Roles of Astrocytes in Radiation-Induced Brain Injury: Pathophysiological Mechanisms and Therapeutic Strategies

Roles of Astrocytes in Radiation-Induced Brain Injury: Pathophysiological Mechanisms and Therapeutic Strategies

Received: 04 February 2026 Revised: 27 February 2026 Accepted: 12 March 2026 Published: 23 March 2026

© 2026 The authors. This is an open access article under the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/).
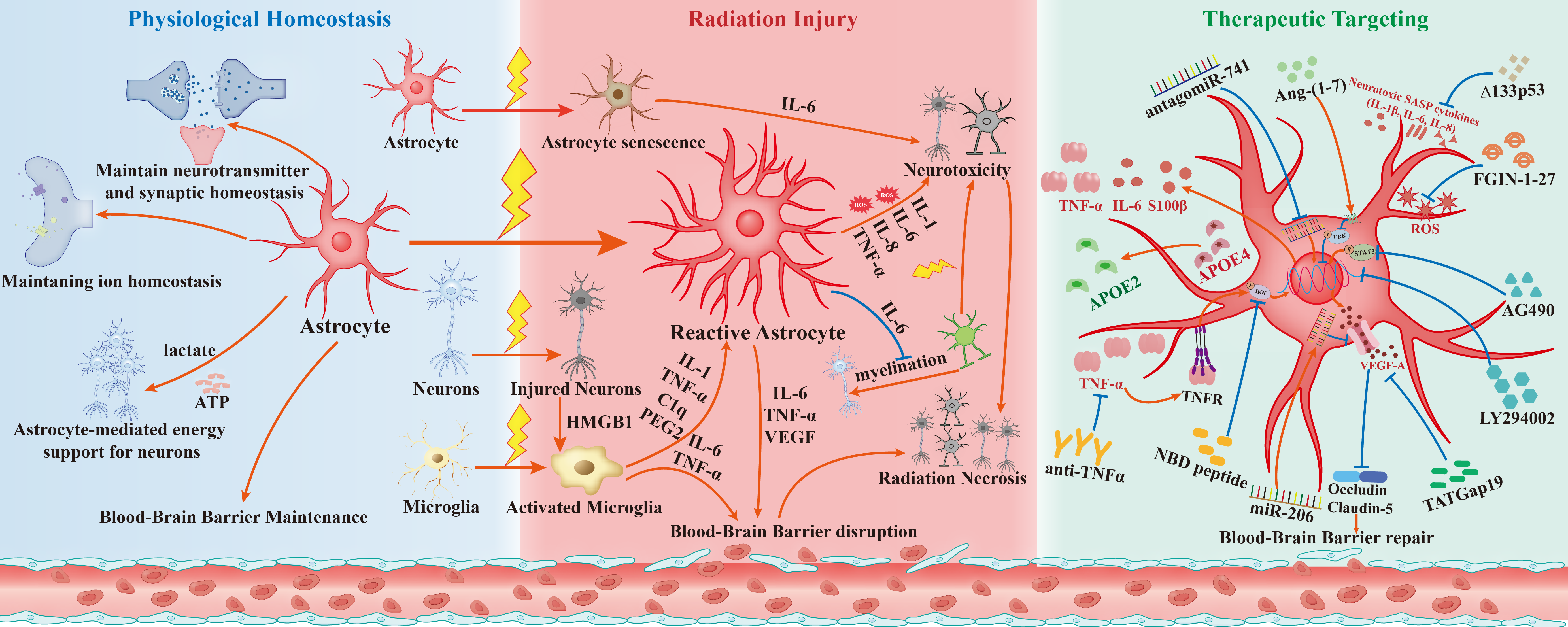
Graphical Abstract
1. Introduction
Head radiotherapy, a crucial therapeutic modality for both primary and metastatic brain malignancies, inevitably affects surrounding normal brain cells during the targeted irradiation of tumor tissues, resulting in RIBI [1,2,3,4]. RIBI is a multifaceted, intricate pathophysiological alteration in neurons, glial cells, and the intracranial vasculature triggered by ionizing radiation. RIBI in clinical radiotherapy is categorized as acute (occurring during irradiation and up to days and weeks post-irradiation), subacute or early-delayed (occurring within four months post-irradiation), and late-delayed (occurring more than 6 months to many years post-irradiation), according to the temporal framework and clinical manifestation [5,6]. In the acute and subacute periods, individuals may exhibit reversible symptoms including headache, fatigue, lethargy, and impaired focus. During this phase, patients may experience pathophysiologic alterations such as DNA damage, oxidative stress, apoptosis, inflammatory symptoms, and compromise of the blood-brain barrier (BBB) [7]. Patients with late-delayed conditions display cerebral white matter reduction, vascular dysfunction, and demyelination, resulting in cognitive decline in memory, learning, processing speed, attention, and executive function [8,9,10]. In the late stage, radiation necrosis (RN) represents one of the most severe complications of RIBI, characterized by irreversible cerebral tissue necrosis caused by endothelial and glial cell damage, a self-reinforcing cycle of inflammation and ischemia, and reactive astrogliosis with aberrant VEGF upregulation [11,12]. Modern radiotherapy techniques, such as intensity-modulated radiation therapy (IMRT), helical tomotherapy (HT), proton therapy, and FLASH radiotherapy (FLASH-RT), can decrease radiation-related damage to normal tissues; however, insufficient comprehension of the cellular response of the CNS to IR constrains the advancement of novel treatments [13].
Among CNS cells, astrocytes are key mediators of IR-induced cellular damage, accounting for 20–40% of brain cells, as their dysfunction exacerbates neuroinflammation and BBB disruption in RIBI [14]. Thus, clarifying astrocyte-related mechanisms is critical for developing targeted therapies. Astrocytes execute several activities in the CNS, such as neurotransmitter uptake and recycling, supplying nutritional support and providing metabolic assistance for the neurons, contributing to synapse development and transmission of synaptic information, maintaining ionic homeostasis, facilitating BBB formation, and executing various other essential brain functions [15,16,17]. There are two primary categories of astrocytes, which are protoplasmic astrocytes and fibrous astrocytes. These astrocytes are primarily found in the gray matter and white matter of the brain, respectively [16,18]. Astrocytes can be activated to differentiate into reactive astrocytes in pathological conditions (e.g., infections, neurodegenerative diseases, RIBI) [19,20,21]. Reactive astrocytes are distinguished by various morphological and functional alterations in response to neurological injury or disease, including increased cell size, thickening and proliferation of protrusions, heightened expression of specific cellular markers, and notably upregulation of glial fibrillary acidic protein (GFAP) [22]. Although reactive astrocytes fall into a number of different categories, they are typically classified into two polarized states during the course of a disease: a neurotoxic or pro-inflammatory phenotype (A1) and a neuroprotective or anti-inflammatory phenotype (A2) [23,24]. Based on the structure, proliferative state, and interacting cell types, reactive astrocytes can be categorized into two groups: those that proliferate and create new, permanent tissue structures, and those that do not proliferate, retaining their original cellular organization, structure, and functional interactions—that is, exhibiting traits of healthy tissues [25]. Additionally, reactive astrocyte type has also been characterized as scar-forming reactive astrocytes and hypertrophic reactive astrocytes [14]. Nonetheless, increasing evidence indicates that a binary polarized classification of reactive astrocytes inadequately represents their phenotypic variability across many illnesses. With the development of single-cell transcriptomes as well as single-cell nuclear transcriptomics, there is a future trend toward determining a more accurate phenotypic classification of astrocyte activation [26]. This review elucidates the regulation of astrocyte activation in RIBI and analyzes the possible therapeutic targets in astrocytes for the treatment of radiation-induced brain injury.
2. Functional Overview of Astrocytes
2.1. Astrocytes: Origin and Classification
Astrocytes are among the most prevalent neuroglial cell types, originating from the primary stem cell type in the nervous system, specifically the neuroepithelial cells within the neural tube. These neuroepithelial cells, which can differentiate into both neurons and glial cells, are crucial for nervous system development [27]. As the cerebral cortex progressively matures, radial glial cells gradually reduce their migratory and neuronal-supporting roles and transform into astroglial precursor cells [28]. Astrocyte precursor cells migrate to different regions of the nervous system, continue to differentiate and mature in specific microenvironments, and eventually develop into mature astrocytes throughout the CNS [29,30]. Mature astrocytes are categorized into two primary types based on their shape and function: protoplasmic astrocytes and fibrous astrocytes. Protoplasmic astrocytes are predominantly situated in the gray matter of the brain and possess an intricate branching network. Their primary function is to maintain synaptic microenvironment stability and regulate neurotransmitter clearance. Fibrous astrocytes are mainly located in the white matter, with long processes that support nerve fiber structure and promote post-injury recovery [31,32].
2.2. Heterogeneity of Astrocytes
Multiple studies have demonstrated the variability of astrocytes, indicating that they do not constitute a uniform cell population. Astrocytes have a significant variety across various dimensions, including shape, developmental origins, gene expression profiles, and pathophysiological roles. Investigating the diversity of astrocytes is crucial for elucidating their many roles in the brain and their involvement in the initiation and advancement of neurological disorders [33]. In recent years, advancements in single-cell RNA, single-cell nuclear transcriptome, and spatial genomics technologies have significantly enhanced the identification of heterogeneous and spatially distinct subpopulations of astrocytes, particularly elucidating their diversity and specific functions across various brain regions [34]. Five astrocyte subtypes with unique transcriptomes in the adult mouse cortex and hippocampus have been found by single-cell RNA sequencing [35]. The variability of astrocytes and their critical significance in neurodegenerative diseases will be elucidated further through advancements in sequencing technologies. This inherent heterogeneity, evidenced by single-cell studies and the discovery of subtype-specific astrogenic pools, means that distinct astrocyte subpopulations likely mediate different aspects of the brain’s response to radiation injury, presenting both challenges and opportunities for developing precise therapeutic strategies [36,37].
2.3. Maintaining BBB Integrity
Astrocyte foot processes are closely encased around cerebral blood vessels, establishing direct contact with the endothelial cells of the BBB and offering structural support for it. Furthermore, astrocytic endfeet are crucial for modulating cerebral blood flow, facilitating nutrient uptake, and contributing to waste elimination [38]. Astrocytes interact with endothelial cells via secretion of growth factors, morphogens, and extracellular vesicles, which regulate BBB integrity by modulating tight junction protein expression [39]. Following nervous system injury, astrocytes undergo a transformation into a complex phenotype that serves a protective function by facilitating the restoration of the BBB. Astrocytes can induce endothelial cell death and reduce the expression of proteins related to BBB integrity [40].
2.4. Metabolic and Nutritional Assistance
Astrocytes are essential for the brain’s energy metabolism because they control the flow and distribution of energy molecules like lactate and glucose, which preserve neuronal function and the intracerebral environment’s homeostasis [41,42]. Through a number of metabolic processes, such as glycolysis, the tricarboxylic acid cycle, and oxidative phosphorylation, astrocytes produce energy-rich ATP molecules. At the same time, astrocytes can also undergo glycolysis of ingested glucose to produce lactic acid and deliver lactic acid to neurons via specific transport proteins, which serve as an additional energy supply for neurons [43]. Astrocytes are essential for the energy provision of the CNS; they function as the principal glycogen-storing cells in the brain, capable of energizing neural activity through glycogen release and supporting axonal function and general viability [44]. Studies have shown that adenosine is able to activate the cAMP signaling pathway in astrocytes by binding to the A2B receptor on astrocytes, thereby affecting glucose metabolism in the nervous system. This process is critical for maintaining brain function during periods of high energy demand. When neuronal activity increases, astrocytes rapidly activate glucose metabolism and release lactate to energize neurons [45]. Furthermore, dysfunction of creatine metabolism in astrocytes, such as downregulation of CKB, represents a key link in the energy crisis and neurodegeneration in AD, underscoring the critical importance of this pathway for maintaining neuronal homeostasis [46]. Therefore, astrocytes are crucial for the energy management of the CNS [47].
2.5. Maintaining Ion Homeostasis
Astrocytes are key glial cells that maintain homeostasis in the central nervous system by absorbing synaptically generated neurotransmitters like glutamate [48]. High densities of Kir4.1 potassium channels, NCX sodium-calcium exchangers, and different chloride channels are expressed in their endfoot structures, and these channels use active uptake and spatial buffering mechanisms to accurately control the extracellular concentrations of K+, Na+, Ca2+, and Cl−. Maintaining neuronal resting potential, regulating synaptic plasticity, and ensuring brain signal transmission all depend on this function [49]. This homeostatic system is tightly linked to pathological processes, including epilepsy and neurodegenerative disorders, and its dysregulation can directly result in aberrant neuronal network excitability [50,51].
2.6. Regulating Neurotransmitter Equilibrium and Sustaining Synaptic Stability
As astrocytes in the CNS take up synaptically produced neurotransmitters such as glutamate, gamma-aminobutyric acid (GABA), and glycine, they are responsible for regulating the homeostasis of neurotransmitters [52,53]. Astrocytes not only eliminate neurotransmitters but also engage in their metabolism and recirculation. Extracellular glutamate induces the release of Ca2+ from calcium reserves inside astrocytes, subsequently triggering the release of glutamate from astrocytes to adjacent neurons through cytokinesis, so coordinating neuronal firing and modulating neuronal excitatory or inhibitory activity [54]. Emerging evidence indicates that astrocytes sense norepinephrine signals via their β-adrenergic receptors and actively modulate synaptic function and plasticity through Ca2+-dependent gliotransmitter release [55]. Additionally, astrocytes have the ability to directly modulate synaptic plasticity and synaptic transmission by releasing gliotransmitters that include glutamate, adenosine triphosphate (ATP), taurine, glycine, and D-serine [56].
