Spontaneous Cell Fusion as the Mechanism of Cancer Progression and Metastasis

Spontaneous Cell Fusion as the Mechanism of Cancer Progression and Metastasis
Ruoxiang Wang
*,†

Received: 17 March 2026 Revised: 23 April 2026 Accepted: 11 May 2026 Published: 22 May 2026

© 2026 The authors. This is an open access article under the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/).
Graphical Abstract
1. Tumor Cell Heterogeneity Plays a Significant Role in PCa Progression and Metastasis
The prostate is not a vital organ. Mortality from PCa often results from widespread metastases to the bone and soft tissues rather than tumor burden at the primary site [1,2]. These metastases damage organ function, leading to cancer wasting or cachexia, and ultimately cancer-related death. Preventing metastasis is a critical therapeutic goal to protect patients from succumbing to cancer.
PCa has a unique racial and familial distribution. Men of Northern European descent and those with a family history are at higher risk [3,4]. This disparity suggests a potential involvement of the germline genome. While individuals at risk may develop a normal, healthy prostate for most of their adult life, the development of PCa in late adulthood is reminiscent of antagonistic pleiotropy observed in aging-related diseases [5,6,7]. Research has identified germline factors that contribute to various predispositions, though not specifically to PCa, but to multiple cancers [8,9]. Although a germline etiology for PCa remains to be identified, understanding the mechanisms of metastasis has long been a focus of cancer research.
PCa can evolve. While primary tumors are often seen in multiple foci [10], the metastasis of PCa is believed to be initiated by a single tumor cell [11]. Despite their single-cell origin, metastatic tumor cells exhibit high diversity, including various genomic abnormalities, asynchronous loss of the glandular epithelial phenotype, and the adoption of behaviors from other cell lineages. This diversity, known as tumor cell heterogeneity, is a primary reason for treatment failure as it ensures that some tumor cells will always survive specific therapies. Heterogeneity may also drive PCa metastasis as some tumor cells become invasive and migratory. The progression and metastasis can be viewed as the evolution of heterogeneity among tumor cells. Understanding tumor cell heterogeneity is essential for uncovering the mechanisms underlying PCa progression and metastasis.
2. The Mechanism of Tumor Cell Heterogeneity Remains Inadequately Explored
Tumor cell heterogeneity was identified almost fifty years ago [12,13,14,15,16,17,18]. This recognition came after the discovery of the central dogma as the molecular mechanism of genetic inheritance. The discovery paved the way for technologies based on nucleotide base-pairing. The central dogma, together with molecular genetic tools, was quickly adopted in cancer research before the recognition of tumor cell heterogeneity.
The central dogma outlines the flow of genetic information from genotype to phenotype. Cancer research has embraced this concept, assuming that mutations in the tumor cell genome lead to malignant behavior, akin to genetic inheritance. Before assessing the relevance of the central dogma to cancer progression, it has been widely accepted that genomic or genetic abnormalities are the probable cause of malignancy, with accumulating mutations propelling the progression of heterogeneity. The acceptance is so complete that there is a dearth of alternative hypotheses to prompt an exploration of a verifiable mechanism for the progression of heterogeneity.
After decades of global efforts and breakthrough discoveries in cancer research, the undeniable cause of tumor cell heterogeneity remains unclear, or at best, contentious. Effective strategies to halt the progression of tumor cell heterogeneity are lacking. This essay explores an alternative mechanism through which cancer cells acquire the ability to progress in heterogeneity.
3. Genotypic and Phenotypic Heterogeneities in PCa Tumor Cells
The heterogeneity of tumor cells appears endless, especially given the wide range of genotypic variation [19,20,21]. In comparison, there are limited criteria for evaluating phenotypic heterogeneity. In clinical studies, changes in prostate-specific antigen (PSA) levels, tumor growth, dormancy, recurrence, metastasis, remission, relapse, resistance to treatment, as well as cancer wasting, patient cachexia, and survival or mortality, serve as indicators of phenotypic heterogeneity. In laboratory investigations, signs of phenotypic heterogeneity include rates of proliferative activity, invasiveness, migratory activity, epithelial to mesenchymal transition, stem cell properties or lineage plasticity, resistance to drugs, and the ability to develop tumors or metastasize in animal models. Unlike genetic inheritance, where the connection between genotype and phenotype is often clear, a similar correlation has not been definitively established in the progression and metastasis of PCa. Discussing genotypic heterogeneity in conjunction with phenotypic diversity in PCa research can be complex. This essay adopts a reductionist approach to exploring tumor cell heterogeneity through genomic heterogeneity, with the expectation that the insights gained from this exploration can be applied to understanding phenotypic heterogeneity within the same cells.
4. Spontaneous PCa Cell Fusion Results in Tumor Cell Heterogeneity
Under the guidance of the late Dr. Leland W. K. Chung, our Molecular Urology and Therapeutics Program, followed by the Uro-Oncology Program, investigated the mechanisms of PCa metastasis by studying the impact of cancer-stromal interactions in the tumor microenvironment. This research was inspired by Chung’s groundbreaking discovery that tissue-specific mesenchymal stromal cells (MSCs) play a pivotal role in determining the fate of glandular epithelial cells [22,23,24,25,26]. Co-inoculation [23,27] or co-culture [28,29,30] with MSCs increased tumor cell heterogeneity in terms of both genotype and phenotype. These findings have led to a search for soluble factors that mediate cancer-stromal interactions. However, the findings of these studies have limited translational significance, as they may not be universally applicable to understanding the progression and metastasis of clinical PCa. After thorough research, it has become clear that the extent of tumor cell heterogeneity is so vast that the abnormalities identified in these studies may only represent a small portion of the irregularities present in PCa patients.
4.1. Cancer Cells Spontaneously Fuse with Adjacent Cells
By using fluorescence proteins to monitor cancer-stromal interactions, we have identified spontaneous cancer cell fusion as a unique feature with the potential for generalization. Commonly used human PCa cells, such as LNCaP, C4-2, C4-2B, ARCaPE, ARCaPM, PC-3, DU145, and even the latent HPE-15 cells [31], all have the ability to spontaneously fuse with prostate-derived MSCs (PrMSCs) [29,32,33], bone marrow-derived MSCs (BMMSCs) [30], or neural stem cells (NSCs) [34]. These cells are considered bystander cells typically found in the tumor microenvironment. Additionally, cells within the same PCa cell line can fuse with each other. Under the same experimental conditions, PCa cells from different cell lines exhibit varying levels of fusogenicity, with some being more fusogenic than others.
The discovery of fusogenicity in PCa cells is significant because cell fusion is one of the theories of cancer etiology [35,36,37]. As discussed later, spontaneous cancer cell fusion may explain clinical cancer progression and metastasis. Regardless of their androgen receptor status, level of PSA production, or aggressiveness in in vitro or in vivo assays, all these PCa cells have the potential for progression and metastasis under established experimental conditions.
4.2. The Fate of Fusion Hybrids Is Determined by the Proliferative Activity of the Cell Being Fused
While studying PCa cell fusion using in vitro models, we have encountered reports of similar fusion occurring in cells of other human cancer types. Interestingly, there is little evidence of cancer cell fusion found in vivo, or ex vivo in clinical tumor specimens. Apart from rare cases, such as patients with a history of allogeneic transplant [38], there are few parameters available for detecting cancer cell fusion in clinical cancer patients. However, mouse tumor models offer the ability to track fusion using fluorescence or xenogeneic markers [39,40].
In addition to explanations provided by other groups [41,42], we speculate that technical challenges in studying cancer cell fusion in vivo may contribute to certain histopathologic observations being open to alternative interpretations. For instance, cancer cell fusion can result in the creation of a hybrid cell with two or more nuclei, resembling the morphology of polyploid giant cancer cells [43]. However, the increased genomic material in such cells is typically attributed to mitotic failure. Similarly, the neuroendocrine property frequently observed during PCa progression and metastasis is usually explained as a result of neuroendocrine differentiation by cancer stem cells, stemming from the lineage plasticity in these cells. Nevertheless, in our studies, PCa cell fusion with NSCs effectively generated hybrid-derived clones, each exhibiting independent genomic hybridization along with distinct neuroendocrine behavior [34].
Until a tracking technology is established, obtaining direct in vivo evidence of cancer cell fusion will be challenging, and evaluating its frequency or clinical correlation will remain uncertain. With this rationale in mind, we utilized PCa cell co-culture with bystander cells to recreate cell fusion during cancer-stromal interaction.
In co-culture studies, long-term assessment of fusion or the fate of fusion hybrids is complex due to the high proliferative activity of cancer cells, which often leads to overgrowth. To address this issue, we focused on fusion with LNCaP or HPE-15 cells, as they have slower growth rates compared to other PCa cells [31]. Their fusion can be tracked with fluorescence proteins and validated by DNA fingerprinting when co-cultured with allogeneic bystander cells. The progression of their heterogeneity can be evaluated through xenograft tumor formation, as both cell lines are non-tumorigenic in athymic mice before fusion.
