Found 6 results
Review
18 February 2025The Intersection between Immune System and Idiopathic Pulmonary Fibrosis—A Concise Review
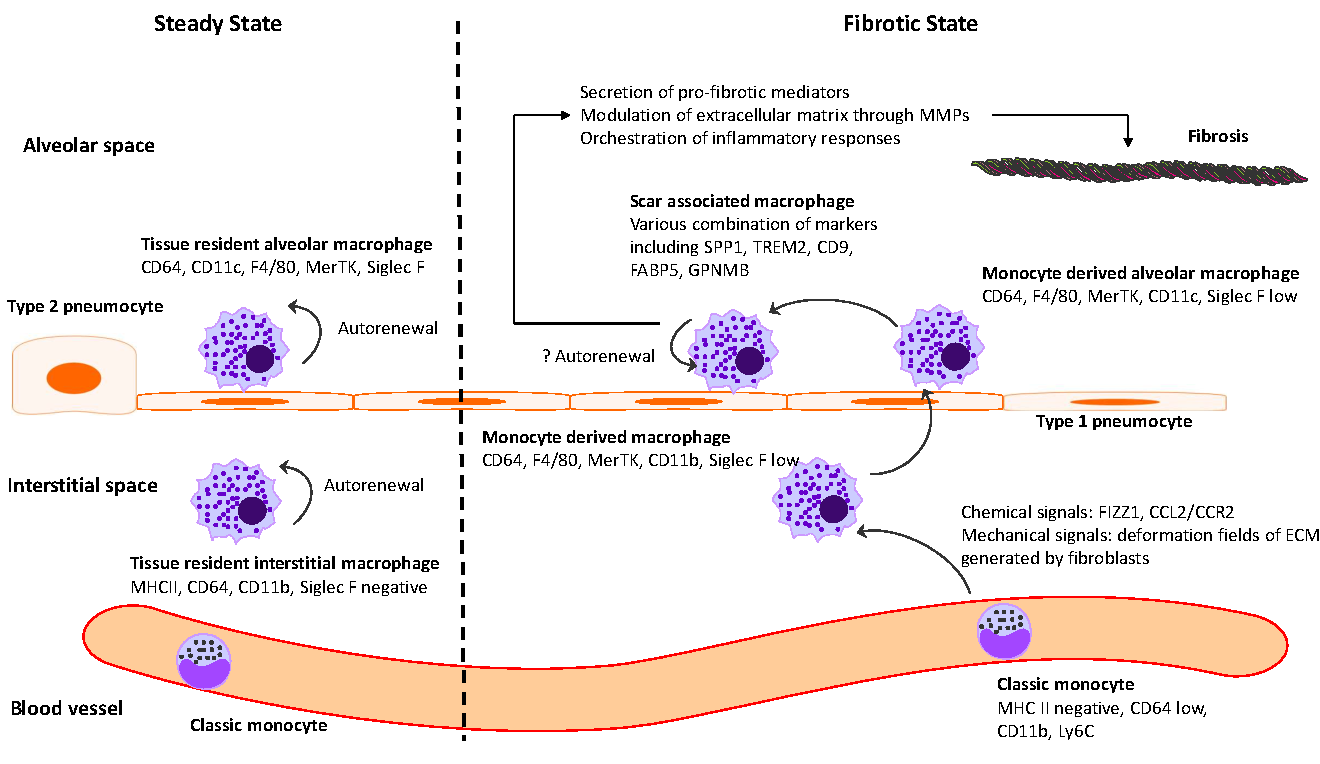
Idiopathic pulmonary fibrosis (IPF) is marked by progressive alveolar destruction, impaired tissue regeneration, and relentless fibrogenesis, culminating in respiratory failure and death. A diverse array of resident and non-resident cells within the lung contribute to disease pathogenesis. Notably, immune cells, both resident and recruited, respond to cues from sites of lung injury by undergoing phenotypic transitions and producing a wide range of mediators that influence, initiate, or dictate the function, or dysfunction, of key effector cells in IPF pathology, such as alveolar epithelial cells, lung fibroblasts, and capillary endothelial cells. The role of the immune system in IPF has undergone an interesting evolution, oscillating from initial enthusiasm to skepticism, and now to a renewed focus. This shift reflects both the past failures of immune-targeting therapies for IPF and the unprecedented insights into immune cell heterogeneity provided by emerging technologies. In this article, we review the historical evolution of perspectives on the immune system’s role in IPF pathogenesis and examine the lessons learned from previous therapeutic failures targeting immune responses. We discuss the major immune cell types implicated in IPF progression, highlighting their phenotypic transitions and mechanisms of action. Finally, we identify key knowledge gaps and propose future directions for research on the immune system in IPF.