3. Roles of Astrocytes in Radiation-Induced Brain Injury
Upon exposure to ionizing radiation, astrocytes undergo reactive activation and engage in complex crosstalk with microglia, oligodendrocytes, neurons, and endothelial cells—these interactions collectively drive the pathogenesis of RIBI via neuroinflammation, oxidative stress, and BBB impairment (Figure 1). Under physiological conditions, astrocytes primarily maintain CNS homeostasis by supporting neurotransmitter balance, ion homeostasis, BBB integrity, and neuronal metabolism; however, after radiation exposure, two key pathological cascades are triggered: (1) Direct radiation stimulation converts astrocytes to reactive astrocytes, which secrete VEGF to disrupt BBB; (2) Activated microglia (induced by radiation) and HMGB1 released from injured neurons further promote astrocyte reactivity by secreting pro-inflammatory factors (e.g., IL-1α, TNF-α, C1q), and the subsequent crosstalk between reactive astrocytes and activated microglia exacerbates neuronal/oligodendrocyte damage via IL-6, TNF-α, and ROS. The following sections will elaborate on these mechanisms based on Figure 1’s cellular interaction framework.
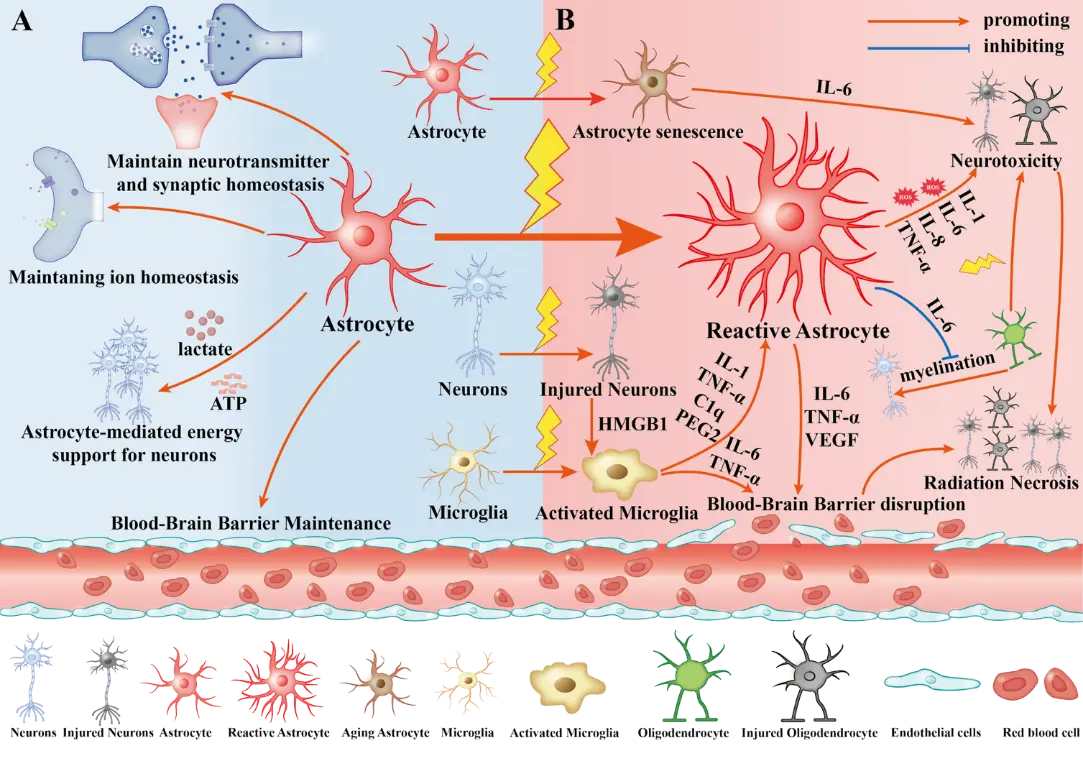
Figure 1. The central role of astrocytes in radiation-induced brain injury (RIBI) and their pathological crosstalk with central nervous system (CNS) cells. Left (A): Physiological state. In the healthy CNS, astrocytes maintain brain homeostasis by uptaking excess neurotransmitters, including glutamate, regulating ion balance, and providing metabolic-nutritional support. Their endfeet interact with endothelial cells to preserve blood-brain barrier (BBB) structural and functional integrity. Right (B): Radiation-induced pathological progression Ionizing radiation (IR) disrupts brain homeostasis and triggers a cascade of reactions: (1) Direct and indirect activation: IR directly converts astrocytes to Reactive Astrocytes; HMGB1 from injured neurons and IR activate microglia, which secrete interleukin-1 (IL-1), tumor necrosis factor-α (TNF-α), complement component 1q (C1q) and prostaglandin E2 (PGE2) to polarize further and expand Reactive Astrocytes. (2) Neurotoxicity and remyelination disorders: Reactive Astrocytes release pro-inflammatory mediators, including IL-1, interleukin-6 (IL-6), interleukin-8 (IL-8), and TNF-α, as well as reactive oxygen species (ROS), directly damaging neurons and oligodendrocytes (OLs). Notably, their secreted IL-6 significantly inhibits oligodendrocyte precursor cell (OPC) differentiation and remyelination. (3) Cellular senescence and inflammation amplification: IR induces astrocytes into a senescent state (Senescent Astrocytes), whose senescence-associated secretory phenotype (SASP) continuously produces IL-6 to exacerbate neurotoxicity. (4) Vasogenic injury: IR induces abnormal VEGF upregulation in Reactive Astrocytes; together with IL-6 and TNF-α, VEGF disrupts endothelial tight junctions, leading to BBB breakdown and cerebral edema. (5) Radiation necrosis (RN) progression: BBB impairment, neuronal and oligodendrocyte death, and astrocyte-microglia-endothelial pathological crosstalk (amplifying inflammation and blocking vascular regeneration) ultimately lead to irreversible RN.
3.1. Reactive Astrocytes
Reactive astrocytes can be classified by different criteria: functional phenotype divides them into A1 (neurotoxic/pro-inflammatory) and A2 (neuroprotective/anti-inflammatory) subtypes, while structural and proliferative characteristics categorize them as scar-forming or hypertrophic reactive astrocytes [14,23]. Astrocytes exhibit ‘reactivity’ under pathological conditions, including inflammation, neurodegenerative diseases, and acute injury, altering the morphology of their elongated protrusions, making them more bifurcated, enlarged, and elongated [57]. This process is accompanied by alterations in gene and protein expression, unlike resting astrocytes. The overexpression of glial fibrillary acidic protein (GFAP) is a prevalent reaction of reactive astrocytes across various species in response to CNS injury, with the extent of upregulation frequently correlating with the lesion’s severity [22]. Scientists have isolated, characterized, and genetically analyzed reactive astrocytes, which can be triggered into two distinct states: a neurotoxic or pro-inflammatory phenotype (A1) and a neuroprotective or anti-inflammatory phenotype (A2). A1 astrocytes secrete pro-inflammatory mediators, including IL-1β, TNF-α, and HMGB1, together with toxic fatty acids, which directly contribute to the destruction of oligodendroglial and neuronal cells [58]. Conversely, A2 astrocytes are protective because they raise cytotrophic and neurotrophic factor levels [59,60]. While the simplistic A1/A2 classification does not cover the full spectrum of astrocyte phenotypes, it improves our understanding of astrocyte responses in various CNS disorders [23].
Radiation therapy for the craniofacial region leads to the direct activation of astrocytes throughout both the acute and late phases of RIBI, primarily evidenced by an increase in GFAP in various brain areas, as documented in several in vivo and in vitro studies. Localized cranial radiation in mice resulted in a marked increase in astrocyte proliferation, vascular permeability, leukocyte adhesion, and vasoconstriction of small arteries within 48 h after a solitary dose of 20 Gy radiation, which was eliminated by TNF suppression [61]. Furthermore, a significant increase in GFAP protein expression was observed in the hippocampus of mice after a single whole-brain irradiation at 30 Gy, and intranasal administration of antagomiR122-5p significantly inhibited radiation-induced activation of hippocampal astrocytes [62]. It was shown that hippocampal astrocyte reactivity was found to increase within 48 h after 9 Gy cranial radiotherapy, and this trend continued until 4 weeks after irradiation [63]. Following the irradiation of the brain with a single dose of 15 Gy of X-rays, astrocytes were activated by the elevated levels of inflammatory markers and overexpression of GFAP [64]. Furthermore, an in vitro experiment revealed that the primary astrocytes derived from the rat cerebral cortex could be activated at 4 h, 12 h, 24 h, or 48 h post-20 Gy radiation, exhibiting reactive hyperplasia and hypertrophy, along with an upregulation of the activation marker of GFAP [65].
On the other hand, X-ray exposure triggered microglia activation and elevated neuronal HMGB1 expression [66,67]. GFAP immunostaining in astrocytes was enhanced by conditioned media derived from irradiated microglia. Subsequent research indicates that activated microglia can induce the formation of reactive astrocytes by the release of pro-inflammatory mediators, including prostaglandin E2 (PGE2), TNF-α, and interleukin-1 (IL-1) [7,21,63].
3.2. Neuroinflammation
Neuroinflammation refers to inflammation in the central nervous system (CNS) or spinal cord induced by infection, trauma, toxicity, and other conditions. Microglia, astrocytes, endothelial cells, and peripheral immune cells can release various cytokines, chemical messengers, reactive oxygen species, and secondary messengers, which induce inflammation and affect the immune system, bodily functions, biochemistry, and mental processes [68,69]. Through traditional inflammatory signaling pathways like NF-κB, reactive astrocytes release a multitude of pro-inflammatory molecules that directly exacerbate neuroinflammation and work in concert with Aβ and Tau pathology to cause neuronal damage and cognitive loss [70,71]. Furthermore, intricate connections among astrocytes and both resident and invading cells inside the CNS are significant in facilitating the onset of neuroinflammation [72].
Specifically, radiation exposure in the brain first elicits an acute inflammatory response, characterized by the activation of astrocytes and microglia. Astrocytes, in collaboration with microglia, secrete an array of pro-inflammatory cytokines (including TNF-α, IL-1β, and IL-6), chemokines, reactive oxygen species (ROS), and reactive nitrogen species (RNS). These compounds harm neuronal cells while simultaneously activating local antigen-presenting cells (APCs), such as microglia and dendritic cells, exacerbating the inflammatory response [73]. The sustained activation of microglia and astrocytes characterizes chronic neuroinflammation, which manifests in the advanced stages of RIBI as enduring cognitive memory impairment [73,74]. Moreover, the secretion of inflammatory agents compromises the BBB, obstructs neurogenesis in the hippocampus, and exacerbates injury to the neurovascular region [75]. In a murine model, cytokines associated with the senescence-associated secretory phenotype (SASP), including IL-1β and IL-6, are elevated subsequent to radiation therapy [76,77]. In the mouse model of Alzheimer’s disease, there was an enhancement of the Nrf2 pathway, a reduction in the formation of neurotoxic reactive astrocytes, amelioration of neuroinflammation-induced cognitive deficits, and a deceleration of aging-related cognitive decline [78]. Conversely, it has been assumed that astrocytes played a dual role in modulating the radiation response. After exposure to radiation, BBB permeability markedly deteriorates before transitioning to a more protective phenotype by reducing oxidative stress during the subacute phase and releasing pro-inflammatory cytokines and chemokines [79]. In addition, astrocytes that were subjected to γ irradiation exhibited a higher exudation of inflammatory cytokines [80]. After irradiation, primary astrocytes exhibited a notable rise in the levels of cytokines, such as IL-1β, IL-6, and IL-8 [81,82]. In summary, both in vitro and in vivo studies demonstrate that inflammatory mediators produced by astrocytes may contribute to neuroinflammation associated with RIBI.
The goal of neuroinflammation is to eliminate harmful metabolite deposits, repair injured tissues, and eradicate pathogenic bacteria. It is the CNS’s reaction to stressful stimuli, including infection, tissue injury, or metabolic abnormalities [83]. Chronic neuroinflammation may be both a cause and a consequence of radiation-induced brain damage. Astrocytes serve as gate-check regulators of innate and adaptive immune responses, significantly influencing neuroinflammation in the radiated brain. Consequently, anti-inflammatory interventions targeting astrocytes for neuroinflammation may be an effective approach for addressing radiation injury in the brain.
3.3. BBB Impairment
The BBB comprises the endfeet of glial cells, the adjacent basement membranes, and the endothelial cells of the brain’s capillaries. The BBB restricts the entry of chemicals from the bloodstream into brain cells, but it also prevents certain therapeutic drugs from penetrating it [84]. Significant secondary damage, including cerebral edema and inflammatory inflammation, occurs following injuries to the head, ischemic stroke, and other disorders of the CNS that compromise the functionality of the BBB. Ionizing radiation impairs the BBB through multiple mechanisms, including DNA damage, endothelial cell death and swelling, local inflammatory responses, and oxygen free radical-induced damage to endothelial tight junctions. IR also increases BBB permeability, thereby facilitating the infiltration of pathogens and inflammatory cells into brain tissue [61,73].