After fusion, the cancer cell becomes part of a cancer-bystander hybrid. An important finding from our observations is that the fate of the hybrid is determined by the proliferative activity of the bystander cell being fused, rather than the fusing cancer cell [32]. Terminally differentiated, most established PrMSC or BMMSC cell lines exhibit minimal proliferative activity. Their fusion with PCa cells results in binucleated or multinucleated hybrids that can survive for extended periods without dividing. By trapping cancer cells through fusion, a stromal cell may serve as a barrier to prevent cancer spread. On the other hand, growth arrest of the hybrids may provide a haven for cancer cells to survive in a dormant state.
In addition to terminally differentiated MSCs, the stromal compartment also contains mesenchymal stem cells or spontaneously transformed MSCs. In our study, some PrMSC cell lines established from clinical tumor specimens contained these cells. This was evidenced by the appearance of distinct colonies with faster growth rates as these cell lines were passaged [44]. Compared to the differentiated PrMSCs, cells from these colonies are smaller, fibroblastic, and faster growing. Surprisingly, many fusion hybrids with these cells are capable of undergoing cell division. Despite the difficulty of division for binuclear cells to produce viable hybrid progeny, nuclear fusion or synkaryon formation was frequently observed. This was followed by successful colony formation among many other hybrids that displayed aborted cell division and death. Proliferative activity in the bystander cell, therefore, plays a determinant role in the fate of the cancer-bystander hybrid.
This conclusion is well supported by additional observations. For instance, primary BMMSCs grow at slow rates. Although they can fuse with cancer cells, the fusion hybrids can barely form derivative hybrid clones [30]. In contrast, virally immortalized BMMSCs, such as HS-5 and HS-27a, have high proliferation rates. Most fusion hybrids with these cells can easily divide to form differently shaped derivative colonies.
For stem cells, self-renewal is supported by proliferative activity, while differentiation occurs after growth arrest. The role of proliferative status has been assessed through cancer cell fusion with rat embryonic NSCs [34]. PCa cells fuse effectively with NSCs on the neurospheres. Upon induction for differentiation, NSCs become growth arrested and adopt neural cell morphology, forming a network of astrocytes, neurons, and oligodendrocytes.
Throughout the process, the fate of the cancer-NSC fusion hybrids is documented through stepwise observation of: (1) accumulation of the hybrids on the neurosphere; (2) widespread death of the hybrids upon differentiation induction; (3) differentiation of the surviving hybrids into neuron-like cells; (4) eventual death in the majority of the differentiated hybrids; and (5) formation of hybrid derivative clones individually displaying markedly diversified neuroendocrine behaviors reminiscent of neuroendocrine differentiation or mimicry in clinical specimens. This study thus corroborates the determinant role of bystander cell proliferative activity from our study with MSCs [32].
In summary, all of our studies on the fate of fusion hybrids indicate that proliferative activity in the tumor microenvironment is either the culprit or accomplice for cancer cell fusion hybrids to form derivative progenies. As discussed in a later section, the generation of hybrid derivative cells is the cause of the ever-changing tumor cell heterogeneity.
4.3. Cancer Cell Fusion Occurs Spontaneously
PCa cells from different cell lines may behave differently when they fuse. For instance, LNCaP cells only show clear fusion with PrMSCs after a 7-day co-culture, while HPE-15 cells fuse within 2 days. The machinery for cell fusion may differ among individual cell lines. However, all PCa cell lines tested have shown the capability to fuse. Importantly, this fusion occurs spontaneously, indicating an inherent trait. The influence of external factors, such as growth factors or fetal bovine serum, is mainly quantitative and cannot completely prevent PCa cell fusion with PrMSCs, BMMSCs, NSCs, or other cancer cells. The spontaneity of fusogenicity carries pathological significance. Since fusion hybrids can produce derivative cells that contribute to tumor cell heterogeneity, the spontaneity of fusogenicity makes this heterogeneity a dynamic and ever-changing process, a hideous aspect of clinical cancer progression.
4.4. Cancer Cell Fusion Is a Common Phenomenon
Biologically, there are two types of cell fusion. The germline fusion of sperm with an oocyte results in hybridization and heterogeneity. Homotypic somatic fusion creates a syncytium with coordinated function. Studies on biological fusion, such as the formation of a zygote, syncytiotrophoblast, osteoclast, and skeletal myofiber; or bioengineering research, such as the development of monoclonal antibodies and the design of artificial cells, have shown that the mechanisms for fusion are encoded in the genome and carried out by dedicated surface proteins, or fusogens [45,46]. Given that cancer progression often involves dysregulated gene expression, it is not surprising that cell-fusion machinery is present in PCa cells.
In addition to PCa, tumor cells from various types of cancers, including breast, ovarian, gastric, lung, colorectal, and others, have been found to possess fusogenicity, capable of fusing spontaneously with bystander cells or with each other. Once improved techniques for detecting tumor cell fusion in clinical specimens are developed, the histopathologic significance of spontaneous cancer cell fusion will be more widely acknowledged. Through cell fusion, cancer cells can literally recruit bystander cells, a phenomenon that has been previously studied [47,48,49,50,51,52,53,54,55].
5. Clinical Relevance of Cancer Cell Fusion
The objective of our research is to clarify the mechanism of PCa progression and metastasis. We propose that this is due to phenotypic heterogeneity caused by the spontaneous fusion of cancer cells with proliferative bystander cells in the tumor microenvironment. This essay aims to demonstrate that the spontaneity of tumor cell fusogenicity is responsible for all pathological aspects of the clinical history of PCa.
5.1. The Random and Dynamic Nature of Tumor Cell Heterogeneity Progression
The study on PCa cell fusion has made important observations relevant to clinical PCa progression and metastasis. Firstly, the fusion process appears random, as PCa cells may fuse with any bystander cell type, leading to heterogeneity. Secondly, the fusion is highly dynamic, as progenies derived from fusion hybrids may inherit fusogenicity, further enhancing heterogeneity. Thirdly, fusion-induced heterogeneity is not directional, as the majority of fusion hybrids either end up in growth arrest without signs of cell division or die out due to failed cell division. Even in fusion with bystander cells of higher proliferative activity, only a minority fraction of hybrids can survive the initial rounds of cell division to form derivative colonies. It is this fraction of hybrids that is consequential to PCa progression and metastasis, because these cells can amplify heterogeneity within a tumor cell population.
5.1.1. Cancer Cells Fuse with Bystander Cells in a Seemingly Random Fashion
In co-culture studies, only a fraction of the input PCa cells fused with PrMSCs or BMMSCs, while other PCa cells in the same co-culture never initiated fusion. When fewer PCa cells were used, individual PCa cells may proliferate to form colonies on a layer of PrMSCs. Still, only a few cells of a colony displayed fusion with the surrounding MSCs, whereas it was impossible to predict which of the cells would eventually initiate a fusion event. Similar unpredictability was observed when co-culture conditions were manipulated to investigate modulating factors. No significant changes in the number of PCa cells participating in fusion were observed upon androgen deprivation, growth factor addition, or cell synchronization by serum starvation.
From other reports on various types of human cancers and animal tumors, it has been observed that cancer cells can spontaneously fuse with each other [56] or with a variety of bystander cells. These bystander cells include epithelial cells [57], endothelial cells [58,59], fibroblasts [60], osteoclasts [61], bone marrow-derived leukocytic cells [38], hematopoietic stem cells [62], monocytes [63], macrophages [64], dendritic cells, as well as myoblasts from either the skeletal or smooth muscle lineage [65]. Fusion with other cell types may also occur, but has not been studied comprehensively, partly due to the challenges in isolating or culturing terminally differentiated cell types for research purposes. Nonetheless, these findings suggest that cancer cells have the ability to randomly fuse with many different types of cells. Interestingly, tumor cells have been found to exhibit behaviors that mimic those of various cell types. It will be intriguing to investigate whether this mimicry is a result of cancer cell fusion with specific types of bystander cells.
5.1.2. Chromosomal Reorganization in Fusion Hybrids Occurs in an Almost Random Fashion
Unlike normal glandular epithelial cells, PCa cells undergo genomic reorganization [66,67,68,69,70,71]. Similarly, their fusion hybrids, heterokaryons or synkaryons, are capable of undergoing genomic reorganization and reducing chromosome numbers [72], likely through a meiosis-like process of crossover and diploidization [73]. Since all PCa cells are aneuploid, there will be a chromosomal mismatch between the fusing cancer cell and the one being fused. In their fusion hybrid, the meiosis-like process will cause deletions, insertions, or chromosomal loss. Genomic recombination occurs at almost random locations [74]. This almost random genomic recombination results in hybrid progenies with a genomic makeup that differs from both the cancer and bystander parents [34].
Because it takes place individually in each fusion hybrid, the almost random genomic recombination creates a unique genomic makeup for each hybrid clone, which is genomically different from the offspring derived from another hybrid. In our co-culture studies, a co-culture between multiple PCa cells and bystander cells has generated many derivative hybrid clones, each exhibiting distinct genotypic and phenotypic traits from the others [32,34]. The survival and proliferation of fusion hybrids therefore amplify tumor cell heterogeneity.
5.1.3. Passing on Fusogenicity Results in Ever-Changing Tumor Cell Heterogeneity
The spontaneity of fusogenicity can be passed down from parental PCa cells to hybrid progenies [32]. This ability allows progenies to fuse with various bystander cells, creating more hybrids. Some of these hybrids will survive and multiply, giving rise to the next-generation of progenies. These progenies can then fuse with even more bystanders, initiating another cycle of progeny production. The transmission of fusogenicity ensures the amplification of heterogeneity throughout the clinical progression of PCa (see Figure 1A). In this sense, the presence of a cancer cell with a specific genotype or phenotype is only temporary, as it is eliminated upon the cyclic fusion.
