Found 11 results
Open Access
Commentary
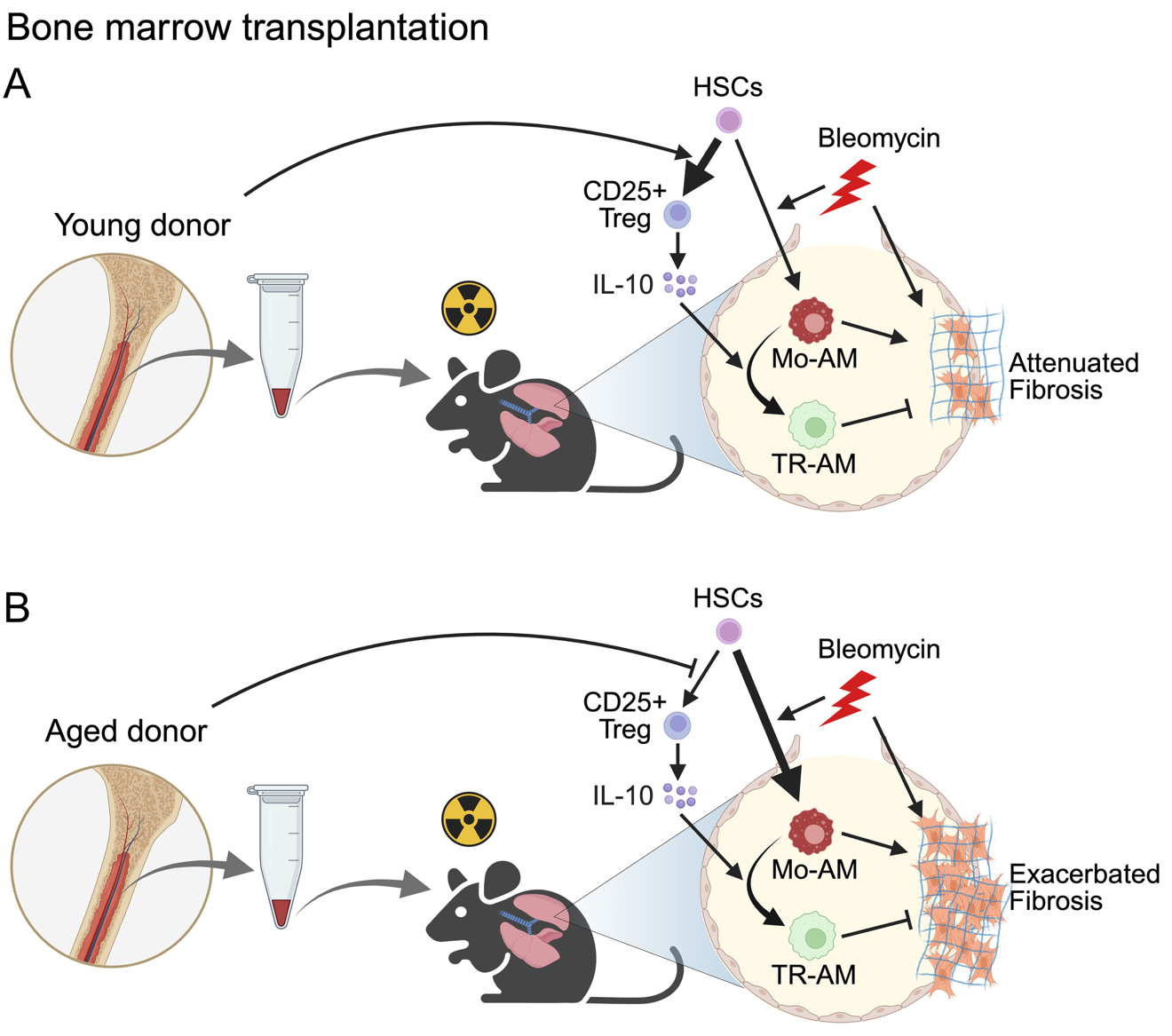
12 June 2025Bone Marrow Transplantation as a Future Therapeutic Strategy for Idiopathic Pulmonary Fibrosis: Lessons from Hematopoietic Aging
Idiopathic pulmonary fibrosis (IPF) is a fatal disease with limited therapeutic options. Lung transplantation is the only curative treatment, but it is rarely available due to a lack of suitable donors. In a recent publication in Science Immunology, Farhat et al. demonstrated that bone marrow transplantation from young donors alleviates fibrosis by restoring immune resolution in aged hosts in animal models. Aged hematopoietic cells exacerbate fibrosis through the persistence of inflammatory macrophages and impaired Treg-derived IL-10, highlighting bone marrow rejuvenation as a potential treatment strategy for IPF.
