Next-Generation Immunotherapy Strategies Driven by Tumor Microenvironment Modulation

Next-Generation Immunotherapy Strategies Driven by Tumor Microenvironment Modulation
Xianfeng Hui
1,2,3,*
 Ge Gao
4
Shihuan Ding
1,3
Shuoxiang Gao
1
Jiaqi Tian
5
Shuochen Xu
1,2,3
Tiesuo Zhao
1,3
Hui Wang
1,2,*
Ge Gao
4
Shihuan Ding
1,3
Shuoxiang Gao
1
Jiaqi Tian
5
Shuochen Xu
1,2,3
Tiesuo Zhao
1,3
Hui Wang
1,2,*
Received: 13 February 2026 Revised: 28 April 2026 Accepted: 25 May 2026 Published: 01 June 2026

© 2026 The authors. This is an open access article under the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/).
1. Introduction
Over the past decade, cancer immunotherapy has transformed the treatment landscape of multiple malignancies [1]. Immune checkpoint inhibitors (ICIs), adoptive cell therapies, cancer vaccines, and bispecific antibodies have demonstrated the potential to induce durable clinical responses, underscoring the promise of harnessing antitumor immunity for long-term disease control [2]. Nevertheless, despite these advances, the majority of patients fail to achieve sustained benefit, and both primary and acquired resistance remain common across cancer types. These limitations indicate that effective immune activation alone is insufficient to guarantee durable therapeutic responses [3].
Increasing evidence suggests that the tumor microenvironment (TME) is a central determinant of immunotherapy efficacy [3,4]. The TME comprises a complex and dynamic network of tumor cells, immune infiltrates, stromal components, vascular structures, and soluble mediators [5]. Rather than acting as a passive background, the TME actively shapes immune cell trafficking, differentiation, metabolic fitness, and effector function [6,7]. Consequently, the outcome of immunotherapy is governed not only by the intrinsic potential of immune cells but also by the permissiveness of the surrounding microenvironment [8].
Early investigations into the TME primarily focused on identifying immunosuppressive cell populations and inhibitory pathways, such as regulatory T cells, tumor-associated macrophages, myeloid-derived suppressor cells, and immune checkpoint molecules [9]. While this reductionist approach yielded critical mechanistic insights and facilitated the clinical translation of ICIs, it has proven inadequate for explaining the heterogeneous and often transient responses observed in patients [10]. Notably, substantial immune infiltration does not always correlate with therapeutic success, and initial responses can be followed by rapid disease progression, indicating that static assessments of immune composition fail to capture the functional complexity of the TME [11,12].
More recent conceptual advances emphasize a functional, state-based view of the TME [3,13]. In this framework, the microenvironment is defined by how immune responses are spatially organized, metabolically supported, and dynamically regulated, rather than by cellular composition alone [3]. Barriers to effective antitumor immunity may include physical exclusion of immune cells from tumor nests, chronic antigen exposure leading to functional exhaustion, competition for nutrients and oxygen, and dysregulated inflammatory signaling that favors tumor growth [14,15]. These features frequently coexist, giving rise to distinct TME states that differentially influence sensitivity or resistance to immunotherapy.
Importantly, the TME is not static but evolves in response to therapeutic pressure [16]. Immunotherapies themselves can drive adaptive remodeling of the microenvironment, including the activation of compensatory immunosuppressive pathways, restructuring of stromal architecture, and rewiring of metabolic and cytokine networks [17]. Such dynamic changes contribute to acquired resistance and underscore the importance of considering temporal context when designing immunotherapeutic interventions. Focusing solely on baseline TME characteristics is therefore insufficient to predict long-term treatment outcomes [18].
These insights have catalyzed a shift toward TME-centered immunotherapy strategies [19]. Rather than relying exclusively on concurrent combination regimens aimed at amplifying immune activation, next-generation approaches prioritize the strategic modulation of TME states to establish conditions conducive to effective and durable immune responses [20]. This paradigm supports the use of sequential or adaptive treatment strategies, in which microenvironmental constraints are first alleviated to create a favorable immunological window, followed by targeted immune engagement [21]. Such approaches reflect an emerging view of immunotherapy as a process of immune ecosystem management rather than isolated target inhibition.
In this review, we integrate current understanding of functional TME states and their dynamic remodeling to highlight how microenvironmental modulation can drive the development of next-generation immunotherapy strategies. By moving beyond static classification schemes and embracing the temporal and contextual complexity of the TME, this perspective aims to provide a conceptual framework for improving the durability and breadth of immunotherapy responses across diverse cancer types.
2. The “State” Rather Than the “Composition” of the Tumor Microenvironment: A New Perspective of Functional Classification
With the rapid advances in tumor immunology, it has become increasingly clear that describing the TME solely at the level of cellular composition is insufficient to explain the striking heterogeneity in responses to immunotherapy [22]. Compared with the question of which immune cells are present within a tumor, the question of whether these immune cells are in a functional state capable of exerting antitumor activity is of greater biological and clinical relevance [12,23]. Consequently, a conceptual shift from a “composition-oriented” to a “state-oriented” framework is emerging as a key paradigm for understanding how TME regulation shapes immunotherapeutic efficacy.
2.1. Limitations of the “Immune-Cold–Immune-Hot” Classification
The dichotomous classification of tumors into “immune-cold” and “immune-hot” categories was initially proposed as a straightforward framework to estimate the potential sensitivity of tumors to immunotherapy [24,25]. This model is typically based on the degree of immune cell infiltration and the expression of inflammation-related gene signatures, and it has provided some guidance for early clinical stratification [26]. However, with the accumulation of clinical and translational data, its limitations have become increasingly apparent.
A growing body of evidence indicates that a subset of tumors classified as “immune-hot”, despite exhibiting abundant T-cell infiltration and pronounced inflammatory signals, display intrinsic or acquired resistance to immune checkpoint inhibitors [27]. This observation underscores the fact that the mere presence of immune cells does not necessarily translate into effective antitumor immune responses [28]. Persistent antigen stimulation, accumulation of inhibitory signals, and metabolic stress frequently drive infiltrating immune cells into dysfunctional or exhausted states [15]. In addition, the “cold–hot” model fails to capture the spatial heterogeneity of immune cell distribution within tumors and cannot adequately reflect the dynamic evolution of the TME during therapeutic intervention [3].
Therefore, reliance on a simplistic “immune-cold–immune-hot” classification is insufficient for accurately predicting immunotherapy outcomes, highlighting the urgent need for more refined, mechanism-oriented systems to evaluate TME immune states.
2.2. Functional State–Based Classification of the TME
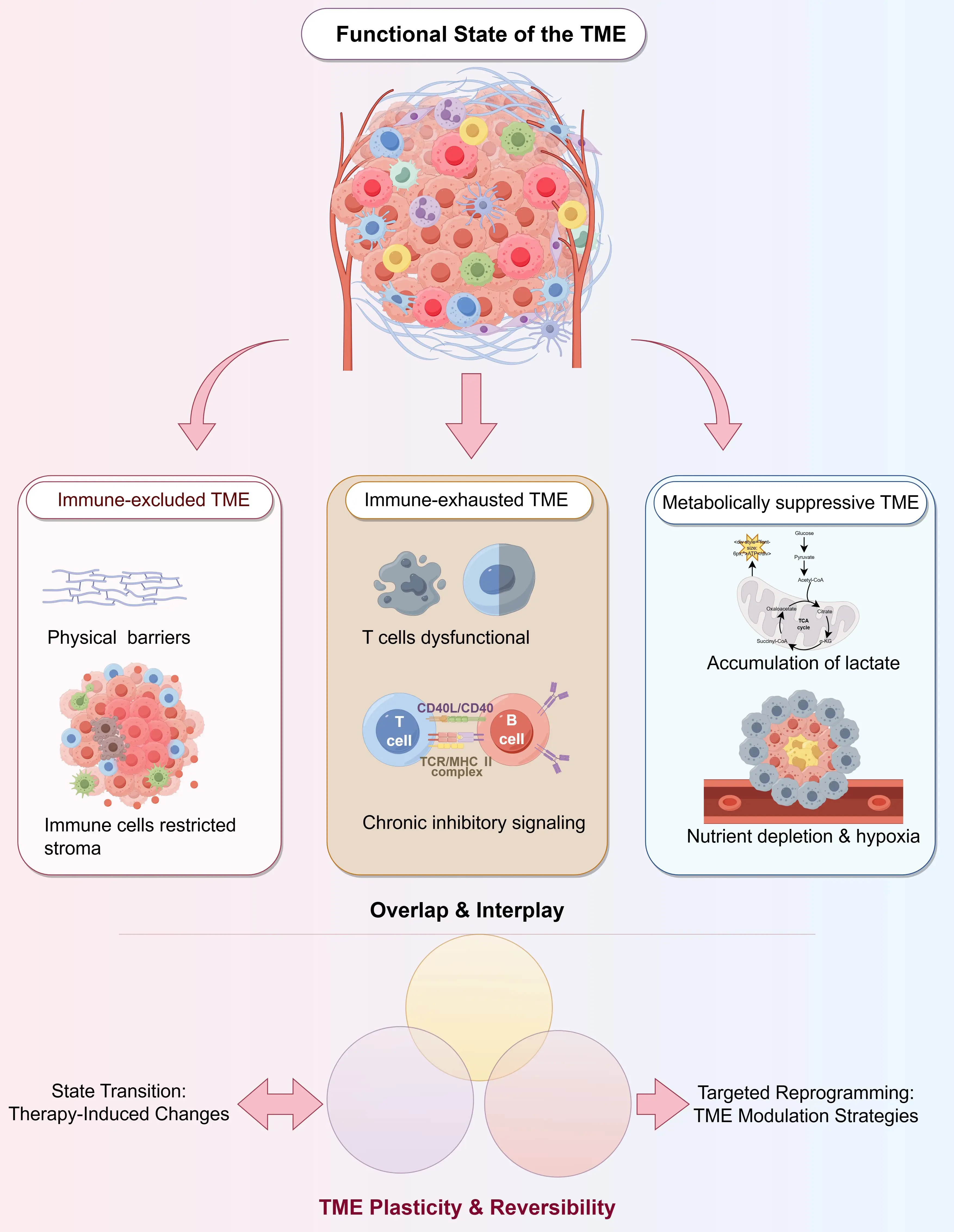
Compared with static descriptions of immune cell types or abundance, a functional interpretation of TME immune states provides deeper insight into the mechanistic basis underlying differential responses to immunotherapy [29,30]. Based on whether immune effector responses can be effectively initiated, executed, and sustained, the TME can be broadly categorized into several representative functional states (Figure 1).