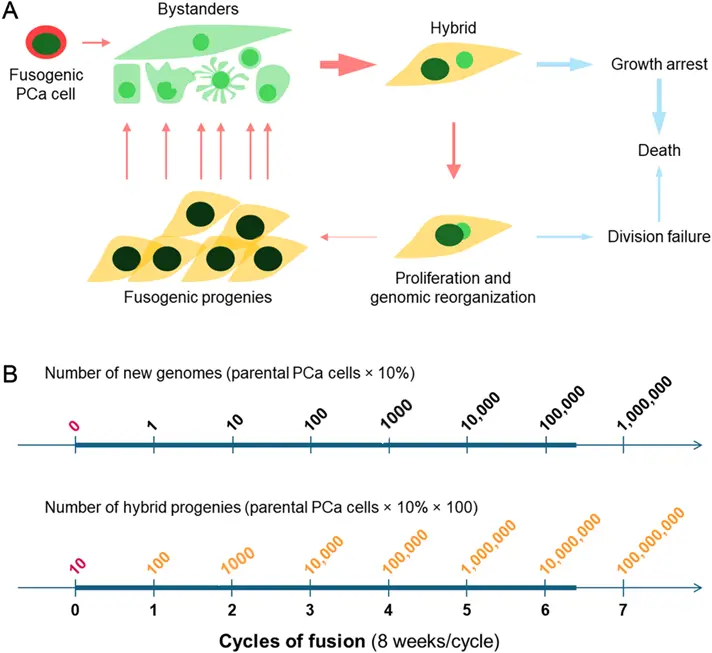
Figure 1. The spontaneity model of tumor cell heterogeneity. This model explores the potential role of PCa cell fusion in creating tumor cell heterogeneity. (A) Starting from the upper left, a fusogenic PCa cell (red) fuses with one of the bystander cells (green) in the tumor microenvironment, resulting in a hybrid with distinct characteristics from both parent cells. This hybrid may experience growth arrest and eventual death due to mitotic catastrophe. However, if the parent cells are synchronal in cell division, the hybrid may continue to proliferate, leading to genomic reorganization. This proliferation gives rise to multiple hybrid progenies, which may retain the fusogenicity of the parental PCa cell, initiating another cycle of fusion with more bystander cells and propagating tumor cell heterogeneity. (B) An estimation of tumor cell heterogeneity introduced through cyclic cancer cell fusion is provided. This estimation is based on the following assumptions: (1) Initially, there are 10 fusogenic PCa cells in the tumor; (2) Although most hybrids may die due to mitotic catastrophe, each fusion event has a 10% chance of producing progenies; (3) Each hybrid generates 100 clonal progenies; (4) Each cycle lasts 8 weeks; and (5) The fusogenicity is carried by all the progenies. The bold line represents a one-year period of disease progression.
The spontaneity of fusogenicity is a highly efficient strategy for creating tumor cell heterogeneity. It takes advantage of proliferative activity in bystander cells to increase the likelihood of generating hybrid progenies. In addition to somatic stem cells, progenitor cells, and precursor cells, bystander cells in the tumor microenvironment can be “re-activated” to proliferate [75]. On the other hand, maintaining fusogenicity from one generation to the next intensifies the heterogeneity (see Figure 1B). The spontaneity of fusogenicity alone can create dynamic PCa cell heterogeneity in both qualitative and quantitative terms. This statement is supported by the observation that the extent of tumor cell heterogeneity is so significant that in each patient, no two cancer cells appear to share the same genotype and phenotype [21,76,77].
In summary, the spontaneity model of tumor cell heterogeneity treats the fusion of PCa cells, which leads to tumor cell heterogeneity, as a random process. While the homotypic fusion of PCa cells may increase malignancy, the heterotypic fusion between PCa and bystander cells creates heterogeneity, which can then be fixed through genomic hybridization and amplified by the formation of hybrid progeny. As long as a cancer cell is capable of fusion, its genotype will be temporary and likely to change with each fusion event. This model argues that there is no programmed mechanism for the directional progression of tumor cell heterogeneity.
5.2. Cancer Cell Fusion Is the Initial Step in the Progression of Tumor Cell Heterogeneity
The fusion of PCa cells with bystander cells leads to cytoplasmic fusion and tumor cell heterogeneity, which is then fixed and propagated through genomic hybridization. Understanding the mechanisms of cell fusion could aid in identifying targets to prevent the progression and metastasis of PCa.
Various proteins act as fusogens [45,78,79]. Some, such as Izumo1, Juno, and SPACA6, are essential for sperm-egg fusion [80,81,82]. Others, such as syncytin-1, syncytin-2, ASCT-1, and ASCT-2, contribute to the formation of syncytiotrophoblast [83,84]. Myomaker and myomerger play a role in myoblast fusion and myofiber formation [85]. In osteoclast formation, proteins like OC-STAMP and DC-STAMP are involved [86]. Certain tetraspanin proteins (TSPAN), including CD9, CD63, and CD81, are present in various cell fusion processes [87,88,89,90,91]. Conversely, CD82 acts as a decoy to prevent cell fusion and cancer metastasis [92,93]. Intracellular proteins like AnxA1, AnxA5 from the annexin family, and actin may also play a role in cell fusion [94].
PCa tumors express fusogens. Unlike CD82, the expression of syncytin-1 and AnxA5 is positively correlated with Gleason Scores [65]. Through transcript analysis, we have found that many of the 33 genes in the TSPAN family are expressed in PCa cell lines. Further research will help pinpoint the fusogen responsible for PCa cell fusion. Importantly, the spontaneity model of tumor cell heterogeneity suggests that genomic or genetic analysis of tumor cell heterogeneity examines the result, rather than the cause, of cancer progression.
5.3. The Lack of Genetic Markers for Cancer Progression
As PCa progresses, the level of tumor cell heterogeneity becomes more prevalent. Genotypically, a variety of mutations, both large (at the chromosomal level) and small (at the DNA sequence level) are present in almost every cancer cell studied. Among individual cells, the mutations are so varied that efforts to determine clonality or establish a unified genetic profile are deemed impossible [19].
The spontaneity of tumor cell heterogeneity is well documented in PCa cell lines. While small mutations require DNA sequencing for detection, chromosomal changes are readily seen through cytogenetic analysis. In each cell line, a high level of chromosomal heterogeneity has been documented among individual cells. For instance, individual LNCaP cells have different chromosomal counts and chromosomal abnormalities [95], which also differ from the LNCaP derivative sublines C4, C5, C4-2, C4-2B2, C4-2B3, C4-2B4, and C4-2B5 [27,96]. Although LNCaP-FGC cells are derived from a single LNCaP cell, cloning did not prevent LNCaP-FGC cells from chromosomal changes. The HPE-15 cell line is established from a single cell of a PCa tumor resection. Although HPE-15 cells contain the least karyotype anomalies among established PCa cell lines, none of the twenty HPE-15 cells examined shares an identical karyotype [31]. Importantly, the anomalies are ever-changing. This is best studied with cells of the PC-3 lineage, in which chromosomal anomalies change with cell passaging [97]. In fact, the changes are so frequent that a numerical range of chromosomal counts, along with a representative group of marker chromosomes, must be used to depict cytogenetic features for each PCa cell line.
The genome in cancer cells is said to be unstable, with erratic cell division and defects in genomic integrity surveillance mechanisms considered the main culprits. However, the exact reason why the cancer genome becomes so unstable remains a topic of debate. It is also unclear whether genomic instability is the root cause of tumor cell heterogeneity. Since cancer cells can fuse, leading to genomic changes, it would be intriguing to determine whether spontaneous fusion is the cause of genomic instability.
In the clinical management of PCa, the progression of tumor cell heterogeneity is primarily evaluated through phenotypic changes, especially when a previously indolent disease becomes aggressive. There is a lack of genetic markers for metastasis, and purported markers are unable to accurately indicate the severity of the disease.
At the translational level, prostate-specific proteins such as PSA, PSMA, and PrLZ, which are typically expressed in glandular epithelium and primary tumors, are often lost asynchronously during metastasis. Proteins from other cell lineages may begin to express in the tumor, often in a sporadic or focalized fashion. The presence of growth- or survival-promoting proteins, whether they are growth factors, cell-cycle regulators, or transcription factors, has limited diagnostic value. On the contrary, the extent of expressional heterogeneity of proteins is a sure sign of cancer progression to a highly advanced stage.
In PCa cells, if protein expressions correlate with transcriptional activity, changes in protein levels should indicate whether the corresponding gene is in an on or off state. Based on this assumption, random protein expression would yield a positivity score close to 50% in a highly heterogeneous tumor cell population. This scenario is often observed in immunohistochemical analyses, in which positivity for candidate markers decreases in advanced tumors.
If genomic heterogeneity is the driving force behind cancer progression, a common pattern of genomic changes is expected to be present in metastatic PCa tumors, with specific tropistic proteins facilitating this process. Despite decades of research, however, a biomarker for PCa metastasis has yet to be identified, even though the basic structure of the human genome and the number of protein-coding genes have long been known. Tumor cell heterogeneity is probably so vast that they do not share common genomic or expression features.