Open Access
Review
07 March 2025Mechanisms and Therapeutic Potential of Myofibroblast Transformation in Pulmonary Fibrosis
Idiopathic pulmonary fibrosis (IPF) is a progressive, irreversible, and fatal disease with an increasing incidence and limited therapeutic options. It is characterized by the formation and deposition of excess extracellular matrix proteins resulting in the gradual replacement of normal lung architecture by fibrous tissue. The cellular and molecular mechanism of IPF has not been fully understood. A hallmark in IPF is pulmonary fibroblast to myofibroblast transformation (FMT). During excessive lung repair upon exposure to harmful stimuli, lung fibroblasts transform into myofibroblasts under stimulation of cytokines, chemokines, and vesicles from various cells. These mediators interact with lung fibroblasts, initiating multiple signaling cascades, such as TGFβ1, MAPK, Wnt/β-catenin, NF-κB, AMPK, endoplasmic reticulum stress, and autophagy, contributing to lung FMT. Furthermore, single-cell transcriptomic analysis has revealed significant heterogeneity among lung myofibroblasts, which arise from various cell types and are adapted to the altered microenvironment during pathological lung repair. This review provides an overview of recent research on the origins of lung myofibroblasts and the molecular pathways driving their formation, with a focus on the interactions between lung fibroblasts and epithelial cells, endothelial cells, and macrophages in the context of lung fibrosis. Based on these molecular insights, targeting the lung FMT could offer promising avenues for the treatment of IPF.

Open Access
Review
18 February 2025The Intersection between Immune System and Idiopathic Pulmonary Fibrosis—A Concise Review
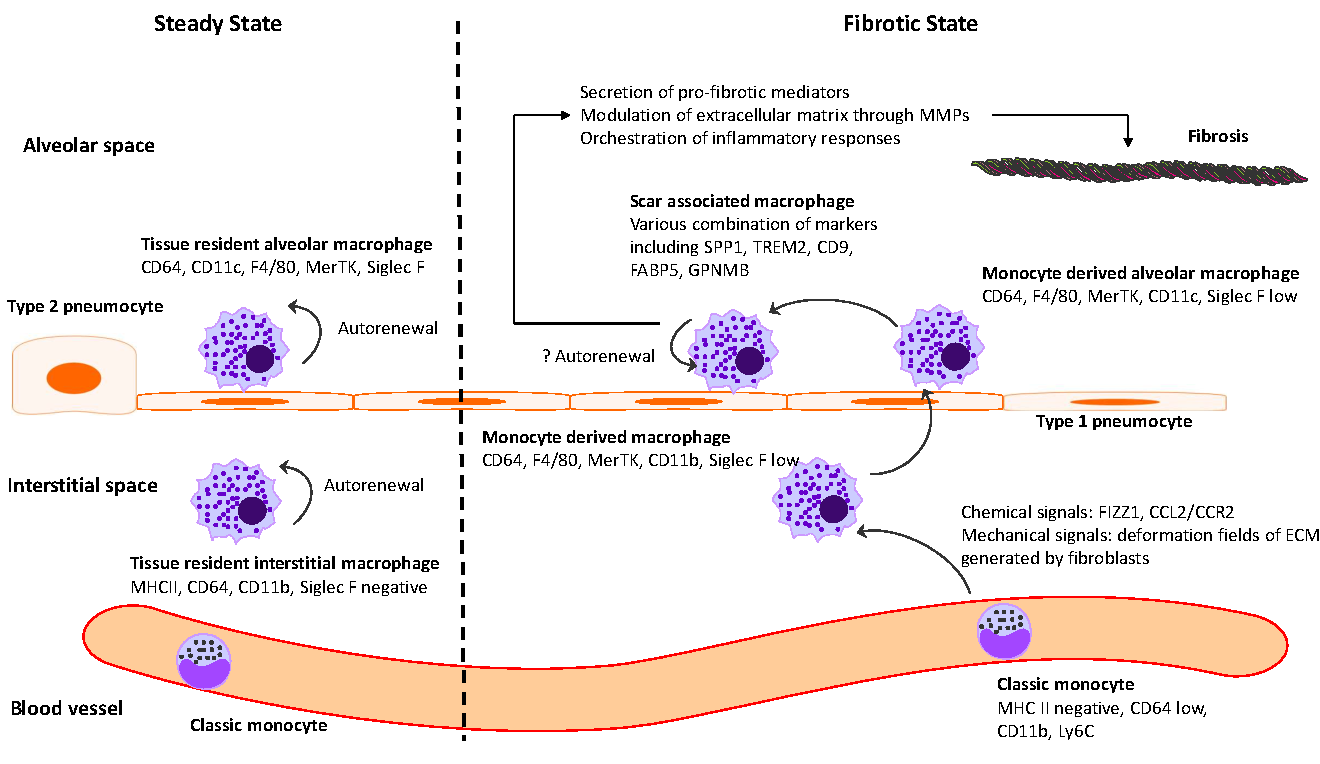
Idiopathic pulmonary fibrosis (IPF) is marked by progressive alveolar destruction, impaired tissue regeneration, and relentless fibrogenesis, culminating in respiratory failure and death. A diverse array of resident and non-resident cells within the lung contribute to disease pathogenesis. Notably, immune cells, both resident and recruited, respond to cues from sites of lung injury by undergoing phenotypic transitions and producing a wide range of mediators that influence, initiate, or dictate the function, or dysfunction, of key effector cells in IPF pathology, such as alveolar epithelial cells, lung fibroblasts, and capillary endothelial cells. The role of the immune system in IPF has undergone an interesting evolution, oscillating from initial enthusiasm to skepticism, and now to a renewed focus. This shift reflects both the past failures of immune-targeting therapies for IPF and the unprecedented insights into immune