Beyond Barrier Function: Tight Junctions as Dynamic Signaling Hubs Orchestrating Tumor Plasticity, Microenvironment Remodeling, and Metastatic Evolution

Beyond Barrier Function: Tight Junctions as Dynamic Signaling Hubs Orchestrating Tumor Plasticity, Microenvironment Remodeling, and Metastatic Evolution
Received: 20 April 2026 Revised: 18 May 2026 Accepted: 27 May 2026 Published: 08 June 2026

© 2026 The authors. This is an open access article under the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/).

Graphical Abstract
1. Introduction
1.1. From Physical Barriers to Signaling Hubs
The studies about tight junctions (TJs) experienced several major changes over the decades [1]. In the early stages of cell biology, researchers viewed TJs as relatively static, sealed intercellular structures, primarily responsible for maintaining barrier integrity through barrier and fence functions [2]. To achieve these functions, TJs need to self-assemble into protein condensates [3]. The assembly of TJ belts is driven by a wetting phenomenon, in which PATJ-mediated extension and fusion of the ZO-1 surface layer provides the physical scaffold for complex signalosomes [4]. Recent biophysical insights suggest that TJs are not merely static physical structures, but have been redefined as hubs of signal transduction and mechanobiology [5]. TJ proteins could act as bidirectional pathways, linking extracellular mechanical signals to intracellular cascade reactions [6]. Studies on early mouse development have shown that atypical TJ proteins localize earlier than mature junctions, indicating an early role in lineage specification [7]. Crucially, emerging paradigms reposition TJs as active signaling platforms that interface with peripheral cascades to modulate gene expression, a concept thoroughly explored in the following sections [8]. Furthermore, atypical regulatory hierarchies, such as interclaudin interference among claudins, demonstrate how TJs actively process biological information to alter the tumor microenvironment (TME) [9]. These insights provide a conceptual framework for understanding how TJ dysregulation in cancer reflects the reactivation of embryonic programs, transforming TJs from passive barriers into active drivers of phenotypic plasticity [10].
1.2. Defining Tumor Plasticity: TJs as Regulators of EMT and MET
Tumor metastasis requires cellular plasticity, enabling cancer cells to adapt their identity [11]. TJs contribute to these transitions through context-dependent modulation [12]. During epithelial–mesenchymal transition (EMT), tumor cells downregulate epithelial junction components, including Claudin-1 and ZO-1, often under the influence of TGF-β [13]. Importantly, TJs are not merely passive targets of EMT; they actively regulate the stability of the epithelial phenotype [14]. For instance, the interclaudin interference mechanism suggests that barrier-forming claudins can actively suppress pore-forming claudins to modulate paracellular flux and cell-cell adhesion strength [15]. Conversely, during mesenchymal–epithelial transition (MET) at secondary sites, re-expression of TJ proteins restores adhesion [16]. The role of TJs in plasticity is further evidenced by Claudin-7’s involvement in colonic stem cell homeostasis via Wnt/Notch signaling, suggesting that TJ proteins maintain the delicate balance between progenitor maintenance and differentiation [17].
1.3. TJs as “Biosensors” Linking Mechanical Forces to Epigenetic Remodeling
An emerging concept in TJ biology is their role as mechanosensitive signaling platforms [18]. Beyond simple structural support, TJ membrane proteins like claudins and JAM-A are essential for the mechanical resistance of the apical junctional complex (AJC), preventing junctional fractures during spontaneous cell stretching [19]. Through structural links to the actin cytoskeleton via ZO-1, TJs sense and transduce mechanical cues, such as matrix stiffness and intercellular tension, into intracellular signals [20].
Mechanical forces induce conformational changes in ZO-1, potentially exposing cryptic binding sites for signaling molecules such as ZONAB [21]. Furthermore, a novel signaling axis has recently been identified where Claudin-mediated adhesion activates SFKs, leading to the phosphorylation and modulation of NRs. This CLDN-SFK-NR pathway provides a direct molecular link between the cell surface and the transcriptional machinery [22]. Recent studies also highlight the role of liquid–liquid phase separation (LLPS) in TJ organization, where ZO-1 forms dynamic, liquid-like condensates [23]. The dissolution or condensation of these scaffolds serves as a biophysical switch, rapidly altering local signaling microenvironments. Together, these mechanisms position TJs as biosensors that integrate mechanical and biochemical cues, coupling extracellular forces to transcriptional regulation and epigenetic remodeling during tumor progression [24].
In summary, the conceptual evolution of TJs from passive physical barriers to dynamic signaling and mechanobiological hubs fundamentally redefines their role in cancer progression. The transition from a structural fence to an active biosensor is underpinned by sophisticated biophysical processes, such as PATJ-mediated surface wetting and ZO-1 phase separation, which establish a plastic scaffold capable of rapid architectural remodeling. By integrating extracellular mechanical tension with intracellular signaling cascades, TJs function as critical gatekeepers of cellular identity. These junctions do not merely facilitate the transitions between EMT and MET states but actively dictate the survival logic of metastatic cells by coupling microenvironmental cues to genomic and epigenetic regulation. Thus, targeting the TJ signalosome and its mechanosensitive properties represents a promising frontier for disrupting the adaptive plasticity that drives advanced malignancies.
However, redefining TJs as independent signaling master regulators introduces significant conceptual challenges. In epithelial biology, TJs do not operate in a vacuum; they are physically and functionally tethered to other junctional complexes, particularly adherens junctions (AJs). Therefore, this review aims to critically evaluate the cross-talk, functional redundancies, and outstanding controversies regarding the relative contributions of TJs versus other cell-cell adhesions during malignant progression.
2. The Molecular Architecture of Tight Junctions: From Structural Scaffolds to Signaling Hubs
TJs are highly organized multiprotein complexes that extend beyond static structural barriers. They are now widely recognized as dynamic and integrated networks that physically and functionally connect the plasma membrane with intracellular signaling machinery. Understanding their molecular architecture is essential for elucidating how cancer cells exploit and remodel these structures during migration and invasion. Collectively, current evidence supports the concept that TJs function as context-dependent structural and signaling platforms in cancer progression [5,10,12,22].
2.1. The Protein–Protein Interaction Network
The protein–protein interaction network of TJs is primarily organized by members of the claudin superfamily, which not only form the structural backbone of the paracellular barrier but also engage in highly specific intermolecular interactions [5]. Beyond their architectural role, recent studies have revealed that the specificity of claudin–claudin interactions is partially governed by an electrostatic code embedded in conserved sequence motifs, particularly the distribution of charged residues within the first extracellular loop (ECL1) [25]. This charge pattern not only determines ion selectivity but also modulates intercellular binding affinity and pairing compatibility between claudin isoforms [26]. For instance, negatively charged residues in ECL1 promote cation-selective permeability. Within the context of the TME, alterations in these interaction interfaces may disrupt claudin pairing and junctional organization, thereby indirectly influencing ionic homeostasis and downstream cellular processes [27]. Furthermore, the assembly and stability of TJ protein complexes are not only strictly dependent on canonical motifs, such as the C-terminal PDZ-binding domain, but also exhibit a high degree of structural plasticity. While the PDZ-binding motif is a primary regulator of strand stability, it mediates interactions with scaffold proteins (ZO family proteins), thereby integrating claudins into a broader multiprotein network [28]. Complementary membrane-anchoring mechanisms allow for robust junctional assembly even in its absence. This multilayered interaction network exhibits intrinsic redundancy and adaptability, which may enable cancer cells to preserve residual junctional integrity and sustain survival signaling even under conditions of regulatory disruption.
2.1.1. Transmembrane Components: Claudin and Occludin
Claudins constitute the backbone of TJ strands, each containing four transmembrane domains and two extracellular loops. ECL1 plays a central role in determining paracellular charge selectivity, as exemplified by Claudin-2, which forms cation-selective pores, whereas Claudin-1 and Claudin-4 function as barrier-forming proteins [28,29]. Importantly, claudins exhibit heterotypic compatibility and a newly identified mechanism, termed interclaudin interference, in which barrier-forming claudins can actively disrupt the higher-order meshworks and channel functions of pore-forming claudins, leading to their endocytosis [15]. Certain claudins can regulate the localization or trafficking of others, thereby modulating junctional composition and permeability [17]. In cancer, such compositional shifts can be driven by aberrant expressions of auxiliary proteins like MUC13, which negatively regulates the levels of claudins and occludin via PKC signaling, thereby impairing barrier integrity [30] (Figure 1).
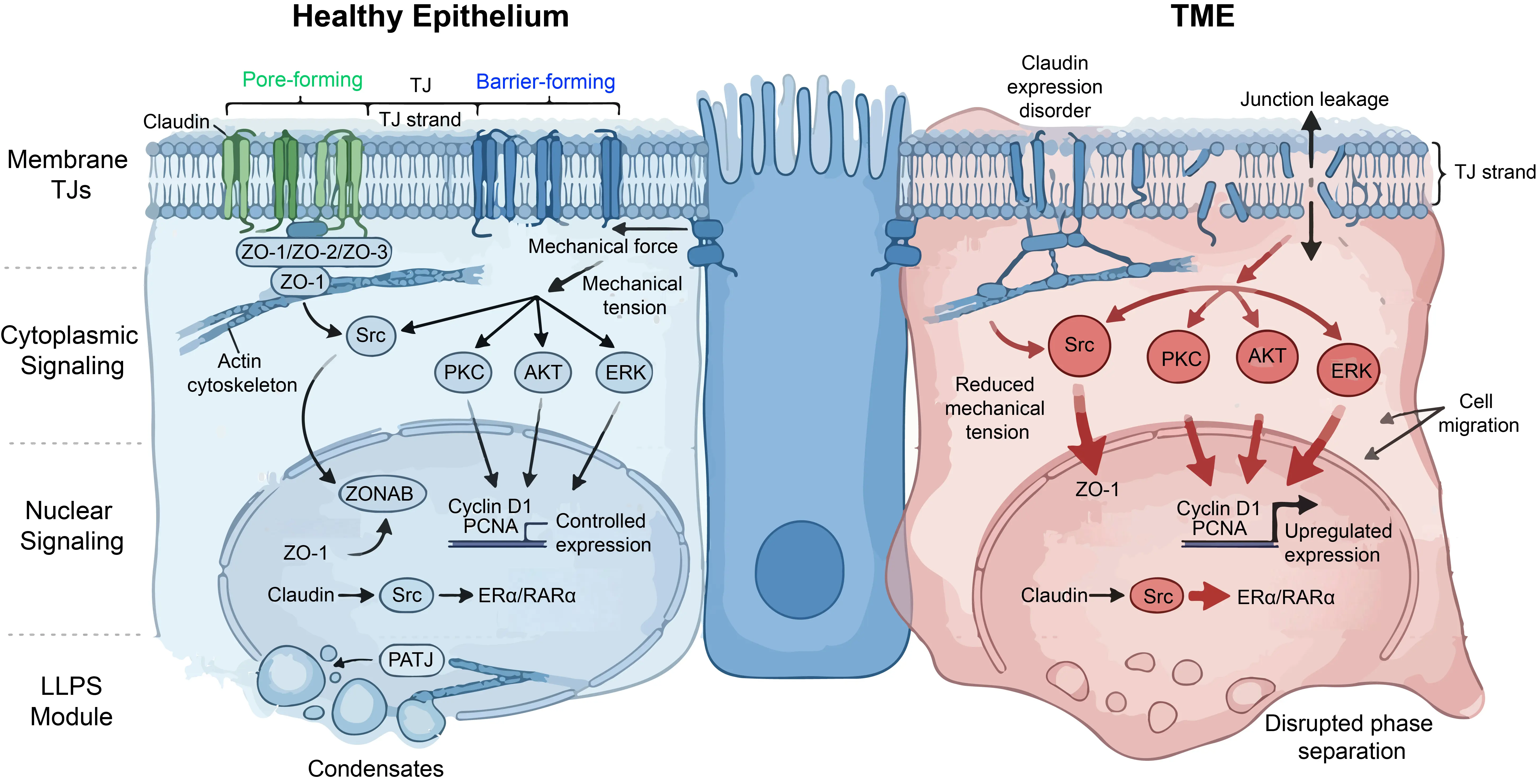
Figure 1. The Molecular Architecture of TJs: From Structural Scaffolds to Signaling Hubs. (Left panel): the TJ function in healthy epithelium as structure scaffolds and signaling hubs. (Right panel): The damaged TJ barrier and the dysregulated signaling pathways in TME.
Occludin, although not essential for strand formation, serves as a regulatory component that facilitates the formation of anastomosing strand networks [2]. It modulates junction stability and permeability through phosphorylation-dependent mechanisms, including PKC and CK2-mediated pathways [31]. In cancer cells and in vascular pathologies driven by BACE1-mediated degradation, occludin phosphorylation and subsequent cleavage are frequently associated with its redistribution from junctions to the cytoplasm, a process correlated with barrier disruption and enhanced metastatic potential [32].
2.1.2. Scaffolding Proteins: The ZO Family
The ZO proteins (ZO-1, ZO-2, ZO-3), members of the MAGUK family, act as central organizers of TJs [1]. They contain PDZ, SH3, and GUK domains, with PDZ domains mediating interactions with claudin C-terminal tails [33]. ZO-1 functions as a molecular bridge linking transmembrane proteins to the actin cytoskeleton. This linkage is essential for maintaining junctional integrity and mechanical resilience [18,34]. Recent findings indicate that ZO-1 is indispensable for the mechanical resistance of the AJC; in its absence, spontaneous cell stretching leads to focal junctional fractures [19]. Emerging evidence further suggests that ZO-1 acts as a mechanosensitive scaffold that undergoes conformational changes in response to actomyosin tension, thereby coupling mechanical forces to junctional signaling [35] (Figure 1).
2.2. Ectopic Functions of TJ Proteins: Beyond the Membrane
Beyond their membrane-localized roles, TJ proteins exhibit ectopic functions in cytoplasmic and nuclear compartments [36]. In terms of biophysical orchestration, the initial scaffolding assembly of certain core TJ components is hypothesized to be facilitated by LLPS [3]. Specifically, empirical evidence supports a model where the co-condensation of ZO-1 with oligomerized adhesion receptors nucleates localized macromolecular clusters [23]. Extending this concept, a recent study proposed a membrane pre-wetting hypothesis mediated by the polarity protein PATJ [4]. According to this theoretical model, PATJ is suggested to promote the transition of ZO-1 from dispersed cytoplasmic clusters into a membrane-associated condensed layer, thereby conceptually facilitating the rapid formation of a continuous TJ belt. While this phase-behavior model offers an attractive explanation for enhancing structural robustness and adaptation to mechanical stress within the TME, further independent studies are required to confirm whether true liquid-like phase transitions, as opposed to classical, high-affinity cooperative protein interactions, are strictly essential for these dynamic properties in vivo.
2.2.1. Nuclear Localization and Transcriptional Regulation
ZO-1 also participates in nuclear signaling. In confluent cells, it sequesters ZONAB at junctions, limiting proliferation. Upon junctional disruption, ZO-1 translocates to the nucleus, releasing ZONAB, which activates genes such as Cyclin D1 and PCNA. Beyond the ZONAB axis, recent literature has hypothesized that a potential TJ-NR signaling pathway has been identified: CLDN-mediated adhesion activates SFKs, which subsequently phosphorylate NRs such as ERα and RARα, directly modulating their transcriptional activity and driving cancer progression [6,21,37,38]. Nevertheless, empirical validation of this specific transmembrane-to-nuclear relay remains limited, and further independent mechanistic studies are required to corroborate the direct interaction kinetics between distinct Claudin isoforms and SFK complexes at the junctional plaque (Figure 1).
2.2.2. Cytoplasmic Signaling Functions
TJ proteins can also function in cytoplasmic signaling. For example, Claudin-1 is frequently mislocalized in metastatic colorectal cancer cells. In this context, it shifts from a structural component to a signaling regulator, activating pathways such as Src and AKT. In parallel, SIRT6 has been shown to protect TJ integrity under inflammatory stress by modulating the ERK1/2 pathway and autophagy, preventing endothelial apoptosis [38]. These pathways promote survival signaling and resistance to anoikis. This functional plasticity underscores the dual role of TJ proteins as both structural gatekeepers and signaling modulators [39,40,41] (Figure 1).
2.3. Liquid–Liquid Phase Separation in TJ Assembly
LLPS offers an emerging biophysical perspective that potentially complements our understanding of how TJs achieve structural stability and dynamic adaptability, with theoretical models suggesting their involvement in partitioning the paracellular space into pore and leak pathways [42].