The immune-excluded TME is characterized by the confinement of immune cells to the tumor periphery or stromal regions, with limited penetration into the tumor parenchyma [31,32]. This state is often associated with aberrant extracellular matrix architecture, dysfunctional vasculature, and activated cancer-associated fibroblasts, which collectively form physical and biochemical barriers [33]. As a result, even when immune cells are successfully recruited, they fail to establish effective contact with tumor cells.
In the immune-exhausted TME, immune cells can infiltrate the tumor core but progressively lose their cytotoxic capacity due to sustained antigen exposure and multiple inhibitory signals [34,35,36]. This state is typically accompanied by the co-expression of multiple inhibitory receptors, reduced production of effector molecules, and impaired formation of immunological memory, which constitute a major basis for secondary resistance to immunotherapy [36,37].
The metabolically suppressive TME restricts immune cell function through nutrient competition, hypoxia, and the accumulation of metabolic byproducts [38,39]. Highly active tumor metabolism deprives immune cells of essential nutrients such as glucose, amino acids, and lipids, while the buildup of metabolites, including lactate and adenosine, further dampens immune effector responses [39].
Importantly, these functional states are not mutually exclusive. Instead, they frequently coexist and intersect within the same tumor, collectively shaping a complex and multilayered immunosuppressive network.
2.3. Plasticity and Reversibility of TME States
It is critical to emphasize that the functional state of the TME is not static. Rather, it continuously evolves under the combined influence of tumor progression, host immune responses, and therapeutic interventions [40,41]. Immunotherapy itself represents a strong selective pressure that can drive the TME toward new equilibrium states—a process that may either enhance antitumor immunity or accelerate immune escape [42].
This high degree of plasticity represents a double-edged sword: on one hand, it underlies immunotherapy failure and the emergence of resistance; on the other hand, it provides a theoretical basis for rational interventions aimed at reprogramming TME states [43]. Strategies that modulate the structural, metabolic, or signaling networks of the TME to convert immune-excluded or immune-exhausted states into conditions more permissive to effective immune responses are increasingly recognized as a central direction for next-generation immunotherapeutic approaches [44,45].
3. Dynamic Remodeling of the TME as a Central Driver of Immunotherapy Failure
Although ICIs have achieved transformative success in multiple solid tumors and hematological malignancies, their clinical benefits remain largely confined to a limited subset of patients [2,46]. Accumulating clinical evidence indicates that even among patients who exhibit objective responses during the initial phase of treatment, a substantial proportion eventually develops acquired resistance during follow-up [47]. These observations suggest that immunotherapy failure is not solely determined by an unfavorable pre-treatment TME, but is also closely linked to the continuous remodeling and dynamic evolution of the TME during therapy [28].
3.1. Therapy-Induced Adaptive Immunosuppression
Immunotherapy does not unidirectionally activate antitumor immunity; rather, it imposes strong selective pressure on the tumor–immune equilibrium [2,19]. Under ICI treatment, such as PD-1/PD-L1 blockade, transient restoration of T-cell function is often followed by adaptive regulatory responses within tumor cells and the surrounding microenvironment, which re-establish immunosuppressive conditions [48].
On the one hand, multiple immunoinhibitory pathways can be compensatorily activated. Clinical specimens and preclinical models have demonstrated that following PD-1/PD-L1 blockade, the expression of alternative checkpoint molecules—including TIM-3, LAG-3, TIGIT, and VISTA—is significantly upregulated, progressively driving T cells into renewed functional suppression or a state of “deep exhaustion” [49,50]. On the other hand, immunosuppressive cell populations within the TME, such as regulatory T cells, myeloid-derived suppressor cells, and immunosuppressive tumor-associated macrophages, may become enriched during treatment [51,52]. These cells further impair effector T-cell activity through the secretion of IL-10 and TGF-β, or via the expression of molecules arginase-1 (ARG1) and indoleamine 2,3-dioxygenase (IDO) [53].
Notably, this form of adaptive immunosuppression is not a stochastic event, but rather the outcome of evolutionary selection driven by immunotherapeutic pressure, underscoring the remarkable plasticity of tumors under immune stress.
3.2. Evolutionary Trajectories of the TME and Immune Escape
From a dynamic perspective, the TME does not simply adopt a binary “responder” or “non-responder” state before and after therapy; instead, it undergoes a continuous evolutionary trajectory [11]. Longitudinal biopsies and multi-omics analyses have revealed that, in a subset of tumors initially sensitive to ICIs, the TME gradually shifts from an inflammatory or immune-active state toward an immunosuppressive, or even immune-excluded, phenotype during later stages of treatment [54,55].
This evolutionary process is typically accompanied by multilayered alterations, including downregulation of antigen presentation pathways, negative feedback inhibition following sustained IFN-γ signaling, metabolic reprogramming of tumor cells that intensifies nutrient competition, and remodeling of stromal components that restrict immune cell infiltration [48,56]. Together, these changes synergistically promote the emergence of immune escape phenotypes.
These findings carry important clinical implications: biomarkers assessed at a single time point—such as PD-L1 expression or baseline immune infiltration—are insufficient to accurately predict long-term therapeutic outcomes and fail to capture the true trajectory of TME evolution during immunotherapy.
3.3. Implications for the Failure of Combination Therapies
To overcome immunotherapy resistance, combination strategies have been widely pursued as a means to enhance efficacy [57,58]. However, in several clinical trials, the simultaneous combination of multiple immunotherapeutic agents has not yielded the anticipated benefits and has, in some cases, increased treatment-related toxicity [59]. These outcomes suggest that the failure of combination therapies may stem not from inadequate target selection but from neglecting the dynamic nature of the TME and the critical importance of treatment timing [60].
Notably, multiple late-phase clinical trials have demonstrated that even rationally designed combinations—such as dual immune checkpoint blockade or metabolic–immune co-targeting—can fail when applied without consideration of TME state transitions (Table 1).
If distinct immunoregulatory pathways are activated or suppressed sequentially at different stages of therapy, indiscriminate simultaneous blockade may fail to align with the real-time immune state of the TME, and may even facilitate immune escape through more complex compensatory mechanisms [42,61]. Therefore, future combination strategies must shift from “static addition” to “dynamic modulation”, in which the sequence, timing, and composition of interventions are rationally tailored according to the evolving states of the TME.