cell heterogeneity provided by emerging technologies. In this article, we review the historical evolution of perspectives on the immune system’s role in IPF pathogenesis and examine the lessons learned from previous therapeutic failures targeting immune responses. We discuss the major immune cell types implicated in IPF progression, highlighting their phenotypic transitions and mechanisms of action. Finally, we identify key knowledge gaps and propose future directions for research on the immune system in IPF.
Open Access
Review
13 January 2025Comparative Analysis of Idiopathic Pulmonary Fibrosis and Progressive Pulmonary Fibrosis: Epidemiology, Pathophysiology, Clinical Features, Diagnosis and Treatment
Idiopathic pulmonary fibrosis (IPF) is a chronic fibrosing interstitial disease of unknown origin, characterized by radiological and histological features consistent with usual interstitial pneumonia (UIP). It is marked by a progressive worsening of dyspnea and a decline in lung function. Both IPF and PPF are comparable because they have poor prognoses with a median survival time from diagnosis of around 2–4 years without antifibrotic therapy. This review shows the main specific characteristics and differences of epidemiology, pathophysiology, clinical and radiological features, treatment, and prognosis of IPF and PPF.

Open Access
Meeting Report
04 December 2024Progress and Gaps in Respiratory Disease Research and Treatment: Highlights of the IRM 2024 in Shanghai
Respiratory diseases pose a major public health challenge globally, necessitating collaborative efforts between basic researchers and clinicians for effective solutions. China, which is heavily impacted by a broad spectrum of respiratory disorders, has made notable strides in both research and clinical management of these diseases. The International Respiratory Medicine (IRM) meeting was organized with the primary goal of facilitating the exchange of recent research developments and promoting collaboration between Chinese and American scientists in both basic and clinical research fields. This article summarizes key insights from IRM2024, held in Shanghai, where a wide range of topics were discussed, including lung tissue development, disease mechanisms, and innovative therapeutic strategies. By integrating perspectives from basic, translational, and clinical research, IRM2024 highlighted recent advancements, addressed persistent challenges, and explored future directions in respiratory science and clinical practice. The insights gained from IRM2024 are poised to be pivotal in shaping future research and therapeutic approaches, further reinforcing the global commitment to enhancing respiratory health and improving patient outcomes.

