.
ACS Omega 2022,
7, 18331–18338.
[Google Scholar]
-
25.
Satowa D, Fujiwara R, Uchio S, Nakano M, Otomo C, Hirata Y, et al.
Metabolic
engineering
of
E.
coli
for
improving
mevalonate
production
to
promote
NADPH
regeneration
and
enhance
acetyl-CoA
supply.
Biotechnol. Bioeng. 2020,
117, 2153–2164.
[Google Scholar]
-
26.
Matsumoto T, Sawamoto S, Sakamoto T, Tanaka T, Fukuda H, Kondo A. Site-specific
tetrameric
streptavidin-protein
conjugation
using
sortase
A.
J. Biotechnol. 2011,
152, 37–42.
[Google Scholar]
-
27.
Lim KH, Hwang I, Park S.
Biotin-assisted
folding
of
streptavidin
on
the
yeast
surface.
Biotechnol. Prog. 2012,
28, 276–283.
[Google Scholar]
-
28.
Bertini I, Jiménez B, Pierattelli R, Wedd AG, Xiao Z.
Protonless
13C
direct
detection
NMR:
Characterization
of
the
37
kDa
trimeric
protein
CutA1.
Proteins 2008,
70, 1196–1205.
[Google Scholar]
-
29.
Gao L, Liu Y, Zhang X, Zhang H.
Efficient
Optimization
of
Gluconobacter oxydans Based
on
Protein
Scaffold-Trimeric
CutA
to
Enhance
the
Chemical
Structure
Stability
of
Enzymes
for
the
Direct
Production
of
2-Keto-L-gulonic
Acid.
J. Chem. 2020,
2020, 5429409.
[Google Scholar]