Idiopathic pulmonary fibrosis (IPF) is a chronic lung disease that progresses over time and is characterized by scar formation in the lung parenchyma, causing stiffness and thickening in peri-alveolar tissues. This results in irreversible lung function damage, leading to increased mortality and reduced quality of life [
1]. The prevalence of IPF is increasing globally, with recent data showing that 12 out of 100,000 individuals develop IPF, affecting a total of 5 million people worldwide [
1]. On average, patients survive around 4 to 5 years after diagnosis, creating a significant socioeconomic burden worldwide. Common symptoms include dry cough, dyspnea, fatigue, pneumonia, heart failure, pulmonary embolism, and pulmonary hypertension [
1].
Nintedanib and Pirfenidone, the two drugs currently authorized for IPF treatment, primarily slow down the progression of the illness [
2]. Despite advancements in therapeutic techniques, there is still a need for improvement in the survival rate of patients. Both Nintedanib and Pirfenidone have issues related to tolerance. It is also important to note that critical patients with the SARS coronavirus in 2003 and coronavirus disease (COVID-19) in 2019, similar to IPF, had irreversible pulmonary fibrosis (PF). Therefore, the use of antifibrotic therapies, whether currently available or in development, may be beneficial for these critical patients [
3]. The effects of PF on these patients will persist after the epidemic, so it is crucial to explore the internal logical relationship of IPF pathogenesis to aid in the theoretical development of new anti-fibrosis therapies.
Lung epithelial cells consist of type I/II alveolar epithelial cells (AECs), basal cells, club cells, ciliated cells, and goblet cells [
4]. AECs and capillary endothelial cells are the main parenchymal cells responsible for gas exchange functions [
5]. The current understanding of IPF is that repetitive epithelial injury and genetic factors work together to cause collagen deposition due to persistent fibroblast/myofibroblast activation [
6]. Repeated micro-injury events lead to the destruction of epithelial cells, resulting in loss of continuity and destruction of the base layer, releasing damaging factors such as TGF-β that activate fibroblasts/myofibroblasts [
7]. PF typically originates in the alveoli, with damage to AECs being the primary cause of PF [
8,
9].
Mitochondria play a crucial role in the pulmonary vascular responses to hypoxia, specifically in hypoxic pulmonary vasoconstriction and hypoxia-induced pulmonary hypertension [
10]. The pathogenesis of chronic lung diseases such as chronic obstructive pulmonary disease and IPF is closely linked to mitochondrial dysfunction, which can be triggered by factors such as excessive reactive oxygen species (ROS), and defects in mitophagy [
11,
12].
The review focuses on the epithelial-mesenchymal transition (EMT) of AECs triggered by mitochondrial damage in the context of IPF. The reciprocal relationship between mitochondrial damage and EMT in AECs contributes to the pathological advancement of IPF, creating a harmful feedback loop. A comprehensive understanding of the cellular and molecular mechanisms involved in the pathogenesis of it has the potential to inform the development of more efficacious therapeutic interventions.
2.1. The EMT Process of AECs Is a Crucial Feature of IPF
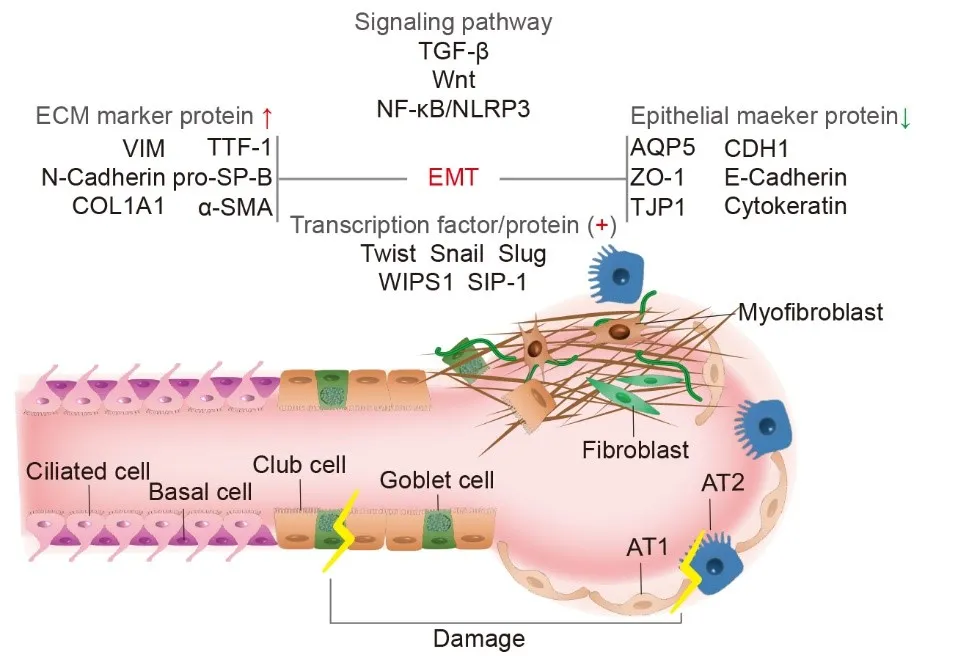
TGF-β is a key fibrogenic cytokine often used as an in vitro model to induce pulmonary fibrosis, and TGF-β is also the target of the clinical anti-pulmonary fibrosis drug pirfenidone [
13]. In RLE-6TN cells derived from the rat type II AECs line, a mesenchymal cell marker and fibroblast-like morphology were detected after six days of TGF-β1 treatment, with TNF-α enhancing this mesenchymal transition. A high number of cells were colocalized with thyroid transcription factor 1, pro-surfactant protein-B, and alpha smooth muscle actin (α-SMA). The mesenchymal marker proteins COL1A1 and vimentin are also increased by TGF-β1, with a simultaneous downregulation of aquaporin 5, zonula occludens-1, and cytokeratins in epithelial cells indicating EMT [
14]. In patients with IPF and bleomycin (BLM) mouse models, the expression of Wnt-inducible signaling pathway protein 1, Twist, and sphingosine 1-phosphate in type II AECs was increased. The lung epithelial cell marker proteins tight junction protein, cadherin 1, and E-cadherin are down-regulated, while mesenchymal marker proteins α-SMA, Recombinant S100 calcium-binding protein A4, COL1A1, vimentin, and N-cadherin are up-regulated, promoting and maintaining the progressive development of fibrosis [
15,
16,
17]. IPF patients and cell experiments showed that a transcription factor called snail family transcriptional repressors 1 and 2 up-regulated a type II AECs mesenchymal transition induced by TGF-β1, enhancing fibroblast differentiation [
18]. In A549 cells derived from the human type II AECs line express fewer mRNAs for fibrosis-related proteins N-cadherin, α-SMA, and transcription factors slug, snail, and twist when treated with Sulforaphane in the TGF-β1-induced model; Sulforaphane treatment also reduces expression of TGF-β1 and collagen I in BLM mouse models [
18]. The natural compound breviscapine inhibits EMT in BLM mouse models via the nuclear transcription kappa-B/NLR Family, Pyrin Domain Containing Protein 3 pathway [
19] ().