Open Access
Article
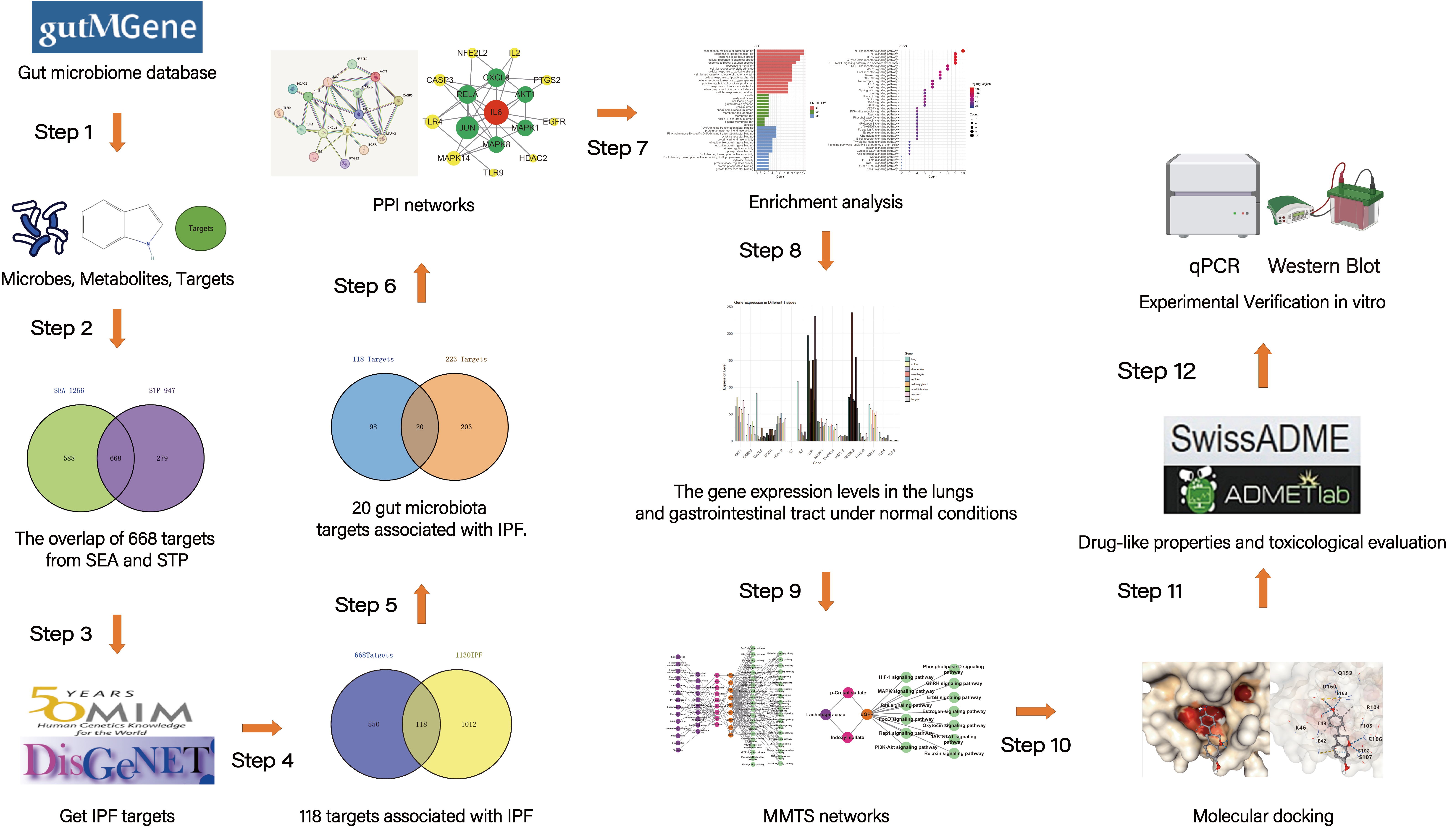
04 November 2024Exploring the Potential Relationship between Gut Microbiome Metabolites and Idiopathic Pulmonary Fibrosis via Network Pharmacology Study
Idiopathic pulmonary fibrosis (IPF) is a chronic, progressive fibrotic lung disease with a poor prognosis. Previous research has revealed that the gut microbiota is associated with human health and immunity, and it interacts with the lung through the “gut-lung axis”. This study explores the potential relationship between Gut Microbiome Metabolites and Idiopathic pulmonary fibrosis via Network Pharmacology Study. The metabolites from gut microbiota were retrieved from the gutMGene database, and gene targets for these metabolites were obtained from previous studies. Gene targets of IPF were obtained from public databases (DisGeNET, OMIM). Subsequently, following the identification of shared targets, IL6 was determined as the core target through protein-protein interaction analysis. Then, a microbiota-metabolite-target-signaling pathway network (MMTS) was constructed using Cytoscape 3.10, and targets with low expression in the lungs and intestines were deleted. The MMTS network revealed that three short-chain fatty acids—acetate, butyrate, propionate, and a flavonoid compound called equol—are IL6-related metabolites. Then, we performed a molecular docking test (MDT) using CB-Dock2 to validate the affinity between core targets and metabolites. MDT confirmed that equol produced by the conversion of isoflavones from Lactobacillus paracasei JS1 was more stable in binding to IL6 than the other three short-chain fatty acids, thereby affecting multiple signaling pathways and influencing the progression of IPF. Finally, we validated this hypothesis through in vitro cell culture experiments, further confirming the effect of Equol.
Open Access
Review
28 October 2024Acute Exacerbations of Interstitial Lung Disease: Evolving Perspectives on Diagnosis and Management