Table 1. Selected clinical examples of limited efficacy in combination immunotherapy.
|
Combination Strategy |
Cancer Type |
Clinical Outcome |
Dominant TME State at Failure |
Proposed Mechanisms Underlying Limited Efficacy |
|---|---|---|---|---|
|
Anti–PD-1 + Anti–CTLA-4 |
Metastatic melanoma |
Improved response rate but no durable OS benefit in a subset; increased grade ≥3 irAEs |
Chronic inflammatory but dysfunctional TME |
T cell exhaustion driven by sustained inhibitory signaling; compensatory upregulation of TIM-3, LAG-3; Treg expansion |
|
Anti–PD-1 + IDO1 inhibitor |
Advanced melanoma |
Phase III trial failure (no PFS/OS benefit) |
Metabolically suppressive TME |
Redundant metabolic suppression pathways; adaptive kynurenine-independent immune inhibition; insufficient target engagement |
|
Anti–PD-L1 + Anti–TGF-β |
Urothelial carcinoma |
Modest clinical activity with heterogeneous responses |
Immune-excluded, stroma-dense TME |
Spatial uncoupling of immune reinvigoration and TGF-β–driven stromal remodeling; delayed immune cell infiltration |
|
Anti–PD-1 + Anti–LAG-3 |
Melanoma (post–PD-1 failure) |
Partial benefit confined to select patient subsets |
Exhausted immune-infiltrated TME |
Late-stage exhaustion limits functional rescue; fixed epigenetic exhaustion programs |
|
Anti–PD-1 + CSF1R inhibitor |
Pancreatic ductal adenocarcinoma |
Minimal objective responses |
Myeloid-dominant immunosuppressive TME |
Myeloid plasticity and compensatory recruitment of alternative suppressive populations |
|
Anti–PD-1 + Anti-angiogenic therapy |
Renal cell carcinoma |
Initial benefit followed by adaptive resistance |
Hypoxic, metabolically stressed TME |
Therapy-induced hypoxia promotes immunosuppressive metabolites and abnormal vascular remodeling |
4. TME State–Oriented Design Principles for Next-Generation Immunotherapy
With an increasingly refined understanding of the dynamic properties and functional heterogeneity of the TME, traditional immunotherapy paradigms centered on single targets or fixed treatment regimens are being fundamentally challenged [57]. A growing body of evidence indicates that the success of immunotherapy depends not only on the intrinsic properties of therapeutic agents, but also on whether these interventions are precisely aligned with the functional state of the TME at specific stages of disease and treatment [62]. In this context, TME state–oriented immunotherapy design has emerged as a central concept guiding the development of next-generation immunotherapeutic strategies [63].
4.1. From “Parallel Combination” to “Sequential Modulation”
Early combination immunotherapy strategies predominantly adopted a “parallel combination” approach, in which multiple immunosuppressive pathways were simultaneously blocked, or several immune effector mechanisms were activated at the same time [64,65]. However, the limited efficacy observed in a number of clinical trials suggests that simple target stacking is insufficient to overcome the highly dynamic and hierarchically regulated nature of the TME [66].
In contrast, the concept of “sequential modulation” highlights the importance of treatment order and temporal windows [43]. Increasing evidence supports the notion that, prior to immune activation, the TME should first undergo functional “preconditioning” to remove key barriers that constrain effective immune responses [3,67]. For example, correcting abnormal tumor vasculature, remodeling the extracellular matrix, or alleviating local metabolic suppression can create permissive conditions for subsequent immune cell infiltration and effector function [68]. Under such conditions, the introduction of immune-activating modalities—such as immune checkpoint inhibitors or cellular therapies—often leads to more durable and stable therapeutic responses [69].
Conceptually, this strategy reframes immunotherapy from a model of “direct immune activation” to one of “environmental conditioning followed by immune activation”, representing a paradigm shift from outcome-oriented interventions to process-oriented immune modulation.
4.2. Therapeutic Matching Based on TME States
Marked heterogeneity in TME states exists not only across tumor types but also within the same tumor at different stages of progression, rendering “one-size-fits-all” immunotherapy regimens inherently suboptimal [70]. Functional classification of the TME provides a rational framework for aligning therapeutic strategies with specific immunological contexts [25].
For instance, in immune-excluded TMEs, effector immune cells are confined to the tumor periphery, and therapeutic efforts should focus on facilitating immune cell access to the tumor parenchyma [57]. Strategies such as modulation of chemokine axes, vascular normalization, or extracellular matrix degradation may be particularly effective in this setting. In contrast, immune-exhausted TMEs are characterized by the presence of tumor-infiltrating T cells that are functionally compromised due to chronic inhibitory signaling and metabolic stress; here, approaches aimed at restoring immune function—such as multi-checkpoint blockade or metabolic interventions—may yield greater benefit [71].
Moreover, in metabolically suppressive or inflammation-imbalanced TMEs, indiscriminate enhancement of immune activity may be counterproductive, potentially accelerating immune exhaustion or exacerbating treatment-related toxicity [72]. Therefore, therapeutic matching guided by the TME state holds promise not only for improving efficacy but also for minimizing ineffective treatments and unnecessary adverse events.
4.3. The Concept of “Adaptive Immunotherapy”
Against the backdrop of continuous TME evolution, future immunotherapy strategies are likely to transition from fixed regimens toward “adaptive immunotherapy” [73]. This concept emphasizes continuous monitoring of key functional indicators within the TME during treatment and the dynamic adjustment of therapeutic combinations, dosing, or sequencing accordingly, with the goal of prolonging effective immune responses [74].
At its core, adaptive immunotherapy treats immunomodulation as an iterative and adjustable process rather than a one-time intervention [43]. Such an approach may prevent overtreatment during early response phases and enable timely intervention when early signs of resistance emerge, thereby redirecting TME evolution and delaying or even reversing immune escape.
Although the clinical implementation of adaptive immunotherapy faces challenges—including biomarker selection, real-time monitoring technologies, and increased decision-making complexity—the conceptual shift itself represents a profound transformation in immunotherapy design logic. It paves the way for more precise and durable clinical benefits [75].
5. Key TME Regulatory Axes Driving Next-Generation Immunotherapy
As immunotherapy transitions from a purely “target-oriented” paradigm toward a “state-oriented” framework, the TME is no longer viewed merely as a passive backdrop for immune responses but is increasingly recognized as an actively modifiable therapeutic target [76]. Accumulating evidence indicates that several non-canonical immunoregulatory dimensions play decisive roles in determining immunotherapy outcomes [77]. Among these, the physical and mechanical properties of the TME, the metabolism–signaling–epigenetic coupling network, and the emerging neuro–immune signaling axis have attracted particular attention. Together, these regulatory layers provide novel intervention points that extend beyond conventional immune checkpoint pathways and underpin the development of next-generation immunotherapeutic strategies.
5.1. Regulation of Immune Responses by Physical and Mechanical Properties
The physical and mechanical attributes of tumor tissues represent a critical yet historically underappreciated regulatory dimension of the TME [78]. Matrix stiffness, tissue tension, and the spatial organization of stromal components not only influence tumor cell invasion and metastatic potential but also profoundly shape immune cell trafficking, spatial distribution, and functional states [79] (Figure 2A).
In most solid tumors, aberrant activation of fibroblasts and excessive collagen deposition lead to pronounced matrix stiffening, creating a physical barrier that limits immune cell infiltration [80]. Moreover, increased matrix tension can directly reprogram immune cell signaling through mechanosensing pathways involving integrins, focal adhesion kinase (FAK), or YAP/TAZ, thereby suppressing T-cell effector functions and promoting the polarization of immunosuppressive macrophage phenotypes [81].
Based on these mechanisms, therapeutic strategies targeting the physical properties of the TME have gained increasing interest [82]. Approaches such as vascular normalization, stromal remodeling, or modulation of mechanotransduction pathways aim to reduce immune exclusion rather than directly activate immune responses. By “unlocking spatial constraints”, these interventions can indirectly enhance immunotherapy efficacy and serve as critical preparatory steps for subsequent immune-activating treatments within a sequential therapeutic framework [57].
5.2. The Coupled Network of Metabolism, Signaling, and Epigenetic Regulation
Metabolic reprogramming in cancer cells not only supports tumor growth but also profoundly influences immune cell fate by reshaping the local nutrient and metabolite landscape [83]. In TMEs characterized by intense competition for glucose, amino acids, and lipids, effector T cells often experience energy deprivation and functional exhaustion, whereas immunosuppressive cell populations display greater adaptability to nutrient-poor, metabolite-rich conditions [84].
Importantly, metabolic alterations are tightly coupled to epigenetic regulation. Numerous metabolic intermediates serve directly as substrates or cofactors for epigenetic enzymes, thereby linking cellular metabolic states to chromatin accessibility and gene expression programs in immune cells. For example, fluctuations in lactate, α-ketoglutarate, or acetyl-CoA levels can modulate histone modifications or DNA methylation patterns, leading to long-lasting changes in immune cell identity and function [85] (Figure 2B).