Astrocytes are critical for BBB integrity, as their endfeet tightly wrap around cerebral capillaries and regulate endothelial cell function via growth factor secretion. The BBB is disrupted when endfeet are lost, blood flow changes, and the body’s ability to get rid of trash changes [40,85,86]. A number of brain diseases can be caused by these changes [73]. Ionizing radiation can activate astrocytes, impede their normal functions, and disrupt the BBB through various mechanisms. Forty-eight hours after administration of a single 20 Gy dose of localized cranial radiation, a marked elevation in astrocyte proliferation, blood vessel permeability, leukocyte adherence, and vasoconstriction of tiny vessels was observed. Inhibition of TNF reversed these effects, suggesting that reactive astrocytes may compromise the BBB by releasing inflammatory mediators after exposure to radiation [61]. Additionally, astrocytes and microglia collaborate to secrete pro-inflammatory cytokines such as TNF-α and IL-6, which compromise the integrity of BBB by prompting surviving endothelial cells to elevate intercellular adhesion molecules on the surface of the arterial lumen [7]. Moreover, radiation induces vesicular VEGF release and disrupts the BBB. However, the impairment to the BBB caused by radiation is alleviated by the blockade of astrocyte junction protein 43 hemichannels [87]. Radiation stimulates excessive VEGF production in astrocytes via the PI3K-AKT pathway, leading to disruption of the BBB [88]. The role of astrocytes in regulating the CNS’s and the BBB’s response to space radiation has been progressively revealed in recent years. In rats subjected to space radiation (SR), enlarged lateral ventricles and exacerbated BBB disruption have been associated with astrocyte depletion and an increase in permeable blood vessels [79].
3.4. Oxidative Stress
Oxidative stress disrupts cellular structure and function by disturbing the balance between the production of reactive oxygen species (ROS) and the antioxidant defense system, thereby altering the intracellular redox state. The brain, utilizing 20% of the body’s oxygen, is particularly susceptible to oxidative stress damage due to its relatively deficient antioxidant defense mechanisms, including catalase, superoxide dismutase, glutathione, and glutathione peroxidase [89]. Astrocytes can express many antioxidant enzymes, such as glutathione peroxidase (GPx), catalase (CAT), and superoxide dismutase (SOD). These enzymes, as pivotal antioxidants within the cell, facilitate the synthesis and regeneration of glutathione and the neutralization of reactive oxygen species (ROS) [90]. Astrocytes have a reactive A1 state triggered by fibrinogen (Fg), resulting in compromised antioxidant function, as indicated by the upregulation of pro-inflammatory cytokines and the generation of NO and ROS, which exacerbates oxidative stress during brain injury [91]. In addition, in some neurodegenerative disorders, such as Alzheimer’s and Parkinson’s diseases, astrocytes produce reactive oxygen species (ROS) and reactive nitrogen species (RNS), which contribute to oxidative stress in the central nervous system and accelerate disease progression [92,93,94].
It has been demonstrated that microglia, astrocytes, and neurons are all capable of continuously producing reactive oxygen species (ROS) and reactive nitrogen species (RNS) molecules as a result of exposure to ionizing radiation [95,96,97]. Excessive accumulation of astrocyte-derived ROS damages peripheral neurons and further induces inflammation and cellular damage [98]. Ionizing radiation serves as an exogenous source of reactive oxygen species (ROS) activation, whereas the mitochondrial respiratory chain and several enzymatic activities act as endogenous sources of ROS. Increased ROS production from mitochondrial dysfunction causes further mitochondrial degradation and overall cellular damage [99]. At 5, 10, or 30 Gy of irradiation, rat astrocytes showed mitochondrial anomalies, such as decreased ATP levels and altered respiratory chain activity, which were suppressed by the EUK-134 SOD/peroxidase mimetic [100]. Moreover, radiation-induced ROS can trigger endoplasmic reticulum (ER) stress, relevant to astrocytes, key mediators of RIBI pathophysiology. Extended ER stress activates the unfolded protein response (UPR), and while direct evidence in RIBI astrocytes is limited, ER stress impairs astrocyte function (e.g., disrupted glutamate uptake, enhanced inflammation) [101]. Notably, SHP aggravates ER stress in mutant astrocytes via XBP1 SUMOylation, reducing reactive astrocyte LCN2 secretion; through autophagic and proteasomal regulation, alleviating neuroinflammation; and luteolin mitigates ER stress-dependent neuroinflammation—all highlighting ER stress pathways as potential targets for astrocyte-mediated RIBI modulation [102,103,104]. Moreover, oxidative stress caused by ionizing radiation exerts various biological consequences on unirradiated bystanders and even remote cells, in addition to its detrimental impacts on irradiated cells [105]. X-ray-induced DNA damage disseminated from irradiated cells to unirradiated bystander cells when a particular segment of brain microvascular endothelial cells was subjected to X-ray radiation, implicating bystander signaling in cellular Ca2+ kinetics and the signaling pathways of IP3, ATP, ROS, and NO [106].
In summary, radiation-induced oxidative stress in astrocytes plays a role in the progression of RIBI. Consequently, the prevention and treatment of RIBI require regulation of reactive oxygen species (ROS) production and scavenging in astrocytes, along with the enhancement of the body’s antioxidant defenses.
3.5. Radiation-Induced Senescence of Astrocytes
Apart from mediating oxidative stress, astrocytes also undergo radiation-induced senescence, another key pathological process that contributes to the progression of RIBI by exacerbating neuroinflammation and neuronal damage. Cellular senescence—defined by persistent, irreversible cell cycle arrest—is a fundamental process contributing to organismal aging and the gradual decline in cellular and tissue function. The accumulation of aged cells in brain tissue is associated with various neurological disorders, including RIBI, Parkinson’s disease (PD), and Alzheimer’s disease (AD) [107,108,109]. In the aging brain, senescence manifests in numerous cell types, including neural precursor cells, neurons, microglia, astrocytes, oligodendrocytes, and brain endothelial cells, among others [110]. The accumulation of senescent cells in the central nervous system can incite local inflammation by secreting pro-inflammatory chemicals, facilitating the gradual infiltration of immune cells into the CNS, and culminating in structural and functional alterations, including a progressive decrease in brain volume and memory deficits [111]. Radiation-induced cellular senescence is a premature aging phenomenon triggered by external stressors, featuring enhanced inflammatory responses, increased oxidative stress, DNA damage accumulation, cell cycle arrest, and secretion of SASP cytokines (e.g., IL-1β, IL-6, IL-8) [99]. Some of the cell types that are found in the nervous system, including neurons, microglia, astrocytes, and brain microvascular endothelial cells, have been shown to undergo senescence as a result of exposure to ionizing radiation [75]. The three primary molecular features of radiation-induced microglial senescence are metabolic alterations, DNA damage, and inflammatory responses; these alterations are intimately linked to long non-coding RNAs (lncRNAs) [112]. Furthermore, prolonged inflammation brought on by microglia and astrocyte activation may exacerbate neuronal aging. While asparagine endopeptidase (AEP) targets microglia-mediated neuronal aging, it plays a significant role in RIBI [113]. Ionizing radiation can cause double-strand DNA breaks (DSBs), which are harmful to genetically fragile DNA and promote oxidative stress, leading to the aging and death of brain endothelial cells [114]. Additionally, a major factor in BBB disruption that causes RIBI and consequent cognitive deterioration is radiation-induced pericyte senescence [115]. Furthermore, pericyte senescence brought on by radiation is a major factor in BBB failure and further facilitates subsequent cognitive deterioration [116].
Patients undergoing cranial radiation therapy exhibit increased astrocyte senescence in brain tissue, and in vitro studies reveal elevated expression of senescence-associated p16INK4A and p21 in primary human astrocytes following radiation exposure [81]. On the other hand, Δ133p53 protected irradiated primary human astrocytes from producing neurotoxic IL-6, promoted DNA repair, and decreased radiation-induced astrocyte senescence [81]. Furthermore, Δ133p53 expression restoration in neurotoxic astrocytes transforms them into neuroprotective astrocytes in various degenerative neuropathies [117]. Senescent astrocytes are therefore considered to be one of the factors contributing to ionizing radiation-induced radiocognitive impairment.
3.6. Astrocytes in Radiation Necrosis
Astrocytes drive RN progression by linking initial radiation injury to irreversible cerebral necrosis through interconnected pathological cascades, with key mechanisms supported by recent studies. Radiation-induced DNA damage and damage-associated molecular patterns from injured cells trigger astrocyte polarization to reactive astrocytes [11]. These reactive astrocytes secrete pro-inflammatory cytokines (IL-6, TNF-α) and chemokines (CXCL12), which amplify neuroinflammation by recruiting immune cells and directly damaging neurons and oligodendrocytes, forming a self-reinforcing inflammatory loop [118]. Hypoxia within RN lesions induces astrocytes to overexpress VEGF [119], which disrupts the blood-brain barrier by downregulating tight junction proteins, leading to vascular leakage and edema while failing to promote functional angiogenesis, thereby exacerbating tissue ischemia [120]. Radiation also triggers a burst of mitochondrial reactive oxygen species (mROS) in astrocytes, accelerating cellular senescence and the secretion of SASP factors that inhibit tissue repair and induce damage in adjacent cells [121]. Additionally, astrocytes engage in pathological crosstalk with microglia and endothelial cells, further amplifying inflammation and blocking vascular regeneration, ultimately leading to irreversible cerebral necrosis [11]. This highlights astrocytes’ pivotal role as central mediators of RN pathogenesis.
3.7. Astrocytes’ Interactions with Other Cells in RIBI
To fully understand how astrocytes orchestrate RIBI, it is essential to dissect their crosstalk with other CNS cells (microglia, neurons, oligodendrocytes, and endothelial cells), as each participates in pathological cascades via distinct molecular signals. The two main cell types found in almost equal amounts in the human central nervous system (CNS) are neurons and glial cells. Astrocytes comprise 20% of the glial cells in the human brain, followed by microglia (5–15%), oligodendrocyte precursor cells (3–10%), and oligodendrocytes (25%) [122]. These cells initiate and respond to an inflammatory process in RIBI, resulting in gradual neurological deterioration.
3.7.1. Astrocytes and Microglia
It has been demonstrated that cytokines generated when radiation-induced microglia activation occurs are important mediators of astrocyte activation, in addition to the direct activation of astrocytes by radiation. Radiation can directly activate astrocytes, which then produce and secrete a variety of cytokines that further activate astrocytes by triggering the signal transducer and activator of transcription 3 (STAT3) signaling pathway. This process eventually keeps astrocytes continuously activated [123]. In the meantime, another crucial mechanism for radiation-induced astrocyte activation is MAP kinase activation [82]. However, the activation of microglia brought on by radiation and the PGE2 that results are important mediators of the rise in reactive astrocytes [21]. Consistent with the cellular crosstalk shown in Figure 1, radiation-activated microglia secrete multiple pro-inflammatory mediators to drive astrocyte reactivity. For example, TNF-α and IL-1α directly induce astrocyte transformation to the neurotoxic A1 phenotype [58], while C1q (another key factor in Figure 1) mediates radiation-induced astrocyte activation—an effect confirmed by in vivo studies where glia-selective C1q deletion prevents astrocyte reactivity and cognitive deficits [63]. As shown in Figure 1, these A1 reactive astrocytes then secrete neurotoxic substances (e.g., ROS, C3) and pro-inflammatory cytokines, which synergize with activated microglia to exacerbate neuronal and oligodendrocyte death [20].
3.7.2. Astrocytes and Neurons
Beyond the crosstalk between astrocytes and microglia, the interaction between astrocytes and neurons also plays a pivotal role in the pathogenesis of RIBI, with irradiated activated astrocytes impairing healthy neurons through multiple distinct mechanisms. Through various mechanisms, irradiation-activated astrocytes harm healthy neurons. When cortical astrocytes and astrocyte-conditioned medium (ACM) are present, the radiosensitivity of cortical neurons is greatly decreased; yet, when astrocyte function is compromised, neuronal sensitivity to radiation and other types of oxidative stress-induced damage may rise [124]. In contrast, after radiation exposure, activated astrocytes, coupled with microglia that are themselves activated, release a collection of pro-inflammatory cytokines, including TNF-α, IL-1β, and IL-6, which collectively increase neuronal damage and cell death [73,81]. Moreover, reactive astrocytes may contribute to the onset of RIBI through the release of chemokine CCL2, complement protein C3, and reactive oxygen and nitrogen species; however, additional research utilizing ex vivo and in vivo radiation models is necessary to validate this.
3.7.3. Astrocytes and Oligodendrocytes
Astrocyte-oligodendrocyte communication is a central determinant of RIBI-associated demyelination. In late-delayed RIBI, radiation-induced astrocytic inflammation directly undermines oligodendrocyte function and myelin stability [73,125]. Beyond direct radiation-induced apoptosis via ceramide-PKB signaling, oligodendrocytes suffer from a loss of astrocytic support [126,127]. While astrocytes normally facilitate myelination via TIMP1 and CXCL1, the post-radiation environment drives them toward a deleterious reactive state. These reactive astrocytes release inhibitory factors, such as IL-6, which act indirectly to suppress OPCs-mediated myelin regeneration and exacerbate white matter injury [128].