2.3.1. ZO-1 Surface Condensation
ZO-1 contains intrinsically disordered regions (IDRs) that enable multivalent interactions and phase separation. These properties drive the formation of liquid-like condensates at the membrane, a process termed surface condensation. These condensates selectively recruit TJ proteins, such as occludin, and signaling molecules, facilitating rapid and efficient junction assembly upon cell–cell contact [43,44].
2.3.2. Coupling with Actin Dynamics
ZO-1 condensates are tightly coupled to actin polymerization. Local actin polymerization and bundling are not only secondary to condensation but also actively drive the elongation and fusion of these condensates into continuous belts. Perturbations in intracellular conditions, such as pH alterations or osmotic stress in mature epithelium, may disrupt this phase behavior, leading to fragmented junctions and increased motility [45].
2.3.3. Evaluating LLPS as a Proposed Signaling Platform
Because LLPS condensates are hypothesized to concentrate multiple proteins, they function as signaling hubs to complement classical, high-affinity molecular interactions. By locally enriching kinases and phosphatases, these structures are suggested to enhance signaling efficiency and spatial specificity. In tumor cells, such condensates have been proposed to facilitate the localized activation of pathways, such as RhoA signaling, thereby promoting cytoskeletal remodeling and migration. While this spatial organization offers an intriguing model for rapid mechanosensitive responses, further empirical validation is required to definitively distinguish true phase separation from canonical macromolecular scaffolding during force-induced calcium signaling and RhoA-mediated TJ remodeling [46,47].
In summary, TJs should be viewed as dynamic, multi-layered systems integrating structural, mechanical, and signaling functions rather than static adhesive barriers.
3. Orchestrating Tumor Plasticity and Stemness
The progression of cancer is not a unidirectional process; rather, tumor cells exhibit significant phenotypic switching to survive under selective pressure. This ability, termed cellular plasticity, is increasingly recognized to be governed by TJs beyond their canonical structural function. TJs act as biochemical hubs and mechanosensors that influence whether a cell maintains an epithelial phenotype or transitions to a mesenchymal state. This section explains how TJs manage this switch and support the stem-like traits of cancer cells [5,12,14,48].
3.1. The Dynamic Switch of EMT and MET
EMT allows cells to detach from the primary tumor, while MET occurs when cancer cells colonize a distant organ. TJs are among the first structures disrupted during EMT and reassembled during MET, contributing to active signaling modulation rather than acting as passive barriers [13,49].
3.1.1. How TJ Loss Triggers Snail and Slug Signaling
In a healthy state, TJs sequester certain signaling molecules at the AJC [1]. For example, some kinases and phosphatases are physically anchored within the TJ complex [6]. When TJs are disrupted, the cell membrane undergoes a major biophysical and biochemical reorganization [18]. Scaffolding proteins like ZO-1 and Occludin are internalized or degraded, a process often driven by the transcriptional repressors Snail and Slug, recognized as master orchestrators of the EMT program [50]. When Snail or Slug levels rise, they downregulate the transcription of CLDN1 and CLDN4, leading to the collapse of the TJ belt [51]. Furthermore, the emerging mechanism of interclaudin interference suggests that the altered composition of claudin isoforms can destabilize the higher-order polymeric strands even before total protein loss [15]. As the belt breaks, the sequestered signaling molecules are released into the cytoplasm and the nucleus [10]. Once free, these molecules activate pathways that further augment Snail and Slug expression, establishing a positive feedback loop. The loss of the physical barrier directly feeds the genetic program of invasion [52]. In many cancers, the dissociation of membrane-bound ZO-1 is a clear molecular hallmark of EMT initiation, as the cell becomes less adhesive and more migratory, transitioning from a cohesive tissue component to a solitary, invasive unit [53] (Figure 2).
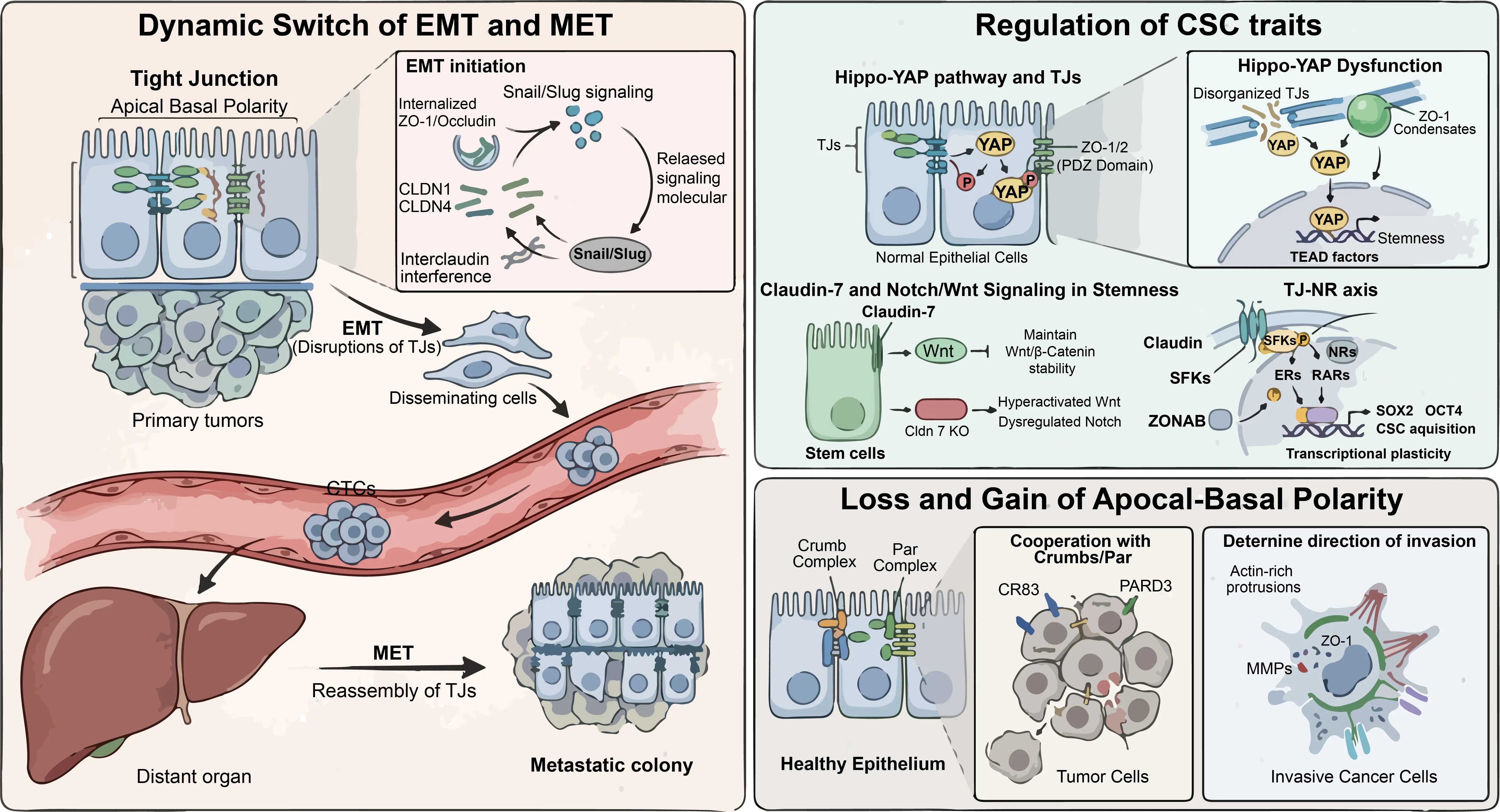
Figure 2. TJs Orchestrate Tumor Plasticity and Stemness. (Left panel): The role of TJs in driving the dynamic switch between EMT and MET. (Right panel) (top): The role of TJs in regulating CSC trait. Right panel (bottom): The role of TJs in directing cancer metastasis.
3.1.2. TJ Re-Expression During Colonization
Metastasis is not successful unless the cancer cell can re-establish a proliferative focus in a distant niche. To do this, the cell must undergo MET, reacquiring epithelial characteristics. This requires the dynamic re-expression of TJ proteins [54,55]. Research shows that successful metastatic colonies often exhibit upregulated levels of TJs [27]. For instance, in breast cancer that spreads to the liver, the cells often regain Claudin-4 and Claudin-7 [56]. This re-expression is crucial because the nascent tumor needs a stable internal environment [33]. TJs provide this stability by reforming the paracellular barrier and establishing the pore and leak pathways necessary for nutrient homeostasis [57]. Also, TJs help the cells to form clusters; collective invasion and colonization are associated with enhanced resistance to anoikis and the host’s immune system [58,59]. Therefore, the plasticity of TJ expression is a key survival skill for metastatic cells.
Crucially, the crosstalk between TJs and integrins extends beyond static spatial co-localization and functions instead as a dynamic bidirectional mechanotransductive relay. During EMT, biochemical disassembly of TJs disrupts apical-basal polarity and redistributes junctional proteins such as claudins and ZO-1 away from the membrane. This structural dissolution relieves spatial constraints on the plasma membrane, facilitating the lateral diffusion and clustering of β1- and β3-integrins. Concurrently, cytoskeletal tension is reprogrammed, promoting the recruitment of talin and paxillin and thereby accelerating focal adhesion (FA) assembly and maturation. Integrin hyper-clustering within nascent FAs subsequently activates Focal Adhesion Kinase (FAK) through Tyr397 autophosphorylation, enabling Src recruitment and formation of the active FAK/Src signaling complex. This mechanosignaling cascade propagates downstream MAPK/ERK signaling and enhances matrix metalloproteinase expression, actomyosin remodeling, and migratory polarization, collectively driving the transition from a stationary epithelial phenotype toward an invasive mesenchymal state [60].
Importantly, the relationship between TJ remodeling and ECM stiffening is increasingly recognized as a bidirectional causal feedback loop rather than a simple correlative association. On one hand, matrix stiffening activates mechanosensitive pathways such as integrin-FAK-RhoA/ROCK signaling, which destabilize TJ architecture and promote junctional disassembly. On the other hand, loss of TJ integrity disrupts epithelial polarity and alters directional secretion of ECM-remodeling mediators, including collagen-associated crosslinking enzymes and matrix metalloproteinases. These changes accelerate aberrant collagen deposition, fiber alignment, and matrix crosslinking, further increasing ECM rigidity and reinforcing pro-invasive mechanotransduction. This reciprocal amplification between TJ dysfunction and ECM remodeling ultimately establishes a mechanically permissive microenvironment that supports tumor invasion and metastatic progression.
3.2. Regulation of CSCs Traits
Cancer Stem Cells (CSCs) are a small subpopulation within a tumor possessing the capacity for self-renewal and multi-lineage differentiation. They are also intrinsically resistant to conventional chemotherapy. Recent studies show that TJ proteins are vital for maintaining the CSC niche [61,62,63].
3.2.1. The Hippo-YAP Pathway and TJ Proteins
The Hippo signaling pathway controls organ size and cell growth, with the transcriptional co-activator YAP serving as the main effector. In normal cells, the Hippo pathway keeps YAP phosphorylated and sequestered in the cytoplasm [64,65].
TJs are essential for sequestering YAP at the cell membrane, thereby preventing its oncogenic activation. Scaffolding proteins, particularly ZO-1 and ZO-2, physically interact with YAP through their PDZ domains to maintain it at the AJC. When TJs are disorganized or lost, often coinciding with the downregulation of barrier-forming Claudins, YAP is released from these junctional anchors and translocates into the nucleus. Once in the nucleus, YAP functions as a co-activator for TEAD transcription factors to upregulate a signature set of genes that promote stemness and survival, such as CTGF, CYR61, AXL, and BIRC5. Crucially, the TJ-mediated regulation of cell volume also feeds into this axis; ZO-1 and ROCK-dependent volume regulation in confluent tissues may set the threshold for YAP activation [66] (Figure 2).
In addition, the biophysical state of the TJ affects YAP. ZO-1 forms condensates through LLPS. These condensates act as rheological sensors of cellular crowding. If the TJs are under low tension, they sequester YAP tightly. If the TJs are stretched or broken, they release YAP [3,23,24]. This means the physical integrity and tension of the TJ directly program the cell’s identity toward a stem-like state.
3.2.2. Claudin-7 and Notch/Wnt Signaling in Stemness
The role of Claudin-7 (CLDN7) is paramount in stem cell homeostasis. CLDN7 is not just a barrier protein; it also functions as a signaling scaffold. In intestinal stem cells, CLDN7 is essential for modulating the Wnt and Notch pathways. In Cldn7 knockout models, these pathways become dysregulated. Specifically, the loss of CLDN7 leads to hyperactivation of Wnt signaling by impairing the stability of the β-catenin destruction complex. High Wnt activity is a hallmark of CSCs, driving symmetric division and expansion. Additionally, CLDN7 affects Notch signaling, which mediates lateral inhibition. This process ensures the proper balance between stem cells and differentiated lineages. When CLDN7 is missing, this homeostatic balance is lost, leading to a poorly differentiated, primitive cellular state. In a tumor, this phenotypic shift renders the whole mass more aggressive and refractory to therapy [17,67,68,69,70] (Figure 2).
3.2.3. The Proposed TJ-NR Axis and Its Interplay with Transcriptional Plasticity
Beyond traditional kinase cascades, recent literature has conceptualized a potential regulatory layer termed the TJ-NR signaling pathway. In this proposed framework, certain transmembrane Claudins are hypothesized to act as upstream scaffolds that recruit and activate SFKs at the cell-cell interface, thereby triggering a signaling relay in which SFKs mediate the non-canonical serine phosphorylation of various NRs, such as ERs and RARs [8,21].
While this claudin-dependent modulation of NRs represents an intriguing model for tumor plasticity, its generalizability and empirical foundation across diverse cancer types require further validation through independent mechanistic studies. In theory, this putative Claudin-SFK-NR axis is suggested to allow tumor cells to bypass classical ligand-dependent activation, potentially influencing transcriptional programs associated with stemness markers, including SOX2 and OCT4 [8]. Importantly, this emerging model must be contextualized alongside well-established junctional relays. A far more consolidated and validated mechanism involves cytoplasmic scaffolding proteins, such as ZONAB (a Y-box factor), which serves as a robust rheostat for cell density. While intact TJs sequester ZONAB to limit the transcription of cell cycle genes like Cyclin D1, their release under low-density or disrupted states drives proliferation. Whether the proposed TJ-NR axis functions universally or acts as a context-dependent amplifier alongside the canonical ZO-1-ZONAB loop to facilitate CSC acquisition remains an open question that necessitates rigorous experimental corroboration [8,21] (Figure 2).
3.3. Loss and Gain of Apical-Basal Polarity
Healthy epithelial cells exhibit established apical-basal polarity, with TJs defining the architectural boundary between these two domains. In cancer, the dissolution of this spatial organization is a major driver of invasion.
3.3.1. Cooperation with Crumbs and Par Complexes
TJs function in concert with the Crumbs complex (defining the apical domain) and the Par complex (regulating junctional positioning). In many tumors, components for Crumbs (like CRB3) or Par (like PARD3) are epigenetically silenced or mutated. When these proteins are deficient, TJs cannot form a continuous circumferential belt. Instead, TJ components such as ZO-1 form ectopic, nonfunctional patches. Without a clear polarity axis, the cell loses its spatial orientation. Consequently, the cell may reorient its protrusive structures toward the underlying basement membrane. This is the critical step of invasion, where the cell uses its disorganized TJ proteins to facilitate the assembly of actin-rich protrusions that secrete matrix metalloproteinases (MMPs) to digest the surrounding matrix [71,72,73,74] (Figure 2).
3.3.2. Determining the Direction of Invasion
The location of the TJ fragments often determines the vector of cellular motility. Research has shown that in some invasive cancers, TJ proteins move to the leading edge of the cell, where ZO-1 coordinates with the perijunctional actin ring to generate contractile force. This force propels the cell forward, while localized TJs create micro-domains of ion signaling. For example, they might concentrate calcium channels at the leading edge, triggering local calcium transients that activate Rho-GTPases and other pro-migratory effectors. Therefore, the ectopic gain of disorganized TJs is just as pathologically significant as the loss of the original barrier [4,6,19] (Figure 2).