The spontaneity model of tumor cell heterogeneity implies that the progression of heterogeneity is a random and dynamic process. Naturally, hybrids formed randomly will experience a purifying selection-like pressure for survival and proliferation. With extensive heterogeneity, some hybrid progenies will always be positively selected. This model implies that genomic mutations are the consequence of cancer cell fusion, and specific or directional genomic mutations are not necessary for the progression of tumor cell heterogeneity.
5.4. Alternative and Non-Genetic Mechanisms Should Be Examined
The progression of tumor cell heterogeneity is believed to be driven by an accumulation of mutations. The spontaneity model of tumor cell heterogeneity offers an alternative mechanism, placing genomic mutations in a passive role, distinct from the inheritance-based scenario.
PCa cells in a tumor are thought to have survival and growth advantages. Counterintuitively, frequent cell deaths are seen within the same tumor before any antitumor treatment is administered, and the high extent of spontaneous cell death is indicative of a poor prognosis [98,99,100,101]. As the objective of antitumor treatments is to eliminate cancer cells, it is puzzling how an increase in PCa cell death in untreated tumors is linked to a worsened disease outcome.
Our observations on the fate of PCa cell fusion suggest that most of the hybrids would eventually die. This is likely because binucleated or multinucleated hybrids, and even some synkaryons, face a spatiotemporal conundrum when entering cell division. The timing of cell division, or the rhythm of the cell cycle, is primarily controlled by factors in the nucleus [102,103], while machineries of karyokinesis and cytokinesis execute the physical division. Unless the rhythms of individual nuclei are synchronal or within a tolerable range, a hybrid is unable to complete a successful cell division and will die through the mechanism of mitotic catastrophe [104,105,106,107]. In cases of fusion with terminally differentiated MSCs, the main fate of the hybrids is death because their proliferative activity is too different. According to this rationale, the death of tumor cells prior to treatment is due to spontaneous cell fusion.
Escaping from mitotic catastrophe is possible when PCa cells fuse with cells that have similar proliferative activity. Besides neighboring cancer cells, many bystander cells have the potential to proliferate. In the mesenchymal tumor microenvironment, terminally differentiated MSCs can become reactive with increased desmoplastic activity. Mesenchymal stem cells maintain cellular homeostasis through proliferation. Fusion with a proliferative neighbor helps the hybrid survive mitotic catastrophe, produce derivative progeny, and initiate a progression of heterogeneity.
This rationale may help explain several puzzling clinical observations. For instance, metastatic tumors are rarely found in cardiac or skeletal muscles, despite these muscles being well perfused with blood, which would facilitate tumor cell homing. A cancer cell fusing with these types of cells would be doomed to mitotic catastrophe, as myofibers are terminally differentiated and lack proliferative activity. However, fusion hybrids can form, and behavioral changes can occur when PCa cells fuse with precursor myoblasts, although the fate of these hybrids remains to be examined [65]. The death from mitotic catastrophe may prevent the formation of metastatic tumors in muscle tissues. On the other hand, chronic bombardment by fusing cancer cells may eventually lead to the loss of myofibers, a typical sign of cancer wasting or cachexia.
For another example, after tumor resection, there may be metastatic tumor formation in the surgical wound [108,109,110]. Mesenchymal stem cells increase their proliferative activity for wound repair, creating a favorable condition for cancer-stem cell hybrids to generate derivative progenies. Cancer progression is described as a wound healing process that never fully heals [111,112].
5.5. PCa Oncogenesis and PCa Metastasis Are Two Distinct Aspects of the Same Disease
If driven by the accumulation of genetic mutations, the progression and metastasis of PCa would be a continuous process. However, findings from pathological epidemiology suggest a different scenario. Although Caucasian men have a much higher chance of having metastatic disease than Asian men, multiple autopsy studies on men who died from causes other than PCa revealed three discordant phenomena. First, these autopsies detected age-related primary tumors in both the Caucasian and Asian groups [113,114,115]. Second, the incidences in both ethnic groups are much higher than the incidence of PCa metastases observed in the clinical setting [113,116]. Third, latent PCa tumors in Asians are found with even higher Gleason scores than in Caucasians, even though PCa metastasis is rare in Asians [113,117]. Results from these studies argue for a distinction between Caucasians and Asians in the metastasis of their PCa tumors.
The racial disparity between local PCa tumor formation and distant metastasis may indicate separate mechanisms for these processes. The author is unsure whether genetic mutations cause initial oncogenesis in the glandular prostate epithelial layer. However, metastasis of PCa tumors is a random and dynamic process, facilitated by various bystander cells in the tumor microenvironment. Investigating the racial disparity, specifically in metastasis, is crucial because, for PCa at least, metastasis is the primary cause of cancer-related deaths, while the majority of local PCa cases can be cured with surgery or hormonal therapy.
In summary, the spontaneity model of tumor cell heterogeneity is based on two crucial elements: the spontaneous fusion of PCa cells to initiate phenotypic heterogeneity, and the proliferation of the hybrid to introduce genomic heterogeneity. Since fusion is mediated by fusogens that are already present on PCa cells, the progression of heterogeneity may not require additional genetic involvement. Cancer oncogenesis and tumor metastasis may have different causes. Our studies strongly suggest that non-genetic mechanisms are responsible for the random and dynamic progression of tumor cell heterogeneity.
5.6. Lessons from Our Molecular Genetic Studies of PCa Tumor Cell Heterogeneity
The concept of randomness and dynamism surrounding tumor cell heterogeneity presents a significant challenge for PCa research. It implies that specimens or cell lines taken from tumors may represent the current state of the disease, but not its future progression. One should not be surprised, then, that cancer studies are often found to have low reproducibility. In the worst scenario, if cancer cell fusion is proven to be the initial step of heterogeneity progression, molecular abnormalities found in tumor cells could be secondary, lacking prognostic value. This concept also raises questions about how we apply our understanding of genetic inheritance to cancer research.
The primary function of the genome is in inheritance. In humans, inheritance is carried out by the genome of germline cells. The germline genome is immortal and is maintained by multiple mechanisms to minimize genomic mutations caused by DNA replication errors [118,119]. The integrity of the germline genome is also under strict purifying selection [120]. In contrast, the somatic genome functions to sustain individual organisms and dissipates as the individual dies. It has little impact on inheritance and is not subject to the pressure of purifying selection [120]. Somatic cells undergo numerous rounds of division to build body mass, while the somatic genome may acquire mutations in the process. In addition to replication error-induced alterations, DNA damage from external risks is common in somatic cells [121,122]. In both the animal and plant kingdoms, there is a clear distinction between the genomes of the germline and the soma [123,124], with the fidelity of the germline genome significantly higher than that of the somatic genome [121].
Importantly, somatic genomes are now known to carry within-individual variations. In addition to replication-induced base-pair changes, many other alterations appear to be programmed for somatic differentiation. Along with frequent mutation and recombination [125,126], somatic genomes experience development-associated ploidy changes [127], programmed DNA elimination or chromatin diminution [128], uniparental genome elimination [129], and even the elimination of entire chromosomes.
The extent of germline/soma distinction in humans has not been fully explored. Tissue-specific genome differences exist [130], while the entire genome is eliminated from mature red blood cells. It is unclear whether within-individual genome variation, especially between germline cells and terminally differentiated somatic cells, is as common as in other species. In this context, a “patchwork” for the human somatic genome has been assembled using DNA sequencing data from peripheral blood mononuclear cells of multiple adult donors from various ethnic backgrounds [131]. The makeup of a human germline genome is yet to be determined.
For decades, research on metastasis has focused on identifying mutations by comparing the cancer genome with the assembled somatic genome, without control samples from the patient. Due to the extensive heterogeneity among PCa tumor cells, it is challenging to determine a consensus profile of genetic abnormalities.
This assertion is based on the results of our molecular genetic investigations in both PCa cell lines and clinical tumor specimens. At the genomic level, clinical tumor cells exhibit a variety of abnormalities, with the quantity increasing as more tumor cells are examined. Similar challenges have been encountered in studies at the transcription or translation level. Few of these findings could be generalized as markers of cancer progression and metastasis.
6. Conclusions
Through examining PCa cell fusion with each other or with bystander cells, we have identified that tumor cell heterogeneity is generated by fusion hybrids in a random and dynamic fashion. This essay aims to highlight the importance of addressing these characteristics rather than the resulting anomalies. Future studies are needed to identify the initiating role of spontaneous fusion in the progression of tumor cell heterogeneity. Intervening in the underlying mechanism may be an evidence-based strategy for limiting PCa progression and metastasis.
Acknowledgments
The text was edited by Michelle Hanna Wang.
Ethics Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Funding
This research was funded by NIH grants CA98912, CA112330, CA132388, and CA256419; by DoD grants CA170974 and PC040578; and by the 2022 Cedars-Sinai Cancer Development Funds for Team-Based Science Award.
Declaration of Competing Interest
The author declares that he has no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
-
Bubendorf L, Schopfer A, Wagner U, Sauter G, Moch H, Willi N, et al. Metastatic patterns of prostate cancer: An autopsy study of 1589 patients. Hum. Pathol. 2000, 31, 578–583. DOI:10.1053/hp.2000.6698 [Google Scholar]
-
Schroder FH. Current concepts in the management of prostatic cancer. Am. J. Clin. Oncol. 1988, 11 (Suppl. S1), S1–S5. Available online: https://journals.lww.com/amjclinicaloncology/citation/1988/12001/Current_Concepts_in_the_Management_of_Prostatic.1.aspx (accessed on 2 Feburary 2026).