. Molecular mechanism of epithelial-mesenchymal transition (EMT) in alveolar epithelial cells (AECs). In the progression of Idiopathic pulmonary fibrosis (IPF), the TGF-β, Wnt, nuclear factor kappa-B /Nuclear transcription kappa-B/Recombinant NLR Family, Pyrin Domain Containing Protein 3 (NF-κB/NLRP3) signaling pathways are activated following AECs injury. This results in the down-regulation of epithelial marker proteins (AQP5: Aquaporin 5; CDH1: Cadherin 1; ZO-1: Zonula occludens-1; TJP1: Recombinant tight junction protein 1; E-Cadherin; Cytokeratin) and the expression of extracellular matrix marker proteins (VIM: vimentin; TTF-1: Thyroid transcription factor 1; N-Cadherin, pro-SP-B: pro-surfactant protein-B; COL1A1; α-SMA: Alpha smooth muscle actin). Transcription factors Slug, Snail, and Twist exhibit increased transcription levels, along with interstitial changes in fibroblast and myofibroblast proliferation. (<named-content style="color:red;">↑</named-content> up-regulation, <named-content style="color:green;">↓</named-content> down-regulaion, <named-content style="color:red;">+</named-content> positive expression).
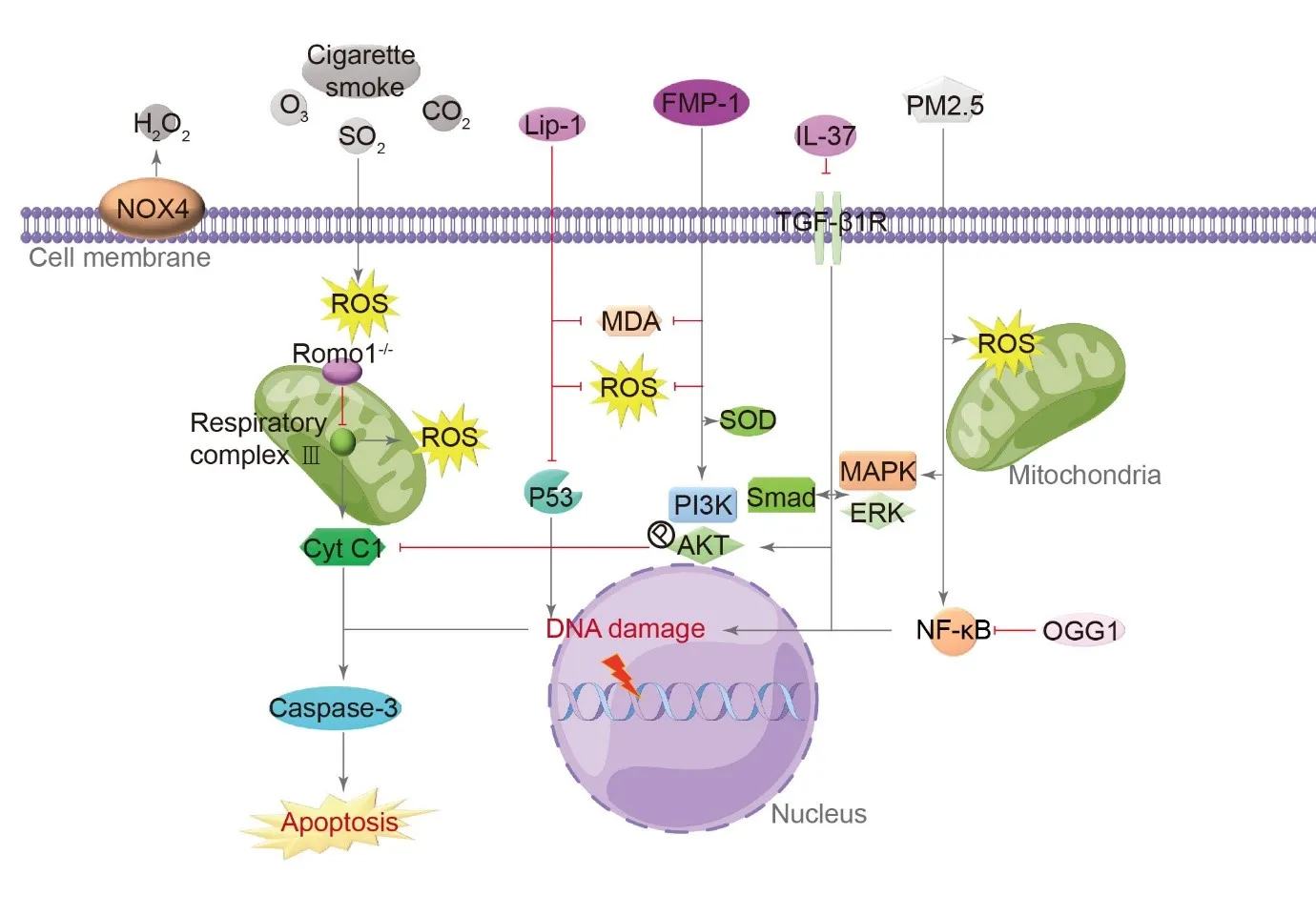
Exogenous and endogenous factors that can cause oxidative stress include O
3, CO
2, SO
2, cigarette smoke, and ROS generated in the airways [
20]. Both in vitro and animal experiments have confirmed that the cellular autoxidation inhibitor Lip-1 protects A549 cells and mice treated with bleomycin. Lip-1 can reduce lipid peroxidation markers malondialdehyde and ROS levels, interfere with the p53 signaling pathway, and attenuate apoptosis of AECs [
21]. ROS regulator 1 (Romo1) mediates ROS production in mitochondria through the electron transport chain’s respiratory complex III. IPF patients express Romo1 mostly on alveolar and bronchial epithelial mitochondria, and TUNEL-positive cells are mainly in alveoli [
22]. Therefore, Romo1 might be responsible for oxidative stress-induced apoptosis in the alveolar epithelium. Knockdown of Romo1 in vitro can inhibit the increase of ROS level and cell death induced by H
2O
2 treatment in A549 cells, playing a protective role [
22]. In A549 cells, FMP-1, a morel polysaccharide exerts significant antioxidant activity in response to H
