In a new study published in European Heart Journal, Chen et al. describe the role of tumour necrosis factor receptor-associated protein 1 (TRAP1) in mediating histone H4 lysine 12 lactylation (H4K12la) via metabolic reprogramming, inducing senescence-associated secretory phenotype and thereby contributing to the pathogenesis of atherosclerosis [
1].
Atherosclerosis is a chronic inflammatory disease, with vascular smooth muscle cells (VSMCs) senescence driving progression and unstable plaque formation [
2]. TRAP1, a mitochondrial isoform of the HSP90 chaperone and a key regulator of mitochondrial energy metabolism, modulates cellular metabolism by altering the conformation, activity, and stability of various substrate proteins [
3].
TRAP1 deficiency mutations are linked to neurological disorders, including Leigh syndrome, while impaired TRAP1 function exacerbates conditions such as myocardial ischemia-reperfusion injury and diabetes. TRAP1 facilitates lactate accumulation through aerobic glycolysis, contributing to metabolic reprogramming that supports tumor progression. Dysregulated TRAP1 affects mitochondrial integrity, lowers toxic reactive oxygen species (ROS) production, and increases cellular resistance to stress-induced cell death [
3]. However, the role of TRAP1 in VSMCs senescence and atherosclerosis remains largely unclear.
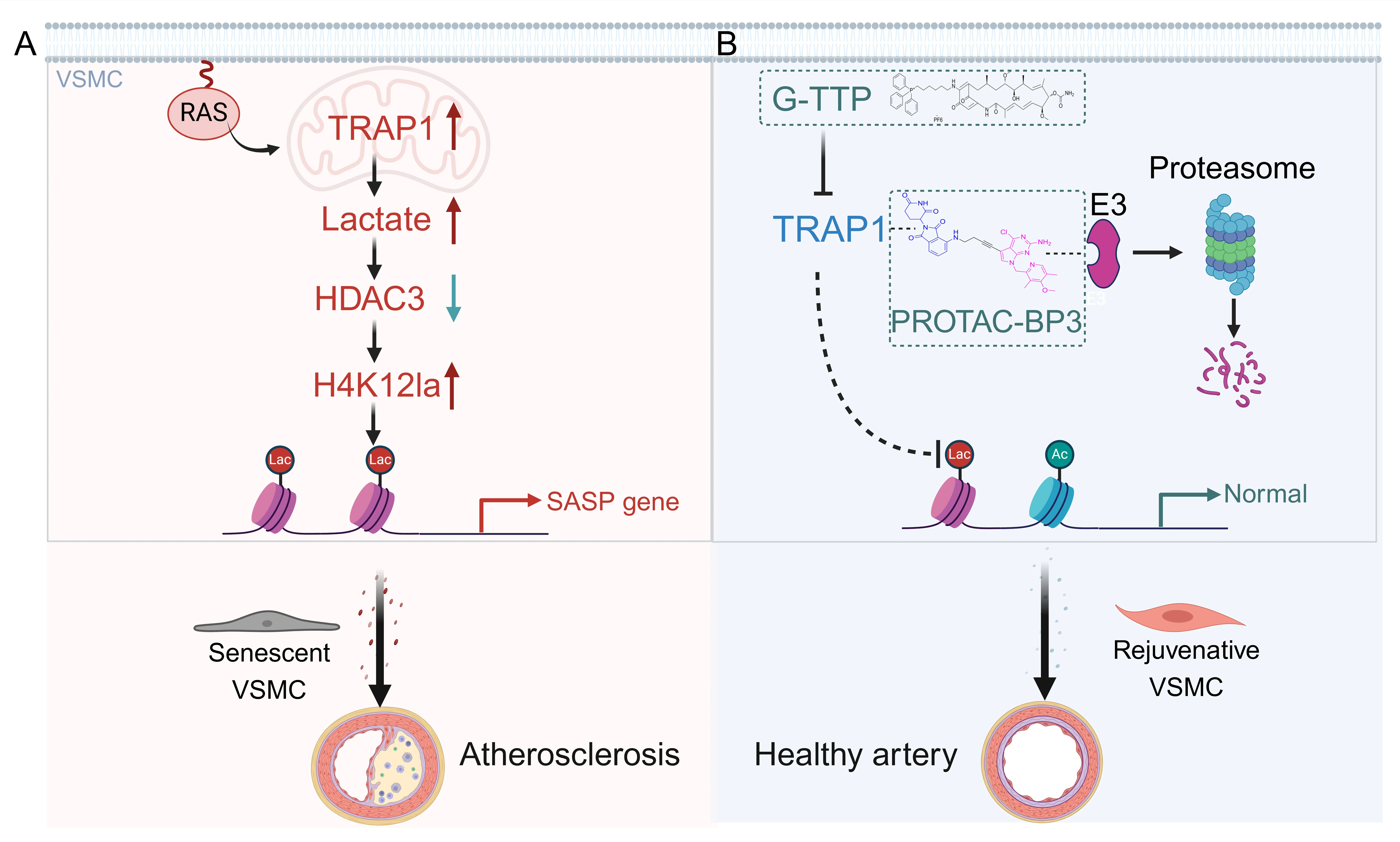
The authors first identified that TRAP1 and senescence-associated molecules were abnormally elevated in VSMCs under senescence-inducing stimuli in atherosclerotic samples. Further assessment revealed that TRAP1 promotes VSMC senescence by regulating metabolic reprogramming, shifting from oxidative phosphorylation to aerobic glycolysis. Through further screening, TRAP1 was found to specifically target and bind to one of the key glycolytic enzymes, phosphofructokinase 1 (PFK1), reducing its ubiquitination and inhibiting mitochondrial complex IV activity, thereby promoting aerobic glycolysis in senescent VSMCs [
1]. Based on this finding, the authors revealed the TRAP1-PFK1-Complex IV loop as a critical pathway for aerobic glycolysis in senescent VSMCs.