-
Bray F, Laversanne M, Sung H, Ferlay J, Siegel RL, Soerjomataram I, et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2024, 74, 229–263. DOI:10.3322/caac.21834 [Google Scholar]
-
Gunderson K, Wang CY, Wang R. Global prostate cancer incidence and the migration, settlement, and admixture history of the Northern Europeans. Cancer Epidemiol. 2011, 35, 320–327. DOI:10.1016/j.canep.2010.11.007 [Google Scholar]
-
Byars SG, Voskarides K. Antagonistic Pleiotropy in Human Disease. J. Mol. Evol. 2020, 88, 12–25. DOI:10.1007/s00239-019-09923-2 [Google Scholar]
-
Troen BR. The biology of aging. Mt. Sinai J. Med. 2003, 70, 3–22. Available online: https://www.academia.edu/download/88001972/Troen_Biolog_of_Aging.pdf (accessed on 2 Feburary 2026).
-
Waters DJ, Shen S, Glickman LT. Life expectancy, antagonistic pleiotropy, and the testis of dogs and men. Prostate 2000, 43, 272–277. DOI:10.1002/1097-0045(20000601)43:4<272::aid-pros6>3.0.co;2-d [Google Scholar]
-
Tuffaha H, Edmunds K, Fairbairn D, Roberts MJ, Chambers S, Smith DP, et al. Guidelines for genetic testing in prostate cancer: A scoping review. Prostate Cancer Prostatic Dis. 2024, 27, 594–603. DOI:10.1038/s41391-023-00676-0 [Google Scholar]
-
Simard J, Dumont M, Labuda D, Sinnett D, Meloche C, El-Alfy M, et al. Prostate cancer susceptibility genes: Lessons learned and challenges posed. Endocr. Relat. Cancer 2003, 10, 225–259. DOI:10.1677/erc.0.0100225 [Google Scholar]
-
Andreoiu M, Cheng L. Multifocal prostate cancer: Biologic, prognostic, and therapeutic implications. Hum. Pathol. 2010, 41, 781–793. DOI:10.1016/j.humpath.2010.02.011 [Google Scholar]
-
Liu W, Laitinen S, Khan S, Vihinen M, Kowalski J, Yu G, et al. Copy number analysis indicates monoclonal origin of lethal metastatic prostate cancer. Nat. Med. 2009, 15, 559–565. DOI:10.1038/nm.1944 [Google Scholar]
-
Calabresi P, Dexter DL, Heppner GH. Clinical and pharmacological implications of cancer cell differentiation and heterogeneity. Biochem. Pharmacol. 1979, 28, 1933–1941. DOI:10.1016/0006-2952(79)90647-6 [Google Scholar]
-
Heppner GH, Dexter DL, DeNucci T, Miller FR, Calabresi P. Heterogeneity in drug sensitivity among tumor cell subpopulations of a single mammary tumor. Cancer Res. 1978, 38, 3758–3763. Available online: https://aacrjournals.org/cancerres/article-abstract/38/11_Part_1/3758/482716 (accessed on 2 Feburary 2026).
-
Dexter DL, Kowalski HM, Blazar BA, Fligiel Z, Vogel R, Heppner GH. Heterogeneity of tumor cells from a single mouse mammary tumor. Cancer Res. 1978, 38, 3174–3181. Available online: https://aacrjournals.org/cancerres/article-abstract/38/10/3174/482372 (accessed on 2 Feburary 2026).
-
Heppner G, Yamashina K, Miller B, Miller F. Tumor heterogeneity in metastasis. Prog. Clin. Biol. Res. 1986, 212, 45–59. PMID:3714732. [Google Scholar]
-
Smith HS, Wolman SR, Hackett AJ. The biology of breast cancer at the cellular level. Biochim. Biophys. Acta 1984, 738, 103–123. DOI:10.1016/0304-419x(84)90009-x [Google Scholar]
-
Miller BE, Miller FR, Heppner GH. Assessing tumor drug sensitivity by a new in vitro assay which preserves tumor heterogeneity and subpopulation interactions. J. Cell. Physiol. 1984, 121, 105–116. DOI:10.1002/jcp.1041210413 [Google Scholar]
-
Heppner GH. Tumor heterogeneity. Cancer Res. 1984, 44, 2259–2265. [Google Scholar]
-
Erickson A, Hayes A, Rajakumar T, Verrill C, Bryant RJ, Hamdy FC, et al. A Systematic Review of Prostate Cancer Heterogeneity: Understanding the Clonal Ancestry of Multifocal Disease. Eur. Urol. Oncol. 2021, 4, 358–369. DOI:10.1016/j.euo.2021.02.008 [Google Scholar]
-
Miyahira AK, Den RB, Carlo MI, de Leeuw R, Hope TA, Karzai F, et al. Tumor cell heterogeneity and resistance; report from the 2018 Coffey-Holden Prostate Cancer Academy Meeting. Prostate 2019, 79, 244–258. DOI:10.1002/pros.23729 [Google Scholar]
-
Yadav SS, Stockert JA, Hackert V, Yadav KK, Tewari AK. Intratumor heterogeneity in prostate cancer. Urol. Oncol. 2018, 36, 349–360. DOI:10.1016/j.urolonc.2018.05.008 [Google Scholar]
-
Chung LW. Implications of stromal-epithelial interaction in human prostate cancer growth, progression and differentiation. Semin. Cancer Biol. 1993, 4, 183–192. Available online: https://europepmc.org/article/med/8318694 accessed on 2 Feburary 2026). [Google Scholar]
-
Gleave ME, Hsieh JT, von Eschenbach AC, Chung LW. Prostate and bone fibroblasts induce human prostate cancer growth in vivo: Implications for bidirectional tumor-stromal cell interaction in prostate carcinoma growth and metastasis. J. Urol. 1992, 147, 1151–1159. DOI:10.1016/s0022-5347(17)37506-7 [Google Scholar]
-
Thompson TC, Chung LW. Regulation of overgrowth and expression of prostatic binding protein in rat chimeric prostate gland. Endocrinology 1986, 118, 2437–2444. DOI:10.1210/endo-118-6-2437 [Google Scholar]
-
Thompson TC, Cunha GR, Shannon JM, Chung LW. Androgen-induced biochemical responses in epithelium lacking androgen receptors: Characterization of androgen receptors in the mesenchymal derivative of urogenital sinus. J. Steroid Biochem. 1986, 25, 627–634. DOI:10.1016/0022-4731(86)90004-x [Google Scholar]
-
Pathak S, Nemeth MA, Multani AS, Thalmann GN, von Eschenbach AC, Chung LW. Can cancer cells transform normal host cells into malignant cells? Br. J. Cancer 1997, 76, 1134–1138. DOI:10.1038/bjc.1997.524 [Google Scholar]
-
Wu HC, Hsieh JT, Gleave ME, Brown NM, Pathak S, Chung LW. Derivation of androgen-independent human LNCaP prostatic cancer cell sublines: Role of bone stromal cells. Int. J. Cancer 1994, 57, 406–412. DOI:10.1002/ijc.2910570319 [Google Scholar]
-
Rhee HW, Zhau HE, Pathak S, Multani AS, Pennanen S, Visakorpi T, et al. Permanent phenotypic and genotypic changes of prostate cancer cells cultured in a three-dimensional rotating-wall vessel. In Vitro Cell Dev. Biol. Anim. 2001, 37, 127–140. DOI:10.1290/1071-2690(2001)037<0127:PPAGCO>2.0.CO;2 [Google Scholar]
-
Sung SY, Hsieh CL, Law A, Zhau HE, Pathak S, Multani AS, et al. Coevolution of prostate cancer and bone stroma in three-dimensional coculture: Implications for cancer growth and metastasis. Cancer Res. 2008, 68, 9996–10003. DOI:10.1158/0008-5472.CAN-08-2492 [Google Scholar]
-
Wang R, Sun X, Wang CY, Hu P, Chu CY, Liu S, et al. Spontaneous cancer-stromal cell fusion as a mechanism of prostate cancer androgen-independent progression. PLoS ONE 2012, 7, e42653. DOI:10.1371/journal.pone.0042653 [Google Scholar]
-
Wang R, Chu GC, Wang X, Wu JB, Hu P, Multani AS, et al. Establishment and characterization of a prostate cancer cell line from a prostatectomy specimen for the study of cellular interaction. Int. J. Cancer 2019, 145, 2249–2259. DOI:10.1002/ijc.32370 [Google Scholar]
-
Wang R, Hu P, Wang F, Lyu J, Ou Y, Edderkaoui M, et al. Spontaneous Fusion with Transformed Mesenchymal Stromal Cells Results in Complete Heterogeneity in Prostate Cancer Cells. Cancers 2024, 16, 951. DOI:10.3390/cancers16050951 [Google Scholar]
-
Wang R, Lewis MS, Lyu J, Zhau HE, Pandol SJ, Chung LWK. Cancer-stromal cell fusion as revealed by fluorescence protein tracking. Prostate 2020, 80, 274–283. DOI:10.1002/pros.23941 [Google Scholar]
-