2O
2-induced oxidative damage, and FMP-1 can inhibit cytochrome C and caspase-3 release through the PI3K/AKT signaling pathway, reducing malondialdehyde and ROS levels and enhancing antioxidant enzymes superoxide dismutase and total antioxidant capacity [
23]. A DNA splicing repair enzyme OGG1 attenuates PM2.5-induced PF by inhibiting signaling pathways related to NF-κB, type II AECs are protected from oxidative damage and death [
24]. AECs apoptosis caused by PM 2.5 oxidative stress also involves MAPK signaling [
25,
26]. The H
2O
2-generating enzyme NOX4 is one of the specific pathological markers of IPF. type II AECs express it highly, and several studies have shown that type II AECs, one of the essential sources of H
2O
2, is associated with PF [
27]. The anti-inflammatory factor IL-37 is significantly down-regulated in AECs of IPF patients [
28]. Mouse AECs are resistant to oxidative stress when treated with IL-37, while knockout of IL-37 increases A549 cell death through inhibiting TGF-β1 receptor, phosphatidyl-inositol 3-kinase/serine-threonine kinase, Smad, and non-Smad (extracellular regulated protein kinases/mitogen-activated protein kinases) signaling pathways [
29] ().
. Molecular mechanism of type II AECs cell apoptosis induced by oxidative stress. In type II AECs, both endogenous and exogenous oxidative stress factors lead to the production of reactive oxygen species (ROS) through mitochondrial respiratory complex III, ultimately promoting apoptosis through Caspase-3. Autooxidation inhibitors such as Lip-1, anti-inflammatory factor IL-37, and ROS regulator 1 (Romo1) can inhibit oxidative stress factors like malondialdehyde (MDA) and ROS, while also enhancing the expression of antioxidant factors like superoxide dismutase (SOD) and total antioxidant capacity (T-AOC). The oxidative damage signaling pathway involves the TGF-β1 receptor, phosphatidyl-inositol 3-kinase/serine-threonine kinase (PI3K/AKT), Smad, and extracellular regulated protein kinases/mitogen-activated protein kinases (ERK/MAPK) signaling pathways. FMP-1is a morel polysaccharide, and OGG1 is a DNA splicing repair enzyme. (→ promotion ┥derepression).