3.7.4. Astrocytes and Endothelial Cells
The interactions between astrocytes and endothelial cells are fundamental to maintaining BBB integrity, a process disrupted throughout RIBI progression. Pro-inflammatory cytokines such as TNF-α and IL-6, secreted by activated microglia and reactive astrocytes, may induce senescence in vascular endothelial cells [73]. After cranial X-ray radiation therapy, astrocytes release key vasoactive substances through their endfeet, including VEGF, Ang II, endothelin-1 (ET-1), and NO, which directly act on capillary endothelial cells to drive BBB damage [73]. VEGF stands out as the most critical mediator owing to its direct, pathway-specific regulation of BBB integrity and central role in astrocyte-endothelial crosstalk during RIBI progression: radiation-induced astrocyte activation promotes excessive VEGF secretion [65,88], and this astrocyte-derived VEGF directly downregulates endothelial tight junction proteins (occludin/claudin-5) a core structural component of the BBB, thereby amplifying vascular leakage and facilitating neuroinflammatory infiltration [65,87,88]. In a study simulating deep space radiation, astrocyte activation and endothelial damage were enhanced 3–7 days post-irradiation, with processes like endothelial permeability and oxidative stress regulated by these vasoactive substances [125]. Moreover, pro-inflammatory cytokines (e.g., TNF-α, IL-6) further upregulate endothelial cell reactivity, amplifying vascular permeability [7,87]. This astrocyte-endothelial crosstalk via VEGF, Ang II, ET-1, and excessive NO, inducing tight junction damage, drives late-delayed RIBI, as sustained BBB leakage and neuroinflammation cause cerebral edema and neuronal loss [88]. Additionally, radiation-induced pro-inflammatory cytokines upregulate ICAM-1 on endothelial cells, increasing vascular permeability and facilitating inflammatory cell infiltration [7,87].
4. Treatment of Radiation Brain Injury by Targeting Astrocytes
Building on the core pathological mechanisms of astrocyte-mediated RIBI (reactive astrocyte polarization, neuroinflammation, BBB impairment, oxidative stress, and cellular senescence) detailed earlier, this section systematically maps each therapeutic strategy to its targeted pathological cascade. It critically compares their translational potential—addressing the key gaps in mechanism-strategy linkage and translational assessment. Key preclinical data and molecular targeting frameworks are summarized in Table 1 and Figure 2, respectively.
4.1. Anti-Inflammatory Therapy
Astrocytes are highly responsive to ionizing radiation, with abnormal activation driving neuroinflammation—a core pathophysiological feature of RIBI. Under pathological conditions like radiation exposure, astrocytes secrete pro-inflammatory cytokines (IL-1β, IL-6, TNF-α) and recruit peripheral immune cells, initiating a cascade that disrupts the BBB and exacerbates neuronal injury [57,72,129]. This sustained inflammatory cascade is a key driver of progressive neurological impairment, highlighting the need for targeted interventions to interrupt astrocyte-mediated neuroinflammation in RIBI [38,130].
Anti-inflammatory strategies targeting the TNF/NF-κB pathway have demonstrated preclinical efficacy in mitigating RIBI. Key interventions include anti-TNF antibodies and the TAT-NBD peptide, a selective NF-κB inhibitor. In C57BL/6J mice subjected to 20 Gy whole-brain irradiation, anti-TNF therapy eliminated astrocyte hyperproliferation, reduced vascular permeability, and attenuated leukocyte adhesion within 48 h—effects attributed to direct inhibition of TNF-mediated NF-κB activation [61]. Similarly, the TAT-NBD peptide suppressed reactive astrogliosis (evidenced by reduced GFAP expression) and diminished pro-inflammatory microglial infiltration in juvenile mice following 5 Gy cranial irradiation, ultimately improving anxiety-like behaviors and exploratory deficits [131]. These findings confirm that blocking the TNF/NF-κB pathway interrupts astrocyte-driven neuroinflammation, reversing both cellular and behavioral hallmarks of RIBI.
Anti-inflammatory therapy holds considerable clinical potential, facilitated by the availability of FDA-approved anti-TNF agents that support expedited clinical advancement [61]. However, systemic NF-κB inhibition carries moderate off-target risks, such as immunosuppression in immunocompromised cancer patients [57,72,129]. Poor BBB penetration of traditional anti-TNF antibodies is a feasibility challenge, but intranasal delivery or GFAP-targeted modification enhances intracerebral concentrations [131]. A critical bottleneck is patient stratification, as efficacy depends on high NF-κB activation (elevated CSF TNF-α/IL-6) [126,127]. Future efforts will focus on developing astrocyte-specific NF-κB inhibitors and validating CSF inflammatory biomarkers for precise patient selection [126,127].
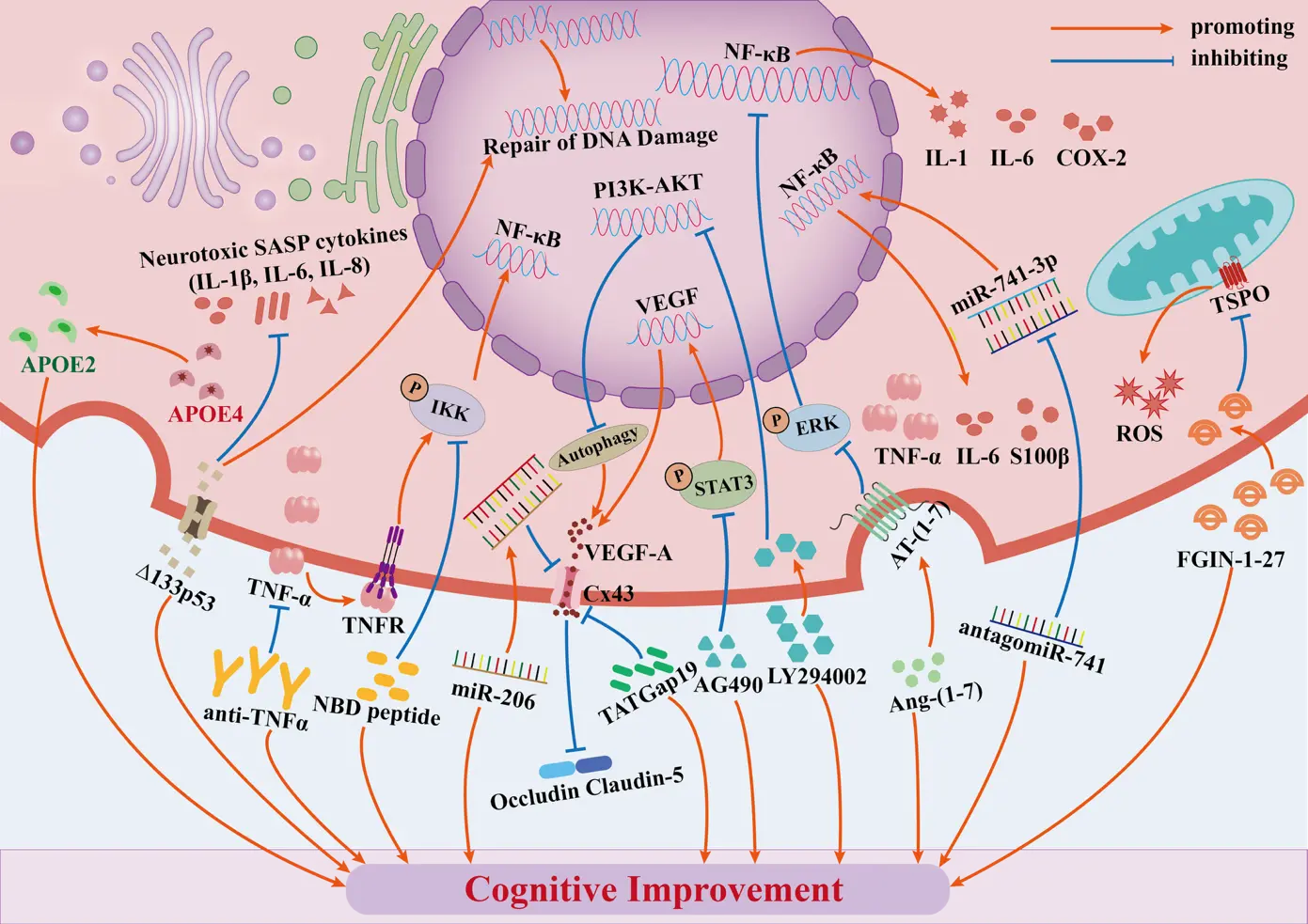
Figure 2. Key astrocyte-targeted therapeutic strategies for radiation-induced brain injury (RIBI). This figure depicts core astrocyte-targeted RIBI therapies acting on specific molecular pathways to alleviate neuroinflammation, blood-brain barrier (BBB) impairment, oxidative stress and cellular senescence, with mechanisms as follows: (1) Anti-inflammatory therapy: Anti-TNF blocks the TNF/NF-κB pathway, and TAT-NBD peptide inhibits NF-κB nuclear translocation; both abrogate NF-κB downstream inflammatory factors (IL-1, IL-6, COX-2) to mitigate neuroinflammation. (2) VEGFA-targeted therapy: TATGap19 (blocks Cx43 hemichannels) and LY294002 (suppresses PI3K-AKT pathway) downregulate VEGFA release and expression, while AG490 inhibits the STAT3 pathway by suppressing JAK2-pSTAT3 activation to block VEGFA transcription (small blue nucleus icon for this transcriptional process); all three relieve VEGFA’s inhibition of claudin-5 and occludin, attenuating radiation-induced BBB damage. (3) TSPO-targeted therapy: FGIN-1-27 binds to TSPO to repair astrocyte mitochondrial function and reduce ROS, alleviating oxidative stress. (4) RAS blocker: Ang-(1-7) targets AT-(1-7) to antagonize Ang II and inhibit the MAPK pathway, diminishing radiation-associated inflammation. (5) ApoE-targeted therapy: Irradiated female APOE2 mice retained spatial memory, while female APOE4 mice did not, reflecting ApoE's isoform-specific RIBI regulation. (6) Δ133p53-targeted therapy: Δ133p53 inhibits astrocyte SASP (IL-1β, IL-6, IL-8) and facilitates DNA double-strand break repair, alleviating senescence and inflammation amplification. (7) Exosome and miRNA-targeted therapy: AntagomiR-741 antagonizes miR-741-3p to inhibit astrocyte NF-κB activation and reduce TNF-α, IL-6 and S100β; miR-206 mimetic targets Cx43 to downregulate its expression, reducing VEGFA release and ameliorating BBB impairment.
Table 1. Radioprotective effect of targeting different molecules in astrocytes in radiation-induced brain injury models.