Yin L, Hu P, Shi X, Qian W, Zhau HE, Pandol SJ, et al. Cancer cell’s neuroendocrine feature can be acquired through cell-cell fusion during cancer-neural stem cell interaction. Sci. Rep. 2020, 10, 1216. DOI:10.1038/s41598-020-58118-z [Google Scholar]
-
Boveri T. Concerning the origin of malignant tumours by Theodor Boveri. Translated and annotated by Henry Harris. J. Cell Sci. 2008, 121 (Suppl. S1), 1–84. DOI:10.1242/jcs.025742 [Google Scholar]
-
Pawelek JM, Chakraborty AK. Fusion of tumour cells with bone marrow-derived cells: A unifying explanation for metastasis. Nat. Rev. Cancer 2008, 8, 377–386. DOI:10.1038/nrc2371 [Google Scholar]
-
Aichel O. About cell fusion with quantitatively abnormal chromosome distribution as cause for tumor formation. In Vortrage und Aufsatze uber Entvickelungsmechanik der Organismen; Roux W, Ed.; Wilhelm Engelmann: Leipzig, Germany, 1911; pp. 92–111. [Google Scholar]
-
Laberge GS, Duvall E, Haedicke K, Pawelek J. Leukocyte(-)Cancer Cell Fusion-Genesis of a Deadly Journey. Cells 2019, 8, 170. DOI:10.3390/cells8020170 [Google Scholar]
-
Nakamura M, Suetsugu A, Hasegawa K, Satake T, Kunisada T, Shimizu M, et al. Color-coded Imaging Distinguishes Cancer Cells, Stromal Cells, and Recombinant Cancer-stromal Cells in the Tumor Microenvironment During Metastasis. Anticancer Res. 2018, 38, 4417–4423. DOI:10.21873/anticanres.12743 [Google Scholar]
-
Sun C, Dai X, Zhao D, Wang H, Rong X, Huang Q, et al. Mesenchymal stem cells promote glioma neovascularization in vivo by fusing with cancer stem cells. BMC Cancer 2019, 19, 1240. DOI:10.1186/s12885-019-6460-0 [Google Scholar]
-
Shultes PV, Weaver DT, Tadele DS, Barker-Clarke RJ, Scott JG. Cell-cell fusion in cancer: The next cancer hallmark? Int. J. Biochem. Cell Biol. 2024, 175, 106649. DOI:10.1016/j.biocel.2024.106649 [Google Scholar]
-
Weiler J, Dittmar T. Cell Fusion in Human Cancer: The Dark Matter Hypothesis. Cells 2019, 8, 132. DOI:10.3390/cells8020132 [Google Scholar]
-
Casalino F, Hossein G, Cohen M. From development to cancer: Wnt/beta-Catenin signaling in cell fusion and polyploid giant cancer cell formation. Cancer Lett. 2026, 643, 218323. DOI:10.1016/j.canlet.2026.218323 [Google Scholar]
-
Sun X, He H, Xie Z, Qian W, Zhau HE, Chung LW, et al. Matched pairs of human prostate stromal cells display differential tropic effects on LNCaP prostate cancer cells. In Vitro Cell Dev. Biol. Anim. 2010, 46, 538–546. DOI:10.1007/s11626-010-9309-z [Google Scholar]
-
Podbilewicz B. Cell fusion. WormBook 2006, 1–32. DOI:10.1895/wormbook.1.52.1 [Google Scholar]
-
Vance TDR, Lee JE. Virus and eukaryote fusogen superfamilies. Curr. Biol. 2020, 30, R750–R754. DOI:10.1016/j.cub.2020.05.029 [Google Scholar]
-
Basheer AS, Abas F, Othman I, Naidu R. Role of Inflammatory Mediators, Macrophages, and Neutrophils in Glioma Maintenance and Progression: Mechanistic Understanding and Potential Therapeutic Applications. Cancers 2021, 13, 4226. DOI:10.3390/cancers13164226 [Google Scholar]
-
Bussolati B, Grange C, Camussi G. Tumor exploits alternative strategies to achieve vascularization. FASEB J. 2011, 25, 2874–2882. DOI:10.1096/fj.10-180323 [Google Scholar]
-
Cirri P, Chiarugi P. Cancer-associated-fibroblasts and tumour cells: A diabolic liaison driving cancer progression. Cancer Metastasis Rev. 2012, 31, 195–208. DOI:10.1007/s10555-011-9340-x [Google Scholar]
-
Dittmar T, Seidel J, Zaenker KS, Niggemann B. Carcinogenesis driven by bone marrow-derived stem cells. Contrib. Microbiol. 2006, 13, 156–169. DOI:10.1159/000092971 [Google Scholar]
-
Gutmann DH. The Sociobiology of Brain Tumors. Adv. Exp. Med. Biol. 2020, 1225, 115–125. DOI:10.1007/978-3-030-35727-6_8 [Google Scholar]
-
Meseure D, Drak Alsibai K, Nicolas A. Pivotal role of pervasive neoplastic and stromal cells reprogramming in circulating tumor cells dissemination and metastatic colonization. Cancer Microenviron. 2014, 7, 95–115. DOI:10.1007/s12307-014-0158-2 [Google Scholar]
-
Ralph SJ, Reynolds MJ. Intratumoral pro-oxidants promote cancer immunotherapy by recruiting and reprogramming neutrophils to eliminate tumors. Cancer Immunol. Immunother. 2023, 72, 527–542. DOI:10.1007/s00262-022-03248-8 [Google Scholar]
-
Silverman DA, Martinez VK, Dougherty PM, Myers JN, Calin GA, Amit M. Cancer-Associated Neurogenesis and Nerve-Cancer Cross-talk. Cancer Res. 2021, 81, 1431–1440. DOI:10.1158/0008-5472.CAN-20-2793 [Google Scholar]
-
Ziaee S, Chu GC, Huang JM, Sieh S, Chung LW. Prostate cancer metastasis: Roles of recruitment and reprogramming, cell signal network and three-dimensional growth characteristics. Transl. Androl. Urol. 2015, 4, 438–454. DOI:10.3978/j.issn.2223-4683.2015.04.10 [Google Scholar]
-
Wiener F, Klein G, Harris H. The analysis of malignancy by cell fusion. VI. Hybrids between different tumour cells. J. Cell Sci. 1974, 16, 189–198. DOI:10.1242/jcs.16.1.189 [Google Scholar]
-
Bhatia B, Multani AS, Patrawala L, Chen X, Calhoun-Davis T, Zhou J, et al. Evidence that senescent human prostate epithelial cells enhance tumorigenicity: Cell fusion as a potential mechanism and inhibition by p16INK4a and hTERT. Int. J. Cancer 2008, 122, 1483–1495. DOI:10.1002/ijc.23222 [Google Scholar]
-
Bjerregaard B, Holck S, Christensen IJ, Larsson LI. Syncytin is involved in breast cancer-endothelial cell fusions. Cell Mol. Life Sci. 2006, 63, 1906–1911. DOI:10.1007/s00018-006-6201-9 [Google Scholar]
-
Mortensen K, Lichtenberg J, Thomsen PD, Larsson LI. Spontaneous fusion between cancer cells and endothelial cells. Cell Mol. Life Sci. 2004, 61, 2125–2131. DOI:10.1007/s00018-004-4200-2 [Google Scholar]
-
Harris H, Wiener F, Klein G. The analysis of malignancy by cell fusion. III. Hybrids between diploid fibroblasts and other tumour cells. J. Cell Sci. 1971, 8, 681–692. DOI:10.1242/jcs.8.3.681 [Google Scholar]
-
Andersen TL, Boissy P, Sondergaard TE, Kupisiewicz K, Plesner T, Rasmussen T, et al. Osteoclast nuclei of myeloma patients show chromosome translocations specific for the myeloma cell clone: A new type of cancer-host partnership? J. Pathol. 2007, 211, 10–17. DOI:10.1002/path.2078 [Google Scholar]
-
Ramakrishnan M, Mathur SR, Mukhopadhyay A. Fusion-derived epithelial cancer cells express hematopoietic markers and contribute to stem cell and migratory phenotype in ovarian carcinoma. Cancer Res. 2013, 73, 5360–5370. DOI:10.1158/0008-5472.CAN-13-0896 [Google Scholar]
-
Aguirre LA, Montalban-Hernandez K, Avendano-Ortiz J, Marin E, Lozano R, Toledano V, et al. Tumor stem cells fuse with monocytes to form highly invasive tumor-hybrid cells. Oncoimmunology 2020, 9, 1773204. DOI:10.1080/2162402X.2020.1773204 [Google Scholar]
-
Shabo I, Midtbo K, Andersson H, Akerlund E, Olsson H, Wegman P, et al. Macrophage traits in cancer cells are induced by macrophage-cancer cell fusion and cannot be explained by cellular interaction. BMC Cancer 2015, 15, 922. DOI:10.1186/s12885-015-1935-0 [Google Scholar]
-
Uygur B, Leikina E, Melikov K, Villasmil R, Verma SK, Vary CPH, et al. Interactions with Muscle Cells Boost Fusion, Stemness, and Drug Resistance of Prostate Cancer Cells. Mol. Cancer Res. 2019, 17, 806–820. DOI:10.1158/1541-7786.MCR-18-0500 [Google Scholar]