This metabolism–signaling–epigenetic network offers a powerful new entry point for immunotherapy [86]. By targeting metabolic pathways to reshape epigenetic landscapes, it may be possible to “reprogram” immune cells toward durable antitumor phenotypes rather than inducing transient immune activation [86]. Such strategies hold promise for enhancing the depth and persistence of immunotherapeutic responses.
5.3. Emerging Roles of the Neuro–Immune–Tumor Axis
In recent years, the role of the nervous system in tumor progression and immune regulation has gained increasing recognition, giving rise to the concept of the neuro–immune–tumor axis [87]. Dense innervation and neurotransmitter signaling within tumor tissues can directly or indirectly influence immune cell differentiation, migration, and functional states [88] (Figure 2C).
Multiple studies have demonstrated that sympathetic and parasympathetic signaling, through the release of neurotransmitters such as norepinephrine and acetylcholine, can modulate the immunophenotypes of tumor-associated macrophages, dendritic cells, and T cells, thereby promoting the establishment of an immunosuppressive microenvironment [89]. In addition, neural signals intersect with tumor metabolic pathways and angiogenic programs, further amplifying their regulatory impact on TME states [90].
Although this field remains in its early stages, neuro–immune regulation introduces a previously underexplored dimension of TME modulation. These findings suggest that future immunotherapeutic strategies may benefit from integrating neuromodulatory approaches to achieve multi-layered remodeling of the tumor immune landscape.
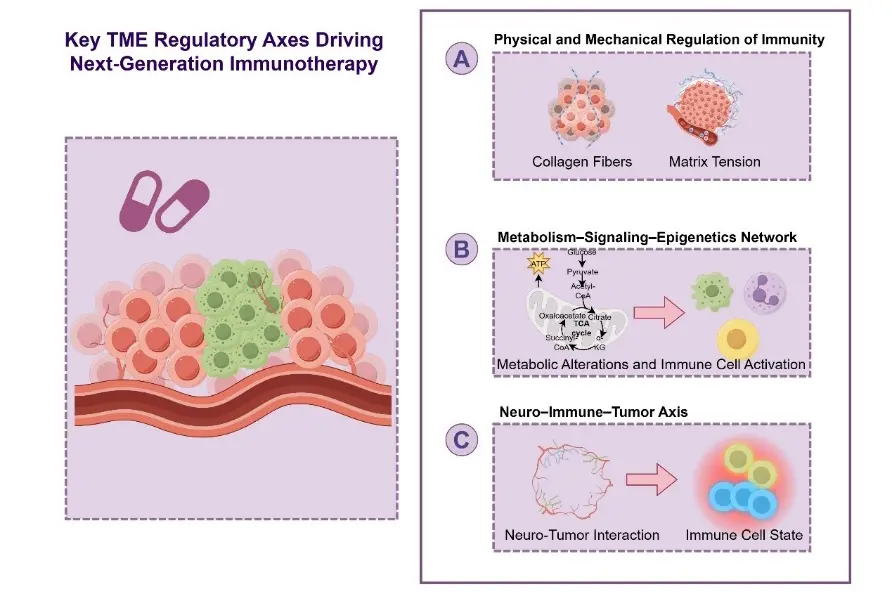
Figure 2. Key TME Regulatory Axes Driving Next-Generation Immunotherapy. (A) Physical and Mechanical Regulation of Immunity; (B) Metabolism–Signaling–Epigenetics Regulatory Network; (C) The Neuro–Immune–Tumor Axis.
6. TME-Driven Biomarkers and Patient Stratification for Immunotherapy
Accurately identifying patient populations most likely to benefit from immunotherapy remains one of the central challenges in tumor immunology [91]. Although a variety of immune-related biomarkers have been proposed and implemented in clinical practice, the pronounced heterogeneity and time dependence of immunotherapeutic responses substantially limit the ability of conventional biomarkers to predict long-term clinical benefit [92]. Biomarker frameworks centered on the TME offer a new conceptual basis for more refined, clinically meaningful patient stratification.
6.1. From Static Indicators to Dynamic Characterization: A Paradigm Shift in TME Biomarkers
Most immunotherapy biomarkers currently used in clinical settings are static in nature, such as baseline PD-L1 expression, tumor mutational burden, or initial levels of immune cell infiltration. While these markers partially capture immune-related tumor features, their fundamental limitation is that they represent only a single snapshot of the TME, thereby failing to reflect its continuous evolution during treatment [93].
A growing body of evidence indicates that immunotherapy responses are not fully determined prior to treatment initiation, but are progressively shaped during early treatment phases and throughout the therapeutic course [11]. Some patients lacking classical “immune-favorable” features at baseline may nevertheless experience functional remodeling of the TME following treatment and derive clinical benefit. Conversely, patients who initially respond well may later develop resistance as a consequence of TME state transitions [19]. These observations highlight that dynamic changes within the TME may be more predictive of long-term outcomes than baseline characteristics alone.
Against this backdrop, the concept of dynamic TME biomarkers has gained increasing attention. Early treatment–induced alterations in immune cell composition, functional states, or metabolic features may serve as informative predictors of durable therapeutic responses [94]. The integration of repeat sampling strategies with liquid biopsy technologies enables longitudinal monitoring of TME dynamics, providing a practical avenue to overcome the inherent limitations of static biomarker assessments [95].
6.2. Precision Immunotherapy Stratification Based on TME State Evolution
As understanding of TME functional states and their evolutionary trajectories deepens, patient stratification strategies are shifting from reliance on single molecular or cellular markers toward an integrated, TME state–oriented approach [96]. Unlike traditional stratification methods, this strategy emphasizes a comprehensive evaluation of multiple TME dimensions—including immune activation, suppressive signaling pathways, metabolic conditions, and spatial organization—to more accurately capture the tumor immune ecosystem [3].
TME state–based stratification not only improves the prediction of immunotherapy efficacy but also provides a rational framework for individualized treatment design. For example, dynamically assessing whether a TME is transitioning from an immunosuppressive to an immune-active state may inform decisions on whether to continue monotherapy or introduce additional interventions to prevent immune escape [97]. Moreover, this stratification paradigm offers critical decision-making support for “adaptive immunotherapy”, enabling timely adjustments of therapeutic strategies in response to evolving TME states [98,99].
From a broader perspective, TME state–oriented biomarker systems have the potential to elevate patient stratification from a binary decision of whether to use immunotherapy to a more nuanced determination of when, how, and in what combinations immunotherapeutic interventions should be applied. In doing so, they pave the way for immunotherapy to fully realize the principles of precision oncology.
7. Challenges and Conceptual Pitfalls in the Clinical Translation of TME-Driven Immunotherapy
Although immunotherapeutic strategies targeting the TME hold substantial theoretical promise, their clinical translation has proven to be far from linear and is accompanied by a series of conceptual misconceptions and practical challenges [100]. A systematic examination of these issues is essential to prevent conceptual oversimplification and to optimize rational treatment design.
7.1. The Oversimplified Assumption That “TME Modulation Equals Therapeutic Benefit”
A common but potentially misleading implicit assumption in current research is that successful modulation of the TME will inevitably enhance immunotherapy efficacy [3]. In reality, the TME represents a highly complex system maintained by finely tuned balances, and the consequences of its modulation are often profoundly context dependent.
On the one hand, not all immunosuppressive features should be indiscriminately regarded as detrimental. Certain inhibitory mechanisms may serve protective roles at specific stages by constraining excessive inflammation or preserving tissue homeostasis. Unrestrained immune activation may transiently amplify antitumor responses, yet ultimately promote immune exhaustion or facilitate the emergence of resistance [101]. On the other hand, substantial heterogeneity exists in the baseline TME state and its plasticity across tumor types, disease stages, and individual patients, such that identical modulation strategies can yield markedly divergent outcomes (Figure 3A).
Equating TME modulation with immune enhancement while overlooking its dual-edged nature may therefore foster overly optimistic expectations of therapeutic efficacy. This conceptual bias may also help explain why some approaches that perform well in preclinical or early-phase studies fail to demonstrate consistent benefit in clinical trials.
7.2. Risks and Translational Barriers Associated with Excessive TME Remodeling
From a clinical perspective, a major additional challenge lies in the potential risks associated with excessive or poorly controlled TME remodeling. When multiple immune-activating or microenvironment-targeting interventions are applied concurrently, the TME may be driven toward a hyperinflammatory state, thereby increasing the incidence and severity of immune-related adverse events (irAEs) [102]. Uncontrolled inflammation not only compromises patient quality of life but may also limit treatment durability or necessitate premature discontinuation.
