Deadline for manuscript submissions: 30 March 2026.



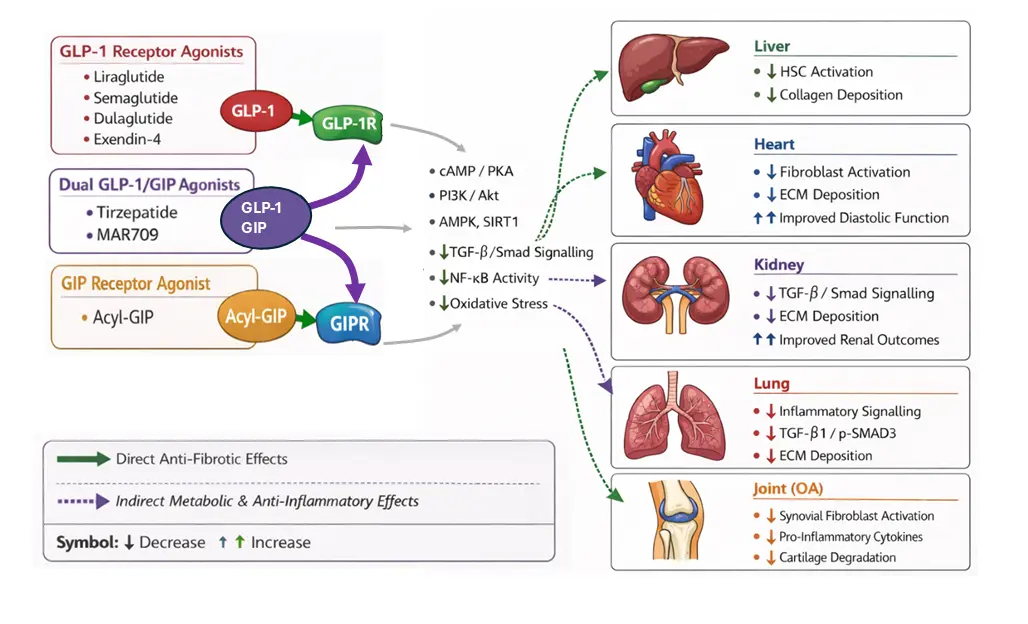
Fibrosis, characterised by the excessive deposition of extracellular matrix via activated fibroblasts, is a pathological feature of several chronic inflammatory disorders, which collectively contribute significantly to global morbidity and mortality. Despite this, current anti-fibrotic therapies are of limited efficacy. However, incretin-based therapies, primarily glucagon-like peptide-1 (GLP-1) receptor agonists, are now emerging as candidate drugs for modulating fibrotic signalling pathways. This review synthesises the growing body of preclinical and clinical evidence that incretin receptor agonists exert direct and indirect anti-fibrotic effects. We detail the molecular mechanisms and survey the promising data across hepatic, cardiac, renal, lung, and joint tissues, which underscore the potential for repurposing of this drug class as a therapeutic strategy for fibro-inflammatory conditions.