The product of glycolysis, lactate, not only plays a role in organ growth and development but also acts as a signaling molecule involved in the pathogenesis of various diseases. The authors found that lactate accumulates abnormally in senescent VSMCs, and TRAP1 promotes VSMC senescence by increasing lactate accumulation. Lactate also serves as a key epigenetic regulator, mediating histone lysine lactylation, and is involved in processes such as cellular metabolism, signal transduction, and gene expression regulation. The TRAP1-lactate signaling axis induces H4K12la, a characteristic lactylation modification in senescent VSMCs, with H4K12la showing perinuclear translocation and condensation, leading to changes in the senescence-associated secretory phenotype (SASP). Lysine lactylation is coordinated by “writer” histone acetyltransferase p300 and “eraser” delactylases histone deacetylase 1 (HDAC1). In Ras-induced VSMCs, screening revealed no changes in the expression of the classical lactylation enzyme p300, but a significant downregulation of the novel delactylase histone deacetylase 3 (HDAC3). Additionally, TRAP1 downregulates HDAC3 expression in senescent VSMCs via lactate. Further experiments demonstrated that the TRAP1-HDAC3-H4K12la axis promotes chromatin conformation changes in SASP, inducing VSMC senescence [
1].
To clarify the role of TRAP1 in VSMC senescence and atherosclerosis, the authors generated smooth muscle-specific TRAP1 knockout ApoeKO mice and induced atherosclerosis. TRAP1 knockout mice exhibited significantly reduced lipid accumulation, plaque size, and necrotic core area in the aortic root, along with increased collagen content. The authors further isolated VSMCs from the model mice to explore the underlying cellular mechanisms, finding that TRAP1 knockout significantly inhibited high-fat-induced metabolic reprogramming, H4K12 lactylation, SASP transcription, and increased senescence in VSMCs. G-TPP, a potent TRAP1 in-hibitor currently undergoing phase I clinical trial as an anti-tumor drug, was found to exhibit novel anti-atherosclerosis activity in this study by delaying VSMC senescence through reducing H4K12la. Further experiments demonstrated that the dosage of G-TPP re-quired for atherosclerosis treatment is much lower than that used in cancer therapy, making it a safe and effective anti-atherosclerosis therapy [
1].