3.4. Deconstructing the Junctional Hierarchy: TJs Versus AJs in Metastasis
A major limitation in current junctional biology is the frequent tendency to ascribe complex signaling networks predominantly to TJs, while underestimating the substantial functional interplay between TJs and Adherens Junctions (AJs). Consequently, the precise division of labor between these two junctional systems during metastatic progression remains incompletely resolved. Historically, the loss of E-cadherin, the central structural component of AJs, has been regarded as the principal initiating event driving EMT and metastatic dissemination [51]. However, increasing evidence indicates that many transcriptional and mechanobiological pathways commonly interpreted as TJ-centric are in fact co-regulated, or even primarily governed, by AJ dynamics [53]. For example, although the sequestration and release of signaling mediators such as β-catenin and YAP/TAZ can be influenced by TJ-associated scaffold proteins, including ZO-1, these processes remain fundamentally dependent on the integrity of the E-cadherin/catenin complex within AJs [53,64]. Moreover, several studies have reported that selective disruption of TJs, in the absence of E-cadherin loss, is insufficient to induce a fully metastatic phenotype in certain carcinoma models. In contrast, E-cadherin ablation frequently leads to a broader collapse of apical–basal polarity, secondarily destabilizing TJ organization and epithelial barrier integrity [51,53].
Taken together, these findings suggest that portraying TJs as autonomous signaling hubs may represent an oversimplification of epithelial junctional biology [5,53]. Rather than functioning independently, TJs are more likely to serve as spatially specialized regulatory platforms that fine-tune, amplify, or contextualize adhesive and signaling inputs primarily orchestrated by AJs [17,53]. Accordingly, disentangling the distinct mechanical and biochemical contributions of TJs and AJs remains a critical methodological and conceptual challenge for future investigations into metastatic progression and oncogenic signaling.
In summary, TJs function as much more than static paracellular seals; they are dynamic signaling hubs that orchestrate tumor plasticity and the acquisition of CSC traits. The loss of TJ integrity facilitates EMT by releasing sequestered signaling molecules and activating Snail/Slug pathways, while their selective re-expression supports metastatic colonization through MET. Furthermore, TJs regulate stemness by modulating the Hippo-YAP axis and Wnt/Notch signaling through both biochemical scaffolding and mechanosensitive phase separation. The newly discovered TJ-NR axis further underscores the direct link between junctional assembly and epigenetic reprogramming. Ultimately, the fragmentation of apicobasal polarity and the ectopic localization of TJ proteins at the leading edge provide the spatial and mechanical cues necessary for directed tumor invasion and survival in hostile microenvironments.
4. Remodeling the TME in Symbiosis and Evasion
The TME is not a passive backdrop but a dynamic space where cancer cells interact with the extracellular matrix (ECM), immune cells, and blood vessels. TJ proteins are now recognized as important mediators of this cross-talk. They do not just hold cells together; they also participate in sensing and integrating environmental cues. Beyond traditional scaffolding, TJs govern the pore and leak paracellular pathways, regulating tumor immunity through a continuum of mechanisms ranging from physical exclusion to phase separation mediated signaling and barrier remodeling [1,5,6,22].
4.1. Mechano-Transduction and Matrix Remodeling
Tumor tissues are often much stiffer than healthy tissues. This stiffness comes from the excessive buildup of collagen and other ECM proteins. TJs contribute significantly to this mechanical sensing process through coordinated molecular condensates [23,75,76].
4.1.1. Sensing Stiffness via the ZO-1-Actin Link
The scaffolding protein ZO-1 functions as a mechanosensitive scaffold linking junctional complexes to the actin cytoskeleton. Recent breakthroughs indicate that TJ assembly is driven by the surface co-condensation of ZO-1 with oligomerized receptors [4,24,45]. As the ECM stiffens, force-induced prewetting of these condensates promotes the growth of a condensed ZO-1 layer around the apical membrane. Elevated tension induces conformational changes in ZO-1, potentially exposing binding sites for signaling molecules. In softer environments, ZO-1 sequesters the transcription factor ZONAB at the membrane, whereas stiffer contexts promote its release [4,24,50]. This mechanosensitive phase transition enables ZONAB to translocate to the nucleus and drive proliferation-associated genes, such as PCNA, providing a biophysical link between tissue stiffness and tumor growth [3] (Figure 3).
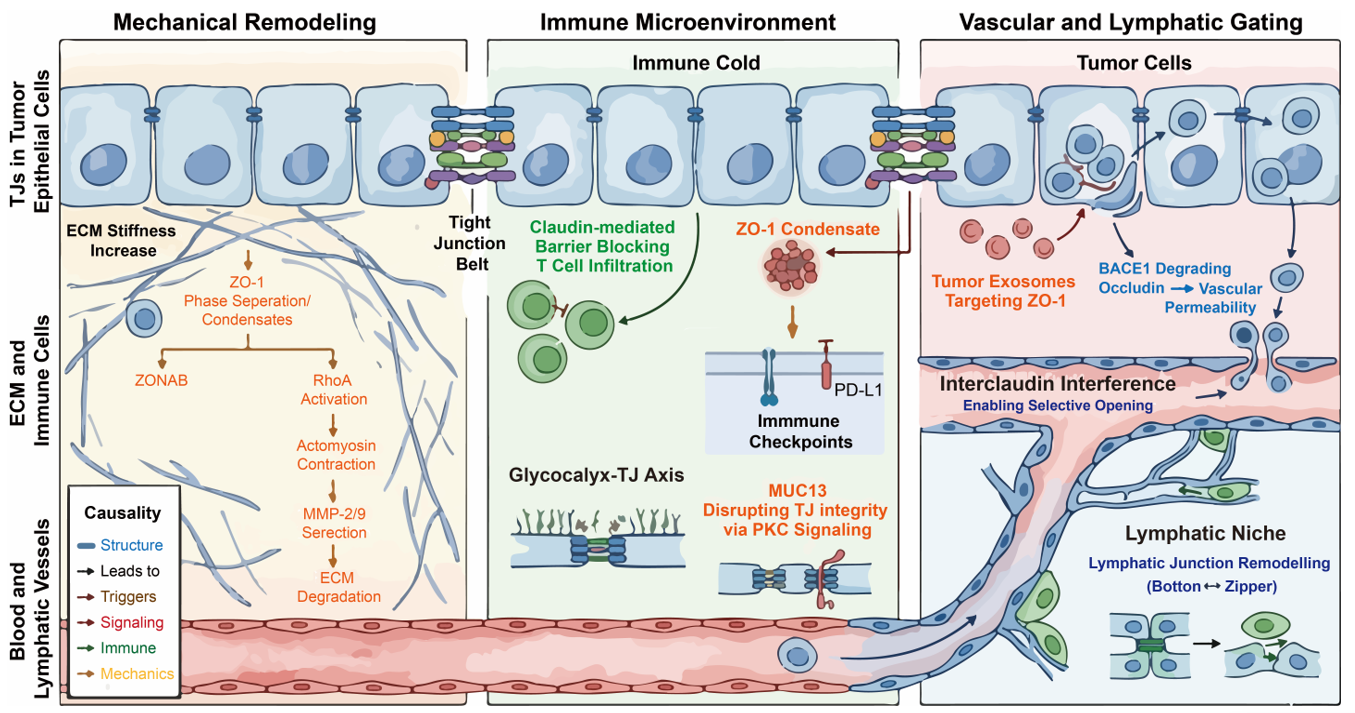
Figure 3. TJs Remodel the TME in Symbiosis and Evasion. (Left panel): The mechanical remodeling in cancer metastasis. (Middle panel): TJs involve in tumor immune microenvironment reprogramming through regulating T cell infiltrating, immune checkpoints, glycocalyx-TJ Axis, and MUC13-PKC pathway. (Right panel): The role of TJs as vascular and lymphatic gating in cancer metastasis. The TJs were indicated among the three panels.
4.1.2. Mechanical Regulation of MMP Release
The remodeling of the ECM requires MMPs. TJs influence the spatial regulation of these enzymes through actomyosin-driven tension [10,19,24,77,78,79]. TJ-associated cytoskeletal tension is linked to the activation of Rho-family GTPases, particularly RhoA. This RhoA activation often occurs at focal breakages of the AJC, especially in claudin-deficient cells. Active RhoA stimulates actomyosin contraction, creating hotspots of mechanical stress that coordinate the localized secretion of MMP-2 and MMP-9 [10,19,24,77,78,79]. Thus, TJs act as mechanical organizers that enable localized ECM degradation to facilitate invasion [10,19,24,77,78,79] (Figure 3).
Crucially, this mechanobiological circuit operates within, and is further reinforced by, the specialized biomechanical architecture of the tumor extracellular matrix (oncomatrix). Rather than representing a simple global increase in tissue stiffness, the oncomatrix is characterized by localized collagen reorganization, particularly involving fibrillar collagens such as Types I and III, together with altered elastic fiber density [80]. These changes transform the ECM from an isotropic, mesh-like structure into a highly aligned, anisotropic network that facilitates directional cancer cell migration.
Mechanistically, such topological remodeling can be quantified using parameters including the fiber alignment index (FAI) and collagen cross-link density, the latter frequently mediated by lysyl oxidase (LOX) activity or advanced glycation end-products [80]. Increased collagen alignment and cross-linking generate mechanically constrained migratory tracks that not only promote invasive movement but also impose compressive and tensile forces on apical junctional complexes. These forces subsequently activate mechanosensitive signaling pathways associated with TJs, thereby enhancing RhoA-dependent actomyosin contractility and further promoting localized MMP secretion [81]. Collectively, these findings support a bidirectional feedback model in which ECM remodeling and TJ-mediated mechanotransduction cooperatively amplify invasive behavior during tumor progression.
4.2. The Immune Microenvironment: Physical and Signaling Filtration
A major challenge for the immune system is to effectively penetrate and function within the core of solid tumors. TJs contribute to the regulation of immune cell infiltration through both physical barrier functions and signaling-mediated modulation of the TME [12,82].
4.2.1. Exclusion of Infiltrating Immune Cells
Metastatic tumors frequently exhibit an immune cold phenotype, characterized by limited effector T-cell infiltration and activity [83]. While stromal density and ECM composition are primary determinants, TJs may contribute as auxiliary physical barriers [84]. In certain carcinomas, the upregulation of Claudin-1 and Claudin-4 correlates with enhanced epithelial cohesion, which may partially impede the trans-epithelial migration of lymphocytes [27,85]. Furthermore, new biophysical evidence suggests that TJs provide critical mechanical resistance to the AJC; the loss of Claudins and JAM-A can lead to focal junctional breakages under spontaneous cell stretching, a process that may be exploited by tumor cells to facilitate invasion while selectively restricting immune cell entry [86] (Figure 3).
Beyond serving as local mechanical barriers at the epithelial interface, TJs also contribute to broader biomechanical remodeling of the TME through the regulation of ECM-modifying programs. Emerging evidence indicates that TJ-associated proteins, including claudins and ZO family members, can indirectly reshape matrix architecture and stiffness by modulating the expression of key ECM cross-linking enzymes, such as lysyl oxidase (LOX) and transglutaminases. Through these downstream enzymatic pathways, TJ-mediated signaling promotes collagen cross-linking, matrix alignment, and tissue stiffening, thereby reinforcing tumor biomechanical integrity and enhancing mechanosensitive oncogenic progression [87].
Importantly, beyond purely structural restriction, TJs may indirectly influence immune exclusion by modulating chemokine gradients and the spatial distribution of adhesion molecules. Experimental observations indicate that T cells recruited to tumor margins can accumulate near junctional regions without efficient transmigration, suggesting that junctional integrity may create a localized retention zone. This retention may be further governed by the liquid-like surface condensation of ZO-1, which acts as a selective filter, enriching or excluding specific membrane-associated signaling components [88] (Figure 3).
Therefore, TJs should be considered modulatory rather than dominant contributors to immune exclusion, acting in concert with stromal, vascular, and biochemical barriers within the TME.
4.2.2. Regulation of PD-L1 and Inhibitory Ligands
TJs also participate in signaling processes associated with immune evasion, although the underlying mechanisms remain incompletely defined. Emerging evidence suggests that certain TJ-associated proteins may influence the expression and stability of immune checkpoint molecules such as PD-L1 [89]. For instance, Claudin-3 and other transmembrane TJ proteins have been implicated in novel signaling pathways where their aberrant expression alters the turnover of NRs and immune-related ligands, possibly through modulation of intracellular trafficking pathways and lysosomal degradation [90] (Figure 3).
Additionally, the scaffolding protein ZO-1 may contribute to the spatial organization of membrane-associated signaling complexes. Recent discoveries regarding the PATJ-mediated prewetting and condensation of ZO-1 layers suggest that the TJ belt functions as a highly organized signaling platform. While direct evidence linking ZO-1 to inhibitory receptor clustering is currently limited, its role in coordinating cytoskeletal tension and membrane domains via localized actin polymerization suggests a plausible indirect influence on receptor distribution and signaling efficiency [90].
Collectively, these findings point to a potential, yet still emerging, role for TJs in modulating immune checkpoint regulation, warranting further mechanistic and in vivo validation.
4.2.3. The Glycocalyx-TJ Axis
The integrity of epithelial and endothelial barriers relies on coordinated interactions between the apical glycocalyx and underlying TJs. Recent work by Zhang et al. suggests that glycocalyx components, particularly heparan sulfate, functionally interact with TJ proteins such as Occludin and ZO-1. This synergy is maintained through STAT3 signaling, which acts as a critical regulator to prevent the shedding of HS and the subsequent impairment of TJ protein expression [91,92] (Figure 3).
Within the TME, enzymatic degradation of the glycocalyx, which is often mediated by tumor-derived heparanase, can precede or accompany alterations in TJ integrity. The disruption of this axis leads to increased endothelial permeability and facilitates inflammatory signaling. Rather than a strictly sequential, top-down failure, current evidence supports a dynamic, reciprocal disruption of glycocalyx and TJ structures. Considering this coordinated dysregulation provides a more integrated framework for understanding how tumors compromise barrier function and promote systemic spread [91,92].
4.2.4. Mucin-Mediated Antagonism: The Role of MUC13
The dynamic remodeling of the TME is further influenced by interactions between transmembrane mucins and TJ assembly. In gastrointestinal and other mucosal malignancies, overexpression of the transmembrane mucin MUC13 has been associated with alterations in epithelial barrier integrity. As reported by Segui-Perez et al., MUC13 acts as a negative regulator of the intestinal barrier; its presence significantly reduces the levels of claudins and occludin in membrane fractions, potentially through activation of Protein Kinase C (PKC) signaling pathways [30] (Figure 3).
This mucin–TJ interplay may represent a characteristic feature of epithelial tumors of the digestive tract. Through PKC-associated modulation of junctional proteins, MUC13 expression has been linked to reduced transepithelial electrical resistance (TEER) and increased paracellular permeability. These changes may facilitate the exchange of soluble factors and contribute to a microenvironment conducive to tumor progression and cell migration. This MUC13-driven remodeling reflects a mechanism in which the tumor actively destabilizes its own junctional network to enhance plasticity, enabling cell migration and metabolic exchange across the compromised epithelial layer [33,93].
4.3. Gating Control of Blood and Lymphatic Vessels
Metastasis requires dynamic remodeling of vascular barriers. Tumor-derived factors can induce changes in endothelial junction integrity, facilitating cancer cell dissemination.
4.3.1. Paracrine Education of Endothelial TJs
Tumor cells secrete extracellular vesicles (exosomes) and growth factors such as VEGF. These factors can target endothelial TJs. For instance, tumor-derived exosomes may contain microRNAs such as miR-105, which has been shown to target TJP1 (ZO-1) mRNA in endothelial cells, particularly in breast cancer models, leading to reduced junctional integrity. Additionally, new mechanisms of vascular impairment involve the upregulation of endothelial BACE1, which has been shown to cleave and degrade the TJ protein Occludin, directly leading to blood-brain barrier (BBB) breakdown and promoting a permissive environment for secondary lesions. This process, often referred to as vascular permeabilization, can increase endothelial permeability and may contribute to pre-metastatic niche formation in organs such as the brain and lungs [94] (Figure 3).
4.3.2. Lymphatic Gating and Nodal Metastasis
In many cancers, lymph nodes are an early site of metastasis. Lymphatic vessels exhibit distinct junctional structures, including discontinuous button-like junctions. Some studies suggest that tumor-derived factors may influence the remodeling of lymphatic junctions, potentially promoting transitions between zipper-like and button-like configurations. This process may involve the redistribution of proteins such as Claudin-5 and Occludin. Moreover, the concept of interclaudin interference, where one claudin disrupts the polymeric strands and channels formed by another, could offer a novel explanation for how tumor factors selectively open lymphatic gates without total barrier collapse. Such changes could facilitate tumor cell entry into lymphatic vessels, although the extent to which tumors actively regulate this process remains under investigation [15,95] (Figure 3).