Furthermore, the clinical translation of TME-targeted strategies is fundamentally constrained by issues of model extrapolation. A substantial proportion of mechanistic insights and therapeutic validations are derived from murine models, yet the TME in these systems differs markedly from that of human tumors in immune system composition, metabolic context, tissue architecture, and evolutionary pressures [103]. Consequently, TME remodeling strategies that appear effective in animal models do not necessarily elicit equivalent immunological outcomes in patients (Figure 3B).
Collectively, these challenges underscore that clinical implementation of TME modulation must move beyond the question of “whether it works” to carefully consider “how much, how broadly, and at what time point” intervention should be applied. Only through a nuanced understanding of human TME–specific features, coupled with a dynamic balance between safety and efficacy, can TME-driven strategies realize their true clinical potential.
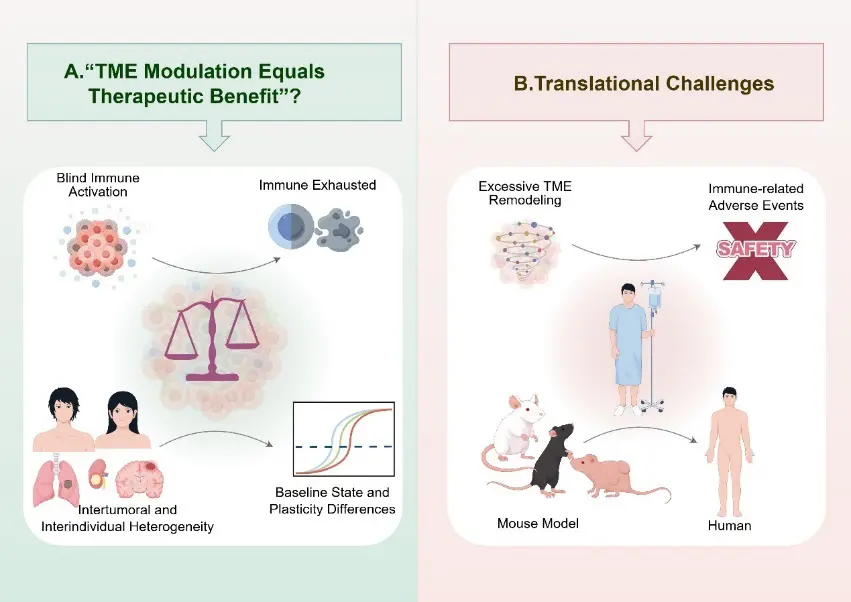
Figure 3. Challenges and Conceptual Pitfalls in the Clinical Translation of TME-Driven Immunotherapy.
8. Conclusions and Perspectives: From “Therapeutic Combinations” to “Immune Ecosystem Management”
The limited and heterogeneous responses to current immunotherapies underscore a fundamental challenge: therapeutic success is not dictated solely by target engagement, but by the broader immunological context in which these therapies operate. Increasing evidence positions the TME as a dynamic and adaptive system that critically shapes treatment outcomes.
In this context, we propose a conceptual shift from conventional “therapeutic combinations” toward “immune ecosystem management”. This framework emphasizes the regulation of immune contexture over time, aiming to achieve a sustained and functional equilibrium between tumor control and immune competence, rather than transient or maximal immune activation.
While much of the current clinical and conceptual focus has centered on immune checkpoint blockade, this framework is not modality-restricted. The same TME-driven principles are highly relevant to adoptive cellular immunotherapies, including chimeric antigen receptor (CAR)-T cells and tumor-infiltrating lymphocyte-based therapies, where microenvironmental factors similarly constrain therapeutic efficacy.
A state-oriented understanding of the TME provides a unifying lens to guide this transition. By focusing on functional immune states and their evolution, therapeutic strategies can be more precisely aligned with disease stage and microenvironmental constraints, enabling context-aware intervention rather than fixed or empirically derived regimens.
In this regard, TME states such as immune exclusion, stromal barriers, and metabolic suppression not only influence endogenous antitumor immunity but also critically affect the trafficking, persistence, and functional durability of adoptively transferred immune cells, underscoring a shared set of biological constraints across immunotherapeutic modalities.
Future progress will rely on the integration of high-resolution technologies and clinically actionable frameworks. Advances in single-cell and spatial multi-omics are expected to refine our understanding of TME heterogeneity, while dynamic biomarker systems may enable real-time monitoring of immune states and guide adaptive treatment strategies. In parallel, emerging therapeutic platforms with improved spatial and temporal control will further enhance our ability to modulate the TME with precision. Such advances may also facilitate the rational coordination of checkpoint-based and cellular therapies within a unified TME-informed framework, rather than treating them as independent or sequential modalities.
Equally important are the implications for regulatory science. As immunotherapy increasingly targets system-level immune dynamics rather than single molecular endpoints, existing potency assessment paradigms for monoclonal antibodies—largely based on reductionist in vitro assays—may prove insufficient. There is a growing need to evolve these frameworks toward context-aware evaluation strategies that incorporate TME-relevant conditions, such as multicellular interactions, stromal constraints, and metabolic pressures. The development of advanced assay systems, including co-culture models, organoid platforms, and other TME-mimetic approaches, alongside state-dependent functional readouts, may improve the translational fidelity of preclinical testing and better align regulatory evaluation with clinical reality.
Ultimately, this shift reflects a broader redefinition of therapeutic objectives. Rather than pursuing maximal immune activation, next-generation strategies may prioritize the induction and maintenance of optimally configured immune states tailored to specific TME contexts.
In summary, embracing the TME as a dynamic, regulatable immune ecosystem provides a coherent framework for overcoming current limitations in cancer immunotherapy. Extending this paradigm across both checkpoint-based and adoptive cellular therapies may further broaden its applicability and impact. By integrating biological insight, technological innovation, and regulatory adaptation, this paradigm holds promise for enabling more precise, durable, and clinically meaningful therapeutic outcomes.
Statement of the Use of Generative AI and AI-Assisted Technologies in the Writing Process
During the preparation of this manuscript, the authors used Deepseek (OpenAI) to assist with language refinement, text editing, content organization, and improvement of readability. After using this tool, the authors critically reviewed, revised, and verified all content and take full responsibility for the accuracy and integrity of the published article.
Acknowledgments
We would like to thank the library staff of Henan Medical University for their support with literature retrieval.
Author Contributions
Conceptualization H.W. and X.H.; Writing—original draft preparation, X.H., G.G., S.G. and H.W.; Writing—review and editing, X.H., H.W., S.D., J.T., S.X. and T.Z.; Supervision X.H. All authors have read and agreed to the published version of this manuscript.
Ethics Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data sharing is not applicable to this article as no data were created or analysed in this study.
Funding
This research was supported by the Key Project of International Scientific and Technological Cooperation of Henan Province (241111520400), the Henan Provincial Key Program for Science and Technology Development (252102310051), the National Natural Science Foundation of China (32300120), the Key Scientific Research Project of Higher Education of Henan Province (25A310013), the 111 Project (D20036, China).