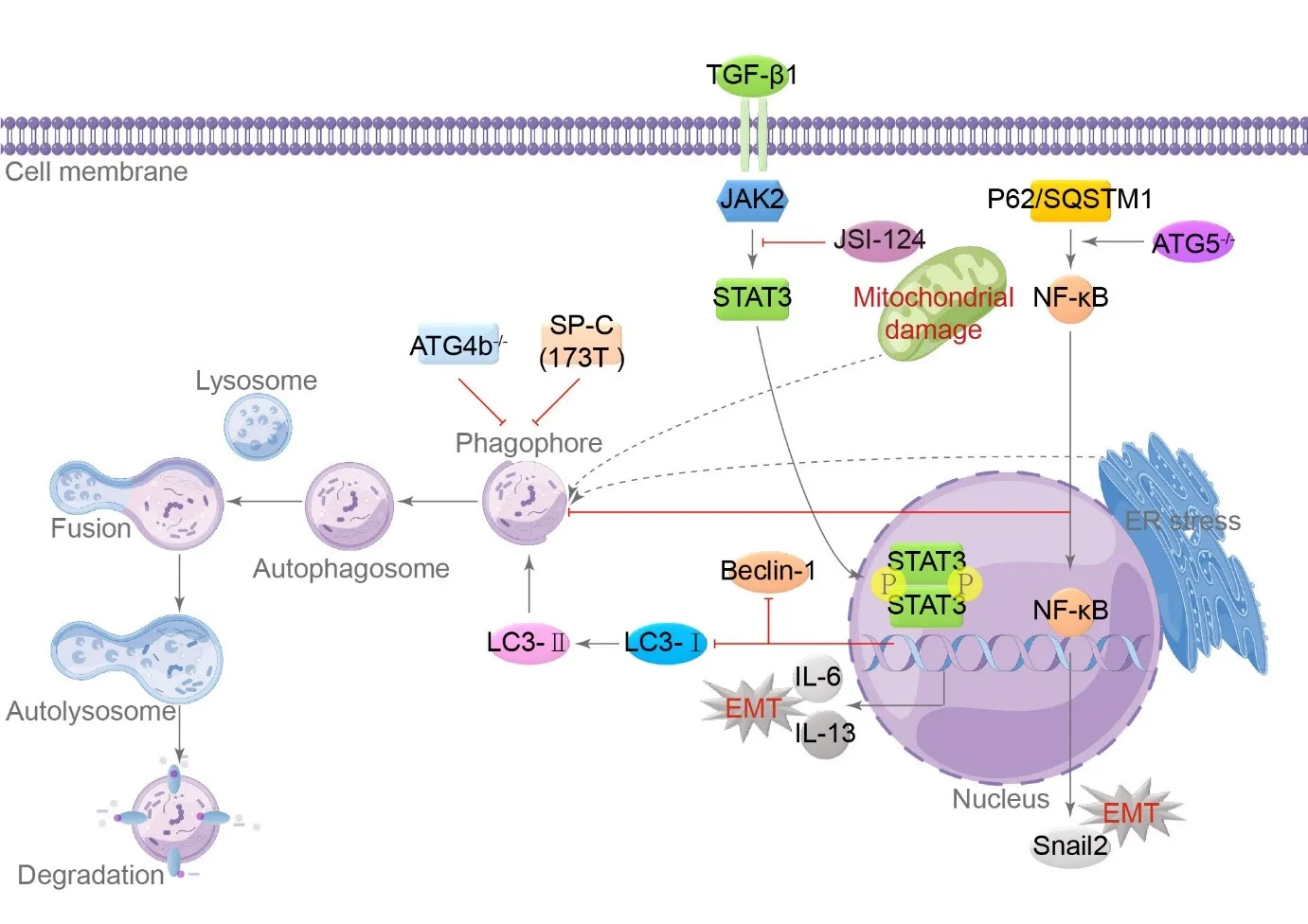
Autophagy is a process in which lysosomes and autophagosomes work together to eliminate misfolded proteins and damaged organelles, such as mitochondria, from cells. Cells can survive on nutrients released by autophagy, such as fatty acids and amino acids released by the lysosomal tract; this is a crucial homeostatic mechanism [
30,
31]. Patients with IPF have significantly lower levels of lung autophagy, and adequate autophagy can lead to cellular senescence [
32]. Decreased autophagy triggers EMT in AECs in the context of IPF [
33], and depletion of autophagy-related gene 5 inhibits autophagy in type II AECs, increasing EMT through the p62/sequestosome 1-NF-κB-Snail2 pathway [
34]. Lung tissue from patients with IPF shows phosphorylation activating the Janus kinase/signal transducer and activator of the transcription 3 signaling pathway [
35]. In vitro experiments with primary type II AECs and A549 cells confirmed that JSI-124, an inhibitor of Janus kinase/signal transducer and activator of transcription 3, can prevent the EMT process caused by TGF-β and the release of IL-6/IL-13, while increasing the expression of autophagy markers Beclin-1 and LC-I/II expression [
30,
36]. Cells that are damaged or stressed show signs of the unfolded protein response (UPR), leading to the accumulation of UPR in endoplasmic reticulum stress (ERS) [
37]. The ER can activate autophagy, and autophagosomes with double membrane sequester cytoplasm and organelles for lysosomal degradation, clearing the enlarged ER and promoting cell survival [
37,
38]. ERS can cause autophagy failure and lead to PF in mice [
39]. Knockout of Atg4b results in reduced autophagy, impairing the pulmonary response to ERS [
37,
40]. The most common SP-C (I73T) mutation found in sporadic and familial cases of IPF is also mechanistically linked to insufficient autophagy in animal models [
37,
41], and the expression level of SP-C (I73T) is directly correlated with the severity of lung injury [
41]. Type II AECs use autophagy to resolve ERS and other types of injuries ().
. Inadequate autophagy promotes the EMT process in type II AECs. In type II AECs, autophagy is hindered by TGF-β1 through Janus kinase/signal transducer and activator of transcription 3 (JAK2/STAT3) signaling, Atg4b deletion via p62/sequestosome 1(SQSTM1)-NF-κB-Snail2, and SP-C (I73T) mutations, leading to the induction of EMT. The classical autophagy process involves phagosome formation, fusion of autophagosome and lysosome, autophagolysosome formation, and final degradation. Autophagic markers include Microtubule-Associated Protein 1 Light Chain 3 (LC3)-I/II and Beclin-1. Both ER and mitochondrial damage can stimulate autophagy, while insufficient autophagy can promote fibrosis. JSI-124 is an inhibitor of JAK2/STAT3, and ATG5 is a autophagy-related gene. (→ promotion ┥derepression).