Interstitial lung diseases (ILDs) are a heterogeneous group of chronic lung diseases caused by several potential etiologies but for many, the cause of a given ILD remains unknown. Accurate epidemiologic data are hard to find because of varying definitions, overlapping characteristics once thought to be unique to specific diseases, and ongoing changes in how ILDs are diagnosed and managed. In addition, there are significant variations in prevalence among different geographic populations, likely reflecting a combination of genetic and environmental differences. Certain risk factors, including exposure to cigarette smoke or environmental toxicants (asbestos, silica, fracking, coal dust, and air pollution), genetic mutations, and single nucleotide polymorphisms, have all been associated with developing interstitial lung disease. Due to the availability of high-resolution computed tomography (CT) scans, earlier and broader recognition of subtle imaging changes, and an aging worldwide population, the incidence and prevalence of ILDs are increasing. While a given cause of particular interstitial lung disease may vary, patients often experience breathlessness and a non-productive cough due to impaired alveolar gas exchange. Patients with ILD are prone to the development of acute exacerbations, marked by acute or chronic respiratory failure because of an acute exacerbation of the underlying lung disease. In this review, we discuss the definition of an acute exacerbation and comment on what is known about the underlying pathophysiology in exacerbations of idiopathic pulmonary fibrosis and other ILDs. We also emphasize the similarities in the clinical presentation of the acute exacerbations regardless of the underlying ILD, highlight key prognostic features of the diagnosis, and underscore the importance of interdisciplinary management of acute interstitial lung disease exacerbations.