|
Intervention |
Model and Dose |
Radiation Site |
Time Post-RT |
Mechanism-Mediated Effects |
Translational Evaluation |
Refs. |
|---|---|---|---|---|---|---|
|
Anti-TNF |
C57BL/6J mouse; X-ray, 20 Gy (1 Gy/min) |
whole brain |
24 h, 48 h |
By blocking radiation-induced NF-κB pathway, Anti-TNF inhibits pro-inflammatory factor (TNF, ICAM-1) expression, preserves BBB integrity, and suppresses reactive astrogliosis |
Clinical Promise: High; Feasibility: Moderate; Bottleneck: Acute-phase targeted intervention |
[61] |
|
TAT-NBD |
C57BL/6J mouse; X-ray, 5 Gy (306 cGy/minute) |
whole brain |
4 h, 3 m |
By blocking radiation-induced NF-κB pathway, TAT-NBD peptide suppresses astrogliosis (GFAP downregulation), reduces neuroinflammation (IBA-1+CD11c+ microglial infiltration and TNFα expression), protects neuronal progenitors, and ameliorates anxiety-like behaviors and exploration deficits |
Clinical Promise: Moderate; Feasibility: Moderate; Bottleneck: Pediatric dose optimization |
[131] |
|
STAT3 Inhibitor (AG490) |
Rat primary astrocyte; X-ray, 20 Gy (400 cGy/min) |
in vitro |
4 h, 12 h, 24 h, 48 h |
By inhibiting radiation-induced STAT3 phosphorylation, AG490 suppresses astrocyte activation, reduces VEGF overexpression (dose/time-dependent), and improves astrocyte viability |
Clinical Promise: Moderate; Feasibility: Moderate; Bottleneck: In vitro-to-in vivo translation (BBB penetration validation needed) |
[65] |
|
TATGap19/Cx43 Blockers |
C57BL/6J mouse and Cx43 MK4 KI/Cx43 GFAP-Cre KO mouse; X-ray, 20 Gy |
whole brain |
6 h, 24 h |
By inhibiting radiation-induced Cx43 hemichannel opening and Ca2+ entry, TATGap19 blocks VAMP3-vesicular VEGF-A transport to astrocytic endfeet, suppresses VEGF-A release and BBB leakage, without altering endothelial tight junction (occludin/claudin-5) expression |
Clinical Promise: Moderate; Feasibility: Moderate; Bottleneck: Lack specific Cx43 inhibitors; late-stage RIBI needs combinatio |
[87] |
|
LY294002 |
C57BL/6 mouse; X-ray, 10 Gy once weekly for 2 weeks (1.22 Gy/min) |
whole brain |
4 w |
Inhibiting astrocytic PI3K-AKT (specific inhibitor) reduced fractionated RT-induced BBB disruption, with restored astrocyte autophagy and lower VEGF |
Clinical Promise: Moderate; Feasibility: Moderate; Bottleneck: Narrow autophagy regulation window |
[88] |
|
Primary astrocytes; X-ray, 30 Gy (4 Gy/min) |
in vitro |
48 h |
PI3K-AKT inhibition restored astrocyte autophagy, alleviated co-cultured endothelial tight junction damage, and reduced endothelial permeability |
[88] |
||
|
TSPO ligands (FGIN-1-27) |
Primary astrocytes; X-ray, 20 Gy (4 Gy/min) |
in vitro |
24 h |
By targeting radiation-impaired mitochondrial function, FGIN-1-27 repairs respiratory chain activity, reduces ROS production, inhibits astrocyte A1 polarization, and normalizes cell proliferation (aligned with oxidative stress axis in RIBI) |
Clinical Promise: Moderate; Feasibility: Low (<5% BBB bioavailability); Bottleneck: Lack non-human primate validation |
[132] |
|
Ang-(1-7) |
Primary astrocytes; γ-ray, 10 Gy (3.57 Gy/min) |
in vitro |
1 h, 7 h |
By binding to AT(1-7) receptor, Ang-(1-7) inhibits radiation-induced MAPK (PKCα/p-MEK/p-ERK) activation, increases DUSP1, suppresses pro-inflammatory cytokines (IL-1β/IL-6) and COX-2/GFAP expression |
Clinical Promise: Moderate (hypertension comorbidity); Feasibility: Moderate; Bottleneck: RAS activation status dependency |
[82] |
|
APOE2 Mimetics |
Human APOE2 targeted replacement mouse; γ-ray, 10 Gy |
whole brain |
3 m |
Irradiated female APOE2-targeted replacement mice (C57BL6/J) retained spatial memory via Morris water maze testing post-irradiation, with reduced astrocyte activation and preserved BBB integrity compared to APOE4 mice |
Clinical Promise: Moderate (subtype-personalized); Feasibility: Moderate (intranasal mimetics bypass BBB); Bottleneck: Sex/isoform dependency |
[133] |
|
Δ133p53 |
Primary astrocytes; X-ray, 2 Gy/4 Gy (3.57 Gy/min) |
in vitro |
4 h, 24 h |
By restoring radiation-diminished Δ133p53, it promotes DNA double-strand break repair (RAD51 upregulation), inhibits astrocyte senescence, and suppresses SASP (IL-6) to reduce neurotoxicity (aligned with senescence-inflammation axis in RBI) |
Clinical Promise: Moderate (endogenous, non-oncogenic); Feasibility: Moderate (lentiviral transduction feasible; no in vivo delivery); Bottleneck: Lack animal model |
[81] |
|
AntagomiR-741 |
C57BL/6 mouse; β-electron beam, 30 Gy (2.5 Gy/min) |
whole brain |
1 w, 6 w |
Intranasal antagomiR-741 reduced hippocampal miR-741-3p in mice post-β-irradiation, improving spatial memory (behavioral tests) and reducing neuronal apoptosis/astrocyte activation |
Clinical Promise: Moderate (nasal delivery non-invasive); Feasibility: Moderate (nose-brain pathway accessible); Bottleneck: Limited target gene validation |
[134] |
|
miR-206 |
HA-1800 normal astrocyte cell line; γ-ray, 2 Gy/4 Gy (3.57 Gy/min) |
in vitro |
12 h, 24 h, 48 h |
By targeting and suppressing radiation-upregulated Cx43, it enhances cell viability, reduces apoptosis (cleaved caspase-3 downregulation), and inhibits secretion of pro-inflammatory cytokines (TNF-α/IL-β/IL-6/IFN-γ) (aligned with miRNA-Cx43-mediated neuroinflammation axis in RBI) |
Clinical Promise: Moderate (target specificity); Feasibility: Moderate (in vitro transfection feasible); Bottleneck: Lack in vivo validation and delivery system optimization |
[80] |
Abbreviations: RT: Radiotherapy; BBB: blood-brain barrier; GFAP: glial fibrillary acidic protein; TNF: tumor necrosis factor; NF-κB: nuclear factor-κB; ICAM-1: intercellular adhesion molecule 1; IBA-1: ionized calcium-binding adapter molecule 1; STAT3: signal transducer and activator of transcription 3; VEGF-A: vascular endothelial growth factor A; Cx43: connexin 43; KI: knock-in; KO: knockout; VAMP3: vesicle-associated membrane protein 3; PI3K-AKT: phosphatidylinositol 3-kinase/protein kinase B; TSPO: translocator protein; ROS: reactive oxygen species; RAS: renin-angiotensin system; Ang-(1-7): angiotensin-(1-7); AT(1-7): AT(1-7) receptor; MAPK: mitogen-activated protein kinase; PKCα: protein kinase C alpha; p-MEK: phosphorylated mitogen-activated protein kinase kinase; p-ERK: phosphorylated extracellular regulated protein kinases; DUSP1: dual specificity phosphatase 1; IL-1β: interleukin-1β; IL-6: interleukin-6; COX-2: cyclooxygenase-2; APOE2/4: apolipoprotein E2/4; Δ133p53: p53 isoform Δ133p53; SASP: senescence-associated secretory phenotype; RAD51: RAD51 recombinase; miR: microRNA; antagomiR-741: miR-741-3p antagonist; caspase-3: cysteine-aspartic acid protease 3; IFN-γ: interferon-γ; RIBI: radiation-induced brain injury; h: hours; m: months; w: weeks.
4.2. VEGF-A Targeted Intervention
VEGF belongs to a family of functional glycoproteins including VEGF-A, VEGF-B, VEGF-C, and PlGF, with VEGF-A (commonly referred to as VEGF) being the key mediator of vascular permeability and angiogenesi [135,136]. Inhibiting VEGF-A signaling has also shown protective effects in inflammatory CNS disorders by mitigating astrocyte-derived VEGF-mediated BBB disruption [137,138]. BBB impairment is driven by aberrant astrocyte-endothelial crosstalk, primarily via the Cx43/PI3K-AKT/STAT3 signaling axis—ionizing radiation triggers Cx43 hemichannel opening, PI3K-AKT-dependent autophagy suppression, and STAT3 phosphorylation in astrocytes, collectively promoting excessive secretion of astrocyte-derived VEGF. This VEGF directly downregulates endothelial tight junction proteins (occludin/claudin-5) and amplifies vascular leakage [65,87,88]. Anti-VEGF therapies, typified by bevacizumab, exert dual effects by specifically targeting astrocyte-secreted VEGF and preserving endothelial barrier integrity, making them particularly effective for RIBI-related complications like radiation necrosis (RN) [139,140,141].
Key interventions validated in preclinical and clinical studies include specific inhibitors and antibodies. TATGap19, a well-characterized and selective Cx43 hemichannel blocker, blocked calcium-dependent VAMP3-vesicular VEGF transport to astrocyte endfeet in C57BL/6J mice, suppressing VEGF release and BBB leakage 24 h post-20 Gy irradiation [87]. Astrocyte-specific Cx43 knockout mice exhibited comparable protective effects [87]. The PI3K-AKT inhibitor LY294002 restored astrocyte autophagy in C57BL/6 mice after fractionated 10 Gy irradiation, reducing VEGF overexpression and reversing BBB disruption [88]. The STAT3 inhibitor AG490 suppressed pSTAT3 binding to the VEGF promoter in irradiated rat primary astrocytes, decreasing VEGF transcription and restoring endothelial tight junctions [65]. Clinically, the humanized anti-VEGF-A antibody bevacizumab exerts its therapeutic effects by specifically targeting astrocyte-derived VEGF—consistent with multi-center retrospective studies, systematic reviews, and clinical case reports. It effectively alleviates neurological symptoms associated with SRS-induced RN and achieves meaningful imaging relief, with long-term efficacy sustained in follow-up observations. The underlying mechanism hinges on bevacizumab’s ability to neutralize astrocyte-secreted VEGF, thereby interrupting the cascade of vascular leakage and tissue damage that drives RN progression [139,140,141].
Anti-VEGF therapy exhibits substantial clinical promise, standing as one of the few astrocyte-targeted approaches with validated clinical effectiveness [141]. It has low off-target effects due to brain-specific VEGF enrichment, which is predominantly derived from activated astrocytes, and moderate-to-high feasibility with small-molecule inhibitors achieving 10–15% BBB penetration [139,140,141]. A key translational bottleneck is compensatory Ang-2 pathway activation, and long-term single-agent use may limit efficacy, necessitating combination with Cx43 blockers for synergistic effects [87]. Future directions include developing astrocyte-specific VEGF siRNA to avoid interfering with normal cerebral angiogenesis and optimizing patient selection via serum VEGF/Ang-2 monitoring.
4.3. TSPO Ligand-Targeted Therapy
TSPO (Translocator Protein), originally named Peripheral Benzodiazepine Receptor (PBR), is a mitochondrial outer membrane protein expressed in multiple cell types and tissues, including immune cells, peripheral organs, and the central nervous system [142,143]. Its core regulatory functions involve mitochondrial processes, including steroidogenesis, cholesterol transport, apoptosis, and oxidative stress responses [144]. TSPO ligands, small molecules that bind and modulate TSPO activity, are valuable tools for investigating neurological disorders, inflammatory responses, and mitochondrial function [144].
Previously thought to be exclusively overexpressed in microglia, TSPO is now confirmed to be upregulated in activated astrocytes, with elevated levels in astrocytes preceding microglia in certain neurodegenerative disorders [145,146,147]. In brain injury-related conditions, increased TSPO expression in glial cells is widely used as a biomarker for neuroinflammation and active brain pathology [148]. For instance, TSPO ligands like PK11195 alleviate neuroinflammation and cognitive impairments in animal models of chronic systemic LPS exposure, while reducing activated microglia in neuroinflammation-associated disorders [149,150]. LPS also upregulates TSPO expression at mRNA and protein levels in astrocytes via a STAT3-dependent mechanism, increasing ROS production and reducing glucose uptake, and these effects are attenuated by the TSPO antagonist FEPPA [151]. Consistent with the astrocyte-mediated pathological cascade of RIBI (as previously discussed), TSPO plays a context-dependent role as a key mediator linking radiation-induced astrocyte dysfunction to RIBI progression. The core pathological mechanism is as follows: radiation induces astrocyte conversion to the neurotoxic A1 phenotype, which triggers abnormal TSPO activation; this activated TSPO disrupts mitochondrial redox homeostasis, leading to mitochondrial hyperfunction, excessive production of oxidized metabolites, and enhanced secretion of pro-inflammatory cytokines and ROS, ultimately amplifying neuroinflammation and BBB impairment [95,152]. In a mouse RIBI model subjected to 8 Gy γ-irradiation, TSPO-expressing brain cells increase alongside enhanced neuroinflammation and M1-type microglia accumulation [153]. At different radiation doses, TSPO behaves differently: high doses promote inflammation, whereas low doses downregulate microglial TSPO, enhancing astrocyte radiation sensitivity via a pro-inflammatory microenvironment that drives A1 phenotype conversion [152,154]. Corresponding to this pathological mechanism, TSPO ligands exert therapeutic effects by directly targeting astrocyte mitochondria to interrupt the pathological cascade from A1 astrocyte conversion to TSPO activation, subsequent mitochondrial dysfunction, and ultimately neuroinflammation. The TSPO ligand FGIN-1-27 reverses radiation-induced astrocyte abnormalities by restoring mitochondrial respiratory chain activity, reducing ROS generation, and inhibiting astrocyte hyperactivation and A1 phenotype conversion. These effects are validated in primary astrocyte cultures, where FGIN-1-27 improves radiation-induced mitochondrial dysfunction and abnormal proliferation [132].
TSPO-targeted therapy has moderate clinical promise for RIBI, with unique advantages and inherent challenges compared to other astrocyte-targeted strategies, such as anti-VEGF therapy. Its key strengths are dual targeting of neuroinflammation and oxidative injury, the core pathologies of RIBI, and accessible small-molecule ligands with manageable pharmacokinetics [132]. Critical limitations include moderate BBB penetration, off-target risks from TSPO expression in normal glia and peripheral tissues, such as disrupted steroidogenesis or mitochondrial function, and an absence of clinical data, in contrast to anti-VEGF therapy, which has proven efficacy in radiation necrosis. Future TSPO-targeted therapy efforts should focus on developing astrocyte-specific ligands, such as GFAP-targeted conjugates, to enhance specificity and BBB penetration, and on validating cerebrospinal fluid TSPO levels as a biomarker for patient stratification [148].
4.4. RAS Pathway Blocking Therapy
RAS critically regulates astrocyte-mediated neuroinflammation and BBB integrity in RIBI [155,156]. RAS blockers include ACEIs such as lisinopril, ARBs such as sartans, and the protective peptide Ang-(1-7), but off-target effects, notably hypotension, limit their direct clinical application, highlighting the need for astrocyte-specific RAS modulators [155,156]. Ionizing radiation activates RAS in astrocytes, triggering MAPK signaling (PKCα/p-MEK/p-ERK) to drive secretion of pro-inflammatory cytokines (IL-1β, IL-6) and upregulation of activation markers (GFAP, COX-2, AP-1, NF-κB), which are key events that amplify neuroinflammation and BBB impairment in RIBI; RAS blockers intervene by inhibiting astrocyte MAPK pathway activation, thereby attenuating these pathological cascades and mitigating RIBI-related deficits. Preclinical evidence validates this link: Initial preclinical studies first demonstrated that ACEIs mitigated RIBI in a rat optic neuropathy model subjected to 30 Gy stereotactic irradiation [157,158]. Additionally, pretreatment with the AT1RA L-158,809 during and after segmental whole-brain irradiation prevented or alleviated radiation-induced cognitive impairments in adult rats [9,159]. However, RAS inhibition with these agents does not prevent or reverse radiation-induced loss of neurogenesis [160].
Ang-(1-7), which exerts physiological effects by binding the Mas receptor and counteracting Ang II, further reinforces this pathway by directly inhibiting MAPK signaling in primary astrocytes irradiated with 10 Gy, attenuating the radiation-induced upsurge of pro-inflammatory cytokines and astrocyte activation markers [82]. To address translational bottlenecks, future efforts should focus on developing astrocyte-specific RAS modulators (e.g., GFAP-promoter targeted conjugates) to enhance cellular specificity and minimize off-target effects, while validating the serum Ang II/Ang-(1-7) ratio as a predictive biomarker may enable stratified patient selection for individuals with astrocyte RAS hyperactivation [155,156]. Collectively, these refinements leverage the well-established anti-inflammatory and BBB-protective actions of RAS blockers, positioning them as promising candidates for RIBI therapy [155,156].