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. DOI:10.1056/NEJMoa1200690 [Google Scholar]
- Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. DOI:10.1126/science.aar4060 [Google Scholar]
- Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. DOI:10.1038/s41591-018-0014-x [Google Scholar]
- Hanahan D, Coussens LM. Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell 2012, 21, 309–322. DOI:10.1016/j.ccr.2012.02.022 [Google Scholar]
- Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. DOI:10.1038/nm.3394 [Google Scholar]
- Giovanelli P, Sandoval TA, Cubillos-Ruiz JR. Dendritic Cell Metabolism and Function in Tumors. Trends Immunol. 2019, 40, 699–718. DOI:10.1016/j.it.2019.06.004 [Google Scholar]
- Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. DOI:10.1038/nri3862 [Google Scholar]
- Chen DS, Mellman I. Elements of cancer immunity and the cancer-immune set point. Nature 2017, 541, 321–330. DOI:10.1038/nature21349 [Google Scholar]
- Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 2013, 14, 1014–1022. DOI:10.1038/ni.2703 [Google Scholar]
- Ozga AJ, Chow MT, Luster AD. Chemokines and the immune response to cancer. Immunity 2021, 54, 859–874. DOI:10.1016/j.immuni.2021.01.012 [Google Scholar]
- Riaz N, Havel JJ, Makarov V, Desrichard A, Urba WJ, Sims JS, et al. Tumor and Microenvironment Evolution during Immunotherapy with Nivolumab. Cell 2017, 171, 934–949.e16. DOI:10.1016/j.cell.2017.09.028 [Google Scholar]
- Huang AC, Postow MA, Orlowski RJ, Mick R, Bengsch B, Manne S, et al. T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature 2017, 545, 60–65. DOI:10.1038/nature22079 [Google Scholar]
- Quail DF, Joyce JA. The Microenvironmental Landscape of Brain Tumors. Cancer Cell 2017, 31, 326–341. DOI:10.1016/j.ccell.2017.02.009 [Google Scholar]
- Salmon H, Franciszkiewicz K, Damotte D, Dieu-Nosjean MC, Validire P, Trautmann A, et al. Matrix architecture defines the preferential localization and migration of T cells into the stroma of human lung tumors. J. Clin. Investig. 2012, 122, 899–910. DOI:10.1172/JCI45817 [Google Scholar]
- Scharping NE, Menk AV, Moreci RS, Whetstone RD, Dadey RE, Watkins SC, et al. The Tumor Microenvironment Represses T Cell Mitochondrial Biogenesis to Drive Intratumoral T Cell Metabolic Insufficiency and Dysfunction. Immunity 2016, 45, 374–388. DOI:10.1016/j.immuni.2016.07.009 [Google Scholar]
- Turner LM, Terhaar H, Jiminez V, Anderson BJ, Grant E, Yusuf N. Tumor Microenvironmental Dynamics in Shaping Resistance to Therapeutic Interventions in Melanoma: A Narrative Review. Pharmaceuticals 2025, 18, 1082. DOI:10.3390/ph18081082 [Google Scholar]
- Hu Q, Zhu Y, Mei J, Liu Y, Zhou G. Extracellular matrix dynamics in tumor immunoregulation: From tumor microenvironment to immunotherapy. J. Hematol. Oncol. 2025, 18, 65. DOI:10.1186/s13045-025-01717-y [Google Scholar]
- Wang B, Pei J, Xu S, Liu J, Yu J. A glutamine tug-of-war between cancer and immune cells: Recent advances in unraveling the ongoing battle. J. Exp. Clin. Cancer Res. 2024, 43, 74. DOI:10.1186/s13046-024-02994-0 [Google Scholar]
- Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. DOI:10.1016/j.cell.2017.01.017 [Google Scholar]
- Hegde PS, Chen DS. Top 10 Challenges in Cancer Immunotherapy. Immunity 2020, 52, 17–35. DOI:10.1016/j.immuni.2019.12.011 [Google Scholar]
- Lv Z, Yang R, Zhang K, Wang R, Shi X, Wu J, et al. The dual immunomodulatory role of B cells in tumorigenesis: Mechanisms, microenvironment crosstalk, and therapeutic implications. Front. Immunol. 2025, 16, 1649812. DOI:10.3389/fimmu.2025.1649812 [Google Scholar]
- Tsahouridis O, Xu M, Song F, Savoldo B, Dotti G. The landscape of CAR-engineered innate immune cells for cancer immunotherapy. Nat. Cancer 2025, 6, 1145–1156. DOI:10.1038/s43018-025-01015-z [Google Scholar]
- Joyce JA, Fearon DT. T cell exclusion, immune privilege, and the tumor microenvironment. Science 2015, 348, 74–80. DOI:10.1126/science.aaa6204 [Google Scholar]
- Galon J, Mlecnik B, Bindea G, Angell HK, Berger A, Lagorce C, et al. Towards the introduction of the ‘Immunoscore’ in the classification of malignant tumours. J. Pathol. 2014, 232, 199–209. DOI:10.1002/path.4287 [Google Scholar]
- Fridman WH, Zitvogel L, Sautes-Fridman C, Kroemer G. The immune contexture in cancer prognosis and treatment. Nat. Rev. Clin. Oncol. 2017, 14, 717–734. DOI:10.1038/nrclinonc.2017.101 [Google Scholar]
- Saltz J, Gupta R, Hou L, Kurc T, Singh P, Nguyen V, et al. Spatial Organization and Molecular Correlation of Tumor-Infiltrating Lymphocytes Using Deep Learning on Pathology Images. Cell Rep. 2018, 23, 181–193.e7. DOI:10.1016/j.celrep.2018.03.086 [Google Scholar]
- Tsujikawa T, Kumar S, Borkar RN, Azimi V, Thibault G, Chang YH, et al. Quantitative Multiplex Immunohistochemistry Reveals Myeloid-Inflamed Tumor-Immune Complexity Associated with Poor Prognosis. Cell Rep. 2017, 19, 203–217. DOI:10.1016/j.celrep.2017.03.037 [Google Scholar]
- Benci JL, Xu B, Qiu Y, Wu TJ, Dada H, Twyman-Saint Victor C, et al. Tumor Interferon Signaling Regulates a Multigenic Resistance Program to Immune Checkpoint Blockade. Cell 2016, 167, 1540–1554.e12. DOI:10.1016/j.cell.2016.11.022 [Google Scholar]
- Zheng S, Wang W, Shen L, Yao Y, Xia W, Ni C. Tumor battlefield within inflamed, excluded or desert immune phenotypes: The mechanisms and strategies. Exp. Hematol. Oncol. 2024, 13, 80. DOI:10.1186/s40164-024-00543-1 [Google Scholar]
- Liu JS, Cai YX, He YZ, Xu J, Tian SF, Li ZQ. Spatial and temporal heterogeneity of tumor immune microenvironment between primary tumor and brain metastases in NSCLC. BMC Cancer 2024, 24, 123. DOI:10.1186/s12885-024-11875-w [Google Scholar]
- Lapuente-Santana O, Sturm G, Kant J, Ausserhofer M, Zackl C, Zopoglou M, et al. Multimodal analysis unveils tumor microenvironment heterogeneity linked to immune activity and evasion. iScience 2024, 27, 110529. DOI:10.1016/j.isci.2024.110529 [Google Scholar]
- Yu J, Kong X, Feng Y. Tumor microenvironment-driven resistance to immunotherapy in non-small cell lung cancer: Strategies for Cold-to-Hot tumor transformation. Cancer Drug Resist. 2025, 8, 21. DOI:10.20517/cdr.2025.14 [Google Scholar]
- Liu YT, Wang YL, Wang S, Li JJ, He W, Fan XJ, et al. Turning cold tumors into hot tumors to ignite immunotherapy. Mol. Cancer 2025, 24, 254. DOI:10.1186/s12943-025-02477-6 [Google Scholar]
- Blank CU, Haining WN, Held W, Hogan PG, Kallies A, Lugli E, et al. Defining ‘T cell exhaustion’. Nat. Rev. Immunol. 2019, 19, 665–674. DOI:10.1038/s41577-019-0221-9 [Google Scholar]
- van der Leun AM, Thommen DS, Schumacher TN. CD8(+) T cell states in human cancer: Insights from single-cell analysis. Nat. Rev. Cancer 2020, 20, 218–232. DOI:10.1038/s41568-019-0235-4 [Google Scholar]
- Beltra JC, Manne S, Abdel-Hakeem MS, Kurachi M, Giles JR, Chen Z, et al. Developmental Relationships of Four Exhausted CD8(+) T Cell Subsets Reveals Underlying Transcriptional and Epigenetic Landscape Control Mechanisms. Immunity 2020, 52, 825–841.e8. DOI:10.1016/j.immuni.2020.04.014 [Google Scholar]
- Yost KE, Satpathy AT, Wells DK, Qi Y, Wang C, Kageyama R, et al. Clonal replacement of tumor-specific T cells following PD-1 blockade. Nat. Med. 2019, 25, 1251–1259. DOI:10.1038/s41591-019-0522-3 [Google Scholar]
