In a recent Immunity publication, Liu et al. demonstrated that cerebral ischemia triggers prolonged activation of Notch1 signaling in peripheral endothelial cell (EC) and increases the expression of adhesion molecules. This chain of events promotes monocyte recruitment and EC adhesion, accelerating plaque development post-stroke. Notably, inhibiting Notch1 activation in ECs or blocking VCAM1-mediated leukocyte adhesion can mitigate these adverse systemic endothelial changes [
1]. The findings of this study reveal mechanisms underlying the increased risk of recurrent vascular events after stroke and identify modifiable processes that may serve as potential therapeutic targets ().
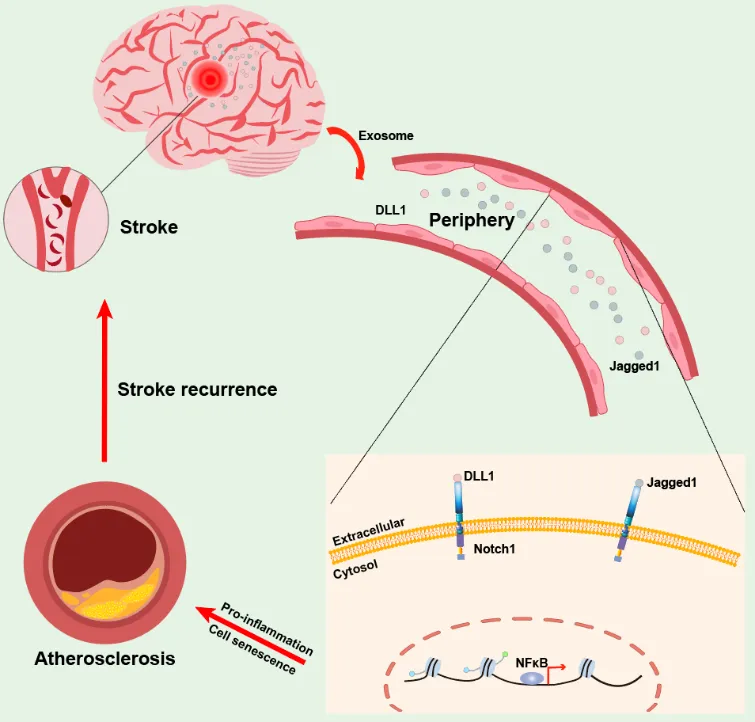
. Stroke elicits persistent activation of peripheral endothelial cells, upregulation of proinflammatory molecules, and increased cellular senescence. This aberrant endothelial activity is caused by sustained Notch1 signaling, which is triggered by increased circulating Notch1 ligands DLL1 and Jagged1 after stroke. Thus, by generating senescent proinflammatory endothelial cells, this leads to increased myeloid cell adhesion and atherosclerotic progression and also increases the risk of recurrent stroke.
Stroke refers to a group of acute cerebrovascular events resulting from brain tissue damage or death due to the rupture or blockage of intracranial blood vessels. Brain injury is known to trigger a systemic inflammatory response, characterized by elevated blood cytokine levels, immune cell mobilization, and significant alterations in immune cell composition and function. Beyond the immediate morbidity and mortality associated with ischemic brain injury, long-term complications are common, including a high incidence of secondary conditions such as cardiac dysfunction, chronic vascular inflammation, and stroke-induced metabolic disorders. Clinically, these adverse cardiovascular events following stroke present significant challenges to patient prognosis and treatment.
A growing body of evidence supports physiological and pathophysiological interactions between the nervous and cardiovascular systems. In recent years, brain-heart interaction has attracted extensive attention. Studies on stroke-induced cardiac injury have shown that stroke triggers the large-scale release of monocytes and neutrophils from the spleen and other organs, along with alterations in lymphocyte production. A similar phenomenon is observed in acute myocardial infarction, where monocytes rapidly accumulate in the heart [
2]. Recently, Simats et al. investigated the effects of brain injury on systemic immunity in the chronic phase. They discovered that the ischemic brain injury leads to developed diastolic dysfunction, potentially driven by IL-1β-mediated epigenetic modifications in the myeloid cells, contributing to cardiac fibrosis [
3]. Despite advances in stroke care, the incidence of recurrent vascular events after stroke remains high. Previous studies have reported that post-stroke accelerates the progression of atherosclerosis, primarily due to the increased recruitment of vascular monocytes [
4]. Liu et al. further explored the chronic inflammatory response of ECs after stroke, which increases susceptibility to atherosclerosis. ECs, serving as the outermost protective barrier of the vessel wall, also function as immune cells. However, the post-stroke pathological response in ECs is not fully understood. As an important carrier, exosomes play a key role in signal transmission after stroke. Using single-cell analysis, Liu et al. identified that exosomes containing Notch1 ligands are released after stroke to continuously activate Notch1 signaling, thereby aggravating the expression of adhesion molecules VCAM1 and cell senescence, which provides a new explanation for the endothelial response after cerebral ischemia.