In summary, the TJ network within the TME has evolved from a static structural seal to a dynamic signaling and mechanical hub. Recent evidence highlights that TJ remodeling is driven by biophysical phenomena such as phase separation and surface condensation of ZO-1, which allow the junctional complex to sense tissue stiffness and activate downstream proliferation via the ZONAB pathway. Furthermore, TJs act as sophisticated filters in the immune landscape, where their integrity, which is regulated by the glycocalyx-STAT3 axis and antagonized by mucins like MUC13, governs the infiltration of immune cells and the stability of checkpoint molecules. The discovery of BACE1-mediated occludin degradation and interclaudin interference underscores a precise gating mechanism by which tumors hijack endothelial and lymphatic barriers. Collectively, these TJ-centric processes coordinate to promote mechanical adaptation, immune evasion, and vascular permeabilization, marking tight junction proteins as important regulators of malignant progression and potential therapeutic targets.
5. Tight Junctions in the Orchestration of Metastatic Evolution
Metastasis is a long and inefficient process during which only a small fraction of cancer cells successfully colonize distant organs. Rather than acting solely as passive structural barriers, TJs should be conceptualized as dynamic mechano-signaling interfaces that coordinate cell cohesion, force transmission, and survival signaling throughout metastatic progression. The assembly of these interfaces is driven by the phase separation of scaffold proteins such as ZO-1, which form surface condensates that facilitate rapid junctional remodeling and belt formation. To succeed, a cancer cell must survive in circulation, adapt to mechanical stress, and eventually colonize a distant niche. Emerging evidence suggests that TJ remodeling is not merely a consequence of metastasis, but an active regulator of multiple steps, including cluster integrity, metabolic adaptation, anoikis resistance, and vascular barrier interaction [3,96].
5.1. The Survival of CTC Clusters
Most research on CTCs has historically focused on single cells. However, accumulating clinical and experimental evidence indicates that CTCs frequently travel as multicellular aggregates, termed CTC clusters, which exhibit markedly enhanced metastatic potential. This enhanced metastatic fitness arises not merely from increased cell number but from collective biomechanical and signaling properties maintained by intercellular junctions, including TJs [57,97].
5.1.1. Mechanical Resistance Against Shear Stress
When a cluster of cells enters the bloodstream, it is exposed to significant hemodynamic shear forces. Recent breakthroughs indicate that the rapid assembly and maintenance of TJ belts in these clusters are driven by ZO-1 surface condensation and membrane prewetting, a process facilitated by the polarity protein PATJ. This phase-separation mechanism allows the TJ scaffold to rapidly capture claudins and form a continuous, elastic barrier. TJ-associated proteins mechanically couple to the actomyosin cytoskeleton, forming a tension-bearing network. This network is reinforced by the prewetting phenomenon, where PATJ-mediated ZO-1 condensation promotes the elongation of TJ belts around the apical interface, ensuring rapid structural sealing [4] (Figure 4). This network distributes intercellular tension and dissipates shear-induced stress across multiple cells, enhancing cluster stability without relying solely on adhesion strength. Furthermore, claudins and JAM-A act as indispensable structural anchors; their absence leads to focal fractures of the AJC under extrinsic stretching [94]. Experimental disruption of TJ components reduces intercellular cohesion and increases cluster fragmentation under flow conditions. Smaller fragments or single cells are consequently more vulnerable to immune clearance and mechanical damage. Thus, TJs contribute to cluster stability as part of a broader junctional complex rather than serving as the sole mechanical adhesive [4,6,22,98].
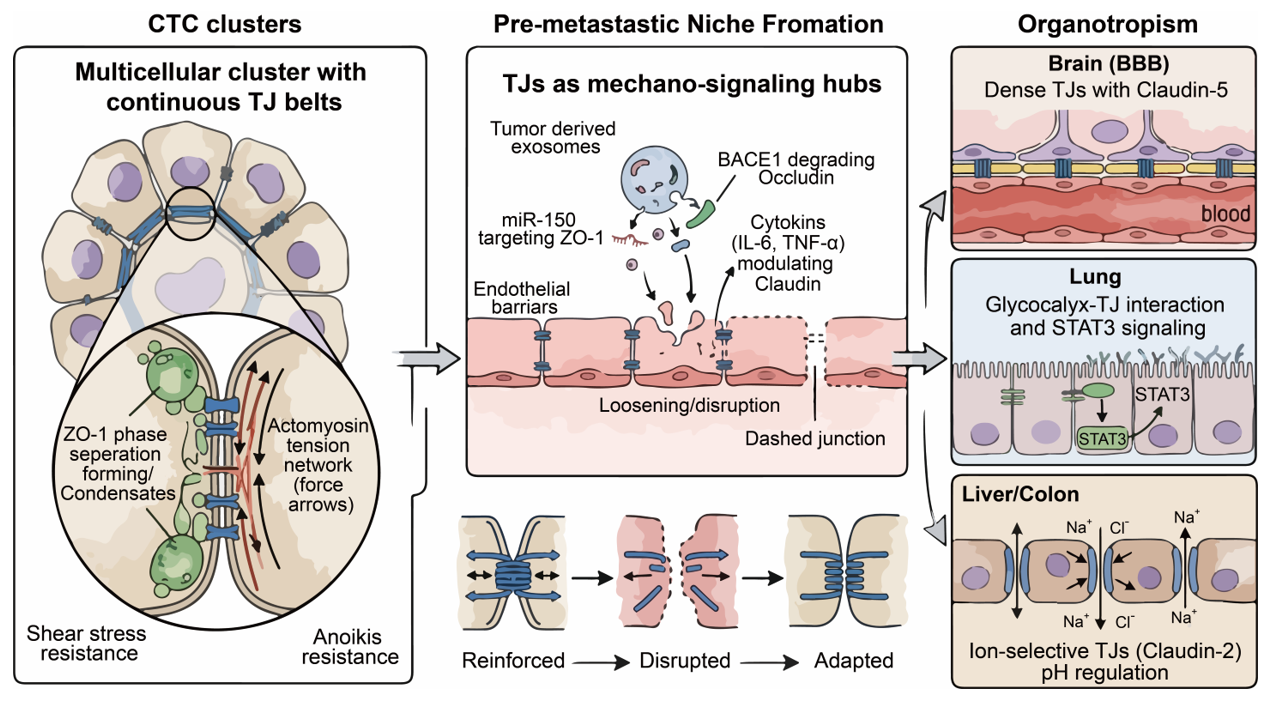
Figure 4. TJs in the Orchestration of Metastatic Evolution. (Left panel): The role of TJs in the survival of CTC Clusters. (Middle panel): The role of TJs in the formation of pre-metastastic niche. (Right panel): TJ components tend to have organotropism during cancer metastasis.
5.1.2. Resistance to Anoikis
Most epithelial cells undergo anoikis upon loss of attachment to the extracellular matrix. For single CTCs, circulation represents a hostile, pro-apoptotic environment. However, in CTC clusters, intercellular contacts are preserved. TJs, together with adherens junctions, maintain a pseudo-epithelial signaling context that actively modulates intracellular survival pathways, thereby suppressing anoikis [8,99]. Beyond classical PI3K/Akt signaling, recent literature has proposed an emerging conceptual framework involving a potential CLDN-NR signaling cross-talk. In this hypothetical model, CLDN-mediated adhesion is postulated to influence SFK activation, which may subsequently modulate the phosphorylation and activity of NRs in specific oncogenic contexts. However, this putative pathway still requires further independent empirical validation to substantiate its role in general cell survival. Comparatively, more established junctional mechanics involve proteins like Claudin-7, which has been reported to associate with EpCAM and integrin-linked signaling complexes to directly activate survival pathways, although the exact molecular hierarchy remains incompletely defined. Additionally, the expression of transmembrane mucins like MUC13 may negatively regulate TJ protein levels via PKC signaling, suggesting a complex internal rheostat for cluster adhesion [30,94].
5.1.3. Mechanical Stability and the AJC
The maintenance of mechanical integrity in CTC clusters is fundamentally dependent on the AJC, which provides structural reinforcement critical for collective migration. Recent landmark studies by Nguyen et al. demonstrated that TJ membrane proteins, specifically the Claudin family and JAM-A, are indispensable for the physical rigidity of the AJC. In the absence of these transmembrane anchors, apical junctions become prone to focal fractures and stochastic breakages under extrinsic stretching forces. This mechanical stabilization is achieved through coordinated interplay with the perijunctional actin cytoskeleton, with Claudins and JAM-A serving as structural anchors that maintain the integrity of the actomyosin ring. Moreover, the presence of interclaudin interference, where barrier-forming claudins like Claudin-4 can selectively inhibit or destabilize pore-forming claudins, adds a layer of regulatory complexity to the AJC’s selective permeability and structural stability [15,23].
5.2. Induction of the Pre-metastatic Niche
Before tumor cells arrive at distant organs, the primary tumor can condition these sites to become more permissive. This process, known as pre-metastatic niche formation, involves systemic signaling, extracellular vesicles, and inflammatory mediators that collectively alter vascular and stromal barriers, including TJs [100,101].
5.2.1. The Role of Tumor-Derived Exosomes
Primary tumors release extracellular vesicles (EVs), including exosomes, into circulation. These vesicles function as long-range signaling mediators that can modulate endothelial barrier properties in distant organs [102]. Studies have shown that tumor-derived exosomes can carry microRNAs such as miR-105 and miR-181 [103,104]. Notably, miR-105 has been experimentally shown to target TJP1 (ZO-1) mRNA, leading to disruption of endothelial TJ integrity and increased vascular permeability. Vascular barrier compromise can also be driven by enzymes like BACE1, which is upregulated in stressed endothelia and promotes the proteolytic cleavage of Occludin, leading to BBB breakdown [33]. When taken up by endothelial cells, these microRNAs reduce TJ protein expression. This results in a partial and regulated increase in paracellular permeability, specifically affecting both the high-capacity pore and leak pathway for larger solutes, rather than complete barrier breakdown. Therefore, it is more precise to describe this process as barrier modulation rather than total TJ destruction [2,3,5,6,10] (Figure 4).
5.2.2. Inflammatory Priming and Claudin-1 Dysfunction
Chronic inflammation contributes to pre-metastatic niche formation [105]. Systemic cytokines such as IL-6, TNF-α, IL-4, and IL-13 have been reported to modulate TJ composition and localization in endothelial and epithelial barriers [106]. Previous studies suggest that cytokine signaling can alter Claudin expression, potentially driving the progression of barrier dysfunction across different organ systems. However, the extent and direction of Claudin-1 regulation appear to be context-dependent, varying by tissue type and inflammatory milieu (Figure 4). Protective factors like SIRT6 may counteract this damage by inhibiting apoptosis and preserving TJ integrity through the ERK1/2 pathway and autophagy activation. In the lung, altered TJ integrity can increase vascular permeability and immune cell infiltration, facilitating ARDS-like pathologies that prime the soil for metastasis. Importantly, inflammatory disruption of TJs is not merely a passive consequence of systemic disease, but may actively cooperate with tumor-derived signals to create a pro-metastatic microenvironment [12,40].
Collectively, TJ remodeling during pre-metastatic niche formation establishes permissive microenvironments that interact with organ-specific junctional characteristics, thereby guiding organotropism of metastatic cells.
5.3. Organotropism: The Selective Pressure of Organ Barriers
Metastasis exhibits strong organ specificity. This organotropism is influenced in part by the molecular composition, biophysical assembly mechanisms, and regulatory dynamics of TJs within different vascular beds [96].
5.3.1. The Brain: The Challenge of the BBB
The BBB is characterized by highly restrictive TJs enriched in Claudin-5, Occludin, and ZO proteins. Recent biophysical evidence suggests that the assembly of these functional belts is driven by ZO-1 surface condensation and membrane prewetting, which ensure a continuous, defect-free seal. These junctions are tightly coupled to low transcytosis rates and high electrical resistance, making the BBB one of the most selective vascular barriers [107] (Figure 4). Studies show that TJs are sensitive to MMPs, whereas other proteases, such as BACE1, have been found to specifically impair cerebral small vessels by mediating the degradation of Occludin. MMP-2 and MMP-9 have been implicated in the cleavage of TJ-associated components and extracellular matrix remodeling, thereby facilitating BBB permeability under pathological conditions. Rather than directly cutting Claudin-5 in all cases, MMP activity and endothelial BACE1 elevation more broadly disrupt the junctional microenvironment, including basement membrane and cytoskeletal anchoring [108,109,110] (Figure 4). Some metastatic cells may exploit paracellular or transcellular routes. The relative contribution of paracellular diapedesis versus transcellular migration in brain metastasis remains an active area of research and is likely context-dependent.
5.3.2. The Lung: The Role of Heparan Sulfate and STAT3
In the lung, endothelial barrier function is jointly regulated by the glycocalyx and TJs. Heparan sulfate–rich glycocalyx interacts with TJ complexes, forming a coupled regulatory axis that coordinates permeability and mechanosensing. Glycocalyx degradation, for example via heparanase, can trigger inflammatory and mechanosensitive pathways such as STAT3, promoting TJ protein internalization (Figure 4). Furthermore, regulatory proteins such as SIRT6 play a protective role in the lung by alleviating TJ injury through the ERK1/2 signaling pathway and autophagy, suggesting that metastatic cells must overcome these endogenous repair mechanisms to colonize lung tissue [91].
5.3.3. The Liver and Colon: Ion Selectivity and pH
In the liver and colon, TJs play a key role in regulating ion and solute permeability. In the biliary system, TJs prevent bile reflux and protect the parenchyma, a barrier that can be compromised in hepatopathies. Claudin family members, such as Claudin-2, form paracellular pores that contribute to tissue-specific permeability profiles (Figure 4). In the intestinal epithelia, transmembrane mucins like MUC13 have been identified as negative regulators of TJ proteins, where MUC13 deletion significantly increases claudin and occludin levels, thereby enhancing barrier integrity [30,93]. Cancer cells colonizing these organs must adapt to unique biochemical environments. Adaptation may involve modulation of claudin expression patterns, thereby altering ionic flux and intracellular pH homeostasis. Studies suggest that interclaudin interference, in which barrier-forming claudins like Claudin-4 selectively inhibit the channels formed by pore-forming claudins, represents a sophisticated mechanism for fine-tuning paracellular permeability during tumor adaptation [2,3,5,6,10].
In summary, TJs function as critical mechanosensitive and signaling hubs throughout the metastatic cascade. The rapid assembly of TJ belts maintains the structural integrity of CTC clusters, a process driven by the phase separation of ZO-1 and membrane prewetting. These TJs, anchored by Claudins and JAM-A, provide essential mechanical resistance against hemodynamic shear stress and prevent anoikis through novel pathways such as the CLDN-SFK-NR axis. During the induction of the pre-metastatic niche, tumor-derived signals and inflammatory cytokines modulate barrier permeability, often via BACE1-mediated proteolytic cleavage or microRNA-induced silencing, to create permissive environments. Finally, metastatic organotropism is dictated by the ability of cancer cells to navigate or manipulate organ-specific TJ profiles, including the highly restrictive BBB or the ion-selective barriers of the gut and liver, often involving sophisticated mechanisms like interclaudin interference and transmembrane mucin regulation.
6. Bridging Intercellular Adhesion and Cancer Metabolism
Metabolic reprogramming is a hallmark of cancer metastasis. Cancer cells must change how they take up nutrients to survive in new environments. Traditionally, metabolism and cell junctions were seen as two separate fields. However, recent evidence shows that TJs are important to metabolic control. When TJs are lost during metastasis, the cell does not just lose its shape. It also loses the ability to manage its energy resources. This section discusses how TJ disruption leads to the spatial redistribution of transporters and the activation of growth-promoting metabolic pathways, driven by the collapse of highly organized molecular condensates [3,5,10,111,112].
6.1. Spatial Control of Nutrient Uptake: Polarity Loss and Transporter Redistribution
In healthy epithelial cells, the cell membrane is divided into two distinct parts: the apical side and the basolateral side. TJs act as the physical fence that keeps these two sides separate. This fence is maintained by the surface condensation of scaffolding proteins like ZO-1, which undergoes membrane prewetting to form a continuous, stable barrier. This separation is vital for the correct placement of nutrient transporters. For example, glucose transporters (GLUTs) and amino acid transporters are usually restricted to specific membrane domains to ensure efficient one-way transport [4,113,114].
6.1.1. The Mislocalization of GLUT1
Glucose transporter 1 (GLUT1) is the primary vehicle for glucose entry into many cells. In a polarized epithelium with intact TJs, GLUT1 is typically localized to the basolateral membrane. This setup allows cells to absorb glucose from the blood supply underneath them. When TJs are disrupted, for instance, through the downregulation of Claudin-1 or ZO-1 during EMT, the fence disappears. This breakdown is often accelerated by the overexpression of transmembrane mucins, such as MUC13, which negatively regulates the stability of TJ complexes via PKC-dependent pathways [30,93]. Without this barrier, GLUT1 molecules begin to diffuse freely across the entire cell surface. This redistribution is a critical turning point for metastatic cells. By spreading GLUT1 to the apical surface, the cell can now capture glucose from all directions. In the context of a tumor mass, this allows cells to survive in nutrient-poor zones where the traditional blood supply is far away [14,107,108]. Furthermore, the loss of TJs is often accompanied by the loss of the CRB3 (Crumbs3) polarity complex. Research shows that CRB3 deficiency directly correlates with increased glucose uptake. Without the TJ-polarity axis, the cell surface becomes a disorganized sponge for glucose (Figure 5). This metabolic advantage supports the high energy demands of cell migration and invasion.