- Hensley CT, Faubert B, Yuan Q, Lev-Cohain N, Jin E, Kim J, et al. Metabolic Heterogeneity in Human Lung Tumors. Cell 2016, 164, 681–694. DOI:10.1016/j.cell.2015.12.034 [Google Scholar]
- Wang J, He Y, Hu F, Hu C, Sun Y, Yang K, et al. Metabolic Reprogramming of Immune Cells in the Tumor Microenvironment. Int. J. Mol. Sci. 2024, 25, 12223. DOI:10.3390/ijms252212223 [Google Scholar]
- McGranahan N, Swanton C. Cancer Evolution Constrained by the Immune Microenvironment. Cell 2017, 170, 825–827. DOI:10.1016/j.cell.2017.08.012 [Google Scholar]
- Shi Q, Chen Y, Li Y, Qin S, Yang Y, Gao Y, et al. Cross-tissue multicellular coordination and its rewiring in cancer. Nature 2025, 643, 529–538. DOI:10.1038/s41586-025-09053-4 [Google Scholar]
- Kalbasi A, Ribas A. Tumour-intrinsic resistance to immune checkpoint blockade. Nat. Rev. Immunol. 2020, 20, 25–39. DOI:10.1038/s41577-019-0218-4 [Google Scholar]
- Sanmamed MF, Chen L. A Paradigm Shift in Cancer Immunotherapy: From Enhancement to Normalization. Cell 2018, 175, 313–326. DOI:10.1016/j.cell.2018.09.035 [Google Scholar]
- Huang AC, Zappasodi R. A decade of checkpoint blockade immunotherapy in melanoma: Understanding the molecular basis for immune sensitivity and resistance. Nat. Immunol. 2022, 23, 660–670. DOI:10.1038/s41590-022-01141-1 [Google Scholar]
- Bian Y, Li J, Cao J, Wang S, Ma C. Advances in Tumor-Derived Exosomal Non-Coding RNAs Regulating M2 Macrophage Polarization: Molecular Mechanisms and Signaling Pathway. Cancer Med. 2025, 14, e71421. DOI:10.1002/cam4.71421 [Google Scholar]
- Zhang F, Liu Z, Liang J, Zhang F, Wu K, Zhou C, et al. The efficacy and safety of immunotherapy targeting the PD-1 pathway for advanced urothelial carcinoma: A meta-analysis of published clinical trials. Clin. Transl. Oncol. 2020, 22, 1750–1761. DOI:10.1007/s12094-020-02316-8 [Google Scholar]
- Patel SP, Kurzrock R. PD-L1 Expression as a Predictive Biomarker in Cancer Immunotherapy. Mol. Cancer Ther. 2015, 14, 847–856. DOI:10.1158/1535-7163.MCT-14-0983 [Google Scholar]
- Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N. Engl. J. Med. 2016, 375, 819–829. DOI:10.1056/NEJMoa1604958 [Google Scholar]
- Koyama S, Akbay EA, Li YY, Herter-Sprie GS, Buczkowski KA, Richards WG, et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat. Commun. 2016, 7, 10501. DOI:10.1038/ncomms10501 [Google Scholar]
- Andrews LP, Marciscano AE, Drake CG, Vignali DA. LAG3 (CD223) as a cancer immunotherapy target. Immunol. Rev. 2017, 276, 80–96. DOI:10.1111/imr.12519 [Google Scholar]
- Veglia F, Sanseviero E, Gabrilovich DI. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat. Rev. Immunol. 2021, 21, 485–498. DOI:10.1038/s41577-020-00490-y [Google Scholar]
- DeNardo DG, Ruffell B. Macrophages as regulators of tumour immunity and immunotherapy. Nat. Rev. Immunol. 2019, 19, 369–382. DOI:10.1038/s41577-019-0127-6 [Google Scholar]
- Batlle E, Massague J. Transforming Growth Factor-beta Signaling in Immunity and Cancer. Immunity 2019, 50, 924–940. DOI:10.1016/j.immuni.2019.03.024 [Google Scholar]
- Sade-Feldman M, Yizhak K, Bjorgaard SL, Ray JP, de Boer CG, Jenkins RW, et al. Defining T Cell States Associated with Response to Checkpoint Immunotherapy in Melanoma. Cell 2018, 175, 998–1013.e20. DOI:10.1016/j.cell.2018.10.038 [Google Scholar]
- Mao Y, Xie H, Lv M, Yang Q, Shuang Z, Gao F, et al. The landscape of objective response rate of anti-PD-1/L1 monotherapy across 31 types of cancer: A system review and novel biomarker investigating. Cancer Immunol. Immunother. 2023, 72, 2483–2498. DOI:10.1007/s00262-023-03441-3 [Google Scholar]
- Bader JE, Voss K, Rathmell JC. Targeting Metabolism to Improve the Tumor Microenvironment for Cancer Immunotherapy. Mol. Cell 2020, 78, 1019–1033. DOI:10.1016/j.molcel.2020.05.034 [Google Scholar]
- Galon J, Bruni D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat. Rev. Drug Discov. 2019, 18, 197–218. DOI:10.1038/s41573-018-0007-y [Google Scholar]
- Fares CM, Van Allen EM, Drake CG, Allison JP, Hu-Lieskovan S. Mechanisms of Resistance to Immune Checkpoint Blockade: Why Does Checkpoint Inhibitor Immunotherapy Not Work for All Patients? Am. Soc. Clin. Oncol. Educ. Book 2019, 39, 147–164. DOI:10.1200/EDBK_240837 [Google Scholar]
- Dolladille C, Ederhy S, Sassier M, Cautela J, Thuny F, Cohen AA, et al. Immune Checkpoint Inhibitor Rechallenge After Immune-Related Adverse Events in Patients With Cancer. JAMA Oncol. 2020, 6, 865–871. DOI:10.1001/jamaoncol.2020.0726 [Google Scholar]
- Liu J, Blake SJ, Yong MC, Harjunpaa H, Ngiow SF, Takeda K, et al. Improved Efficacy of Neoadjuvant Compared to Adjuvant Immunotherapy to Eradicate Metastatic Disease. Cancer Discov. 2016, 6, 1382–1399. DOI:10.1158/2159-8290.CD-16-0577 [Google Scholar]
- Wei SC, Duffy CR, Allison JP. Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discov. 2018, 8, 1069–1086. DOI:10.1158/2159-8290.CD-18-0367 [Google Scholar]
- Bai R, Lv Z, Xu D, Cui J. Predictive biomarkers for cancer immunotherapy with immune checkpoint inhibitors. Biomark. Res. 2020, 8, 34. DOI:10.1186/s40364-020-00209-0 [Google Scholar]
- Galluzzi L, Humeau J, Buque A, Zitvogel L, Kroemer G. Immunostimulation with chemotherapy in the era of immune checkpoint inhibitors. Nat. Rev. Clin. Oncol. 2020, 17, 725–741. DOI:10.1038/s41571-020-0413-z [Google Scholar]
- Sharma P, Allison JP. The future of immune checkpoint therapy. Science 2015, 348, 56–61. DOI:10.1126/science.aaa8172 [Google Scholar]
- Wei SC, Levine JH, Cogdill AP, Zhao Y, Anang NAS, Andrews MC, et al. Distinct Cellular Mechanisms Underlie Anti-CTLA-4 and Anti-PD-1 Checkpoint Blockade. Cell 2017, 170, 1120–1133.e17. DOI:10.1016/j.cell.2017.07.024 [Google Scholar]
- Nagasaki J, Ishino T, Togashi Y. Mechanisms of resistance to immune checkpoint inhibitors. Cancer Sci. 2022, 113, 3303–3312. DOI:10.1111/cas.15497 [Google Scholar]
- Lin Z, Zhou Y, Liu Z, Nie W, Cao H, Li S, et al. Deciphering the tumor immune microenvironment: Single-cell and spatial transcriptomic insights into cervical cancer fibroblasts. J. Exp. Clin. Cancer Res. 2025, 44, 194. DOI:10.1186/s13046-025-03432-5 [Google Scholar]
- Wang G, Xu J, Zhao J, Yin W, Liu D, Chen W, et al. Arf1-mediated lipid metabolism sustains cancer cells and its ablation induces anti-tumor immune responses in mice. Nat. Commun. 2020, 11, 220. DOI:10.1038/s41467-019-14046-9 [Google Scholar]
- Litchfield K, Reading JL, Puttick C, Thakkar K, Abbosh C, Bentham R, et al. Meta-analysis of tumor- and T cell-intrinsic mechanisms of sensitization to checkpoint inhibition. Cell 2021, 184, 596–614.e14. DOI:10.1016/j.cell.2021.01.002 [Google Scholar]
- Bejarano L, Jordao MJC, Joyce JA. Therapeutic Targeting of the Tumor Microenvironment. Cancer Discov. 2021, 11, 933–959. DOI:10.1158/2159-8290.CD-20-1808 [Google Scholar]
- Chowell D, Yoo SK, Valero C, Pastore A, Krishna C, Lee M, et al. Improved prediction of immune checkpoint blockade efficacy across multiple cancer types. Nat. Biotechnol. 2022, 40, 499–506. DOI:10.1038/s41587-021-01070-8 [Google Scholar]
- Frlic T, Pavlin M. Metabolic reprogramming of CAR T cells: A new frontier in cancer immunotherapy. Front. Immunol. 2025, 16, 1688995. DOI:10.3389/fimmu.2025.1688995 [Google Scholar]