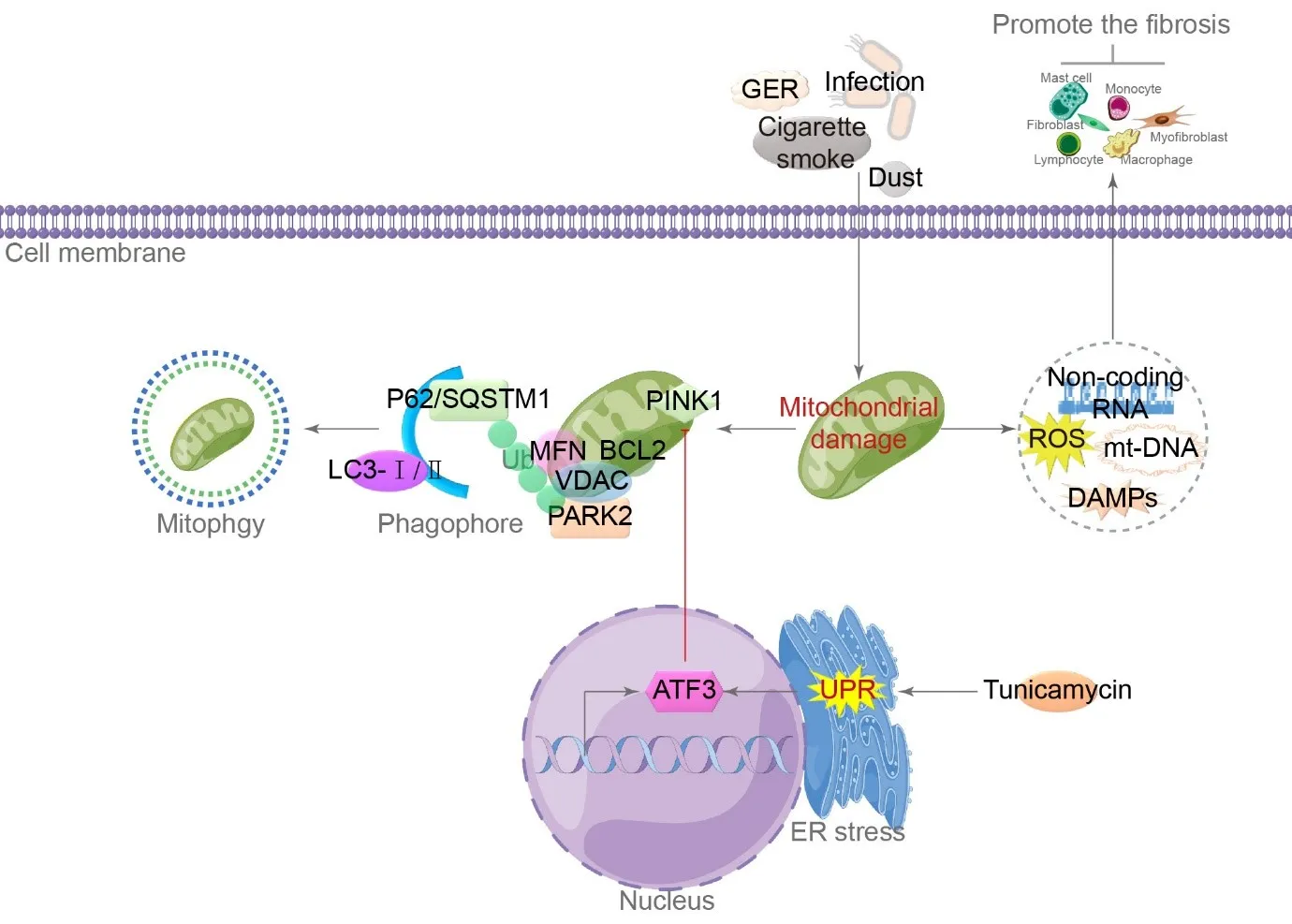
Genetic susceptibility and repeated microinjury from environmental exposures such as cigarette smoke, inhalated dust, infection, and gastroesophageal reflux lead to damage in type II AECs mitochondria [
42,
43,
44,
45]. Damaged mitochondria release a plethora of bioactive molecules, namely damage-associated molecular patterns (DAMPs), which include mtDNA, ROS, and non-coding RNA, which activate fibroblasts/myofibroblasts, macrophages, and immune cell recruitment. These cells secrete inflammatory and growth factors that stimulate their growth and specialization to promote fibrosis [
46]. Mitophagy, a selective process that degrades impaired mitochondria, occurs when autophagy targets mitochondria degraded by lysosomes [
37,
46]. At the mitochondrial outer membrane, the degradation machinery recognizes PTEN-induced putative kinase 1, which can recruit the E3 ubiquitin ligase PARK2 [
37,
47]. Substances such as B-cell lymphoma 2, mitofusin, and voltage-dependent anion channel, are ubiquitinated by p62/SQSTM1 and activate mitophagy through interaction with the LC3 protein [
37,
48]. In PF areas, type II AECs accumulate enlarged and misshapen mitochondria, correlating with markers of ERS [
49]. In a mouse model, tunicamycin-induced ERS is sufficient to induce type II AECs mitochondrial enlargement, mediated by the UPR’s PERK arm. The production of activating transcription factor 3 represses the transcription of PINK1, resulting in higher mitochondrial amounts and spontaneous collagen deposition in alveolar walls in mice [
37,
50] ().
. Mechanism of mitochondrial damage in type II AECs. Both internal and external factors contribute to mitochondrial damage. On one hand, the release of ROS and damage-associated molecular patterns (DAMPs) activates the process of fibrosis. On the other hand, the PTEN-induced putative kinase 1/ E3 ubiquitin ligase (PNK1/PARK2) signaling pathway activates mitochondrial autophagy. ERS increases the expression of the activating transcription factor 3 (ATF3) transcription factor, inhibiting mitochondrial autophagy and promoting PF. GER is gastroesophageal reflux, VDAC is voltage-dependent anion channel, MFN is mitofusin, BCL2 is B-cell lymphoma 2, and UPR is unfolded protein response. (→ promotion ┥derepression).