Review
13 January 2025Comparative Analysis of Idiopathic Pulmonary Fibrosis and Progressive Pulmonary Fibrosis: Epidemiology, Pathophysiology, Clinical Features, Diagnosis and Treatment
Idiopathic pulmonary fibrosis (IPF) is a chronic fibrosing interstitial disease of unknown origin, characterized by radiological and histological features consistent with usual interstitial pneumonia (UIP). It is marked by a progressive worsening of dyspnea and a decline in lung function. Both IPF and PPF are comparable because they have poor prognoses with a median survival time from diagnosis of around 2–4 years without antifibrotic therapy. This review shows the main specific characteristics and differences of epidemiology, pathophysiology, clinical and radiological features, treatment, and prognosis of IPF and PPF.

Article
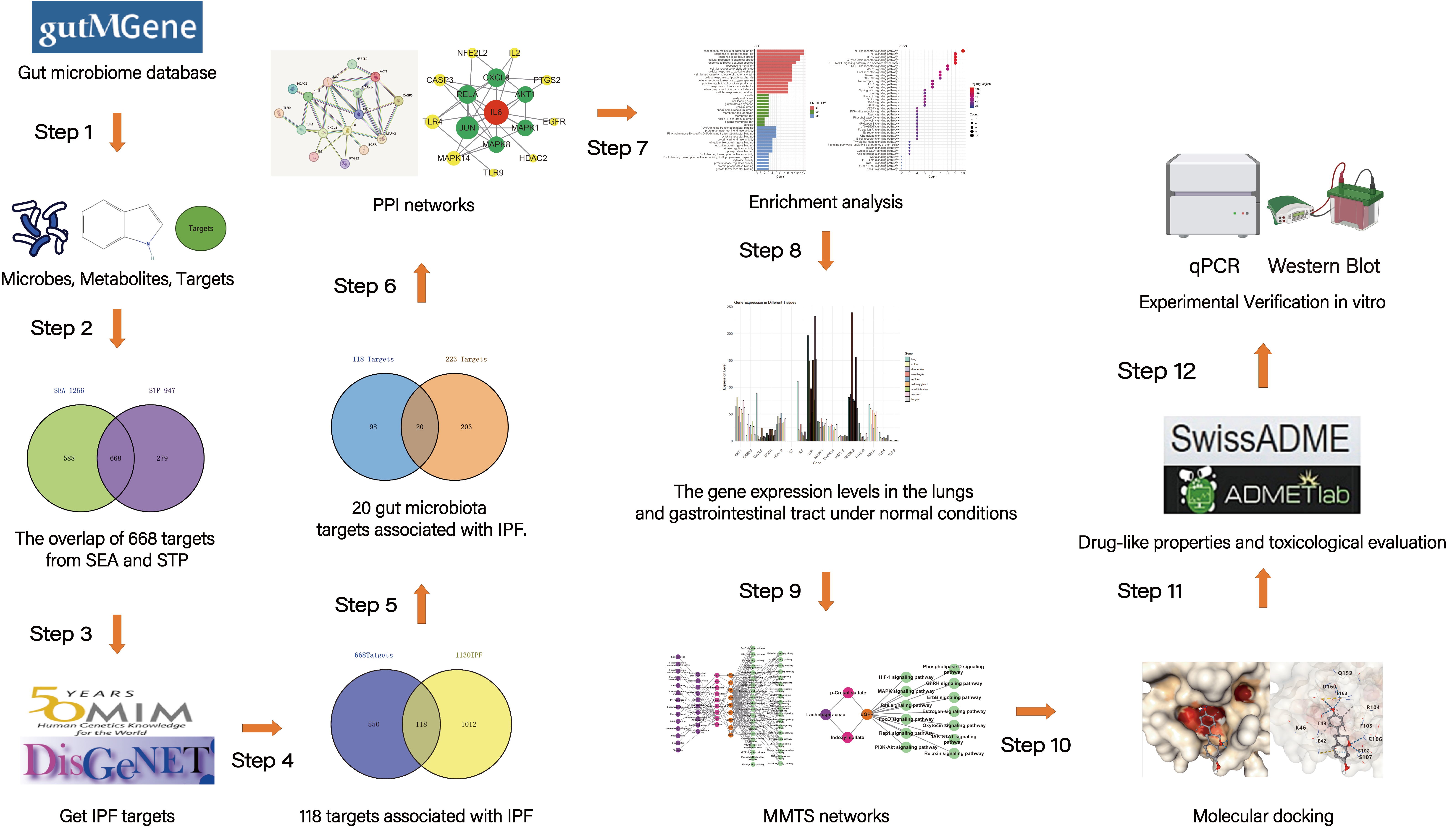
04 November 2024Exploring the Potential Relationship between Gut Microbiome Metabolites and Idiopathic Pulmonary Fibrosis via Network Pharmacology Study
Idiopathic pulmonary fibrosis (IPF) is a chronic, progressive fibrotic lung disease with a poor prognosis. Previous research has revealed that the gut microbiota is associated with human health and immunity, and it interacts with the lung through the “gut-lung axis”. This study explores the potential relationship between Gut Microbiome Metabolites and