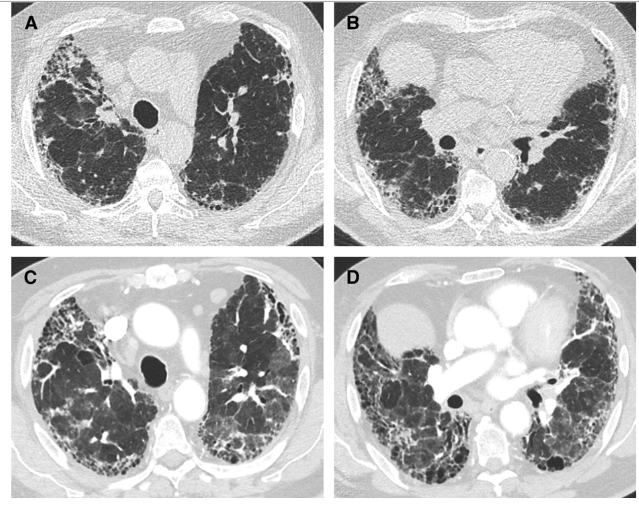
Open Access
Review
16 July 2024Mitochondrial Damage and Epithelial-Mesenchymal Transition as Major Triggers of the Development of Idiopathic Pulmonary Fibrosis
Idiopathic pulmonary fibrosis (IPF) is a type of interstitial pneumonia with an unknown cause that progresses gradually, primarily affecting the elderly. The presence of fibrosis has significant implications for individuals with reduced lung compliance, resulting in decreased quality of life and limited survival. Although the exact mechanism remains unclear, researchers have investigated various factors, such as senescent telomerase replication and abnormal lung stem cell differentiation, to understand the root cause. Extensive research has consistently shown that IPF is closely linked to the dysfunction of alveolar epithelial cells. Current scientific studies on IPF cover a range of aspects including oxidative stress, endoplasmic reticulum stress, mitochondrial damage, and iron-induced apoptosis. By examining these mechanisms, a comprehensive model has been developed that explains the process of IPF. Oxidative stress is identified as the primary trigger, followed by mitochondrial damage as a central component, leading to the mesenchymal transformation of alveolar epithelial cells as the ultimate outcome. This model is expected to serve as a valuable reference for understanding the mechanism of IPF and guiding future drug development efforts.
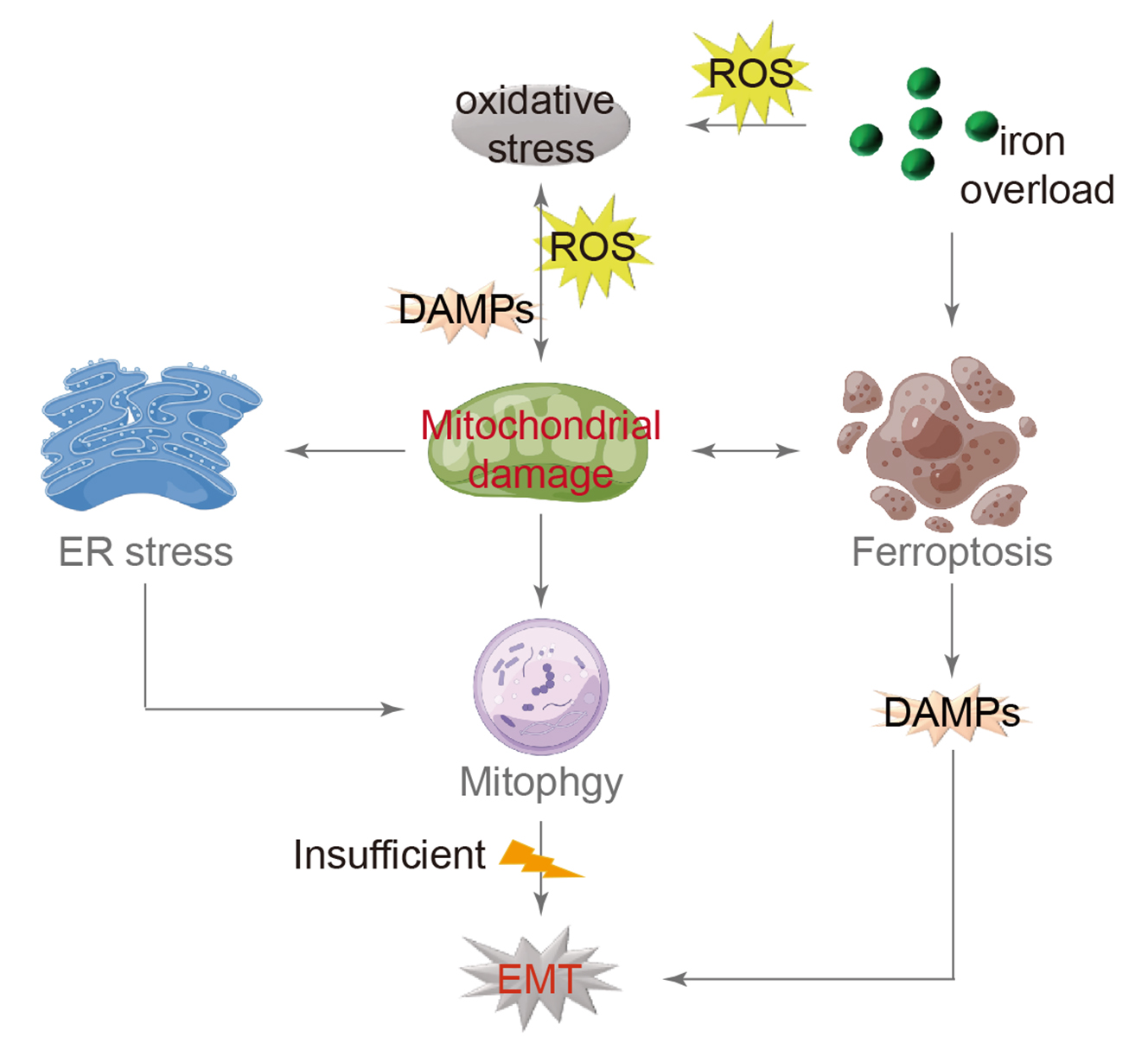
Open Access
Article