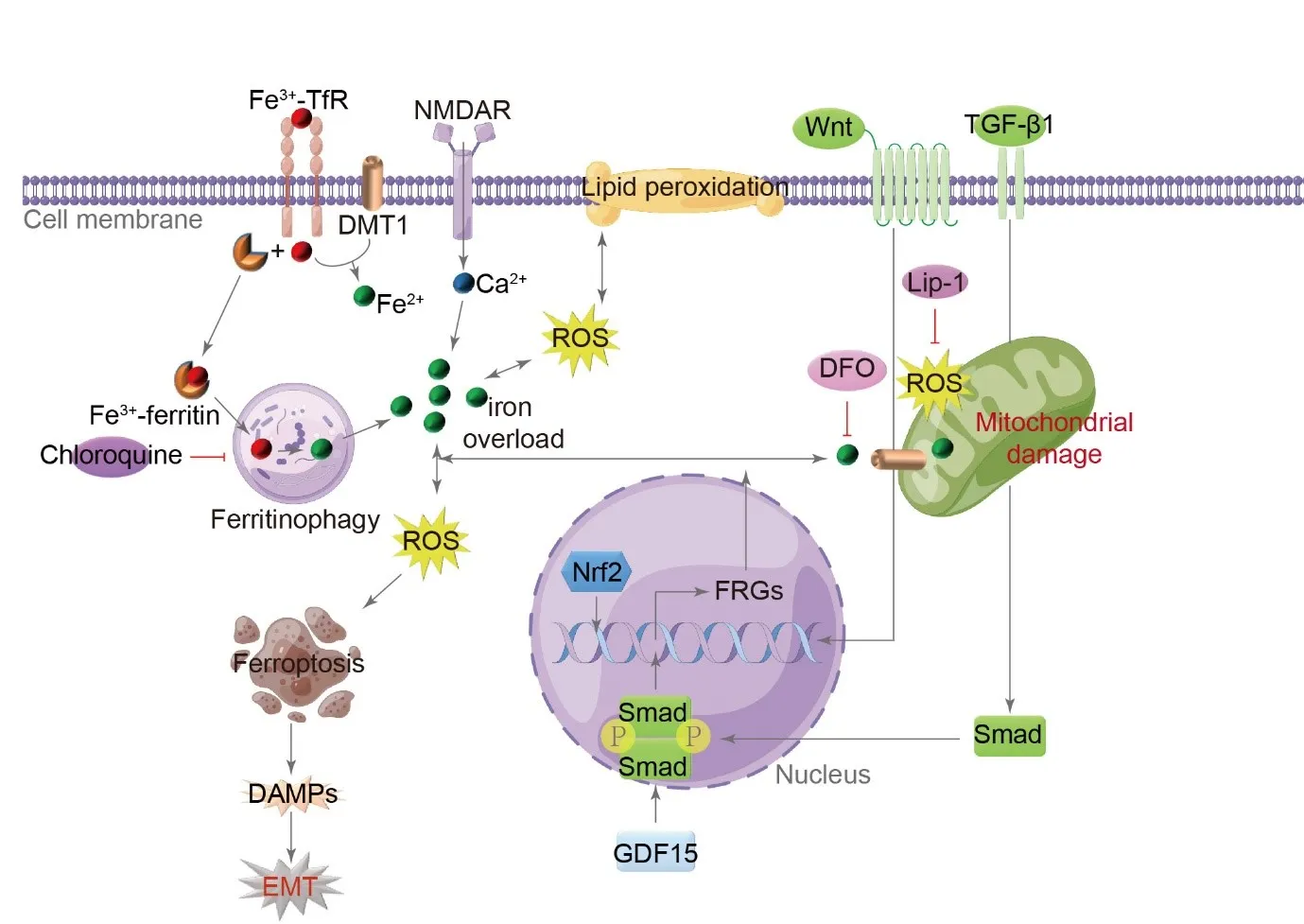
Iron is essential for cellular functions, but excessive levels can be fatal. Iron overload leads to oxidative stress, lipid imbalances, and specific pathological manifestations in mitochondria, resulting in ferroptosis, a form of programmed cell death [
51]. Ferroptotic cells display necrotic morphology, triggering inflammatory reactions and the release of DAMPs from the cells [
52]. Eight ferroptosis-related gene signatures in bronchoalveolar lavage fluid samples from patients with IPF were associated with a poor prognosis in IPF [
53]. In MLE-12 cells derived from the mouse lung epithelial cell line, a cell model of iron accumulation demonstrated that iron accumulation induced cytoplasmic oxidative stress, which subsequently triggers autophagy and the expression of profibrotic factors. In response to repetitive injury, lung epithelial cells undergo prolonged inflammation and sustained EMT [
54]. The regulation of ferroptosis in EMT is significantly influenced by the maintenance of iron homeostasis and oxidation homeostasis [
55].
The precise mechanism by which iron accumulation contributes to the development of IPF remains unknown [
56]. Recent research has identified five ferroptosis-related genes, namely aconitase 1, NRAS proto-oncogene, ectonucleotide pyrophosphatase/phosphodiesterase 2, Mucin 1, and ZFP36 ring finger protein, as potential risk markers for IPF and predictors of unfavorable outcomes [
56]. Additionally, the regulation of ferroptosis through
caveolin 1, nitric oxide synthase 2, growth differentiation factor 15, and
cyclin dependent kinase inhibitor 2A has been found to influence the progression of PF. These genes are involved in processes such as organic anion transport, hypoxia response, oxygen deficiency response, hypoxia-inducible factor 1 signaling pathway, and arachidonic acid metabolism [
57].