In the study of cardiovascular diseases, Notch signaling pathway is implicated in a variety of disease progression processes, including atherosclerosis, angiogenesis, and intimal neogenesis after vascular injury. Several genes are directly regulated by activated Notch1, with key targets including members of the NF-κB family, CYCLIN D1, p21, GATA3, MYC, and dtx1. In mammals, Notch signaling is typically activated upon interaction with delta-like ligands (DLL1, DLL3, DLL4) and Jagged ligands (Jagged 1, Jagged 2), initiating a series of proteolytic cleavage events culminating in release of the Notch1-intracellular domain from its membrane receptor and leading to its nuclear translocation. Using a combination of single-cell sequencing analysis and proteomic microarray, Liu et al. found that the Notch1 receptor ligands DLL1 and Jagged1 could be persistently released from exosomes from the injured brain tissue after stroke. The secretion of IL-1β in the early stage after stroke can mediate the innate immune memory of monocytes and macrophages [
3], indicating that, beyond the role of exosome-carried substances in distal vascular ECs, chemokines and inflammatory factors released may cause inflammatory reactions and immune responses of ECs. In addition, there are other signals in the blood that may mediate the aggravation of atherosclerosis after cerebral ischemia that need to be further explored.
In summary, Liu et al. found that luminal narrowing due to atherosclerosis, embolism due to plaque shedding, and occlusion of small vessels are the main factors leading to recurrence of stroke within 3 months after onset. They also demonstrated that cerebral ischemia triggers continuous activation and senescence of peripheral vascular ECs, up-regulating the immune adhesion molecule VCAM1, which increase monocytes and neutrophils adhesion. The study further revealed that Notch1 receptor ligands DLL1 and Jagged1 activate the Notch1 pathway in peripheral vascular ECs, promoting vascular inflammation and accelerating atherosclerosis progression. Blocking the adhesion molecules VCAM1 or Notch1 receptor using antibody therapies could potentially delay the progression of atherosclerosis post-stroke. This study reveals a systemic mechanism driving sustained activation of peripheral ECs after stroke, offering novel insights for therapeutic intervention or prevention of recurrent vascular events after stroke.
The authors found that the expression of notch1 ligands DLL1 and jagged1 was increased in exosomes of patients with ischemic stroke. The clinical prognostic significance relevance of the increased expression of Notch1 ligands after stroke requires further analysis within our clinical cohort. Additionally, the role of epigenetic mechanisms in ECs has not been explained. As a special immune cell, do ECs produce inflammatory memory at the onset of stroke? Neuronal death caused by stroke induces localized brain inflammation, primarily characterized by activation of microglia and production of cytokines and chemokines. The changes of cytokines and chemokines in microglia after stroke and their subsequent effects on ECs, remain an area of exploration. It is worth mentioning that post-stroke pan-vascular disease, including coronary artery disease, cerebrovascular disease and peripheral artery disease, is also an important cause of myocardial infarction in patients. Currently, there is no specific treatment to prevent cardiovascular complications following stroke. In the future, there is a need for in-depth investigation after stroke into the regulatory mechanisms governing EC homeostasis, as many aspects remain poorly understood.
K.L., C.G., S.Y., Z.X. and M.Z. wrote the manuscript. K.L. prepared the figure. All the authors read and approved the manuscript.
Not applicable.
Not applicable.
This work was supported by grants of the National Natural Science Foundation of China (No. 82270487); the Shandong Provincial Natural Science Foundation (ZR2023JQ030, 2021ZDSYS05); Non-profit Central Research Institute Fund of Chinese Academy of Medical Sciences (2023-PT320-06) and the Fundamental Research Funds for the Central Universities (2023QNTD003).
The authors declare that they have no known competing financial interests.