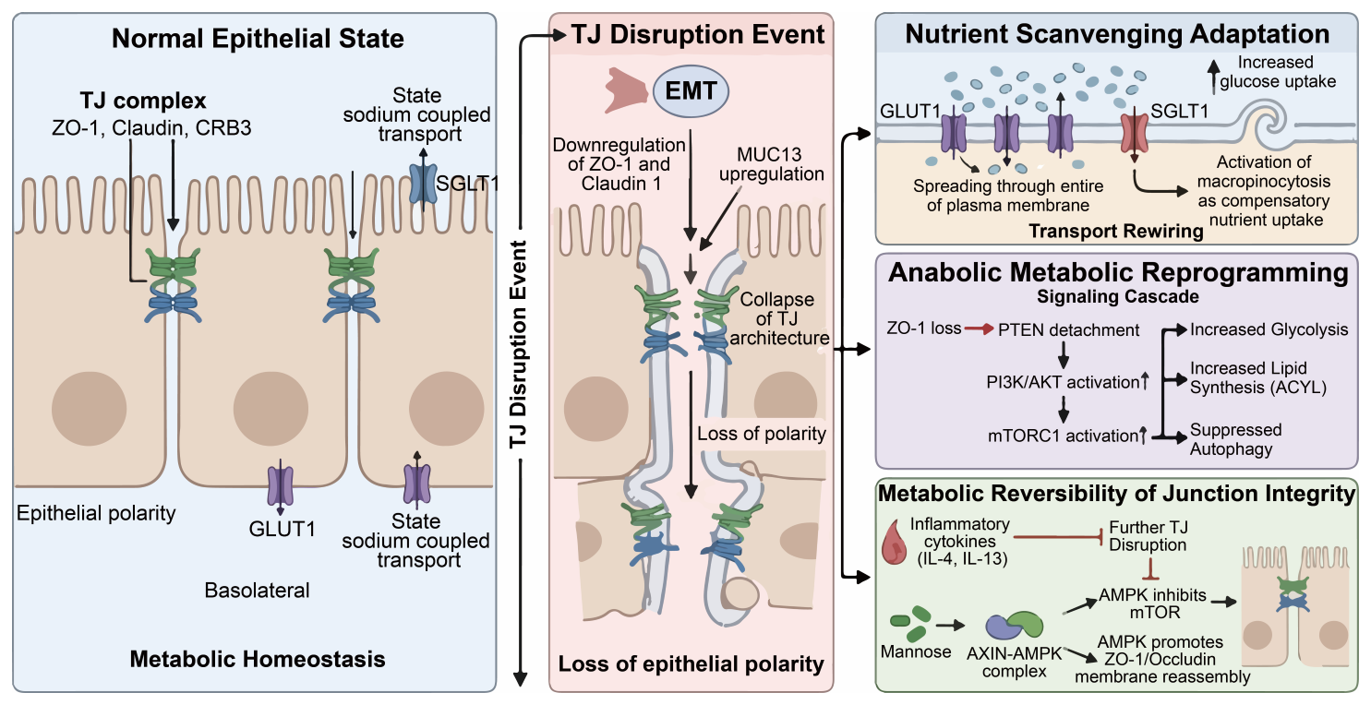
Figure 5. Bridging Intercellular Adhesion and Cancer Metabolism. (Left panel): The role of TJs maintaining metabolic homeostasis in normal epithelial cells. (Middle panel): The disruption fo TJs results in loss of epithelial polarity. (Right panel): During cancer metastasis, the tumor epithelial cells undergo nutrient scanvenging adaptation, anabolic metabolic reprogramming, and netabolic reversibility of TJs integrity.
6.1.2. Impact on Sodium-Dependent Transporters
The impact of TJs extends to sodium-dependent transporters, such as SGLT1. These transporters rely on the ion gradients maintained by the TJ barrier [57]. TJs regulate the paracellular flux of sodium ions via charge-selective channels formed by specific claudins, which can be further modulated by interclaudin interference where barrier-forming claudins (e.g., CLDN4) disrupt pore-forming ones. If the junctions become leaky, the sodium gradient across the membrane collapses [15] (Figure 5). In metastatic colorectal cancer, the loss of Claudin-7 has been shown to disrupt these gradients [15,67,68,69,70]. When the gradient is lost, the cell can no longer effectively use sodium-coupled transport to bring in essential nutrients like amino acids or vitamins. To compensate for this loss, the cancer cell often switches to a more primitive form of metabolism, including increased reliance on micropinocytosis [115]. This switch is a direct metabolic consequence of TJ failure. It shows that TJs do not just hold cells together; they define the metabolic logic of the cell membrane.
6.2. Signaling Feedback Loops: The TJ-AKT/mTOR Axis in Anabolism
Beyond physical barriers, TJs serve as scaffolds for signaling molecules that control cell growth. The most important of these is the PI3K/AKT/mTOR pathway. Moreover, recent studies suggest a novel TJ-NR signaling pathway, where claudin-mediated adhesion activates SFKs to regulate nuclear hormone receptors, directly linking the cell surface to metabolic gene transcription.
6.2.1. Activation of AKT via Junctional Scaffolding Loss
The scaffolding protein ZO-1 does more than just link claudins to actin. It also binds several phosphatase proteins, including PTEN [116]. PTEN is a famous tumor suppressor that acts as a brake on the AKT pathway. In healthy cells, the TJ complex recruits PTEN to the plasma membrane. This localization allows PTEN to effectively inhibit PI3K signaling at the cell-cell contact site. However, when TJs break down during the early stages of metastasis, ZO-1 is internalized or degraded. In vascular contexts, such disruption can be triggered by elevated BACE1 levels, which cleave Occludin and impair endothelial homeostasis [8,32]. This causes PTEN to lose its anchor at the membrane. Without PTEN at the membrane, the AKT pathway becomes hyperactivated. Activated AKT moves to the cytoplasm and nucleus, where it triggers several metabolic changes, including (i) increased glycolysis: AKT stimulates hexokinase and other enzymes in the glycolytic pathway; (ii) lipid synthesis: AKT activates ACLY (ATP citrate lyase), which is essential for building new cell membranes during rapid cell division; (iii) suppression of autophagy: AKT inhibits the cell’s self-eating process, forcing the cell to focus on building new biomass instead of recycling old parts [8,32,116] (Figure 5).
6.2.2. mTOR and the Chronic Inflammation of Metabolic Stress
The relationship between TJ integrity and mTOR activity is particularly evident in lung and intestinal injuries. Chronic inflammation leads to the release of cytokines like IL-4 and IL-13. These cytokines target Claudin-1, leading to TJ dysfunction. Experimental data show that when the epithelial barrier is compromised, the cells experience metabolic stress. In models of Acute Respiratory Distress Syndrome (ARDS), loss of TJs is often accompanied by heparan sulfate shedding, which activates STAT3 signaling and further exacerbates permeability. To counter this, protective factors like SIRT6 work to ameliorate TJ injury through the ERK1/2 pathway and autophagy regulation. In metastatic lung cancer, this TJ-mTOR link is used to prepare the pre-metastatic niche. When the endothelial barrier becomes leaky, the surrounding cells experience a surge in nutrient availability and mTORC1 activation, making the tissue a fertile ground for tumor growth [117] (Figure 5).
6.2.3. AMPK as a Potential Reversal Mechanism
Interestingly, the relationship between TJs and metabolism is a two-way street. Just as TJ loss activates growth pathways, activating metabolic sensors can restore TJs. The studies highlight the role of Mannose and the AMPK pathway. AMPK is the cell’s energy sensor that is activated when energy is low. When Mannose is added to the system, it activates the AXIN-AMPK complex. This complex does two things. First, it inhibits mTOR, slowing down the tumor’s anabolic growth. Second, it directly promotes the phosphorylation of ZO-1 and Occludin. This phosphorylation helps the TJ proteins move back to the cell membrane and rebuild the barrier [118]. This therapeutic potential is complemented by the discovery that TJ assembly is also dependent on cytoskeletal tension and volume regulation, suggesting that biophysical cues can be harnessed alongside metabolic activators (Figure 5).
This finding is highly significant for cancer therapy. It suggests that by targeting the cell’s metabolism using Mannose or AMPK activators, we can repair the broken TJs. Repairing the junctions could effectively lock the cancer cells in place and prevent them from spreading. It proves that the TJ-Metabolism axis is a dynamic system that can be manipulated to stop metastasis (Figure 5).
In summary, TJs are invisible regulators of tumor metabolism. Their role goes far beyond being a simple seal. By maintaining the spatial organization of GLUT transporters, they prevent the disorganized uptake of glucose. By anchoring proteins like PTEN and modulating NR activity, they keep the growth-promoting pathways under control. The loss of TJs is therefore a double blow to the body: it allows cancer cells to physically escape, and it gives them the metabolic fuel they need to survive the journey. Understanding this hidden role offers new hope for metabolic-based therapies to combat tumor metastasis.
7. Clinical Translation: From Molecular Scaffolding to Precision Oncology
The clinical landscape of TJ research has shifted from observing barrier leakiness to manipulating TJ-mediated signaling for therapeutic benefit. Recent evidence underscores that TJ proteins are not merely structural; they are dynamic controllers of the TME and neurovascular homeostasis. This section evaluates the transition of these proteins into the clinical sphere, focusing on their utility as predictive indicators and their role as high-precision therapeutic targets.
7.1. Predictive Biomarkers: The Metastasis Risk Score and Biofluid Diagnostics
TJ proteins provide a measurable spatial signature of tumor progression. The expression and localization of these proteins provide higher predictive value than traditional histological grading alone, particularly when quantified via immunofluorescence intensity and subcellular localization scoring of the AJC.
7.1.1. CLDN1 and the Chronic Inflammation of Metastasis
In cutaneous and mucosal malignancies, the downregulation of Claudin-1 (CLDN1) serves as a primary driver of chronic inflammation. Beyond cancer, CLDN1 deficiency is a functional trigger for the systemic propagation, where skin barrier breakdown facilitates systemic allergic sensitization [119]. In oncology, CLDN1 depletion is not just a marker of poor differentiation; it is a functional prerequisite for the activation of the Snail/Slug transcription factors. When CLDN1 levels drop, the paracellular resistance of the epithelium is significantly reduced, allowing tumor-secreted factors to enter the lymphatic system. Consequently, a low CLDN1 score in early-stage biopsies is now utilized to identify patients at a high risk for occult lymph node metastasis.
7.1.2. CLDN4 as a Guardian of Ovarian and Pancreatic Genome Stability
In contrast to CLDN1, Claudin-4 (CLDN4) is frequently overexpressed in aggressive carcinomas. Based on recent studies, CLDN4 does more than seal the paracellular space. It translocates to the nucleus and associates with the cell-cycle machinery to support genome stability. Furthermore, CLDN4 acts as a potent regulator of barrier tightness by inducing interclaudin interference, where it disrupts the pore-forming channels of CLDN2/15, thereby selectively reducing cation flux [8,119]. High CLDN4 levels allow cancer cells to survive the rapid proliferation required for metastatic colonization. Therefore, CLDN4 serves as a survival biomarker. High CLDN4 expression levels in pancreatic and ovarian tissues correlate with resistance to DNA-damaging agents and a higher recurrence rate.
7.1.3. Circulating TJ Fragments
Advancements in biofluid analysis have enabled the detection of circulating TJ components. When CTCs or metabolic insults compromise the BBB or lung endothelial barriers, specific fragments of Occludin, ZO-1, and CLDN5 are shed. Notably, the upregulation of endothelial BACE1 has been identified as a key driver of Occludin degradation, leading to cerebral small vessel injury [8,24,32,120]. These fragments, often regulated by miR-155 or miR-130a signaling, act as early warning signals. Detecting these proteins via high-sensitivity assays allows clinicians to predict barrier failure and subsequent brain colonization before secondary lesions are detectable by MRI.
7.2. Disrupting the Signaling Signalosome: The SFK-NR Axis as a Therapeutic Frontier
Traditional therapeutic strategies targeting TJs have primarily focused on monoclonal antibodies (such as Zolbetuximab) or ADCs designed to eliminate cells expressing specific claudins. However, recent discoveries of the claudin-mediated Claudin-SFK-NR signaling axis reveal a new opportunity: to therapeutically interfere with the “adhesion-dependent” signalosome that drives tumor malignancy, potentially without fully dismantling junctional structures [8,15].
7.2.1. Targeting the Transmembrane-to-Nuclear Relay
The TJ signalosome functions as a high-fidelity conduit where claudin-mediated adhesion activates the SFKs by recruiting them to the plasma membrane. This recruitment actively promotes the non-canonical serine phosphorylation of NRs, including estrogen receptors (ERs) and retinoic acid receptors (RARs), linking membrane events directly to transcriptional regulation. In precision oncology, this axis represents a targetable vulnerability. Small-molecule SFK inhibitors (such as Dasatinib or Saracatinib) can be repurposed to specifically intercept this membrane-to-nucleus relay, thereby suppressing pro-stemness transcriptional programs driven by SOX2 and OCT4 that are aberrantly activated by dysregulated TJ signaling [8,15,24,32].
7.2.2. Contextualizing Potential Roles in Endocrine and Retinoid Resistance
A speculative but intriguing clinical implication of the proposed TJ-NR axis lies in its hypothesized role in therapy resistance. The conceptual framework presented by Sugimoto et al. suggests a model wherein claudin-associated states might promote NR phosphorylation, potentially sustaining transcriptional activity in a ligand-independent manner [8,24]. While this perspective offers a novel hypothetical insight, suggesting that tumors overexpressing certain claudins might theoretically exploit non-canonical signaling to evade standard endocrine or retinoid therapies, robust empirical validation across independent, mechanistic studies remains indispensable to substantiate this link.
Should this pathway be rigorously validated in future functional assays, patients exhibiting elevated junctional SFK activity or specific NR serine phosphorylation profiles might eventually be considered for tailored combination strategies. Conceptually, synergistic intervention, such as pairing exploratory TJ-modulating agents with established SFK inhibitors, is hypothesized to help restore treatment sensitivity or restrict phenotypic de-differentiation [8,24]. However, given the pleiotropic and essential homeostatic functions of both TJs and Src family kinases, translating this preliminary model into safe, stratified clinical therapies demands cautious, multi-model empirical verification.
7.2.3. Beyond Scaffolding: Pharmacological Dissolution of Condensates
Scaffolding proteins such as ZO-1 and PATJ facilitate TJ-mediated signaling via LLPS, a biophysical process forming concentrated signaling hubs at the apical-lateral interface. Targeting these hubs can involve interfering with the co-condensation of ZO-1 and adhesion receptors or disrupting the PATJ-mediated prewetting transition that promotes the continuous growth of the TJ belt [4].
This approach may neutralize tumor adaptive plasticity at its biophysical source, providing a novel avenue for therapeutic intervention beyond classical kinase inhibition by targeting the assembly of the mechanical signaling platform itself.
7.3. Evolution of Targeting Strategies: A Three-Generation Framework
Understanding these biomarker-driven insights provides a foundation for designing therapeutic interventions targeting TJ-mediated signaling. The pharmacological targeting of TJs has evolved from simple blockade to complex immune-recruitment and signaling disruption, transitioning from targeting the protein's presence to its dynamic assembly state.
7.3.1. First Generation: Naked Monoclonal Antibodies (mAbs)
The first generation utilized the unique expression profiles of CLDNs. In healthy tissues, TJ proteins are often buried within the junctional complex and are inaccessible to circulating antibodies. In cancer, the loss of cell polarity exposes these proteins to the interstitial space. Zolbetuximab (IMAB362) represents the gold standard for this generation. Targeting CLDN18.2 induces antibody-dependent cellular cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC) specifically in gastric and pancreatic tumor cells. Its clinical success stems from the high tumor-to-normal exposure ratio of the CLDN18.2 epitope and the high density of TJ protein clusters on the apical membrane of malignant cells [121].