A novel protein degradation technology, Proteolysis Targeting Chimeras (PROTAC), has garnered widespread attention. PROTAC is a bifunctional molecule that relies on the ubiquitin-proteasome system to induce the degradation of specific target proteins, thereby eliminating their function to treat diseases, offering advantages such as better selectivity, safety, and lower drug resistance compared to traditional inhibitors. The authors used PROTAC-BP3 to degrade TRAP1, effectively reducing high-fat-induced H4K12la and senescence in smooth muscle cells, decreasing plaque size, enhancing stability, and alleviating atherosclerosis, indicating its potential as a treatment for VSMC senescence and atherosclerosis. In addition, the researchers evaluated H4K12la expression in atherosclerosis patients and found significantly elevated levels in atherosclerotic samples, which suggest that TRAP1 and its mediated H4K12la are clinically significant and represent promising therapeutic targets for atherosclerosis treatment [
1]. Collectively, the authors’ work represents the identification of the TRAP1-HDAC3-H4K12 lactylation regulatory axis as a novel modification pathway that regulates metabolic reprogramming, induces epigenetic remodeling of the chromatin microenvironment surrounding SASP genes, and promotes VSMC senescence and atherosclerosis progression ().
. Protective effects of TRAP1 inhibitor G-TPP and PROTAC-BP3 against VSMC senescence in atherosclerosis. (<b>A</b>). The proto-oncogene Ras in VSMCs upregulates TRAP1, which through the TRAP1-HDAC3-H4K12 axis, leads to epigenetic remodeling and alteration of the chromatin environment at SASP gene loci. This process promotes VSMC senescence and contributes to the progression of atherosclerosis. (<b>B</b>). TRAP1 inhibitor G-TPP and PROTAC-BP3, significantly reduce H4K12ac and VSMC senescence, indicating the potential as therapeutic agents for the treatment of atherosclerosis. Green arrows indicate downregulation; red arrows indicate upregulation of genes.
Chen and colleagues comprehensively investigated the mechanism by which TRAP1 reprograms energy metabolism, leading to VSMC aging and atherosclerosis. TRAP1 upregulation enhances glycolysis, increases lactate, and suppresses HDAC3 expression, promoting H4K12la and SASP activation, thereby accelerating VSMC aging and atherosclerosis. This study provides new insights into the link between energy metabolism reprogramming and epigenetic modifications, uncovering novel mechanisms of cellular aging. HDAC3 enhances metabolism by promoting fatty acid oxidation and circadian histone deacetylation. Previous studies in myocardial infarction models show that arginase 1 (Arg1) suppression and macrophage reprogramming are independent of its deacetylase activity [
4]. This study identifies HDAC3 as a novel histone delactylase regulating VSMC aging, which is crucial for pinpointing histone lactylation-modifying enzymes and sites linked to cell aging and atherosclerosis [
1]. The application of PROTAC technology in the vascular system could enhance drug targeting efficiency. However, TRAP1 degradation via PROTAC may affect its homologs, Hsp90α/β and Grp94, potentially disrupting cellular processes. For instance, inhibiting the Grp94-Trim28-Eomes axis could impair NK cell functions, raising infection risks [
5]. TRAP1 also regulates oxidative stress and lysosomal recycling, essential for cellular homeostasis [
6]. Given its role in diseases like Parkinson’s and Alzheimer’s, further refinement of PROTAC molecules is needed to improve cell specificity and avoid off-target effects.
We thank all members of the Zhang lab for their helpful comments and discussion.
C.M. and W.Z. wrote the manuscript. C.M. prepared the figure. The authors read and approved the manuscript.
Not applicable.
Not applicable.
This research was funded by the National Natural Science Foundation of China (No. 82470499, 82270457), the Taishan Scholar Project of Shandong Province of China (No. tstp20240852).
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.