4.5. Precision Regulation of ApoE Isoforms
Astrocytes are the primary source of ApoE in the brain, where ApoE regulates lipid transport, neuronal nutrition, and synaptic plasticity, and these key functions are closely linked to RIBI pathophysiology [161]. ApoE in humans encompasses three isoforms (ApoE2, ApoE3, ApoE4) that exert isoform-specific effects in RIBI: ApoE2 mitigates RIBI by suppressing astrocyte overactivation, preserving BBB integrity, and maintaining cognitive function post-radiation [133], while ApoE4 exacerbates RIBI through promoting reactive astrogliosis, increasing BBB permeability, and amplifying neuroinflammation [162]. This subtype-specific discrepancy directly ties to astrocyte-mediated pathological cascades: radiation-induced astrocyte dysfunction is amplified by ApoE4, which impairs astrocyte-derived trophic support and enhances pro-inflammatory cytokine secretion, whereas ApoE2 counteracts these effects by stabilizing astrocyte homeostasis [133].
Preclinical data across genetic and radiation models further validate astrocyte-derived ApoE as a critical regulator of RIBI progression. ApoE-deficient mice exhibit impaired learning and memory, age-related synaptic degeneration, compromised BBB integrity, and elevated CNS pro-inflammatory cytokines [163,164]. Their phosphorylated proteome also shows dysregulated synaptic plasticity and calcium signaling following chronic low-dose radiation [165]. In wild-type mice, relative to ApoE-deficient counterparts, miR-9 and miR-let7b show elevated expression, a phenomenon associated with the retention of radial glia-like (RGL) cells, suggesting ApoE-mediated regulation of neural progenitor pools during the radiation response. Notably, ApoE deficiency exacerbates radiation-induced behavioral deficits [166], and conditional deletion of ApoE3 in astrocytes leads to significant post-radiation cognitive decline [167]. This underscores the cell-specific relevance of astrocyte-derived ApoE. Sex and isoform dependency further shape RIBI outcomes: galactic cosmic radiation simulation impairs memory in male ApoE3 mice [168], while irradiated female ApoE2 mice retain spatial memory, in contrast to their ApoE3 and ApoE4 counterparts [133]. Beyond radiation-specific models, traumatic brain injury studies confirm astrocyte-derived ApoE is essential for neurogenesis during functional impairment, a mechanism plausibly translatable to RIBI-related neural repair [169]. Humanized ApoE4 animals also show elevated BBB permeability, which is reversed within one month of astrocyte-specific ApoE4 knockdown, reinforcing ApoE4’s role in astrocyte-mediated BBB disruption [162]. The core mechanism-strategy mapping for ApoE-targeted therapy in RIBI centers on addressing isoform imbalance, with preclinical strategies including upstream targeting of ApoE4, conversion to protective isoforms, modulation of ApoE lipidation, anti-ApoE4 immunotherapy, antisense oligonucleotide therapy, and non-pharmacological interventions [170,171]. ApoE plays a key role in the metabolism and redistribution of lipoproteins and cholesterol, and its deficiency exacerbates behavioral deficits following radiation exposure, supporting a protective role for astrocyte-derived ApoE against radiation-induced cognitive impairment [166]. In addition, specialized techniques, including anti-APOE antibodies and the targeted regulation of ApoE gene transcription and release, enhance ApoE2’s protective biological functions while mitigating the pathological cellular and molecular responses induced by ApoE4.
ApoE-targeted therapies exhibit notable translational potential for RIBI management, with isoform-specific targeting reducing off-target risks as ApoE2 and ApoE4 exert opposing roles in RIBI, and this strategy aligns with precision medicine trends [162]. Non-invasive delivery routes such as intranasal ApoE mimetic peptides bypass BBB challenges, and there is potential to repurpose ApoE-targeted agents validated in neurodegenerative disease trials [170,171]. Sex and isoform dependency limit the universal efficacy of ApoE-targeted therapies, and large-scale clinical data in RIBI patients remain scarce [133]. Delivery systems struggle to achieve consistent therapeutic concentrations in damaged brain regions, and current strategies fail to address ApoE’s dynamic functional shifts across RIBI phases [170,171]. Elucidating the isoform-specific mechanisms of astrocyte-derived ApoE is vital for developing targeted therapies, as it lays the foundation for precise intervention [161]. Future efforts should focus on optimizing astrocyte-specific delivery, validating serum ApoE isoform ratios as biomarkers, and designing genotype- and sex-stratified clinical trials to overcome existing bottlenecks [166].
4.6. Δ133p53-Targeted Restoration
The Δ133p53 and p53β isoforms, which are isoforms of the p53 protein, are important regulators of the accelerated senescence feature of cells, and the p53 signaling pathway is a fundamental regulator of senescence [172]. Mechanistically, Δ133p53 promotes DNA double-strand break (DSB) repair, inhibits apoptosis, and enhances antioxidant function, thereby suppressing cellular senescence [117,173,174]. These combined actions make Δ133p53 a critical modulator of cellular homeostasis under pathological conditions. Enhancing neuroprotection in aging astrocytes is a significant function of Δ133p53. In vitro studies have provided direct evidence for this regulatory role: primary astrocytes undergoing cellular senescence had higher p53β expression and lower Δ133p53 expression. Similarly, brain tissue from patients with Alzheimer’s disease (AD) showed increased senescent astrocytes, accompanied by reduced Δ133p53 expression and elevated p53β expression. These findings underscore the crucial importance of p53β and Δ133p53 in preserving astrocyte functionality and neuroprotection, along with their potential role in neurodegenerative diseases [117].
Δ133p53 holds considerable therapeutic promise for RIBI. It specifically inhibits X-ray-induced senescence in primary human astrocytes and promotes DSB repair in irradiated cells, mitigating radiation-induced genomic damage. Preclinical experiments using 2–4 Gy X-rays confirm that Δ133p53 overexpression reduces senescence rates and decreases pro-inflammatory cytokine secretion, such as IL-6, limiting astrocyte-mediated neuroinflammation and neuronal damage, which are core RIBI pathological cascades. Furthermore, Δ133p53-targeted strategies offer unique merits: their endogenous regulatory role minimizes off-target risks, and dual actions on DNA repair and senescence address multiple RIBI pathological cascades [81]. However, key challenges remain: absence of astrocyte-specific delivery systems, unvalidated in vivo efficacy in RIBI animal models, ambiguous optimal intervention timing, and dose-dependent tumor progression risks necessitating precise expression modulation [172]. Future priorities involve developing astrocyte-specific activators such as GFAP-promoter-driven gene therapy, validating serum Δ133p53/p53β ratios as predictive biomarkers, and performing RIBI animal trials to confirm cognitive protection and long-term safety [166,167]. These measures will bridge preclinical-clinical gaps, establishing Δ133p53 as a novel astrocyte-targeted strategy for RIBI.
4.7. Exosomes and miRNAs
Exosomes are cell-secreted vesicles that mediate intercellular communication by transporting bioactive cargo, including microRNAs (miRNAs), and play pivotal roles in RIBI pathophysiology [175]. As small endogenous RNAs, miRNAs post-transcriptionally regulate gene expression, and their aberrant expression in astrocytes and other CNS cells directly drives core RIBI pathological cascades, including neuroinflammation, BBB impairment, and reactive astrocyte activation [176,177]. Exosomes further amplify these effects by shuttling pathogenic miRNAs between cells and hold potential as targeted delivery vehicles for therapeutic miRNAs [178].
Radiation-induced dysregulation of exosomal miRNAs directly exacerbates astrocyte-mediated RIBI through two core pathways. First, miRNA-dependent astrocyte activation: Radiation upregulates miR-741-3p in hippocampal astrocytes, promoting the transition to neurotoxic A1 astrocytes by enhancing GFAP expression and IL-6/TNF-α secretion, key mediators of neuroinflammation and neuronal apoptosis [125,177]. Second, miRNA-mediated BBB impairment: Radiation-induced downregulation of miR-206 in astrocytes increases Cx43 expression, triggering excessive VEGF secretion and disruption of endothelial tight junctions (occludin/claudin-5), thereby amplifying BBB leakage [80]. Beyond these core pathways, exosomal miRNAs are common contributors to RIBI progression: dysregulated miR-181b-2-3p modulates microglial activation and neural stem cell dysfunction to indirectly regulate astrocyte activation, while downregulated miR-23a-3p mediates irradiation-induced neuronal apoptosis, and these alterations synergize with astrocyte-driven neuroinflammation to amplify brain injury [179,180]. Notably, exosomes from irradiated astrocytes and oligodendrocytes shuttle miR-7 to distant tissues, such as the lung, mediating off-target radiation-induced bystander effects (RIBE), including autophagy, highlighting the broader systemic pathological role of exosomal miRNAs in RIBI [181].
To counteract exosomal miRNA-driven pathological mechanisms in RIBI, intranasal delivery of antagomiR-741 precisely targets hippocampal astrocytes to inhibit miR-741-3p-mediated A1 polarization and neuroinflammation, offering high translational promise due to non-invasive, off-target effect-avoiding delivery and robust preclinical cognitive improvement in irradiated mice, with moderate feasibility yet unvalidated long-term safety and limited tissue specificity as key challenges [134]. For radiation-induced miR-206 downregulation-driven aberrant Cx43/VEGF pathway activation, miR-206 mimics downregulate astrocyte Cx43 to reduce excessive VEGF secretion and alleviate BBB impairment, providing moderate translational promise via a well-validated RIBI pathway and high astrocyte specificity but low feasibility due to reliance on liposomal carriers with poor in vivo BBB penetration and systemic inflammation risks, with the lack of astrocyte-specific delivery systems as a critical bottleneck [80]. Exosome-based delivery systems derived from healthy astrocytes or engineered to carry therapeutic miRNAs offer natural biocompatibility and reduced immunogenicity [175]. They hold significant translational promise due to their ability to encapsulate miRNAs, protecting them from degradation. Their lipid bilayer also enables partial penetration of the BBB. However, feasibility is constrained by low brain accumulation of unmodified exosomes [182]. Engineering strategies to enhance BBB penetration may cause unintended off-target disruption of the BBB. Technical challenges in the scalable production of uniform exosome batches further limit their clinical application.
Across these strategies, intranasal antagomiR-741 stands out with the greatest near-term translational potential, while common hurdles include insufficient delivery specificity, limited in vivo miRNA stability, and inadequate BBB penetration [134]. Future efforts should prioritize validating astrocyte-specific delivery systems such as GFAP-targeted lipid nanoparticles, optimizing miRNA chemical modifications to enhance stability, and conducting preclinical trials to confirm cognitive protection and long-term safety. These steps will strengthen the translational narrative by systematically linking astrocyte-related exosome/miRNA mechanisms to actionable therapies [181,183,184].
4.8. Physical Targeting of Astrocytes
Beyond molecular and pharmacological strategies, physical interventions provide a complementary strategy to target reactive astrocytes in RIBI. These approaches leverage the unique biological features of reactive astrocytes and their sensitivity to physical stimuli, enabling selective modulation of pathological astrocytes while protecting normal neural cells. Subsequently, we discuss two representative physical modalities, FLASH radiotherapy and LITT, that mitigate or eliminate reactive astrocytes to prevent and treat RIBI, respectively.
4.8.1. FLASH Radiotherapy
FLASH radiotherapy employs ultra-high dose-rate irradiation (>40 Gy/s) to eradicate tumors while protecting normal brain tissue [185]. By reducing ROS production, FLASH-RT inhibits NF-κB pathway activation and downregulates GFAP expression, thereby mitigating reactive astrocytes [186]. This mechanism alleviates neuroinflammation, BBB disruption, and persistent cognitive deficits, serving as a preventive strategy that targets astrocyte-mediated pathological initiation in RIBI [187]. While FLASH-RT affects multiple cell types, its suppression of reactive astrocytes represents a core neuroprotective component.
4.8.2. Laser Interstitial Thermal Therapy (LITT)
Laser interstitial thermal therapy is a minimally invasive image-guided ablation technique for brain metastases and RN [188]. It precisely ablates reactive astrocytes, the primary source of VEGF and pro-inflammatory cytokines (IL-6, TNF-α) in RN lesions, interrupting the inflammation-ischemia-necrosis cycle [189]. For steroid/anti-VEGF refractory RN, LITT yields notable radiological improvement and facilitates steroid withdrawal, validating its role in targeting reactive astrocytes as a key pathological component of late-stage RIBI [12]. While LITT impacts multiple cell populations, reactive astrocytes are central to its therapeutic efficacy.
5. Conclusions and Prospects
Radiation therapy frequently results in radiation-induced brain injury (RIBI), particularly in individuals with brain and head malignancies. A key factor in the pathological development of RIBI is abnormal astrocyte activation and dysfunction [21,61]. It is anticipated that functional astrocyte regulation will benefit patients with RIBI by reducing oxidative stress, preventing inflammation, preserving the integrity of the BBB, and promoting neuronal survival [20,78,87,131]. Modulating certain molecular pathways in astrocytes, including NF-κB, JAK/STAT3, Nrf2, and PI3K/AKT, can substantially reduce neurological side effects induced by aberrant astrocyte activation [65,78,88,131]. In general, targeting astrocytes for RIBI therapy is more specific and less toxic than non-targeted strategies.