7.3.2. Second Generation: Payload Delivery and Immune Recruitment
The second generation increases potency by adding functional groups to the antibody backbone: Antibody-Drug Conjugates (ADCs): Drugs like those targeting CLDN6 or CLDN18.2 utilize the internalizing nature of TJ proteins. Once the ADC binds to the extracellular loop, the entire complex is endocytosed via the constitutive TJ recycling pathway. Inside the lysosome, the payload (such as MMAE or DM1) is released to induce mitotic arrest.
CAR-T Therapy: Chimeric Antigen Receptor T-cells are engineered to recognize specific TJ motifs. The CLDN6-CAR-T (such as BNT211) has shown a remarkable ability to infiltrate solid tumors. Because CLDN6 is an oncofetal protein (absent in healthy adult tissues), these cells exhibit minimal off-target toxicity while maintaining high specificity for the claudin-rich tumor architecture [122].
7.3.3. Third Generation: Signaling Hub and Phase Separation Inhibitors
The third generation targets the multivalent signalosome associated with TJs. Research on ZO-1 surface condensation and membrane prewetting by polarity proteins such as PATJ has opened the door to a new class of condensate disruptors. These small molecules interfere with the multivalent interactions between PDZ domains or the C-terminal tails of claudins. Instead of just killing the cell, these inhibitors dissolve the liquid-like signaling platforms that activate Wnt, STAT3, and RhoA [4,47,92]. By disrupting the biophysical-signaling logic of the cell and modulating the mechanical resistance of the AJC, these drugs can reverse the EMT process and re-sensitize metastatic cells to the host’s immune system.
7.4. Overcoming Resistance and Enhancing Tissue Penetration
A significant barrier to chemotherapy is the physical resistance of the tumor mass. TJs within the tumor core prevent the deep penetration of large-molecule drugs by creating a high-pressure paracellular seal.
7.4.1. Pharmacological Gate Opening
Researchers are developing transient TJ modulators, including C-CPE or claudin-mimetic peptides. These agents temporarily reverse the “interclaudin interference” to destabilize higher-order meshworks, opening the gates of the tumor. Administering these modulators shortly before chemotherapy increases the intratumoral concentration of drugs such as paclitaxel or doxorubicin by over 400%, effectively overcoming physical drug resistance by converting the paracellular leak pathway into an open conduit [8,15].
7.4.2. The AXIN-AMPK Axis and Barrier Normalization
While opening the tumor is important, protecting the host is equally critical. Recent data indicate that Mannose can be used as a metabolic modulator of TJs. Mannose triggers the AXIN-AMPK pathway, which promotes the precise assembly of ZO-1 and Occludin. In the context of immunotherapy, this has a normalizing effect. It strengthens leaky blood vessels in the tumor to allow T cells to enter more efficiently, while simultaneously reinforcing the gut and lung barriers via STAT3-mediated signaling. This prevents the systemic leaky-barrier induced translocation of microbial products and inflammatory cytokines, reducing the side effects of immune-checkpoint inhibitors and enhancing the efficacy of the neurovascular barrier against pathological insults [4,47,92].
7.5. Navigating the Bottlenecks: PK/PD Challenges, Toxicity, and Druggability Hurdles
Despite the therapeutic allure of modulating the TJ signalosome, translating these concepts into clinical practice presents formidable pharmacological bottlenecks. A critical evaluation of historical attempts reveals a narrow therapeutic window, primarily owing to the dual role of TJs in maintaining systemic physiological homeostasis.
First, targeting intracellular scaffolding proteins such as ZO-1 or its transient LLPS represents a classic undruggable paradigm. Unlike cell-surface claudins, ZO-1 possesses vast, flat protein-protein interaction domains without defined hydrophobic pockets suitable for small-molecule binding. Furthermore, executing in vivo ablation of liquid condensates remains highly experimental. Because LLPS of junctional plaques relies on transient, low-affinity multivalent interactions that dynamically overlap with generalized cellular macromolecular crowding, any small molecule attempting to dissolve TJ selectively condensates risks causing extensive off-target disruption to other vital biomolecular condensates in normal cells.
Second, severe pharmacokinetic/pharmacodynamic (PK/PD) liabilities and systemic toxicity pose significant barriers. Systemic delivery of non-selective TJ-disrupting peptides or antibodies has historically faced failure due to generalized epithelial hyperpermeability. The systemic disassembly of tight junctions in healthy tissues inevitably disrupts the BBB, alveolar-capillary barriers, and intestinal epithelial sheets, leading to severe neurotoxicity, pulmonary edema, and intractable diarrhea. Consequently, previous clinical or preclinical candidates designed to enhance drug delivery by transiently breaking down junctions often failed due to the inability to restrict structural disruption exclusively to the tumor microenvironment.
To overcome these severe translation hurdles, future efforts must pivot away from global junctional disruption. Instead, strategic focus must be placed on highly context-dependent targets, such as oncofetal claudin isoforms (CLDN18.2 and CLDN6) whose physiological expression is heavily restricted, or the deployment of tumor-cleavable antibody-drug conjugates (ADCs) and conditional prodrugs that selectively activate only within the acidic, protease-rich tumor milieu (Table 1).
In summary, the transition of TJ research into clinical application marks a paradigm shift from viewing these structures as static barriers to recognizing them as dynamic signalosomes. Emerging predictive biomarkers, such as the expression profiles of CLDN1 and CLDN4 and circulating fragments of Occludin, provide high-resolution insights into metastatic risk and barrier integrity. The discovery of the CLDN-SFK-NR axis and the biophysical principles of LLPS involving ZO-1 and PATJ have paved the way for third-generation therapeutics. These novel strategies aim to disrupt the mechanical and signaling hubs of cancer cells or metabolically normalize host barriers through the AXIN-AMPK and STAT3 pathways. Ultimately, these advancements offer a sophisticated framework for precision oncology and the management of neurovascular and mucosal diseases.
Table 1. TJs—From Biomarkers to Therapies.
|
Drug Code/Name |
Target |
Drug Class |
Clinical Indication/Status |
Molecular Mechanism |
|---|---|---|---|---|
|
Zolbetuximab [123] |
CLDN18.2 |
Monoclonal Ab (1st Gen) |
Gastric/Pancreatic Cancer (Phase III) |
Induces ADCC/CDC via epitope binding |
|
BNT211 [122] |
CLDN6 |
CAR-T (2nd Gen) |
Ovarian/Testicular Cancer (Phase I/II) |
T-cell mediated lysis of CLDN6+ cells |
|
SYSA1801 [124] |
CLDN18.2 |
ADC (2nd Gen) |
Solid Tumors (Phase I) |
Endocytosis followed by toxin release |
|
C-CPE Derivatives [125] |
CLDN3/CLDN4 |
Peptide Modulator |
Advanced Carcinomas (Pre-clinical) |
Binds ECL2 to transiently open the paracellular gate |
|
AMG 910 [126] |
CLDN18.2 |
Bispecific T-cell Engager |
Gastric Cancer (Phase I) |
Recruits CD3+ T-cells to CLDN18.2+ cells |
|
Mannose (Metabolic) [127] |
AXIN-AMPK |
TJ Stabilizer (3rd Gen) |
Colitis/TME Normalization (Research) |
Enhances ZO-1/Occludin membrane docking |
|
PDZ-Inhibitors [128] |
ZO-1/CLDN C-term |
Small Molecule (3rd Gen) |
Metastatic Invasion (Pre-clinical) |
Disrupts LLPS-mediated signaling hubs |
8. Challenges and Future Perspectives
TJs are fundamental in regulating cell–cell adhesion and maintaining tissue integrity. Their dysregulation contributes to cancer progression and metastasis. Recent studies show that TJ proteins not only form physical barriers but also engage in signaling that influences tumor behavior. However, translating this knowledge into mechanistic understanding and clinical practice faces major challenges. Key hurdles include the spatiotemporal heterogeneity of metastatic processes and the complexity of multi‑omic data integration required to map junctional dynamics and signaling networks at high resolution. Addressing these issues will deepen our understanding of metastasis and guide future therapeutic strategies.
8.1. Navigating Spatiotemporal Heterogeneity in Metastasis
Metastasis is a highly dynamic process. TJ proteins change their expression and localization constantly. This variability is known as spatiotemporal heterogeneity. It makes TJs difficult to study and even harder to target.
8.1.1. Precision Mapping of Tight Junction Dynamics Across Metastatic Stages
Tumor cells adjust their junctional profiles at every step of the metastatic cascade. During the early stage, cells often downregulate TJs to undergo EMT. This loss of adhesion allows cells to leave the primary tumor. Recent studies on Claudins and ZO-1 confirm this trend. However, the story changes when cells enter the bloodstream. CTCs frequently form clusters. These clusters rely on the re-expression of certain TJs for survival. TJs protect these clusters from mechanical shear stress in the blood. They also help cells resist death. When cells reach a distant organ, they must re-establish TJs to colonize the new tissue. This is part of MET. Mapping these changes with high precision is essential. We need to know which proteins are active at each specific moment. Current research shows that a single Claudin can act as a tumor suppressor in one stage but as a promoter in another.
8.1.2. Methodological Hurdles in Capturing Transient Junctional States
Most clinical samples provide only a snapshot of the tumor. They do not show the fluid nature of junctional remodeling. TJs are not just present or absent. They move between the cell membrane, the cytoplasm, and even the nucleus. Internalization of TJ proteins is a key transient event. Proteins like Occludin can be endocytosed during growth factor signaling. Traditional immunohistochemistry often fails to distinguish between functional membrane TJs and inactive cytoplasmic pools. Furthermore, the assembly of TJ strands is a rapid process. We lack the tools to watch these strands form in live human tumors. Animal models help, but they do not always reflect human metastatic biology. We need better live-imaging techniques to track these transient states in real time.
8.2. Multi-Omic Integration of the Junctional Interactome
To solve these problems, we must look at the junctional interactome. This refers to the entire network of proteins and genes connected to TJs. Multi-omic technologies offer a way to see this big picture.
8.2.1. Spatial Transcriptomics and Single-Cell Proteomic Profiling
High-resolution spatial transcriptomics and single-cell proteomic profiling are redefining our understanding of TJ heterogeneity within the TME. Spatial technologies now map the distinct TJ expression gradients between the hypoxic tumor core and the invasive leading edge, revealing how localized biomechanical and metabolic stressors drive junctional remodeling during metastasis.
Complementarily, single-cell proteomics bypasses the limitations of bulk sequencing by resolving the critical post-translational modifications (PTMs), such as site-specific phosphorylation of Claudins and Occludin, that govern barrier stability and scaffold-mediated signaling. These granular datasets identify rare “metastasis-initiating” sub-populations characterized by specific junctional signatures and phase-separated ZO-1 condensates. Integrating spatial architecture with single-cell protein network dynamics enables the discovery of high-fidelity biomarkers that reflect the functional, rather than merely numerical, state of the epithelial barrier.
8.2.2. System-Level Modeling of Tight Junction Signaling Networks
Modern oncology is transitioning from analyzing individual proteins to mapping TJs signaling networks. TJs function as integrated input-output hubs that transduce extracellular mechanical stress into intracellular biochemical cues. These structures are cross-linked with core pathways, including Hippo-YAP, Wnt/β-catenin, and the AXIN-AMPK axis, creating complex regulatory feedback loops.
Mathematical and systems biology modeling is essential to predict emergent behaviors in the TME. For instance, such models can simulate how the junctional network compensates for the targeted inhibition of CLDN18.2 or CLDN4. By identifying bottleneck proteins, which are nodes that simultaneously control multiple oncogenic pathways, researchers can develop multi-target strategies more effective than single-claudin blockade. Integrating spatial transcriptomics with system-level modeling allows for the decoding of the “junctional interactome”. This approach shifts the paradigm of TJs from static barriers to dynamic clinical targets, providing a foundation for neutralizing metastatic evolution through precise network interference.
While the reconceptualization of TJs from static epithelial seals to dynamic signaling hubs has substantially expanded their relevance in cancer biology, future progress will depend on moving beyond descriptive network mapping toward rigorous mechanistic and causal validation. Although system-level modeling provides powerful tools for predicting junctional behaviors within the TME, these models remain constrained by an incomplete understanding of the hierarchical interplay between TJs and AJs. Future investigations must therefore distinguish signaling events that are directly driven by TJ-specific mechanisms from those that arise secondarily from broader disruption of epithelial cohesion and polarity. Resolving these mechanistic ambiguities will be essential for refining computational models, improving target prioritization, and ultimately translating junctional network biology into clinically effective therapeutic strategies against metastatic progression.
9. Conclusions
TJs have shifted from being viewed as static paracellular barriers to dynamic master regulators of the metastatic cascade. Beyond promoting EMT through barrier disruption, TJs are essential for the survival of circulating tumor cell clusters, which depend on TJ-mediated mechanical integrity, coordinated by proteins such as ZO-1 and JAM-A, to resist hemodynamic shear stress and thereby gain enhanced metastatic competence. Alterations in transmembrane components, including loss of mucins such as MUC13 or dysregulation of Claudin-7 expression, contribute to organotropic colonization by enabling adaptation to specific metastatic niches. Furthermore, the spatiotemporal redistribution of TJ components from apical-lateral junctions to cytoplasmic or nuclear compartments influences tumor evolution from local detachment to distant metastatic seeding. Beyond structural roles, TJs function as mechanosensitive signaling hubs that integrate extracellular mechanical forces with intracellular pathways such as Wnt/β-catenin, Hippo–YAP, and STAT3. Emerging evidence suggests Claudin-mediated signaling interfaces with Src-family kinases to regulate nuclear receptor phosphorylation, directly linking cell adhesion to transcriptional control. Scaffold proteins like ZO-1 exhibit phase-separation behavior that organizes actin dynamics and enables mechanotransduction. Additionally, interclaudin regulatory mechanisms, in which pore-forming and barrier-forming claudins reciprocally modulate endocytosis, reshape ionic homeostasis and the TME. Collectively, these multilayered functions redefine TJs as active signal-processing and mechanobiological units, and their dysregulation provides a framework for advanced metastatic behavior and potential precision therapeutic targeting.
Statement of the Use of Generative AI and AI-Assisted Technologies in the Writing Process
During the preparation of this manuscript, the authors used AI-assisted language editing tools in order to improve language clarity and readability. After using these tools, the authors carefully reviewed, edited, and validated the content, and take full responsibility for the content of the published article.
Author Contributions
Conceptualization, J.J., T.A.M. and W.J.; Methodology, J.J., T.A.M. and W.J.; Software, J.J., T.A.M. and W.J.; Validation, J.J., T.A.M. and W.J.; Formal Analysis, J.J., T.A.M. and W.J.; Investigation, J.J., T.A.M. and W.J.; Resources, J.J., T.A.M. and W.J.; Data Curation, J.J., T.A.M. and W.J.; Writing—Original Draft Preparation, J.J., T.A.M. and W.J.; Writing—Review & Editing, J.J., T.A.M. and W.J.; Visualization, J.J., T.A.M. and W.J.; Supervision, J.J., T.A.M. and W.J.; Project Administration, J.J., T.A.M. and W.J.
Ethics Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Funding
This research received no external funding.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Anderson JM, Van Itallie CM. Physiology and function of the tight junction. Cold Spring Harb. Perspect. Biol. 2009, 1, a002584. DOI:10.1101/cshperspect.a002584 [Google Scholar]
- Tsukita S, Furuse M, Itoh M. Multifunctional strands in tight junctions. Nat. Rev. Mol. Cell Biol. 2001, 2, 285–293. DOI:10.1038/35067088 [Google Scholar]
- Beutel O, Maraspini R, Pombo-García K, Martin-Lemaitre C, Honigmann A. Phase Separation of Zonula Occludens Proteins Drives Formation of Tight Junctions. Cell 2019, 179, 923–936.e11. DOI:10.1016/j.cell.2019.10.011 [Google Scholar]
- Shin K, Straight S, Margolis B. PATJ regulates tight junction formation and polarity in mammalian epithelial cells. J. Cell Biol. 2005, 168, 705–711. DOI:10.1083/jcb.200408064 [Google Scholar]
- Zihni C, Mills C, Matter K, Balda MS. Tight junctions: from simple barriers to multifunctional molecular gates. Nat. Rev. Mol. Cell Biol. 2016, 17, 564–580. DOI:10.1038/nrm.2016.80 [Google Scholar]
- Citi S. The mechanobiology of tight junctions. Biophys. Rev. 2019, 11, 783–795. DOI:10.1007/s12551-019-00582-7 [Google Scholar]
- Stephenson RO, Rossant J, Tam PP. Intercellular interactions, position, and polarity in establishing blastocyst cell lineages and embryonic axes. Cold Spring Harb. Perspect. Biol. 2012, 4, a008235. DOI:10.1101/cshperspect.a008235 [Google Scholar]
- Sugimoto K, Chiba H. A Novel Tight Junction-Nuclear Receptor Signaling Pathway Regulating Cancer Progression. Pathol. Int. 2025, 75, 443–450. DOI:10.1111/pin.70040 [Google Scholar]
- Arumugam P, Saha K, Nighot P. Intestinal Epithelial Tight Junction Barrier Regulation by Novel Pathways. Inflamm. Bowel Dis. 2025, 31, 259–271. DOI:10.1093/ibd/izae232 [Google Scholar]
- Martin TA, Jiang WG. Tight junctions and their role in cancer metastasis. Histol. Histopathol. 2001, 16, 1183–1195. Available online: https://digitum.um.es/items/a37dcd3a-0cc9-4165-be46-7cee1155687d (accessed on 18 September 2020).