The activation of
N-methyl-
d-aspartate receptors impacts iron homeostasis in type II AECs, leading to increased calcium influx. This, in turn, enhances the expression of neuronal nitric oxide synthase and iron regulator protein 1 genes related to iron metabolism, ultimately causing mitochondrial damage, dysfunction, and ferroptosis. This process ultimately contributes to the development of IPF [
58].
Lip-1 and the iron chelator deferoxamine have been found to alleviate PF induced by bleomycin or LPS through iron chelation or reduction of lipid peroxidation in murine models [
59,
60]. Additionally, chloroquine, an autophagy inhibitor, effectively reduces iron-driven oxidative damage-induced EMT in AECs [
54]. The regulation of EMT-induced PF through TGF-β/Smad, nuclear factor, erythroid derived 2, and Wnt signaling pathways is mediated by ferroptosis [
55] ().
. Role of ferroptosis in the pathogenesis of IPF. Endogenous and exogenous injury factors activate various signaling pathways, including TGF-β/Smad, nuclear factor, erythroid derived 2 (Nrf2), and Wnt, resulting in the transcription of ferroptosis-related genes (FRGs). This activation also leads to enhanced binding of Fe<sup>3+</sup> with transferrin receptor (TfR) and subsequent transformation into Fe<sup>2+</sup> through the action of bivalent metal transporter 1 (DMT1) during endocytosis. The stored Fe<sup>3+</sup>, in combination with ferritin, triggers iron autophagy, releasing Fe<sup>2+</sup> and causing its accumulation in the cytoplasm. Furthermore, activating N-methyl-D-aspartate receptors (NMDAR) promotes an influx of calcium and contributes to iron overload. The excessive presence of Fe<sup>2+</sup> leads to mitochondrial damage, the generation of ROS, the promotion of lipid peroxidation, the induction of ferroptosis, the release of DAMPs, and the facilitation of EMT. In lung epithelial cells, the iron-chelating agent deferoxamine (DFO), the autophagy inhibitor chloroquine, and lip1 effectively inhibit EMT induced by iron-driven oxidative damage. GDF15 is growth differentiation factor 15. (→ promotion ┥derepression).
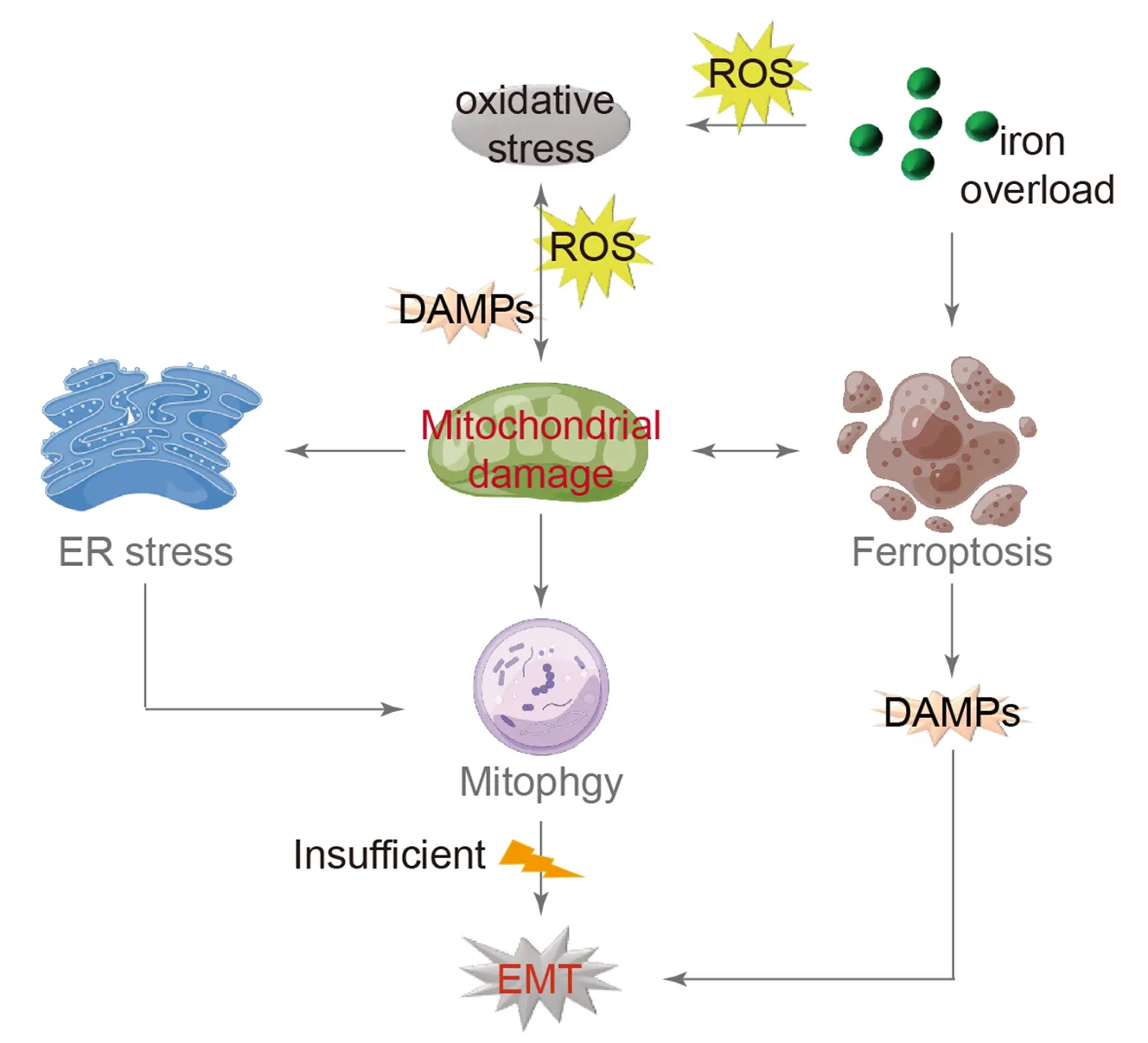
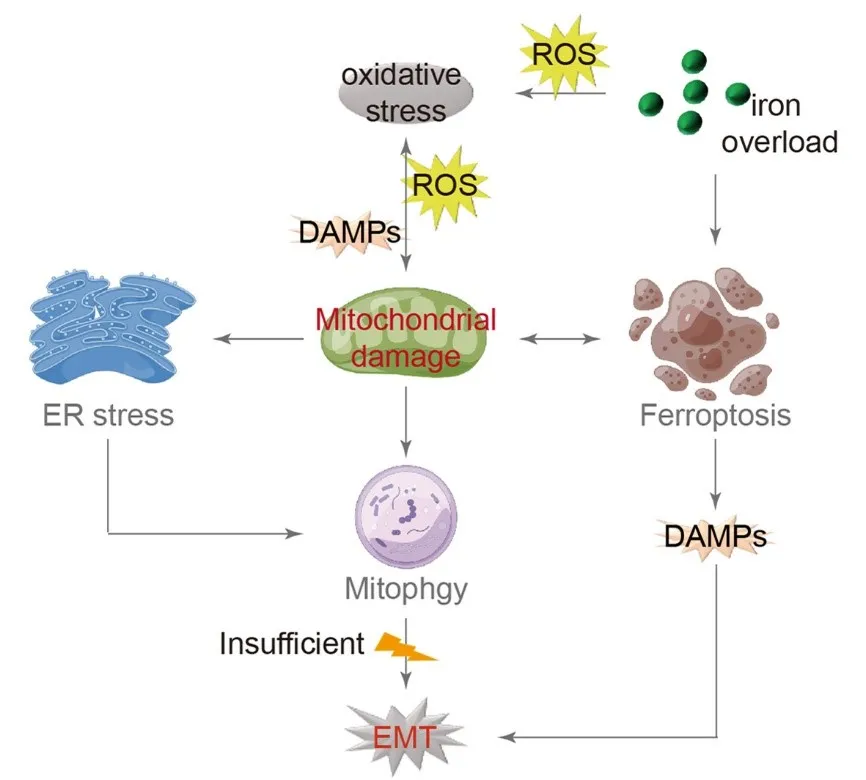
As previously mentioned, the EMT concept in the approach shed light on various characteristics of IPF lungs. The occurrence of oxidative stress and mitochondrial damage is significant, establishing AEC damage as a fundamental model for the development and progression of IPF. Both endogenous and exogenous oxidative stress factors contribute to inducing mitochondrial damage, releasing additional DAMPs and ROS, which worsens oxidative stress. Consequently, damaged mitochondria undergo autophagy for metabolic purposes while also contributing to ERS, which can activate autophagy as well. Inadequate autophagy can lead to EMT.