01 February 2024Molecular Regulation of Transforming Growth Factor-β1-induced Thioredoxin-interacting Protein Ubiquitination and Proteasomal Degradation in Lung Fibroblasts: Implication in Pulmonary Fibrosis
Thioredoxin-interacting protein (TXNIP) plays a critical role in regulation of cellular redox reactions and inflammatory responses by interacting with thioredoxin (TRX) or the inflammasome. The role of TXNIP in lung fibrosis and molecular regulation of its stability have not been well studied. Therefore, here we investigated the molecular regulation of TXNIP stability and its role in TGF-β1-mediated signaling in lung fibroblasts. TXNIP protein levels were significantly decreased in lung tissues from bleomycin-challenged mice. Overexpression of TXNIP attenuated transforming growth factor-β1 (TGF-β1)-induced phosphorylation of Smad2/3 and fibronectin expression in lung fibroblasts, suggesting that decrease in TXNIP may contribute to the pathogenesis of lung fibrosis. Further, we observed that TGF-β1 lowered TXNIP protein levels, while TXNIP mRNA levels were unaltered by TGF-β1 exposure. TGF-β1 induced TXNIP degradation via the ubiquitin-proteasome system. A serine residue mutant (TNXIP-S308A) was resistant to TGF-β1-induced degradation. Furthermore, downregulation of ubiquitin-specific protease-13 (USP13) promoted the TGF-β1-induced TXNIP ubiquitination and degradation. Mechanistic studies revealed that USP13 targeted and deubiquitinated TXNIP. The results of this study revealed that the decrease of TXNIP in lungs apparently contributes to the pathogenesis of pulmonary fibrosis and that USP13 can target TXNP for deubiquitination and regulate its stability.