Idiopathic pulmonary fibrosis via Network Pharmacology Study. The metabolites from gut microbiota were retrieved from the gutMGene database, and gene targets for these metabolites were obtained from previous studies. Gene targets of IPF were obtained from public databases (DisGeNET, OMIM). Subsequently, following the identification of shared targets, IL6 was determined as the core target through protein-protein interaction analysis. Then, a microbiota-metabolite-target-signaling pathway network (MMTS) was constructed using Cytoscape 3.10, and targets with low expression in the lungs and intestines were deleted. The MMTS network revealed that three short-chain fatty acids—acetate, butyrate, propionate, and a flavonoid compound called equol—are IL6-related metabolites. Then, we performed a molecular docking test (MDT) using CB-Dock2 to validate the affinity between core targets and metabolites. MDT confirmed that equol produced by the conversion of isoflavones from Lactobacillus paracasei JS1 was more stable in binding to IL6 than the other three short-chain fatty acids, thereby affecting multiple signaling pathways and influencing the progression of IPF. Finally, we validated this hypothesis through in vitro cell culture experiments, further confirming the effect of Equol.
Review
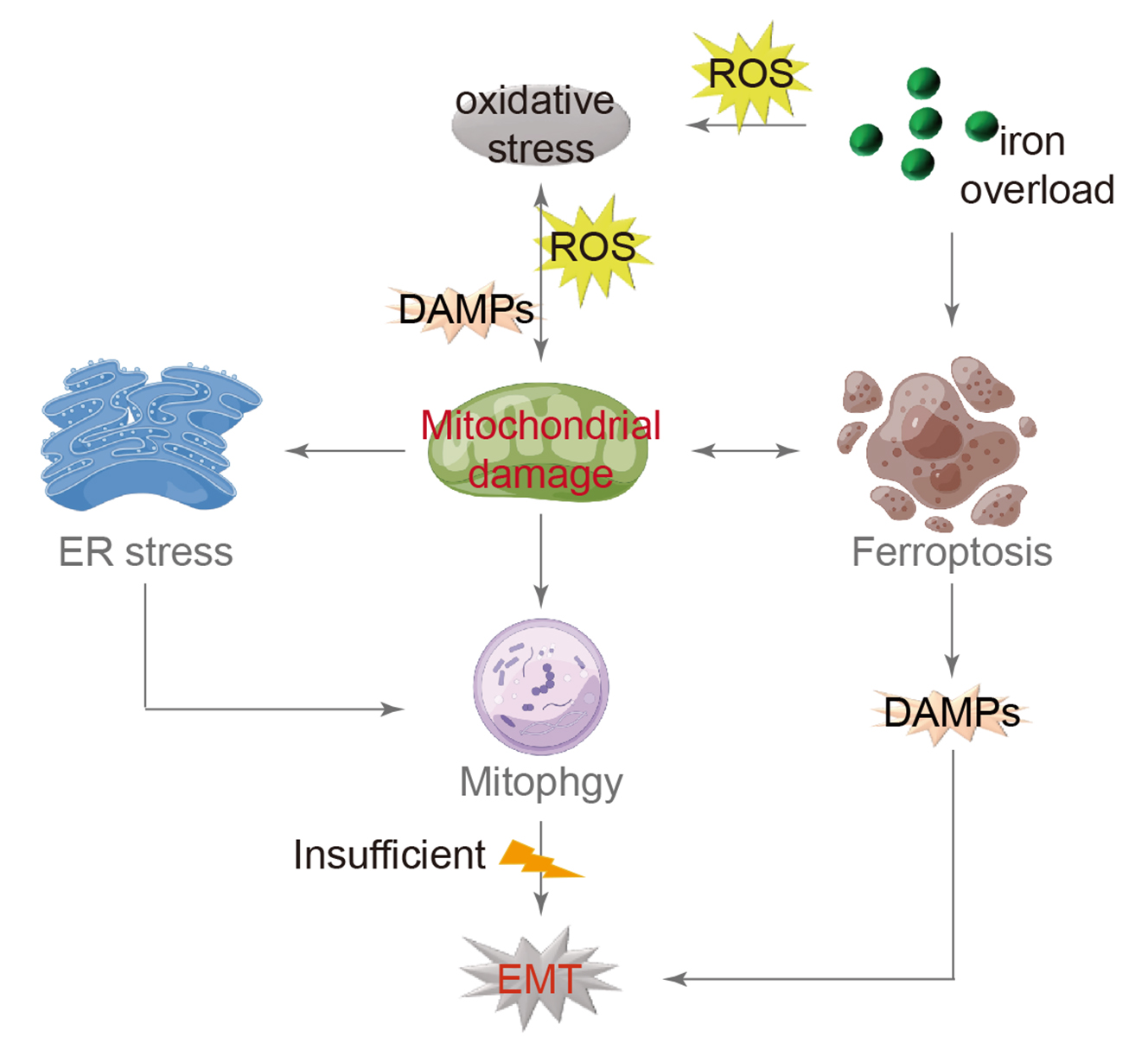
16 July 2024Mitochondrial Damage and Epithelial-Mesenchymal Transition as Major Triggers of the Development of Idiopathic Pulmonary Fibrosis
Idiopathic pulmonary fibrosis (IPF) is a type of interstitial pneumonia with an unknown cause that progresses gradually, primarily affecting the elderly. The presence of fibrosis has significant implications for individuals with reduced lung compliance, resulting in decreased quality of life and limited survival. Although the exact mechanism remains unclear, researchers have investigated various factors, such as senescent telomerase replication and abnormal lung stem cell differentiation, to understand the root cause. Extensive research has consistently shown that IPF is closely linked to the dysfunction of alveolar epithelial cells. Current scientific studies on IPF cover a range of aspects including oxidative stress, endoplasmic reticulum stress, mitochondrial damage, and iron-induced apoptosis. By examining these mechanisms, a comprehensive model has been developed that explains the process of IPF. Oxidative stress is identified as the primary trigger, followed by mitochondrial damage as a central component, leading to the mesenchymal transformation of alveolar epithelial cells as the ultimate outcome. This model is expected to serve as a valuable reference for understanding the mechanism of IPF and guiding future drug development efforts.