- Imani S, Farghadani R, Roozitalab G, Maghsoudloo M, Emadi M, Moradi A, et al. Reprogramming the breast tumor immune microenvironment: Cold-to-hot transition for enhanced immunotherapy. J. Exp. Clin. Cancer Res. 2025, 44, 131. DOI:10.1186/s13046-025-03394-8 [Google Scholar]
- Wu TD, Madireddi S, de Almeida PE, Banchereau R, Chen YJ, Chitre AS, et al. Peripheral T cell expansion predicts tumour infiltration and clinical response. Nature 2020, 579, 274–278. DOI:10.1038/s41586-020-2056-8 [Google Scholar]
- Krishna C, DiNatale RG, Kuo F, Srivastava RM, Vuong L, Chowell D, et al. Single-cell sequencing links multiregional immune landscapes and tissue-resident T cells in ccRCC to tumor topology and therapy efficacy. Cancer Cell 2021, 39, 662–677.e6. DOI:10.1016/j.ccell.2021.03.007 [Google Scholar]
- Mengistu BA, Demessie Y, Kinde MZ, Getnet K, Bitew AB, Berrie K, et al. Biology of stem cell paradox: A double-edged sword-implications for cancer therapy. Cancer Cell Int. 2025, 25, 380. DOI:10.1186/s12935-025-03981-x [Google Scholar]
- Oura K, Morishita A, Tani J, Masaki T. Tumor Immune Microenvironment and Immunosuppressive Therapy in Hepatocellular Carcinoma: A Review. Int. J. Mol. Sci. 2021, 22, 5801. DOI:10.3390/ijms22115801 [Google Scholar]
- Nia HT, Liu H, Seano G, Datta M, Jones D, Rahbari N, et al. Solid stress and elastic energy as measures of tumour mechanopathology. Nat. Biomed. Eng. 2016, 1, 0004. DOI:10.1038/s41551-016-0004 [Google Scholar]
- Pickup MW, Mouw JK, Weaver VM. The extracellular matrix modulates the hallmarks of cancer. EMBO Rep. 2014, 15, 1243–1253. DOI:10.15252/embr.201439246 [Google Scholar]
- Kalluri R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. DOI:10.1038/nrc.2016.73 [Google Scholar]
- Canel M, Slawinska AD, Lonergan DW, Kallor AA, Upstill-Goddard R, Davidson C, et al. FAK suppresses antigen processing and presentation to promote immune evasion in pancreatic cancer. Gut 2023, 73, 131–155. DOI:10.1136/gutjnl-2022-327927 [Google Scholar]
- Stylianopoulos T, Martin JD, Chauhan VP, Jain SR, Diop-Frimpong B, Bardeesy N, et al. Causes, consequences, and remedies for growth-induced solid stress in murine and human tumors. Proc. Natl. Acad. Sci. USA 2012, 109, 15101–15108. DOI:10.1073/pnas.1213353109 [Google Scholar]
- Lou W, Gong C, Ye Z, Hu Y, Zhu M, Fang Z, et al. Lipid metabolic features of T cells in the Tumor Microenvironment. Lipids Health Dis. 2022, 21, 94. DOI:10.1186/s12944-022-01705-y [Google Scholar]
- Corrado M, Pearce EL. Targeting memory T cell metabolism to improve immunity. J. Clin. Investig. 2022, 132, e148546. DOI:10.1172/JCI148546 [Google Scholar]
- Tiggeler A, Coffer PJ. Acetyl-CoA subcellular compartmentalization regulates T cell adaptation. Cell Immunol. 2025, 414, 105000. DOI:10.1016/j.cellimm.2025.105000 [Google Scholar]
- Singh V, Nandi S, Ghosh A, Adhikary S, Mukherjee S, Roy S, et al. Epigenetic reprogramming of T cells: Unlocking new avenues for cancer immunotherapy. Cancer Metastasis Rev. 2024, 43, 175–195. DOI:10.1007/s10555-024-10167-w [Google Scholar]
- Zahalka AH, Frenette PS. Nerves in cancer. Nat. Rev. Cancer 2020, 20, 143–157. DOI:10.1038/s41568-019-0237-2 [Google Scholar]
- Kuol N, Stojanovska L, Apostolopoulos V, Nurgali K. Role of the nervous system in cancer metastasis. J. Exp. Clin. Cancer Res. 2018, 37, 5. DOI:10.1186/s13046-018-0674-x [Google Scholar]
- Aquino-Acevedo AN, Knochenhauer H, Castillo-Ocampo Y, Ortiz-Leon M, Rivera-Lopez YA, Morales-Lopez C, et al. Stress hormones are associated with inflammatory cytokines and attenuation of T-cell function in the ascites from patients with high grade serous ovarian cancer. Brain Behav. Immun. Health 2022, 26, 100558. DOI:10.1016/j.bbih.2022.100558 [Google Scholar]
- Battaglin F, Jayachandran P, Strelez C, Lenz A, Algaze S, Soni S, et al. Neurotransmitter signaling: A new frontier in colorectal cancer biology and treatment. Oncogene 2022, 41, 4769–4778. DOI:10.1038/s41388-022-02479-4 [Google Scholar]
- Pavelescu LA, Enache RM, Rosu OA, Profir M, Cretoiu SM, Gaspar BS. Predictive Biomarkers and Resistance Mechanisms of Checkpoint Inhibitors in Malignant Solid Tumors. Int. J. Mol. Sci. 2024, 25, 9659. DOI:10.3390/ijms25179659 [Google Scholar]
- Hou W, Zhao Y, Zhu H. Predictive Biomarkers for Immunotherapy in Gastric Cancer: Current Status and Emerging Prospects. Int. J. Mol. Sci. 2023, 24, 15321. DOI:10.3390/ijms242015321 [Google Scholar]
- Bruni D, Angell HK, Galon J. The immune contexture and Immunoscore in cancer prognosis and therapeutic efficacy. Nat. Rev. Cancer 2020, 20, 662–680. DOI:10.1038/s41568-020-0285-7 [Google Scholar]
- Cho E, Choi W, Lim JH, Son JW, Jang SH, Lee SH, et al. Temporal changes in tongue color during immune checkpoint inhibitor therapy in patients with non-small-cell lung cancer: A prospective observational study using digital tongue diagnosis. Oncol. Rev. 2025, 19, 1697252. DOI:10.3389/or.2025.1697252 [Google Scholar]
- Wan JCM, Massie C, Garcia-Corbacho J, Mouliere F, Brenton JD, Caldas C, et al. Liquid biopsies come of age: Towards implementation of circulating tumour DNA. Nat. Rev. Cancer 2017, 17, 223–238. DOI:10.1038/nrc.2017.7 [Google Scholar]
- Bagaev A, Kotlov N, Nomie K, Svekolkin V, Gafurov A, Isaeva O, et al. Conserved pan-cancer microenvironment subtypes predict response to immunotherapy. Cancer Cell 2021, 39, 845–865.e7. DOI:10.1016/j.ccell.2021.04.014 [Google Scholar]
- Tong M, Wang J, He W, Wang Y, Pan H, Li D, et al. Predictive biomarkers for tumor immune checkpoint blockade. Cancer Manag. Res. 2018, 10, 4501–4507. DOI:10.2147/CMAR.S179680 [Google Scholar]
- Du W, Hu S, Cai S, Wang Y, Wu L, Sun D, et al. Dynamic testing of stimulative and suppressive biomarkers on peripheral blood cells at early stages of immunotherapy predicts response in advanced cancer patients. Discov. Med. 2018, 25, 277–290. Available online: http://www.discoverymedicine.com/Wushuang-Du/2018/06/biomarkers-on-peripheral-blood-cells-at-early-stages-of-cancer-immunotherapy-predicts-response/ (accessed on 1 February 2026).
- Hui X, Tian X, Ding S, Sun A, Zhao T, Wang H. Reprogramming the tumor microenvironment to overcome immunotherapy resistance in pancreatic cancer. Front. Immunol. 2025, 16, 1717062. DOI:10.3389/fimmu.2025.1717062 [Google Scholar]
- Lin YY, Ding JL, Shen HT, Lin YM, Adhidjaja EC, Chang SC. PD-1/PD-L1 Cancer Immunotherapeutics Reshape Tumor Microenvironment—Clinical Evidence and Molecular Mechanisms for AI-based Precision Medicine. Clin. Rev. Allergy Immunol. 2025, 68, 91. DOI:10.1007/s12016-025-09105-7 [Google Scholar]
- Morana O, Wood W, Gregory CD. The Apoptosis Paradox in Cancer. Int. J. Mol. Sci. 2022, 23, 1328. DOI:10.3390/ijms23031328 [Google Scholar]
- Postow MA, Sidlow R, Hellmann MD. Immune-Related Adverse Events Associated with Immune Checkpoint Blockade. N. Engl. J. Med. 2018, 378, 158–168. DOI:10.1056/NEJMra1703481 [Google Scholar]
- Allen BM, Hiam KJ, Burnett CE, Venida A, DeBarge R, Tenvooren I, et al. Systemic dysfunction and plasticity of the immune macroenvironment in cancer models. Nat. Med. 2020, 26, 1125–1134. DOI:10.1038/s41591-020-0892-6 [Google Scholar]