-
van Bokhoven A, Caires A, Maria MD, Schulte AP, Lucia MS, Nordeen SK, et al. Spectral karyotype (SKY) analysis of human prostate carcinoma cell lines. Prostate 2003, 57, 226–244. DOI:10.1002/pros.10291 [Google Scholar]
-
Strefford JC, Lillington DM, Young BD, Oliver RT. The use of multicolor fluorescence technologies in the characterization of prostate carcinoma cell lines: A comparison of multiplex fluorescence in situ hybridization and spectral karyotyping data. Cancer Genet. Cytogenet. 2001, 124, 112–121. DOI:10.1016/s0165-4608(00)00339-3 [Google Scholar]
-
Pan Y, Kytola S, Farnebo F, Wang N, Lui WO, Nupponen N, et al. Characterization of chromosomal abnormalities in prostate cancer cell lines by spectral karyotyping. Cytogenet. Cell Genet. 1999, 87, 225–232. DOI:10.1159/000015432 [Google Scholar]
-
Greene SB, Dago AE, Leitz LJ, Wang Y, Lee J, Werner SL, et al. Chromosomal Instability Estimation Based on Next Generation Sequencing and Single Cell Genome Wide Copy Number Variation Analysis. PLoS ONE 2016, 11, e0165089. DOI:10.1371/journal.pone.0165089 [Google Scholar]
-
Seim I, Jeffery PL, Thomas PB, Nelson CC, Chopin LK. Whole-Genome Sequence of the Metastatic PC3 and LNCaP Human Prostate Cancer Cell Lines. G3 Genes|Genomes|Genet. 2017, 7, 1731–1741. DOI:10.1534/g3.117.039909 [Google Scholar]
-
Sienkiewicz K, Yang C, Paschal BM, Ratan A. Genomic analyses of the metastasis-derived prostate cancer cell lines LNCaP, VCaP, and PC3-AR. Prostate 2022, 82, 442–451. DOI:10.1002/pros.24290 [Google Scholar]
-
Gordon S. Cell fusion and some subcellular properties of heterokaryons and hybrids. J. Cell Biol. 1975, 67, 257–280. DOI:10.1083/jcb.67.2.257 [Google Scholar]
-
Wolfe KH. Yesterday’s polyploids and the mystery of diploidization. Nat. Rev. Genet. 2001, 2, 333–341. DOI:10.1038/35072009 [Google Scholar]
-
Walmsley RH. The general theory of mapping functions for random genetic recombination. Biophys. J. 1969, 9, 421–431. DOI:10.1016/S0006-3495(69)86394-0 [Google Scholar]
-
Rowley DR. What might a stromal response mean to prostate cancer progression? Cancer Metastasis Rev. 1998, 17, 411–419. DOI:10.1023/a:1006129420005 [Google Scholar]
-
Kreten F, Buttner R, Peifer M, Harder C, Hillmer AM, Abedpour N, et al. Tumor architecture and emergence of strong genetic alterations are bottlenecks for clonal evolution in primary prostate cancer. Cell Syst. 2024, 15, 1061–1074.e7. DOI:10.1016/j.cels.2024.10.005 [Google Scholar]
-
Berglund E, Maaskola J, Schultz N, Friedrich S, Marklund M, Bergenstrahle J, et al. Spatial maps of prostate cancer transcriptomes reveal an unexplored landscape of heterogeneity. Nat. Commun. 2018, 9, 2419. DOI:10.1038/s41467-018-04724-5 [Google Scholar]
-
Oren-Suissa M, Podbilewicz B. Cell fusion during development. Trends Cell Biol. 2007, 17, 537–546. DOI:10.1016/j.tcb.2007.09.004 [Google Scholar]
-
Podbilewicz B, Chernomordik LV. Membrane fusion: Conserved and diverse. Semin. Cell Dev. Biol. 2016, 60, 63–64. DOI:10.1016/j.semcdb.2016.11.009 [Google Scholar]
-
Klinovska K, Sebkova N, Dvorakova-Hortova K. Sperm-egg fusion: A molecular enigma of mammalian reproduction. Int. J. Mol. Sci. 2014, 15, 10652–10668. DOI:10.3390/ijms150610652 [Google Scholar]
-
Inoue N, Hagihara Y, Wright D, Suzuki T, Wada I. Oocyte-triggered dimerization of sperm IZUMO1 promotes sperm-egg fusion in mice. Nat. Commun. 2015, 6, 8858. DOI:10.1038/ncomms9858 [Google Scholar]
-
Lorenzetti D, Poirier C, Zhao M, Overbeek PA, Harrison W, Bishop CE. A transgenic insertion on mouse chromosome 17 inactivates a novel immunoglobulin superfamily gene potentially involved in sperm-egg fusion. Mamm. Genome 2014, 25, 141–148. DOI:10.1007/s00335-013-9491-x [Google Scholar]
-
Larsson LI, Bjerregaard B, Talts JF. Cell fusions in mammals. Histochem. Cell Biol. 2008, 129, 551–561. DOI:10.1007/s00418-008-0411-1 [Google Scholar]
-
Larsson LI, Bjerregaard B, Wulf-Andersen L, Talts JF. Syncytin and cancer cell fusions. Sci. World J. 2007, 7, 1193–1197. DOI:10.1100/tsw.2007.212 [Google Scholar]
-
Leikina E, Gamage DG, Prasad V, Goykhberg J, Crowe M, Diao J, et al. Myomaker and Myomerger Work Independently to Control Distinct Steps of Membrane Remodeling during Myoblast Fusion. Dev. Cell 2018, 46, 767–780.e7. DOI:10.1016/j.devcel.2018.08.006 [Google Scholar]
-
Ishii T, Ruiz-Torruella M, Ikeda A, Shindo S, Movila A, Mawardi H, et al. OC-STAMP promotes osteoclast fusion for pathogenic bone resorption in periodontitis via up-regulation of permissive fusogen CD9. FASEB J. 2018, 32, 4016–4030. DOI:10.1096/fj.201701424R [Google Scholar]
-
Neumann E, Schwarz MC, Hasseli R, Hulser ML, Classen S, Sauerbier M, et al. Tetraspanin CD82 affects migration, attachment and invasion of rheumatoid arthritis synovial fibroblasts. Ann. Rheum. Dis. 2018, 77, 1619–1626. DOI:10.1136/annrheumdis-2018-212954 [Google Scholar]
-
Prabhu VV, Devaraj SN. KAI1/CD82, Metastasis Suppressor Gene as a Therapeutic Target for Non-Small-Cell Lung Carcinoma. J. Environ. Pathol. Toxicol. Oncol. 2017, 36, 269–275. DOI:10.1615/JEnvironPatholToxicolOncol.2017024619 [Google Scholar]
-
Fanaei M, Monk PN, Partridge LJ. The role of tetraspanins in fusion. Biochem. Soc. Trans. 2011, 39, 524–528. DOI:10.1042/BST0390524 [Google Scholar]
-
Fujihara Y, Ikawa M. GPI-AP release in cellular, developmental, and reproductive biology. J. Lipid Res. 2016, 57, 538–545. DOI:10.1194/jlr.R063032 [Google Scholar]
-
Lefevre B, Wolf JP, Ziyyat A. Sperm-egg interaction: Is there a link between tetraspanin(s) and GPI-anchored protein(s)? Bioessays 2010, 32, 143–152. DOI:10.1002/bies.200900159 [Google Scholar]
-
Pique C, Lagaudriere-Gesbert C, Delamarre L, Rosenberg AR, Conjeaud H, Dokhelar MC. Interaction of CD82 tetraspanin proteins with HTLV-1 envelope glycoproteins inhibits cell-to-cell fusion and virus transmission. Virology 2000, 276, 455–465. DOI:10.1006/viro.2000.0538 [Google Scholar]
-
Dong JT, Lamb PW, Rinker-Schaeffer CW, Vukanovic J, Ichikawa T, Isaacs JT, et al. KAI1, a metastasis suppressor gene for prostate cancer on human chromosome 11p11.2. Science 1995, 268, 884–886. DOI:10.1126/science.7754374 [Google Scholar]
-
Brukman NG, Uygur B, Podbilewicz B, Chernomordik LV. How cells fuse. J. Cell Biol. 2019, 218, 1436–1451. DOI:10.1083/jcb.201901017 [Google Scholar]
-
Horoszewicz JS, Leong SS, Kawinski E, Karr JP, Rosenthal H, Chu TM, et al. LNCaP model of human prostatic carcinoma. Cancer Res. 1983, 43, 1809–1818. Available online: https://aacrjournals.org/cancerres/article-abstract/43/4/1809/487457 (accessed on 2 Feburary 2026).
-
Thalmann GN, Sikes RA, Wu TT, Degeorges A, Chang SM, Ozen M, et al. LNCaP progression model of human prostate cancer: Androgen-independence and osseous metastasis. Prostate 2000, 44, 91–103. DOI:10.1002/1097-0045(20000701)44:2<91::AID-PROS1>3.0.CO;2-L [Google Scholar]
-
Ohnuki Y, Marnell MM, Babcock MS, Lechner JF, Kaighn ME. Chromosomal analysis of human prostatic adenocarcinoma cell lines. Cancer Res. 1980, 40, 524–534. Available online: https://aacrjournals.org/cancerres/article-abstract/40/3/524/484646 (accessed on 2 Feburary 2026).