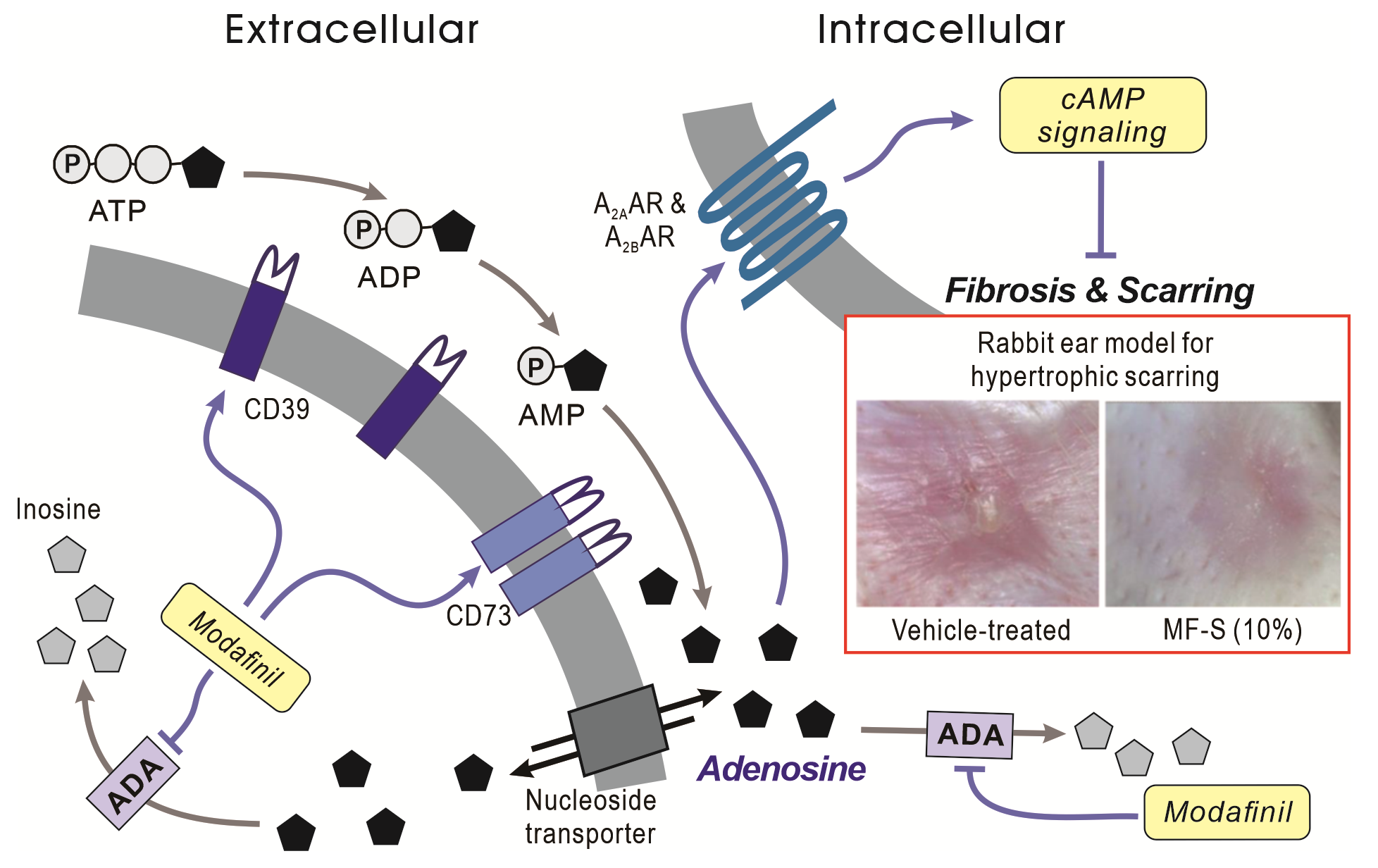
Modafinil (MF) is a clinically approved wake-promoting agent with emerging anti-inflammatory and anti-fibrotic effects, although its upstream molecular target has remained undefined. Here, we identify adenosine deaminase (ADA) as a previously unrecognized target mediating the therapeutic actions of MF. Its S- and R-isomers (MF-S and MF-R) robustly increased intracellular cAMP levels in fibroblasts with efficacy comparable to NECA, despite minimal direct binding to adenosine receptors, and suppressed KCa3.1 channel activity via a PKA–dependent mechanism. MF-S markedly upregulated CD39 and CD73, leading to increased adenosine availability. Pharmacological inhibition of CD73 with AB680 abolished MF-S–induced increases in cAMP and Epac levels and reversed suppression of TGFβ–induced collagen expression. Consistently, MF-S attenuated canonical profibrotic signaling by inhibiting TGFβ–induced Smad4 upregulation. In vivo, MF-S significantly reduced hypertrophic scarring in a rabbit ear model, with efficacy comparable to Contratubex. Mechanistically, MF-S directly inhibited purified ADA at subnanomolar concentrations and suppressed cellular ADA activity in fibroblast and immune cells. Collectively, these findings establish ADA inhibition as a key upstream mechanism by which MF enhances adenosine–cAMP signaling to suppress inflammation and fibrosis, highlighting MF and its isomers as promising therapeutic candidates for inflammatory and fibrotic diseases.