Iron overload has become a prominent topic of investigation in current research, mainly due to excessive external supplements or internal metabolic dysregulation. This issue is especially common in individuals aged 65 and older. Iron, acting as a powerful catalyst, helps produce oxygen free radicals, which can lead to the peroxidation of the phospholipid membrane in lung epithelial cells. This process notably causes oxidative damage to the mitochondrial membrane and triggers ferroptosis. After apoptosis, AECs release DAMPs, which worsen the progression of EMT ().
. Model of IPF with mitochondrial damage as the central mechanism. Oxidative stress is the initial trigger for mitochondrial damage, leading to the release of additional DAMPs and ROS, further exacerbating oxidative stress. Mitochondria respond to this damage by initiating autophagy for metabolic purposes. Moreover, mitochondrial damage induces ERS, which can also activate autophagy. Inadequate autophagy may ultimately lead to EMT. Iron overload, a known contributor to oxidative stress, enhances DAMPs production, which is lined to mitochondrial damage and EMT. (→ promotion).
IPF is a chronic and progressive respiratory condition characterized by the destruction of alveolar structures and the excessive growth of fibrotic tissue in the extracellular matrix. It is now recognized that the process of transforming AECs into fibroblasts, known as EMT, contributes to the development of PF. Furthermore, both in vivo and in vitro experiments have shown the significant role of EMT in type II AECs. The IPF model discussed in this review establishes a link between mitochondrial damage, oxidative stress, autophagy, and iron-induced cell death, providing a more comprehensive understanding of the EMT process in AECs. Initially, mitochondrial damage is not only caused by oxidative stress but also acts as an intrinsic factor contributing to oxidative stress. Additionally, iron accumulation is identified as the root cause of oxidative stress, and the interaction between iron-induced cell death and mitochondrial damage is mutually influential. Furthermore, autophagy and UPR may play crucial roles in regulating normal cellular stress responses, and any disruption in these pathways negatively affects type II AECs in the pulmonary system. This evidence highlights the significant role of mitochondrial damage in the alveolar epithelium and the persistent nature of oxidative stress in the development of IPF.
We use Figdraw software to create figures.
G.Y., X.G.: Writing-original draft preparation, funding acquisition; X.G., R.W.: Writing—review and editing; X.G., Y.Z., Y.W., X.P., C.X., Z.L., H.L.: Figure preparation and proofreading. All authors have read and agreed to the published version of the manuscript.
No humans or animals were used in the present research.
Supported by Ministry of Science and Technology, PR China grant 2019YFE0119500; Key R&D Program of Henan province grant 231111310400; Zhongyuan scholar 244000510009; Henan Project of Science and Technology grants 232102521025, and GZS2023008.
The authors have declared that no competing interest exists.