-
Haussler O, Epstein JI, Amin MB, Heitz PU, Hailemariam S. Cell proliferation, apoptosis, oncogene, and tumor suppressor gene status in adenosis with comparison to benign prostatic hyperplasia, prostatic intraepithelial neoplasia, and cancer. Hum. Pathol. 1999, 30, 1077–1086. DOI:10.1016/s0046-8177(99)90226-5 [Google Scholar]
-
Voelkel-Johnson C, Voeks DJ, Greenberg NM, Barrios R, Maggouta F, Kurtz DT, et al. Genomic instability-based transgenic models of prostate cancer. Carcinogenesis 2000, 21, 1623–1627. DOI:10.1093/carcin/21.5.623 [Google Scholar]
-
Stapleton AM, Zbell P, Kattan MW, Yang G, Wheeler TM, Scardino PT, et al. Assessment of the biologic markers p53, Ki-67, and apoptotic index as predictive indicators of prostate carcinoma recurrence after surgery. Cancer 1998, 82, 168–175. DOI:10.1002/(SICI)1097-0142(19980101)82:1<168::AID-CNCR21>3.0.CO;2-%23 [Google Scholar]
-
Tu H, Jacobs SC, Borkowski A, Kyprianou N. Incidence of apoptosis and cell proliferation in prostate cancer: Relationship with TGF-beta1 and bcl-2 expression. Int. J. Cancer 1996, 69, 357–363. DOI:10.1002/(SICI)1097-0215(19961021)69:5<357::AID-IJC1>3.0.CO;2-4 [Google Scholar]
-
Afanzar O, Buss GK, Stearns T, Ferrell JE, Jr. The nucleus serves as the pacemaker for the cell cycle. Elife 2020, 9, e59989. DOI:10.7554/eLife.59989 [Google Scholar]
-
Matson JP, Cook JG. Cell cycle proliferation decisions: The impact of single cell analyses. FEBS J. 2017, 284, 362–375. DOI:10.1111/febs.13898 [Google Scholar]
-
Canman CE. Replication checkpoint: Preventing mitotic catastrophe. Curr. Biol. 2001, 11, R121–R124. DOI:10.1016/s0960-9822(01)00057-4 [Google Scholar]
-
Castedo M, Perfettini JL, Roumier T, Andreau K, Medema R, Kroemer G. Cell death by mitotic catastrophe: A molecular definition. Oncogene 2004, 23, 2825–2837. DOI:10.1038/sj.onc.1207528 [Google Scholar]
-
Castedo M, Perfettini JL, Roumier T, Kroemer G. Cyclin-dependent kinase-1: Linking apoptosis to cell cycle and mitotic catastrophe. Cell Death Differ. 2002, 9, 1287–1293. DOI:10.1038/sj.cdd.4401130 [Google Scholar]
-
King KL, Cidlowski JA. Cell cycle and apoptosis: Common pathways to life and death. J. Cell Biochem. 1995, 58, 175–180. DOI:10.1002/jcb.240580206 [Google Scholar]
-
Otsuka I. Cutaneous Metastasis after Surgery, Injury, Lymphadenopathy, and Peritonitis: Possible Mechanisms. Int. J. Mol. Sci. 2019, 20, 3286. DOI:10.3390/ijms20133286 [Google Scholar]
-
Baum M. Does surgery disseminate or accelerate cancer? Lancet 1996, 347, 260. DOI:10.1016/s0140-6736(96)90433-x [Google Scholar]
-
Coffey JC, Wang JH, Smith MJ, Bouchier-Hayes D, Cotter TG, Redmond HP. Excisional surgery for cancer cure: Therapy at a cost. Lancet Oncol. 2003, 4, 760–768. DOI:10.1016/s1470-2045(03)01282-8 [Google Scholar]
-
Deyell M, Garris CS, Laughney AM. Cancer metastasis as a non-healing wound. Br. J. Cancer 2021, 124, 1491–1502. DOI:10.1038/s41416-021-01309-w [Google Scholar]
-
Ganesh K, Basnet H, Kaygusuz Y, Laughney AM, He L, Sharma R, et al. L1CAM defines the regenerative origin of metastasis-initiating cells in colorectal cancer. Nat. Cancer 2020, 1, 28–45. DOI:10.1038/s43018-019-0006-x [Google Scholar]
-
Zlotta AR, Egawa S, Pushkar D, Govorov A, Kimura T, Kido M, et al. Prevalence of prostate cancer on autopsy: Cross-sectional study on unscreened Caucasian and Asian men. J. Natl. Cancer Inst. 2013, 105, 1050–1058. DOI:10.1093/jnci/djt151 [Google Scholar]
-
Jahn JL, Giovannucci EL, Stampfer MJ. The high prevalence of undiagnosed prostate cancer at autopsy: Implications for epidemiology and treatment of prostate cancer in the Prostate-specific Antigen-era. Int. J. Cancer 2015, 137, 2795–2802. DOI:10.1002/ijc.29408 [Google Scholar]
-
Kimura T, Sato S, Takahashi H, Egawa S. Global Trends of Latent Prostate Cancer in Autopsy Studies. Cancers 2021, 13, 359. DOI:10.3390/cancers13020359 [Google Scholar]
-
Siegel RL, Miller KD, Wagle NS, Jemal A. Cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. DOI:10.3322/caac.21763 [Google Scholar]
-
Jeong IG, Dajani D, Verghese M, Hwang J, Cho YM, Hong JH, et al. Differences in the aggressiveness of prostate cancer among Korean, Caucasian, and African American men: A retrospective cohort study of radical prostatectomy. Urol. Oncol. 2016, 34, 3.e9–3.e14. DOI:10.1016/j.urolonc.2015.08.004 [Google Scholar]
-
Pray LA. DNA replication and causes of mutation. Nat. Educ. 2008, 1, 214. Available online: https://www.nature.com/scitable/topicpage/dna-replication-and-causes-of-mutation-409 (accessed on 2 Feburary 2026)
-
Reijns MAM, Kemp H, Ding J, de Proce SM, Jackson AP, Taylor MS. Lagging-strand replication shapes the mutational landscape of the genome. Nature 2015, 518, 502–506. DOI:10.1038/nature14183 [Google Scholar]
-
Seidel GE, Jr. Maintaining integrity of germline DNA: Individuals age, species do not. Reprod. Fertil. Dev. 2015, 27, 865–871. DOI:10.1071/RD14514 [Google Scholar]
-
Milholland B, Dong X, Zhang L, Hao X, Suh Y, Vijg J. Differences between germline and somatic mutation rates in humans and mice. Nat. Commun. 2017, 8, 15183. DOI:10.1038/ncomms15183 [Google Scholar]
-
Larkin PJ, Scowcroft WR. Somaclonal variation—A novel source of variability from cell cultures for plant improvement. Theor. Appl. Genet. 1981, 60, 197–214. DOI:10.1007/BF02342540 [Google Scholar]
-
Suh A, Dion-Cote AM. New Perspectives on the Evolution of Within-Individual Genome Variation and Germline/Soma Distinction. Genome Biol. Evol. 2021, 13, evab095. DOI:10.1093/gbe/evab095 [Google Scholar]
-
Acuna-Hidalgo R, Bo T, Kwint MP, van de Vorst M, Pinelli M, Veltman JA, et al. Post-zygotic Point Mutations Are an Underrecognized Source of De Novo Genomic Variation. Am. J. Hum. Genet. 2015, 97, 67–74. DOI:10.1016/j.ajhg.2015.05.008 [Google Scholar]
-
Nicolas G, Veltman JA. The role of de novo mutations in adult-onset neurodegenerative disorders. Acta Neuropathol. 2019, 137, 183–207. DOI:10.1007/s00401-018-1939-3 [Google Scholar]
-
Kaeser G, Chun J. Brain cell somatic gene recombination and its phylogenetic foundations. J. Biol. Chem. 2020, 295, 12786–12795. DOI:10.1074/jbc.REV120.009192 [Google Scholar]
-
Darmasaputra GS, van Rijnberk LM, Galli M. Functional consequences of somatic polyploidy in development. Development 2024, 151, dev202392. DOI:10.1242/dev.202392 [Google Scholar]
-
Asalone KC, Takkar AK, Saldanha CJ, Bracht JR. A Transcriptomic Pipeline Adapted for Genomic Sequence Discovery of Germline-Restricted Sequence in Zebra Finch, Taeniopygia guttata. Genome Biol. Evol. 2021, 13, evab088. DOI:10.1093/gbe/evab088 [Google Scholar]
-
Majtanova Z, Dedukh D, Choleva L, Adams M, Rab P, Unmack PJ, et al. Uniparental Genome Elimination in Australian Carp Gudgeons. Genome Biol. Evol. 2021, 13, evab030. DOI:10.1093/gbe/evab030 [Google Scholar]
-
Schatz DG, Ji Y. Recombination centres and the orchestration of V(D)J recombination. Nat. Rev. Immunol. 2011, 11, 251–263. DOI:10.1038/nri2941 [Google Scholar]
-
The National Human Genome Research Institute. Fact Sheet, Human Genome Project, The Most Important Biochemical Research Undertaking of the 20th Century; NHGRI: Bethesda, MD, USA, 2024. [Google Scholar]