Ionizing radiation can cause astrocytes to become pathologically activated, which can exacerbate brain injury by causing neuroinflammation and disrupting the BBB. Thus, regulating astrocyte activity and preventing over-activation could be a novel strategy to lessen RIBI. Future research may focus on astrocyte-targeting treatment approaches for RIBI. According to newly available data, cfDNA, whose concentration tends to rise sharply under a variety of pathological and stressful circumstances, may be a useful biomarker and therapeutic mechanism for astrocyte targeting in the treatment of radiation brain injury because it reflects the level of BBB damage and the state of astrocyte activation [20]. FLASH-RT is an emerging radiation therapy technique characterized by irradiating the tumor target area with an ultra-high dose rate (≥40 Gy/s) in a very short period of time (usually within microseconds to hundreds of milliseconds), which can effectively kill tumor cells while significantly reducing damage to normal tissues. When compared to traditional dose-rate radiation, FLASH-RT has been demonstrated to dramatically reduce the generation of reactive astrocytes; nevertheless, additional research is required to clarify the precise biological mechanisms of astrocytes involved in the FLASH effect [190,191,192]. Furthermore, the mitogen-activated protein kinase (MAPK) pathway is crucial for regulating cell proliferation, differentiation, and death, as well as for the targeted control of radiative damage. Activated JNK and p38 signaling may mitigate the inflammatory response and death in astrocytes, hence exerting a protective impact on brain tissue [193,194]. Additionally, radiotherapy to the head and neck may influence astrocyte autophagy via the MAPK/mTOR pathway. By focusing on important molecules that block this pathway, astrocyte autophagy may be increased, eliminating cellular waste products from radiation damage and reducing the cellular stress response [195,196,197]. Lastly, it is still technically difficult to accurately modulate the distinct responses of astrocytes in the early and late phases of injury since their function varies greatly among clinical situations and time points. Therapeutic strategies that are universally applicable may be difficult to implement due to the variability of astrocytes in various brain areas, each of which may have distinct activities. We anticipate that new precision-targeted astrocyte therapies and approaches will soon be available for the treatment of patients receiving head and neck radiotherapy, and future research should focus on developing astrocyte-specific delivery systems (e.g., BBB-penetrating nanoparticles) and clarifying the role of astrocyte subtypes in RIBI.
Acknowledgments
The authors would like to thank their colleagues for the insightful academic discussions and valuable suggestions, which greatly facilitated the conceptualization and drafting of this review. Schematic diagrams were generated in Adobe Illustrator.
Author Contributions
Conceptualization, W.L. and C.S.; Writing—Original Draft Preparation, W.L.; Writing—Review and Editing, W.L. and C.S.; Figure Preparation, W.L.; Supervision, C.S.; Funding Acquisition, C.S. All authors have read and agreed to the published version of the manuscript.
Ethics Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
This is a Review article with no original data generated; all analyzed data are from previously published studies in the References.
Funding
This research was funded by the National Natural Science Foundation of China (Nos. 12235004, 32171235).
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
-
Soubéran A, Jiguet-Jiglaire C, Toutain S, Morando P, Baeza-Kallee N, Appay R, et al. Brain tumoroids: Treatment prediction and drug development for brain tumors with fast, reproducible, and easy-to-use personalized models. Neuro-Oncol. 2025, 27, 415–429. DOI:10.1093/neuonc/noae184 [Google Scholar]
-
Schaff LR, Mellinghoff IK. Glioblastoma and other primary brain malignancies in adults: A review. JAMA 2023, 329, 574–587. DOI:10.1001/jama.2023.0023 [Google Scholar]
-
Shah N, Mohammad AS, Saralkar P, Sprowls SA, Vickers SD, John D, et al. Investigational chemotherapy and novel pharmacokinetic mechanisms for the treatment of breast cancer brain metastases. Pharmacol. Res. 2018, 132, 47–68. DOI:10.1016/j.phrs.2018.03.021 [Google Scholar]
-
van den Bent MJ, Geurts M, French PJ, Smits M, Capper D, Bromberg JEC, et al. Primary brain tumours in adults. Lancet 2023, 402, 1564–1579. DOI:10.1016/S0140-6736(23)01054-1 [Google Scholar]
-
Balentova S, Adamkov M. Molecular, Cellular and Functional Effects of Radiation-Induced Brain Injury: A Review. Int. J. Mol. Sci. 2015, 16, 27796–27815. DOI:10.3390/ijms161126068 [Google Scholar]
-
Alterio D, Marvaso G, Ferrari A, Volpe S, Orecchia R, Jereczek-Fossa BA. Modern radiotherapy for head and neck cancer. Semin. Oncol. 2019, 46, 233–245. DOI:10.1053/j.seminoncol.2019.07.002 [Google Scholar]
-
Liu Q, Huang Y, Duan M, Yang Q, Ren B, Tang F. Microglia as Therapeutic Target for Radiation-Induced Brain Injury. Int. J. Mol. Sci. 2022, 23, 8286. DOI:10.3390/ijms23158286 [Google Scholar]
-
Makale MT, McDonald CR, Hattangadi-Gluth J, Kesari S. Mechanisms of radiotherapy-associated cognitive disability in patients with brain tumours. Nat. Rev. Neurol. 2018, 13, 52–64. DOI:10.1038/nrneurol.2016.185 [Google Scholar]
-
Greene-Schloesser D, Robbins ME. Radiation-induced cognitive impairment—From bench to bedside. Neuro Oncol. 2012, 14 (Suppl. S4), iv37–iv44. DOI:10.1093/neuonc/nos196 [Google Scholar]
-
Wilke C, Grosshans D, Duman J, Brown P, Li J. Radiation-induced cognitive toxicity: Pathophysiology and interventions to reduce toxicity in adults. Neuro Oncol. 2018, 20, 597–607. DOI:10.1093/neuonc/nox195 [Google Scholar]
-
Furuse M, Nonoguchi N, Kawabata S, Miyatake S-I, Kuroiwa T. Delayed brain radiation necrosis: Pathological review and new molecular targets for treatment. Med. Mol. Morphol. 2015, 48, 183–190. DOI:10.1007/s00795-015-0123-2 [Google Scholar]
-
Vellayappan B, Lim-Fat MJ, Kotecha R, De Salles A, Fariselli L, Levivier M, et al. A systematic review informing the management of symptomatic brain radiation necrosis after stereotactic radiosurgery and international stereotactic radiosurgery society recommendations. Int. J. Radiat. Oncol.*Biol.*Phys. 2024, 118, 14–28. DOI:10.1016/j.ijrobp.2023.07.015 [Google Scholar]
-
Lin B, Gao F, Yang Y, Wu D, Zhang Y, Feng G, et al. FLASH Radiotherapy: History and Future. Front. Oncol. 2021, 11, 644400. DOI:10.3389/fonc.2021.644400 [Google Scholar]
-
Khakh BS, Sofroniew MV. Diversity of astrocyte functions and phenotypes in neural circuits. Nat. Neurosci. 2015, 18, 942–952. DOI:10.1038/nn.4043 [Google Scholar]
-
Baldwin KT, Murai KK, Khakh BS. Astrocyte morphology. Trends Cell Biol. 2024, 34, 547–565. DOI:10.1016/j.tcb.2023.09.006 [Google Scholar]
-
Zhou B, Zuo Y-X, Jiang R-T. Astrocyte morphology: Diversity, plasticity, and role in neurological diseases. CNS Neurosci. Ther. 2019, 25, 665–673. DOI:10.1111/cns.13123 [Google Scholar]
-
Zengeler KE, Hollis A, Deutsch TCJ, Samuels JD, Ennerfelt H, Moore KA, et al. Inflammasome signaling in astrocytes modulates hippocampal plasticity. Immunity 2025, 58, 1519–1535.e11. DOI:10.1016/j.immuni.2025.04.007 [Google Scholar]
-
Khakh BS, Deneen B. The Emerging Nature of Astrocyte Diversity. Annu. Rev. Neurosci. 2019, 42, 187–207. DOI:10.1146/annurev-neuro-070918-050443 [Google Scholar]
-
Liddelow SA, Barres BA. Reactive Astrocytes: Production, Function, and Therapeutic Potential. Immunity 2017, 46, 957–967. DOI:10.1016/j.immuni.2017.06.006 [Google Scholar]
-
Patani R, Hardingham GE, Liddelow SA. Functional roles of reactive astrocytes in neuroinflammation and neurodegeneration. Nat. Rev. Neurol. 2023, 19, 395–409. DOI:10.1038/s41582-023-00822-1 [Google Scholar]
-
Hwang S-Y, Jung J-S, Kim T-H, Lim S-J, Oh E-S, Kim J-Y, et al. Ionizing radiation induces astrocyte gliosis through microglia activation. Neurobiol. Dis. 2006, 21, 457–467. DOI:10.1016/j.nbd.2005.08.006 [Google Scholar]
-
Yang Z, Wang KKW. Glial fibrillary acidic protein: From intermediate filament assembly and gliosis to neurobiomarker. Trends Neurosci. 2015, 38, 364–374. DOI:10.1016/j.tins.2015.04.003 [Google Scholar]
-
Fan Y-Y. A1/A2 astrocytes in central nervous system injuries and diseases: Angels or devils? Neurochem. Int. 2021, 148, 105080. DOI:10.1016/j.neuint.2021.105080 [Google Scholar]
-
Yun SP, Kam T-I, Panicker N, Kim S, Oh Y, Park J-S, et al. Block of A1 astrocyte conversion by microglia is neuroprotective in models of Parkinson’s disease. Nat. Med. 2018, 24, 931–938. DOI:10.1038/s41591-018-0051-5 [Google Scholar]
-
Sofroniew MV. Astrocyte Reactivity: Subtypes, States, and Functions in CNS Innate Immunity. Trends Immunol. 2020, 41, 758–770. DOI:10.1016/j.it.2020.07.004 [Google Scholar]
-
Escartin C, Galea E, Lakatos A, O’Callaghan JP, Petzold GC, Serrano-Pozo A, et al. Reactive astrocyte nomenclature, definitions, and future directions. Nat. Neurosci. 2021, 24, 312–325. DOI:10.1038/s41593-020-00783-4 [Google Scholar]
-
Farhy-Tselnicker I. Astrocytes, neurons, synapses: A tripartite view on cortical circuit development. Neural Dev. 2018, 13, 7. DOI:10.1186/s13064-018-0104-y [Google Scholar]
-
Li X, Liu G, Yang L, Li Z, Zhang Z, Xu Z, et al. Decoding Cortical Glial Cell Development. Neurosci. Bull. 2021, 37, 440–460. DOI:10.1007/s12264-021-00640-9 [Google Scholar]
-
Hasel P, Liddelow SA. Astrocytes. Curr. Biol. 2021, 31, R326–R327. DOI:10.1016/j.cub.2021.01.056 [Google Scholar]
-
Torres-Ceja B, Olsen ML. A closer look at astrocyte morphology: Development, heterogeneity, and plasticity at astrocyte leaflets. Curr. Opin. Neurobiol. 2022, 74, 102550. DOI:10.1016/j.conb.2022.102550 [Google Scholar]
-
Tabata H. Diverse subtypes of astrocytes and their development during corticogenesis. Front. Neurosci. 2015, 7, 114. DOI:10.3389/fnins.2015.00114 [Google Scholar]
-
Zhou J, Vitali I, Roig-Puiggros S, Javed A, Cantando I, Puglisi M, et al. Dual lineage origins contribute to neocortical astrocyte diversity. Nat. Commun. 2025, 16, 6992. DOI:10.1038/s41467-025-61829-4 [Google Scholar]
-
Spurgat MS, Tang S-J. Single-Cell RNA-Sequencing: Astrocyte and Microglial Heterogeneity in Health and Disease. Cells 2022, 11, 2021. DOI:10.3390/cells11132021 [Google Scholar]
-
Batiuk MY, Martirosyan A, Wahis J, de Vin F, Marneffe C, Kusserow C, et al. Identification of region-specific astrocyte subtypes at single cell resolution. Nat. Commun. 2020, 11, 1220. DOI:10.1038/s41467-019-14198-8 [Google Scholar]
-
Bayraktar OA, Bartels T, Holmqvist S, Kleshchevnikov V, Martirosyan A, Polioudakis D, et al. Astrocyte layers in the mammalian cerebral cortex revealed by a single-cell in situ transcriptomic map. Nat. Neurosci. 2020, 23, 500–509. DOI:10.1038/s41593-020-0602-1 [Google Scholar]
-
Bocchi R, Thorwirth M, Simon-Ebert T, Koupourtidou C, Clavreul S, Kolf K, et al. Astrocyte heterogeneity reveals region-specific astrogenesis in the white matter. Nat. Neurosci. 2025, 28, 457–469. DOI:10.1038/s41593-025-01878-6 [Google Scholar]