- Yuan S, Norgard RJ, Stanger BZ. Cellular Plasticity in Cancer. Cancer Discov. 2019, 9, 837–851. DOI:10.1158/2159-8290.CD-19-0015 [Google Scholar]
- Bhat AA, Uppada S, Achkar IW, Hashem S, Yadav SK, Shanmugakonar M, et al. Tight Junction Proteins and Signaling Pathways in Cancer and Inflammation: A Functional Crosstalk. Front. Physiol. 2019, 9, 1942. DOI:10.3389/fphys.2018.01942 [Google Scholar]
- Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. DOI:10.1038/nrm3758 [Google Scholar]
- Kyuno D, Takasawa A, Kikuchi S, Takemasa I, Osanai M, Kojima T. Role of tight junctions in the epithelial-to-mesenchymal transition of cancer cells. Biochim. Biophys. Acta. 2021, 1868, 183503. DOI:10.1016/j.bbamem.2020.183503 [Google Scholar]
- Shashikanth N, France MM, Xiao R, Haest X, Rizzo HE, Yeste J, et al. Tight junction channel regulation by interclaudin interference. Nat. Commun. 2022, 13, 3780. DOI:10.1038/s41467-022-31587-8 [Google Scholar]
- Bakir B, Chiarella AM, Pitarresi JR, Rustgi AK. EMT, MET, Plasticity, and Tumor Metastasis. Trends Cell Biol. 2020, 30, 764–776. DOI:10.1016/j.tcb.2020.07.003 [Google Scholar]
- Wang K, Liu Y, Li H, Liang X, Hao M, Yuan D, et al. Claudin-7 is essential for the maintenance of colonic stem cell homoeostasis via the modulation of Wnt/Notch signalling. Cell Death Dis. 2024, 15, 284. DOI:10.1038/s41419-024-06658-x [Google Scholar]
- Hartsock A, Nelson WJ. Adherens and tight junctions: Structure, function and connections to the actin cytoskeleton. Biochim. Biophys. Acta. 2008, 1778, 660–669. DOI:10.1016/j.bbamem.2007.07.012 [Google Scholar]
- Nguyen TP, Otani T, Tsutsumi M, Kinoshita N, Fujiwara S, Nemoto T, et al. Tight junction membrane proteins regulate the mechanical resistance of the apical junctional complex. J. Cell Biol. 2024, 223, e202307104. DOI:10.1083/jcb.202307104 [Google Scholar]
- Balda MS, Garrett MD, Matter K. The ZO-1-associated Y-box factor ZONAB regulates epithelial cell proliferation and cell density. J. Cell Biol. 2003, 160, 423–432. DOI:10.1083/jcb.200210020 [Google Scholar]
- Balda MS, Matter K. The tight junction protein ZO-1 and an interacting transcription factor regulate ErbB-2 expression. EMBO J. 2000, 19, 2024–2033. DOI:10.1093/emboj/19.9.2024 [Google Scholar]
- Otani T, Furuse M. Tight junction structure and function revisited. Trends Cell Biol. 2020, 30, 805–817. DOI:10.1016/j.tcb.2020.08.004 [Google Scholar]
- Schwayer C, Shamipour S, Pranjic-Ferscha K, Schauer A, Balda M, Tada M, et al. Mechanosensation of Tight Junctions Depends on ZO-1 Phase Separation and Flow. Cell 2019, 179, 937–952.e18. DOI:10.1016/j.cell.2019.10.006 [Google Scholar]
- Nitta Y, Urayama S, Kawashima M, Nakao H, Ikeda T, Higashiyama K, et al. Context-dependent interactions among afadin, ZO-1, and actin filaments. Cell Struct. Funct. 2025, 50, 223–238. DOI:10.1247/csf.25019 [Google Scholar]
- Suzuki H, Nishizawa T, Tani K, Yamazaki Y, Tamura A, Ishitani R, et al. Crystal structure of a claudin provides insight into the architecture of tight junctions. Science 2014, 344, 304–307. DOI:10.1126/science.1248571 [Google Scholar]
- Samanta P, Wang Y, Fuladi S, Zou J, Li Y, Shen L, et al. Molecular determination of claudin-15 organization and channel selectivity. J. Gen. Physiol. 2018, 150, 949–968. DOI:10.1085/jgp.201711868 [Google Scholar]
- Tabariès S, Siegel PM. The role of claudins in cancer metastasis. Oncogene 2017, 36, 1176–1190. DOI:10.1038/onc.2016.289 [Google Scholar]
- Rodgers LS, Beam MT, Anderson JM, Fanning AS. Epithelial barrier assembly requires coordinated activity of multiple domains of the tight junction protein ZO-1. J. Cell Sci. 2013, 126, 1565–1575. DOI:10.1242/jcs.113399 [Google Scholar]
- Van Itallie CM, Anderson JM. Claudins and epithelial paracellular transport. Annu. Rev. Physiol. 2006, 68, 403–429. DOI:10.1146/annurev.physiol.68.040104 [Google Scholar]
- Segui-Perez C, Stapels DAC, Ma Z, Su J, Passchier E, Westendorp B, et al. MUC13 negatively regulates tight junction proteins and intestinal epithelial barrier integrity via protein kinase C. J. Cell Sci. 2024, 137, jcs261468. DOI:10.1242/jcs.261468 [Google Scholar]
- Clarke H, Marano CW, Peralta Soler A, Mullin JM. Modification of tight junction function by protein kinase C isoforms. Adv. Drug Deliv. Rev. 2000, 41, 283–301. DOI:10.1016/s0169-409x(00)00047-8 [Google Scholar]
- Park JH, Choi JY, Kim GY, Kim M, Park SH, Koh YH. Endothelial BACE1, regulating the endothelial-to-mesenchymal transition (EndMT) induced by particulate matter (PM), is associated with cerebrovascular disease: Involvement of endothelial BACE1 in BBB disruption caused by diabetes. Free Radic. Biol. Med. 2026, 243, 96–110. DOI:10.1016/j.freeradbiomed.2025.11.021 [Google Scholar]
- Itoh M, Furuse M, Morita K, Kubota K, Saitou M, Tsukita S. Direct binding of three tight junction-associated MAGUKs, ZO-1, ZO-2, and ZO-3, to the COOH termini of claudins. J. Cell Biol. 1999, 147, 1351–1363. DOI:10.1083/jcb.147.6.1351 [Google Scholar]
- Brooks JW, Parton RG, Yap AS, Duszyc K. Epithelial mechanosensing at cell-cell contacts and tight junctions. In Tight Junctions; Springer International Publishing: Cham, Switzerland, 2022; pp. 27–50. DOI:10.1007/978-3-030-97204-2_3 [Google Scholar]
- Haas AJ, Zihni C, Krug SM, Maraspini R, Otani T, Furuse M, et al. ZO-1 Guides Tight Junction Assembly and Epithelial Morphogenesis via Cytoskeletal Tension-Dependent and -Independent Functions. Cells 2022, 11, 3775. DOI:10.3390/cells11233775 [Google Scholar]
- Hagen SJ. Non-canonical functions of claudin proteins: Beyond the regulation of cell-cell adhesions. Tissue Barriers 2017, 5, e1327839. DOI:10.1080/21688370.2017.1327839 [Google Scholar]
- Balda MS, Matter K. Tight junctions and the regulation of gene expression. Biochim. Biophys. Acta. 2009, 1788, 761–767. DOI:10.1016/j.bbamem.2008 [Google Scholar]
- Liu G, Fang Y, Zhang Y, Zhu M. Dihydroquercetin improves the proliferation of porcine intestinal epithelial cells via the Wnt/β-catenin pathway. Biochem. Biophys. Res. Commun. 2024, 734, 150460. DOI:10.1016/j.bbrc.2024.150460 [Google Scholar]
- Kim NY, Pyo JS, Kang DW, Yoo SM. Loss of claudin-1 expression induces epithelial-mesenchymal transition through nuclear factor-kappaB activation in colorectal cancer. Pathol. Res. Pract. 2019, 215, 580–585. DOI:10.1016/j.prp.2019.01.015 [Google Scholar]
- Liu H, Wang S, Gong L, Shen Y, Xu F, Wang Y, et al. SIRT6 ameliorates LPS-induced apoptosis and tight junction injury in ARDS through the ERK1/2 pathway and autophagy. Int. J. Med. Sci. 2023, 20, 581–594. DOI:10.7150/ijms.80920 [Google Scholar]
- Martin TA. The role of tight junctions in cancer metastasis. In Seminars in Cell & Developmental Biology; Academic Press: Cambridge, MA, USA, 2014; Volume 36, pp. 224–231. DOI:10.1016/j.semcdb.2014.09.008 [Google Scholar]
- Hyman AA, Weber CA, Jülicher F. Liquid-liquid phase separation in biology. Annu. Rev. Cell Dev. Biol. 2014, 30, 39–58. DOI:10.1146/annurev-cellbio-100913-013325 [Google Scholar]
- Banani SF, Lee HO, Hyman AA, Rosen MK. Biomolecular condensates: organizers of cellular biochemistry. Nat. Rev. Mol. Cell Biol. 2017, 18, 285–298. DOI:10.1038/nrm.2017.7 [Google Scholar]
- Case LB, Zhang X, Ditlev JA, Rosen MK. Stoichiometry controls activity of phase-separated clusters of actin promoters. Science 2019, 363, 1093–1097. DOI:10.1126/science.aau6313 [Google Scholar]
- Sun D, Zhao X, Wiegand T, Martin-Lemaitre C, Borianne T, Kleinschmidt L, et al. Assembly of tight junction belts by ZO1 surface condensation and local actin polymerization. Dev. Cell. 2025, 60, 1234–1250.e6. DOI:10.1016/j.devcel.2024 [Google Scholar]
- Xie CC, Wang T, Liu XR, Wang Y, Dang Q, Ding T, et al. Liquid-Liquid Phase Separation in Major Hallmarks of Cancer. Cell Prolif. 2026, 59, e70122. DOI:10.1111/cpr.70122 [Google Scholar]
- Varadarajan S, Chumki SA, Stephenson RE, Misterovich ER, Wu JL, Dudley CE, et al. Mechanosensitive calcium flashes promote sustained RhoA activation during tight junction remodeling. J. Cell Biol. 2022, 221, e202105107. DOI:10.1083/jcb.202105107 [Google Scholar]
- Pérez-González A, Bévant K, Blanpain C. Cancer cell plasticity during tumor progression, metastasis and response to therapy. Nat. Cancer. 2023, 4, 1063–1082. DOI:10.1038/s43018-023-00595-y [Google Scholar]
- Lambert AW, Pattabiraman DR, Weinberg RA. Emerging biological principles of metastasis. Cell 2017, 168, 670–691. DOI:10.1016/j.cell.2016 [Google Scholar]
- Peinado H, Olmeda D, Cano A. Snail, Zeb and bHLH factors in tumour progression: An alliance against the epithelial phenotype? Nat. Rev. Cancer. 2007, 7, 415–428. DOI:10.1038/nrc2131 [Google Scholar]
- Cano A, Pérez-Moreno MA, Rodrigo I, Locascio A, Blanco MJ, del Barrio MG, et al. The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat. Cell Biol. 2000, 2, 76–83. DOI:10.1038/35000025 [Google Scholar]
- Awad K, Barmeyer C, Bojarski C, Nagel O, Lee IM, Schweiger MR, et al. Epithelial Barrier Dysfunction in Diarrhea-Predominant Irritable Bowel Syndrome (IBS-D) via Downregulation of Claudin-1. Cells 2023, 12, 2846. DOI:10.3390/cells12242846 [Google Scholar]
- Tornavaca O, Chia M, Dufton N, Almagro LO, Conway DE, Randi AM, et al. ZO-1 controls endothelial adherens junctions, cell-cell tension, and angiogenesis. J. Cell Biol. 2015, 208, 821–838. DOI:10.1083/jcb.201404140 [Google Scholar]
- Demirkan B. The Roles of Epithelial-to-Mesenchymal Transition (EMT) and Mesenchymal-to-Epithelial Transition (MET) in Breast Cancer Bone Metastasis: Potential Targets for Prevention and Treatment. J. Clin. Med. 2013, 2, 264–282. DOI:10.3390/jcm2040264 [Google Scholar]
- Chiba H, Osanai M, Murata M, Kojima T, Sawada N. Transmembrane proteins of tight junctions. Biochim Biophys Acta. 2008, 1778, 588–600. DOI:10.1016/j.bbamem.2007.08.017 [Google Scholar]
- Okui N, Kamata Y, Sagawa Y, Kuhara A, Hayashi K, Uwagawa T, et al. Claudin 7 as a possible novel molecular target for the treatment of pancreatic cancer. Pancreatology 2019, 19, 88–96. DOI:10.1016/j.pan.2018.10.009 [Google Scholar]
- Horowitz A, Chanez-Paredes SD, Haest X, Turner JR. Paracellular permeability and tight junction regulation in gut health and disease. Nat. Rev. Gastroenterol. Hepatol. 2023, 20, 417–432. DOI:10.1038/s41575-023-00766-3 [Google Scholar]
- Aceto N, Bardia A, Miyamoto DT, Donaldson MC, Wittner BS, Spencer JA, et al. Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell 2014, 158, 1110–1122. DOI:10.1016/j.cell.2014.07.013 [Google Scholar]
- Follain G, Osmani N, Azevedo AS, Allio G, Mercier L, Karreman MA, et al. Hemodynamic forces tune the arrest, adhesion, and extravasation of circulating tumor cells. Dev. Cell 2018, 45, 33–52.e12. DOI:10.1016/j.devcel.2018.02.015 [Google Scholar]
- Murphy KN, Brinkworth AJ. Manipulation of Focal Adhesion Signaling by Pathogenic Microbes. Int. J. Mol. Sci. 2021, 22, 1358. DOI:10.3390/ijms22031358 [Google Scholar]
- Batlle E, Clevers H. Cancer stem cells revisited. Nat. Med. 2017, 23, 1124–1134. DOI:10.1038/nm.4409 [Google Scholar]
- Nassar D, Blanpain C. Cancer stem cells: Basic concepts and therapeutic implications. Annu. Rev. Pathol. 2016, 11, 47–76. DOI:10.1146/annurev-pathol-012615-044438 [Google Scholar]
- Lu Z, Ding L, Lu Q, Chen YH. Claudins in intestines: Distribution and functional significance in health and diseases. Tissue Barriers 2013, 1, e24978. DOI:10.4161/tisb.24978 [Google Scholar]
- Yu FX, Zhao B, Guan KL. Hippo pathway in organ size control, tissue homeostasis, and cancer. Cell 2015, 163, 811–828. DOI:10.1016/j.cell.2015.10.044 [Google Scholar]
- Zhao B, Li L, Tumaneng K, Wang CY, Guan KL. A coordinated phosphorylation by Lats and CK1 determines YAP stability through SCF(beta-TRCP). Genes. Dev. 2010, 24, 72–85. DOI:10.1101/gad.1843810 [Google Scholar]
- Ma S, Meng Z, Chen R, Guan KL. The Hippo pathway: Biology and pathophysiology. Annu. Rev. Biochem. 2019, 88, 577–604. DOI:10.1146/annurev-biochem-013118-111829 [Google Scholar]
- Naser AN, Xing T, Tatum R, Lu Q, Boyer PJ, Chen Y. Colonic crypt stem cell functions are controlled by tight junction protein claudin-7 through Notch/Hippo signaling. Ann. N. Y Acad. Sci. 2024, 1535, 92–108. DOI:10.1111/nyas.15137 [Google Scholar]
- Xu C, Ding YH, Wang K, Hao M, Li H, Ding L. Claudin-7 deficiency promotes stemness properties in colorectal cancer through Sox9-mediated Wnt/beta-catenin signalling. J. Transl. Med. 2021, 19, 311. DOI:10.1186/s12967-021-02983-3 [Google Scholar]
- Lai X, Yan Y, Sun L, Lei Z, Yang Y. CLDN7: Epithelial gatekeeper from physiology to pathology-roles in cancer and epithelial-related diseases (Review). Int. J. Mol. Med. 2026, 57, 110. DOI:10.3892/ijmm.2026.5781 [Google Scholar]