Article
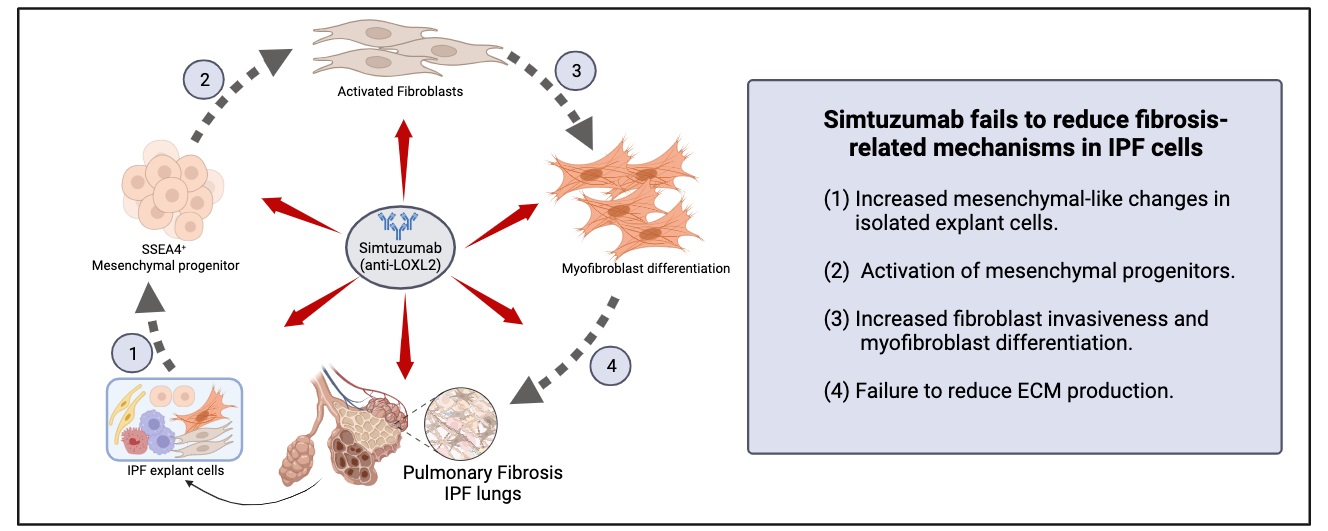
28 November 2023Translational Studies Reveal the Divergent Effects of Simtuzumab Targeting LOXL2 in Idiopathic Pulmonary Fibrosis
The composition of extracellular matrix (ECM) is altered during pathologic scarring in damaged organs including the lung. One major change in the ECM involves the cross-linking of collagen, which promotes fibroblast to myofibroblast differentiation. We examined the role of lysyl oxidase (LOX)-like 2 in lung progenitors and fibroblasts cultured from normal or IPF lung samples and in a humanized mouse model of IPF using a monoclonal antibody (Simtuzumab). Primary lung fibroblasts from normal donor lungs and IPF lung explants were examined for expression of LOXL2. Targeting LOXL2 with Simtuzumab on normal and IPF fibroblasts was examined both in vitro and in vivo for synthetic, functional, and profibrotic properties. LOXL2 was increased at transcript and protein level in IPF compared with normal lung samples. In a dose-dependent manner, Simtuzumab enhanced differentiation of fibroblasts into myofibroblasts. Inhibition of LOXL2 also enhanced fibroblast invasion and accelerated the outgrowth of fibroblasts from dissociated human lung cell preparations. Finally, preventative or delayed delivery of Simtuzumab enhanced lung fibrosis in a humanized mouse model of pulmonary fibrosis. Consistent with its failure in a Phase 2 clinical trial, Simtuzumab exhibited no therapeutic efficacy in translational in vitro and in vivo assays.