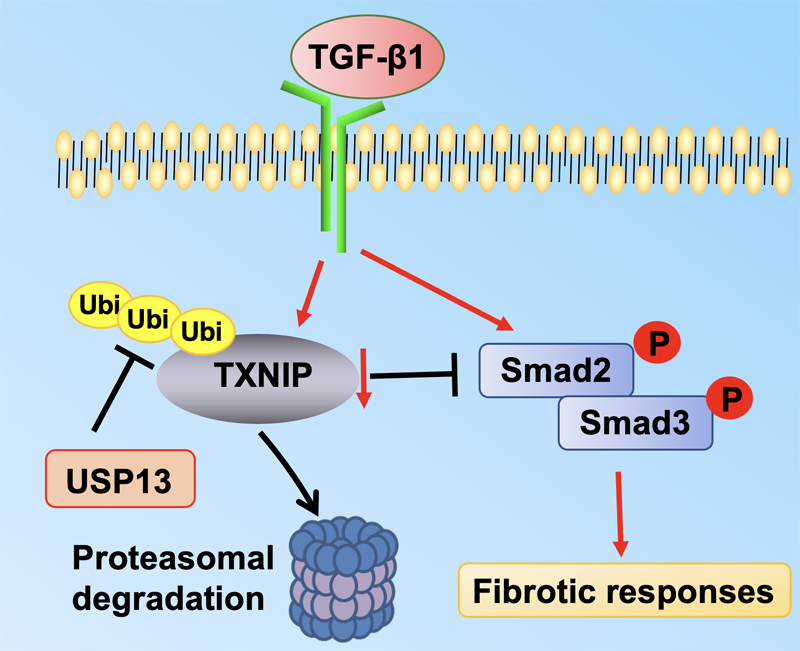
Open Access
Article
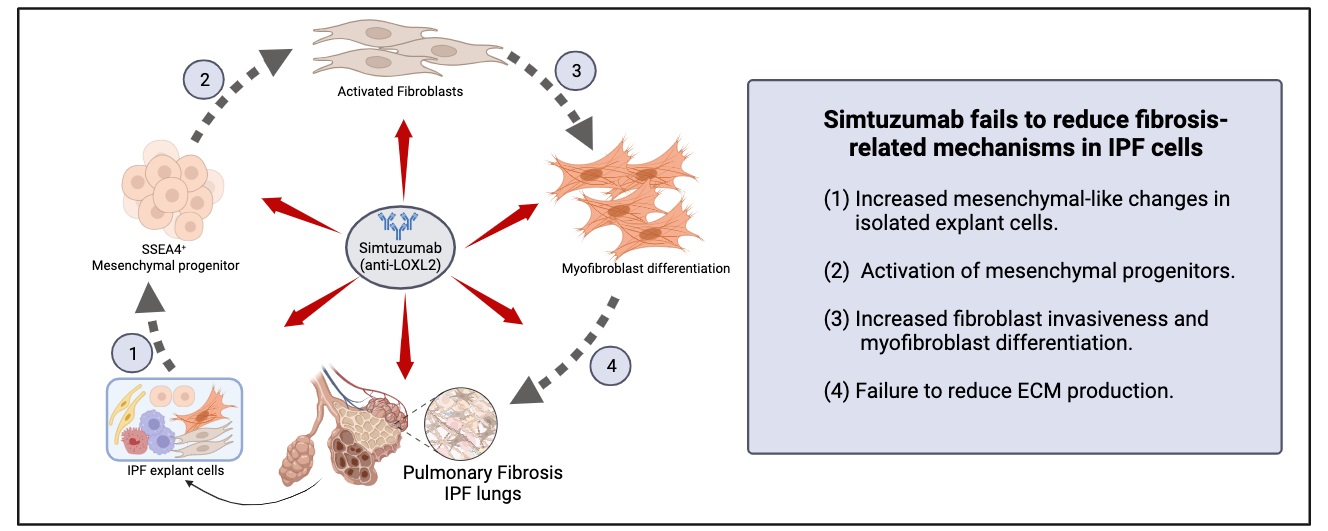
28 November 2023Translational Studies Reveal the Divergent Effects of Simtuzumab Targeting LOXL2 in Idiopathic Pulmonary Fibrosis
The composition of extracellular matrix (ECM) is altered during pathologic scarring in damaged organs including the lung. One major change in the ECM involves the cross-linking of collagen, which promotes fibroblast to myofibroblast differentiation. We examined the role of lysyl oxidase (LOX)-like 2 in lung progenitors and fibroblasts cultured from normal or IPF lung samples and in a humanized mouse model of IPF using a monoclonal antibody (Simtuzumab). Primary lung fibroblasts from normal donor lungs and IPF lung explants were examined for expression of LOXL2. Targeting LOXL2 with Simtuzumab on normal and IPF fibroblasts was examined both in vitro and in vivo for synthetic, functional, and profibrotic properties. LOXL2 was increased at transcript and protein level in IPF compared with normal lung samples. In a dose-dependent manner, Simtuzumab enhanced differentiation of fibroblasts into myofibroblasts. Inhibition of LOXL2 also enhanced fibroblast invasion and accelerated the outgrowth of fibroblasts from dissociated human lung cell preparations. Finally, preventative or delayed delivery of Simtuzumab enhanced lung fibrosis in a humanized mouse model of pulmonary fibrosis. Consistent with its failure in a Phase 2 clinical trial, Simtuzumab exhibited no therapeutic efficacy in translational in vitro and in vivo assays.