- Lu Z, Ding L, Hong H, Hoggard J, Lu Q, Chen YH. Claudin-7 inhibits human lung cancer cell migration and invasion through ERK/MAPK signaling pathway. Exp. Cell Res. 2011, 317, 1935–1946. DOI:10.1016/j.yexcr.2011.05.019 [Google Scholar]
- Martin-Belmonte F, Perez-Moreno M. Epithelial cell polarity, stem cells and cancer. Nat. Rev. Cancer. 2011, 12, 23–38. DOI:10.1038/nrc3169 [Google Scholar]
- Bera S, Loeffler D. Cell polarity: Cell type-specific regulators, common pathways, and polarized vesicle transport. Leukemia 2025, 39, 1558–1570. DOI:10.1038/s41375-025-02601-x [Google Scholar]
- Xue B, Krishnamurthy K, Allred DC, Muthuswamy SK. Loss of Par3 promotes breast cancer metastasis by compromising cell-cell cohesion. Nat. Cell Biol. 2013, 15, 189–200. DOI:10.1038/ncb2663 [Google Scholar]
- Bulgakova NA, Knust E. The Crumbs complex: from epithelial-cell polarity to whole-organ architecture. J. Cell Sci. 2009, 122, 2587–2596. DOI:10.1242/jcs.023648 [Google Scholar]
- Pickup MW, Mouw JK, Weaver VM. The extracellular matrix modulates the hallmarks of cancer. EMBO Rep. 2014, 15, 1243–1253. DOI:10.15252/embr.201439246 [Google Scholar]
- Northcott JM, Dean IS, Mouw JK, Weaver VM. Feeling Stress: The Mechanics of Cancer Progression and Aggression. Front. Cell Dev. Biol. 2018, 6, 17. DOI:10.3389/fcell.2018.00017 [Google Scholar]
- Kessenbrock K, Plaks V, Werb Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell 2010, 141, 52–67. DOI:10.1016/j.cell.2010.03.015 [Google Scholar]
- Citi S, Guerrera D, Spadaro D, Shah J. Epithelial junctions and Rho family GTPases: The zonular signalosome. Small GTPases 2014, 5, e973760. DOI:10.4161/21541248.2014.973760 [Google Scholar]
- Oku N, Sasabe E, Ueta E, Yamamoto T, Osaki T. Tight junction protein claudin-1 enhances the invasive activity of oral squamous cell carcinoma cells by promoting cleavage of laminin-5 γ2 chain via MMP-2 and MT1-MMP. Cancer Res. 2006, 66, 5251–5257. DOI:10.1158/0008-5472.CAN-05-4478 [Google Scholar]
- Kim DH, Ewald AJ, Park J, Kshitiz, Kwak M, Gray RS, et al. Biomechanical interplay between anisotropic re-organization of cells and the surrounding matrix underlies transition to invasive cancer spread. Sci. Rep. 2018, 8, 14210. DOI:10.1038/s41598-018-32010-3 [Google Scholar]
- Sun B. The mechanics of fibrillar collagen extracellular matrix. Cell Rep. Phys. Sci. 2021, 2, 100515. DOI:10.1016/j.xcrp.2021.100515 [Google Scholar]
- Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, et al. Understanding the tumor immune microenvironment (TIME) for effective cancer immunotherapy. Nat. Med. 2018, 24, 541–550. DOI:10.1038/s41591-018-0014-x [Google Scholar]
- Bonaventura P, Shekarian T, Alcazer V, Valladeau-Guilemond J, Valsesia-Wittmann S, Amigorena S, et al. Cold Tumors: A Therapeutic Challenge for Immunotherapy. Front. Immunol. 2019, 10, 168. DOI:10.3389/fimmu.2019.00168 [Google Scholar]
- Salmon H, Franciszkiewicz K, Damotte D, Dieu-Nosjean MC, Validire P, Trautmann A, et al. Matrix architecture defines the preferential localization and migration of T cells into the stroma of human lung tumors. J. Clin. Investig. 2012, 122, 899–910. DOI:10.1172/JCI45817 [Google Scholar]
- Shang X, Lin X, Alvarez E, Manorek G, Howell SB. Tight junction proteins claudin-3 and claudin-4 control tumor growth and metastases. Neoplasia 2012, 14, 974–985. DOI:10.1593/neo.12942 [Google Scholar]
- Ebnet K, Aurrand-Lions M. Junctional adhesion molecule-C: A multifunctional mediator of cell adhesion. Cell Mol. Life Sci. 2025, 82, 312. DOI:10.1007/s00018-025-05829-z [Google Scholar]
- Klabukov I, Smirnova A, Yakimova A, Kabakov AE, Atiakshin D, Petrenko D, et al. Oncomatrix: Molecular Composition and Biomechanical Properties of the Extracellular Matrix in Human Tumors. J. Mol. Pathol. 2024, 5, 437–453. DOI:10.3390/jmp5040029 [Google Scholar]
- Schrope JH, Robertson TF, Sarris M, Huttenlocher A. Chemical and Mechanical Regulation of Leukocyte Migration. Cold Spring Harb. Perspect. Biol. 2026, 18, a041752. DOI:10.1101/cshperspect.a041752 [Google Scholar]
- Hu X, Ouyang W, Chen H, Liu Z, Lai Z, Yao H. Claudin-9 (CLDN9) promotes gastric cancer progression by enhancing the glycolysis pathway and facilitating PD-L1 lactylation to suppress CD8+ T cell anti-tumor immunity. Cancer Pathog. Ther. 2024, 3, 253–266. DOI:10.1016/j.cpt.2024.09.006 [Google Scholar]
- Lim SH, Kuwata T, An M, Hong JY, Kim ST, Matsubara Y, et al. Dynamic modulation of claudin18.2 expression and remodeling of the tumor microenvironment in gastric cancer during chemo-immunotherapy. J. Immunother. Cancer 2025, 13, e012683. DOI:10.1136/jitc-2025-012683 [Google Scholar]
- Yilmaz O, Afsar B, Ortiz A, Kanbay M. The role of endothelial glycocalyx in health and disease. Clin. Kidney J. 2019, 12, 611–619. DOI:10.1093/ckj/sfz042 [Google Scholar]
- Zhang D, Qi B, Peng Z, Huang X, Chen Y, Sun T, et al. Heparan sulfate acts in synergy with tight junction through STAT3 signaling to maintain the endothelial barrier and prevent lung injury development. Int. Immunopharmacol. 2025, 147, 113957. DOI:10.1016/j.intimp.2024.113957 [Google Scholar]
- Kumari S, Khan S, Gupta SC, Kashyap VK, Yallapu MM, Chauhan SC, et al. MUC13 contributes to rewiring of glucose metabolism in pancreatic cancer. Oncogenesis 2018, 7, 19. DOI:10.1038/s41389-018-0031-0 [Google Scholar]
- Peters JJ, Teng C, Peng K, Li X. Deciphering the Blood-Brain Barrier Paradox in Brain Metastasis Development and Therapy. Cancers 2025, 17, 298. DOI:10.3390/cancers17020298 [Google Scholar]
- Roth KS, Pabst R. The Functional Unit of the Lymphatic System: Towards Understanding the Importance of a Well-Rehearsed Interaction of Lymphatic Capillaries, Collecting Vessels, and Lymph Nodes. J. Vasc. Res. 2025, 62, 183–192. DOI:10.1159/000545084 [Google Scholar]
- Ji W, Zhuang X, Jiang WG, Martin TA. Tight junctional protein family, Claudins in cancer and cancer metastasis. Front. Oncol. 2025, 15, 1596460. DOI:10.3389/fonc.2025.1596460 [Google Scholar]
- Bangarh R, Saini RV, Saini AK, Singh T, Joshi H, Ramniwas S, et al. Dynamics of epithelial-mesenchymal plasticity driving cancer drug resistance. Cancer Pathog. Ther. 2024, 3, 120–128. DOI:10.1016/j.cpt.2024.07.002 [Google Scholar]
- Gawryś K, Bednarek R, Świątkowska M. The role of Junctional Adhesion Molecule-A (JAM-A) in breast cancer metastasis. Mol. Biol. Rep. 2026, 53, 488. DOI:10.1007/s11033-026-11675-4 [Google Scholar]
- Shaw P, Dey Bhowmik A, Gopinatha Pillai MS, Robbins N, Dwivedi SKD, Rao G. Anoikis resistance in Cancer: Mechanisms, therapeutic strategies, potential targets, and models for enhanced understanding. Cancer Lett. 2025, 624, 217750. DOI:10.1016/j.canlet.2025.217750 [Google Scholar]
- Amare YE, Vuerich R, Zacchigna S. VEGFR1 as a Target for Cardiovascular Gene Therapy. J. Cardiovasc. Transl. Res. 2025, 18, 1485–1502. DOI:10.1007/s12265-025-10672-5 [Google Scholar]
- Wang Y, Jia J, Wang F, Fang Y, Yang Y, Zhou Q, et al. Pre-metastatic niche: formation, characteristics and therapeutic implication. Signal Transduct. Target. Ther. 2024, 9, 236. DOI:10.1038/s41392-024-01937-7 [Google Scholar]
- Dunbar KJ, Efe G, Cunningham K, Esquea E, Navaridas R, Rustgi AK. Regulation of metastatic organotropism. Trends Cancer 2025, 11, 216–231. DOI:10.1016/j.trecan.2024.11.012 [Google Scholar]
- Wong FC, Rak J. The Role of Extracellular Vesicles in the Control of Vascular Checkpoints for Cancer Metastasis. Cancers 2025, 17, 1966. DOI:10.3390/cancers17121966 [Google Scholar]
- Zhou W, Fong MY, Min Y, Somlo G, Liu L, Palomares MR, et al. Cancer-secreted miR-105 destroys vascular endothelial barriers to promote metastasis. Cancer Cell. 2014, 25, 501–515. DOI:10.1016/j.ccr.2014.03.007 [Google Scholar]
- Chee YJ, Dalan R, Cheung C. The Interplay Between Immunity, Inflammation and Endothelial Dysfunction. Int. J. Mol. Sci. 2025, 26, 1708. DOI:10.3390/ijms26041708 [Google Scholar]
- Capaldo CT, Nusrat A. Cytokine regulation of tight junctions. Biochim. Biophys. Acta. 2009, 1788, 864–871. DOI:10.1016/j.bbamem.2008.08.027 [Google Scholar]
- Daneman R, Prat A. The blood-brain barrier. Cold Spring Harb. Perspect. Biol. 2015, 7, a020412. DOI:10.1101/cshperspect.a020412 [Google Scholar]
- Obermeier B, Daneman R, Ransohoff RM. Development, maintenance and disruption of the blood-brain barrier. Nat. Med. 2013, 19, 1584–1596. DOI:10.1038/nm.3407 [Google Scholar]
- Peng X, Liu X, Kim JY, Nguyen A, Leal J, Ghosh D. Brain-Penetrating Peptide Shuttles across the Blood-Brain Barrier and Extracellular-like Space. Bioconjugate Chem. 2023, 34, 2319–2336. DOI:10.1021/acs.bioconjchem.3c00446 [Google Scholar]
- Blanchette M, Bajc K, Gastfriend BD, Profaci CP, Ruderisch N, Dorrier CE, et al. Regional heterogeneity of the blood-brain barrier. Nat. Commun. 2025, 16, 7332. DOI:10.1038/s41467-025-61841-8 [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011, 144, 646–674. DOI:10.1016/j.cell.2011.02.013 [Google Scholar]
- Pavlova NN, Thompson CB. The emerging hallmarks of cancer metabolism. Cell Metab. 2016, 23, 27–47. DOI:10.1016/j.cmet.2015.12.006 [Google Scholar]
- Rodriguez-Boulan E, Macara IG. Organization and execution of the epithelial polarity programme. Nat. Rev. Mol. Cell Biol. 2014, 15, 225–242. DOI:10.1038/nrm3775 [Google Scholar]
- Wang Y, Sun Z, Zhao Z, Pang J, Chen J. Recent Progress in the Development of Glucose Transporter (GLUT) Inhibitors. J. Med. Chem. 2025, 68, 1033–1050. DOI:10.1021/acs.jmedchem.4c02717 [Google Scholar]
- Manning BD, Toker A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. DOI:10.1016/j.cell.2017.04.001 [Google Scholar]
- Wu X, Hepner K, Castelino-Prabhu S, Do D, Kaye MB, Yuan XJ, et al. Evidence for regulation of the PTEN tumor suppressor by a membrane-localized multi-PDZ domain containing scaffold protein MAGI-2. Proc. Natl. Acad. Sci. USA 2000, 97, 4233–4238. DOI:10.1073/pnas.97.8.4233 [Google Scholar]
- Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293. DOI:10.1016/j.cell.2012.03.017 [Google Scholar]
- Zhang CS, Li M, Ma T, Zong Y, Cui J, Feng JW, et al. Metformin Activates AMPK through the Lysosomal Pathway. Cell Metab. 2016, 24, 521–522. DOI:10.1016/j.cmet.2016.09.003 [Google Scholar]
- Therachiyil L, Bhat AA, Uddin S. Claudins in ovarian cancer: Emerging biomarkers and therapeutic targets. Tissue Barriers 2026, 14, 2539026. DOI:10.1080/21688370.2025.2539026 [Google Scholar]
- Osanai M, Takasawa A, Murata M, Sawada N. Claudins in cancer: Bench to bedside. Pflugers Arch. 2017, 469, 55–67. DOI:10.1007/s00424-016-1877-7 [Google Scholar]
- Shitara K, Lordick F, Bang YJ, Enzinger P, Ilson D, Shah MA, et al. Zolbetuximab plus mFOLFOX6 in patients with CLDN18.2-positive, HER2-negative, untreated, locally advanced unresectable or metastatic gastric or gastro-oesophageal junction adenocarcinoma (SPOTLIGHT). Lancet 2023, 401, 1655–1668. DOI:10.1016/S0140-6736(23)00620-7 [Google Scholar]
- Shah MA, Shitara K, Ajani JA, Bang YJ, Enzinger P, Ilson D, et al. Zolbetuximab plus CAPOX in CLDN18.2-positive gastric or gastroesophageal junction adenocarcinoma: The randomized, phase 3 GLOW trial. Nat. Med. 2023, 29, 2133–2141. DOI:10.1038/s41591-023-02465-7 [Google Scholar]
- Mackensen A, Haanen JBAG, Koenecke C, Alsdorf W, Wagner-Drouet E, Borchmann P, et al. CLDN6-specific CAR-T cells plus amplifying RNA vaccine in relapsed or refractory solid tumors: The phase 1 BNT211-01 trial. Nat. Med. 2023, 29, 2844–2853. DOI:10.1038/s41591-023-02612-0 [Google Scholar]
- Du H, Hao XF, Lin BW, Zhang Y, Rui L, Wang J, et al. Comparative analysis of the efficacy and safety of antibody-drug conjugates, radionuclide-drug conjugates and their combination targeting claudin 18.2 in gastric cancer treatment. Oncol. Rep. 2026, 55, 4. DOI:10.3892/or.2025.9009 [Google Scholar]
- Hosomi K, Shimoyama A, Hinenoya A, Hatanaka N, Noguchi T, Ebina H, et al. Endotoxin-Free Stx2B-C-CPE Vaccine and Its Optimized Adjuvant Regimen for Preventing Food Poisoning. Front. Biosci. 2023, 28, 15. DOI:10.31083/j.fbl2801015 [Google Scholar]
- Xu Y, Fu J, Henderson M, Lee F, Jurcak N, Henn A, et al. CLDN18.2 BiTE Engages Effector and Regulatory T Cells for Antitumor Immune Response in Preclinical Models of Pancreatic Cancer. Gastroenterology 2023, 165, 1219–1232. DOI:10.1053/j.gastro.2023.06.037 [Google Scholar]
- Liu W, Xie J, Jiang H, Zhou J, Lu X, Zuo D, et al. Mannose attenuates intestinal epithelial tight junction damage in experimental colitis mice by activating the AXIN-AMPK pathway. Int. Immunopharmacol. 2024, 127, 111319. DOI:10.1016/j.intimp.2023.111319 [Google Scholar]
- Hoffer L, Roche P, Morelli X. Rational Design of PDZ Domain Inhibitors: Discovery of Small Organic Compounds Targeting PDZ Domains. Methods Mol. Biol. 2021, 2256, 277–289. DOI:10.1007/978-1-0716-1166-1_16 [Google Scholar]