Article
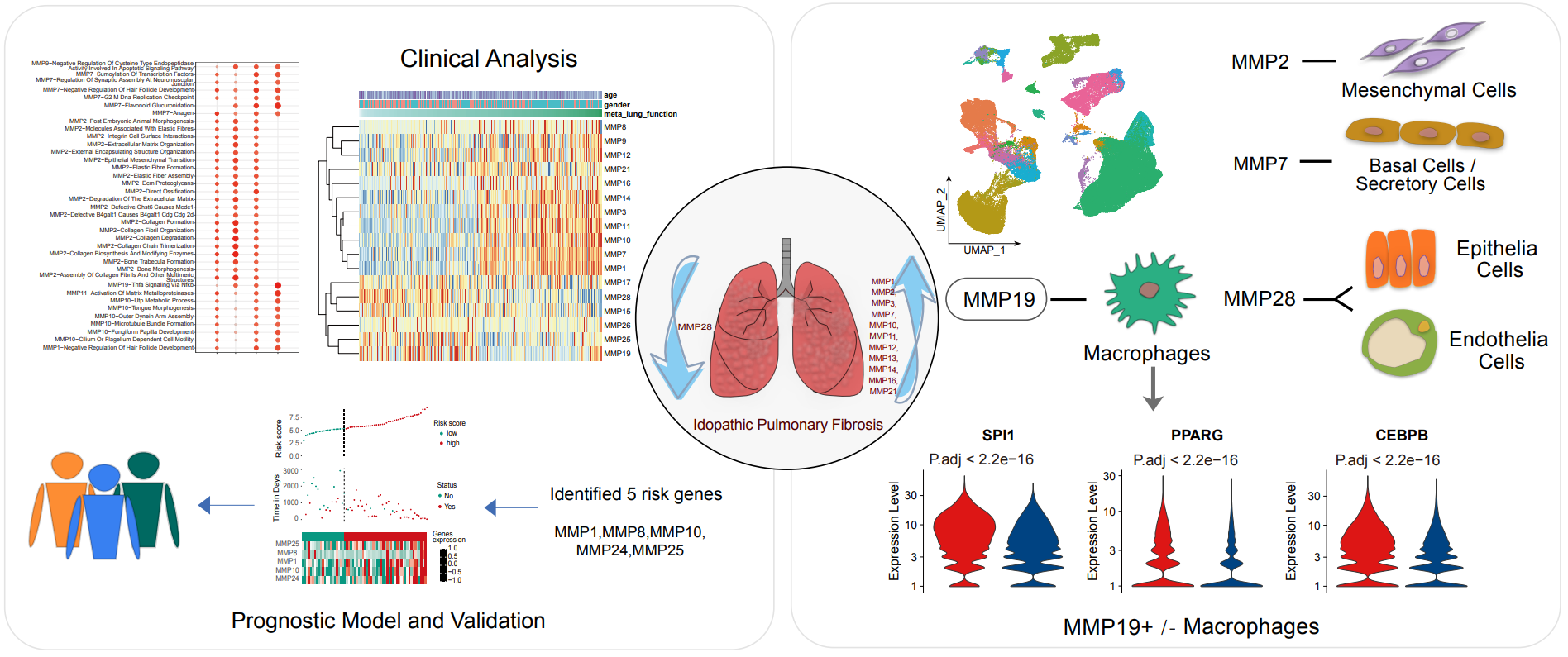
19 May 2023Comprehensive Landscape of Matrix Metalloproteinases in the Pathogenesis of Idiopathic Pulmonary Fibrosis
Idiopathic pulmonary fibrosis (IPF) is a progressive, chronic interstitial lung disease with unknown etiology. Matrix metalloproteinases (MMPs) are involved in fibrotic lung tissues, contributing to the initiation, progression, or resolution of chronic inflammatory disease. In present study, comprehensive changes of MMPs expressions were investigated in IPF by integrative analysis of single-cell transcriptome and bulk transcriptome data. 24 of MMPs were altered and the changes could significantly distinguish IPF from normal subjects and other lung diseases. Among them, MMP1, MMP7 and MMP19 were closely associated to lung functions, susceptibility and alveolar surface density. MMP1 and MMP7 as potential diagnostic indicators, MMP1 and MMP19 as prognostic markers in IPF could accurately predict disease progression. Devolution of MMPs at single-cell resolution, MMP19 was highly expressed in macrophages and markedly interfered with TNF signaling pathway which synchronizes fibrotic microenvironment. MMP19+ macrophages were significantly different from MMP19- macrophages in energy metabolism and immune function. The interaction of MMP19+ macrophages with hyperplastic AT2 was mediated by TNFSF12-TNFRSF12A, and further activated the TNFRSF12A receptor to affect cell glucose metabolism and mitochondrial function. In summary, MMPs has great application potential in the diagnosis, treatment, and prognosis of IPF.