-
O’Dea MR, Hasel P. Are we there yet? Exploring astrocyte heterogeneity one cell at a time. Glia 2025, 73, 619–631. DOI:10.1002/glia.24621 [Google Scholar]
-
Díaz-Castro B, Robel S, Mishra A. Astrocyte Endfeet in Brain Function and Pathology: Open Questions. Annu. Rev. Neurosci. 2023, 46, 101–121. DOI:10.1146/annurev-neuro-091922-031205 [Google Scholar]
-
Pivoriūnas A, Verkhratsky A. Astrocyte-Endotheliocyte Axis in the Regulation of the Blood-Brain Barrier. Neurochem. Res. 2021, 46, 2538–2550. DOI:10.1007/s11064-021-03338-6 [Google Scholar]
-
Manu DR, Slevin M, Barcutean L, Forro T, Boghitoiu T, Balasa R. Astrocyte Involvement in Blood-Brain Barrier Function: A Critical Update Highlighting Novel, Complex, Neurovascular Interactions. Int. J. Mol. Sci. 2023, 24, 17146. DOI:10.3390/ijms242417146 [Google Scholar]
-
Bentivegna M, Pomilio C, Bellotto M, Pérez NG, Rossi SP, Gregosa A, et al. Amyloid Beta Regulates Astrocytic Glucose Metabolism and Insulin Signaling in Experimental Models of Alzheimer’s Disease. Aging Dis. 2025, 17, 4. DOI:10.14336/AD.2025.0484 [Google Scholar]
-
Lei P, Walker T, Ayton S. Neuroferroptosis in health and diseases. Nat. Rev. Neurosci. 2025, 26, 497–511. DOI:10.1038/s41583-025-00930-5 [Google Scholar]
-
Magistretti PJ, Allaman I. Lactate in the brain: From metabolic end-product to signalling molecule. Nat. Rev. Neurosci. 2018, 19, 235–249. DOI:10.1038/nrn.2018.19 [Google Scholar]
-
Xiong X-Y, Tang Y, Yang Q-W. Metabolic changes favor the activity and heterogeneity of reactive astrocytes. Trends Endocrinol. Metab. 2022, 33, 390–400. DOI:10.1016/j.tem.2022.03.001 [Google Scholar]
-
Theparambil SM, Kopach O, Braga A, Nizari S, Hosford PS, Sagi-Kiss V, et al. Adenosine signalling to astrocytes coordinates brain metabolism and function. Nature 2024, 632, 139–146. DOI:10.1038/s41586-024-07611-w [Google Scholar]
-
Zheng T, Kotol D, Sjöberg R, Mitsios N, Uhlén M, Zhong W, et al. Characterization of reduced astrocyte creatine kinase levels in Alzheimer’s disease. Glia 2024, 72, 1590–1603. DOI:10.1002/glia.24569 [Google Scholar]
-
Almeida A, Jimenez-Blasco D, Bolaños JP. Cross-talk between energy and redox metabolism in astrocyte-neuron functional cooperation. Essays Biochem. 2023, 67, 17–26. DOI:10.1042/EBC20220075 [Google Scholar]
-
Verkhratsky A, Nedergaard M. Physiology of Astroglia. Physiol. Rev. 2018, 98, 239–389. DOI:10.1152/physrev.00042.2016 [Google Scholar]
-
Abbasian V, Davoudi S, Vahabzadeh A, Maftoon-Azad MJ, Janahmadi M. Astroglial Kir4.1 and AQP4 Channels: Key Regulators of Potassium Homeostasis and Their Implications in Autism Spectrum Disorders. Cell Mol. Neurobiol. 2025, 45, 56. DOI:10.1007/s10571-025-01574-w [Google Scholar]
-
Lee H-G, Wheeler MA, Quintana FJ. Function and therapeutic value of astrocytes in neurological diseases. Nat. Rev. Drug Discov. 2022, 21, 339–358. DOI:10.1038/s41573-022-00390-x [Google Scholar]
-
van der Knaap MS, Min R. Multiple sclerosis: An immune attack on astrocyte-mediated ion and water homeostasis. Nat. Rev. Neurol. 2025, 21, 283–289. DOI:10.1038/s41582-025-01081-y [Google Scholar]
-
Sofroniew MV, Vinters HV. Astrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 7–35. DOI:10.1007/s00401-009-0619-8 [Google Scholar]
-
Tong K, Song Y-T, Jing S-Q, You Y, Wang S-J, Wu T, et al. Reactive astrocytes mediate postoperative surgery-induced anxiety through modulation of GABAergic signalling in the zona incerta of mice. Br. J. Anaesth. 2025, 134, 111–123. DOI:10.1016/j.bja.2024.08.043 [Google Scholar]
-
Mahmoud S, Gharagozloo M, Simard C, Gris D. Astrocytes Maintain Glutamate Homeostasis in the CNS by Controlling the Balance between Glutamate Uptake and Release. Cells 2019, 8, 184. DOI:10.3390/cells8020184 [Google Scholar]
-
Lefton KB, Wu Y, Dai Y, Okuda T, Zhang Y, Yen A, et al. Norepinephrine signals through astrocytes to modulate synapses. Science 2025, 388, 776–783. DOI:10.1126/science.adq5480 [Google Scholar]
-
Liu J, Feng X, Wang Y, Xia X, Zheng JC. Astrocytes: GABAceptive and GABAergic Cells in the Brain. Front. Cell Neurosci. 2022, 16, 892497. DOI:10.3389/fncel.2022.892497 [Google Scholar]
-
Han RT, Kim RD, Molofsky AV, Liddelow SA. Astrocyte-immune cell interactions in physiology and pathology. Immunity 2021, 54, 211–224. DOI:10.1016/j.immuni.2021.01.013 [Google Scholar]
-
Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. DOI:10.1038/nature21029 [Google Scholar]
-
Williamson MR, Fuertes CJA, Dunn AK, Drew MR, Jones TA. Reactive astrocytes facilitate vascular repair and remodeling after stroke. Cell Rep. 2021, 35, 109048. DOI:10.1016/j.celrep.2021.109048 [Google Scholar]
-
Chang J, Qian Z, Wang B, Cao J, Zhang S, Jiang F, et al. Transplantation of A2 type astrocytes promotes neural repair and remyelination after spinal cord injury. Cell Commun. Signal. 2023, 21, 37. DOI:10.1186/s12964-022-01036-6 [Google Scholar]
-
Wilson CM, Gaber MW, Sabek OM, Zawaski JA, Merchant TE. Radiation-Induced Astrogliosis and Blood-Brain Barrier Damage Can Be Abrogated Using Anti-TNF Treatment. Int. J. Radiat. Oncol.*Biol.*Phys. 2009, 74, 934–941. DOI:10.1016/j.ijrobp.2009.02.035 [Google Scholar]
-
Zhou H, Sun F, Ou M, Zhang Y, Lin M, Song L, et al. Prior nasal delivery of antagomiR-122 prevents radiation-induced brain injury. Mol. Ther. 2021, 29, 3465–3483. DOI:10.1016/j.ymthe.2021.06.019 [Google Scholar]
-
Markarian M, Krattli RP, Baddour JD, Alikhani L, Giedzinski E, Usmani MT, et al. Glia-Selective Deletion of Complement C1q Prevents Radiation-Induced Cognitive Deficits and Neuroinflammation. Cancer Res. 2021, 81, 1732–1744. DOI:10.1158/0008-5472.CAN-20-2565 [Google Scholar]
-
Zhang C, Zheng J, Chen W, Yang W, Tan X, Fan X, et al. Mitochondrial-targeting fluorescent small molecule IR-780 alleviates radiation-induced brain injury. Brain Res. 2023, 1805, 148285. DOI:10.1016/j.brainres.2023.148285 [Google Scholar]
-
Zhou G, Xu Y, He B, Ma R, Wang Y, Chang Y, et al. Ionizing radiation modulates vascular endothelial growth factor expression through STAT3 signaling pathway in rat neonatal primary astrocyte cultures. Brain Behav. 2020, 10, e01529. DOI:10.1002/brb3.1529 [Google Scholar]
-
Xu L, Huang H, Liu T, Yang T, Yi X. Exposure to X-rays Causes Depression-like Behaviors in Mice via HMGB1-mediated Pyroptosis. Neuroscience 2022, 481, 99–110. DOI:10.1016/j.neuroscience.2021.11.023 [Google Scholar]
-
Marinelli S, Basilico B, Marrone MC, Ragozzino D. Microglia-neuron crosstalk: Signaling mechanism and control of synaptic transmission. Semin. Cell Dev. Biol. 2019, 94, 138–151. DOI:10.1016/j.semcdb.2019.05.017 [Google Scholar]
-
Leng F, Edison P. Neuroinflammation and microglial activation in Alzheimer disease: Where do we go from here? Nat. Rev. Neurol. 2021, 17, 157–172. DOI:10.1038/s41582-020-00435-y [Google Scholar]
-
DiSabato DJ, Quan N, Godbout JP. Neuroinflammation: The devil is in the details. J. Neurochem. 2016, 139 (Suppl. S2), 136–153. DOI:10.1111/jnc.13607 [Google Scholar]
-
Al-Ghraiybah NF, Wang J, Alkhalifa AE, Roberts AB, Raj R, Yang E, et al. Glial Cell-Mediated Neuroinflammation in Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 23, 10572. DOI:10.3390/ijms231810572 [Google Scholar]
-
Linnerbauer M, Wheeler MA, Quintana FJ. Astrocyte Crosstalk in CNS Inflammation. Neuron 2020, 108, 608–622. DOI:10.1016/j.neuron.2020.08.012 [Google Scholar]
-
Lee H-G, Lee J-H, Flausino LE, Quintana FJ. Neuroinflammation: An astrocyte perspective. Sci. Transl. Med. 2023, 15, eadi7828. DOI:10.1126/scitranslmed.adi7828 [Google Scholar]
-
Lumniczky K, Szatmári T, Sáfrány G. Ionizing Radiation-Induced Immune and Inflammatory Reactions in the Brain. Front. Immunol. 2017, 8, 517. DOI:10.3389/fimmu.2017.00517 [Google Scholar]
-
Constanzo J, Midavaine É, Fouquet J, Lepage M, Descoteaux M, Kirby K, et al. Brain irradiation leads to persistent neuroinflammation and long-term neurocognitive dysfunction in a region-specific manner. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2020, 102, 109954. DOI:10.1016/j.pnpbp.2020.109954 [Google Scholar]
-
Turnquist C, Harris BT, Harris CC. Radiation-induced brain injury: Current concepts and therapeutic strategies targeting neuroinflammation. Neurooncol Adv. 2020, 2, vdaa057. DOI:10.1093/noajnl/vdaa057 [Google Scholar]
-
Lee WH, Sonntag WE, Mitschelen M, Yan H, Lee YW. Irradiation induces regionally specific alterations in pro-inflammatory environments in rat brain. Int. J. Radiat. Biol. 2010, 86, 132–144. DOI:10.3109/09553000903419346 [Google Scholar]
-
Dong X, Luo M, Huang G, Zhang J, Tong F, Cheng Y, et al. Relationship between irradiation-induced neuro-inflammatory environments and impaired cognitive function in the developing brain of mice. Int. J. Radiat. Biol. 2015, 91, 224–239. DOI:10.3109/09553002.2014.988895 [Google Scholar]
-
Nakano-Kobayashi A, Canela A, Yoshihara T, Hagiwara M. Astrocyte-targeting therapy rescues cognitive impairment caused by neuroinflammation via the Nrf2 pathway. Proc. Natl. Acad. Sci. USA 2023, 120, e2303809120. DOI:10.1073/pnas.2303809120 [Google Scholar]
-
Verma SD, Passerat de la Chapelle E, Malkani S, Juran CM, Boyko V, Costes SV, et al. Astrocytes regulate vascular endothelial responses to simulated deep space radiation in a human organ-on-a-chip model. Front. Immunol. 2022, 13, 864923. DOI:10.3389/fimmu.2022.864923 [Google Scholar]
-
Zeng W, Fu L, Xu H. MicroRNA-206 relieves irradiation-induced neuroinflammation by regulating connexin 43. Exp. Ther. Med. 2021, 22, 1186. DOI:10.3892/etm.2021.10620 [Google Scholar]
-
Turnquist C, Beck JA, Horikawa I, Obiorah IE, Von Muhlinen N, Vojtesek B, et al. Radiation-induced astrocyte senescence is rescued by Δ133p53. Neuro Oncol. 2019, 21, 474–485. DOI:10.1093/neuonc/noz001 [Google Scholar]
-
Moore ED, Kooshki M, Metheny-Barlow LJ, Gallagher PE, Robbins ME. Angiotensin-(1-7) prevents radiation-induced inflammation in rat primary astrocytes through regulation of MAP kinase signaling. Free Radic. Biol. Med. 2013, 65, 1060–1068. DOI:10.1016/j.freeradbiomed.2013.08.183 [Google Scholar]
-
Heneka MT, Kummer MP, Latz E. Innate immune activation in neurodegenerative disease. Nat. Rev. Immunol. 2014, 14, 463–477. DOI:10.1038/nri3705 [Google Scholar]
-
Lacoste B, Prat A, Freitas-Andrade M, Gu C. The Blood-Brain Barrier: Composition, Properties, and Roles in Brain Health. Cold Spring Harb. Perspect. Biol. 2025, 17, a041422. DOI:10.1101/cshperspect.a041422 [Google Scholar]
-
Spampinato SF, Bortolotto V, Canonico PL, Sortino MA, Grilli M. Astrocyte-Derived Paracrine Signals: Relevance for Neurogenic Niche Regulation and Blood-Brain Barrier Integrity. Front. Pharmacol. 2019, 10, 1346. DOI:10.3389/fphar.2019.01346 [Google Scholar]
-
Michinaga S, Koyama Y. Dual Roles of Astrocyte-Derived Factors in Regulation of Blood-Brain Barrier Function after Brain Damage. Int. J. Mol. Sci. 2019, 20, 571. DOI:10.3390/ijms20030571 [Google Scholar]
-
Schumacher S, Tahiri H, Ezan P, Rouach N, Witschas K, Leybaert L. Inhibiting astrocyte connexin-43 hemichannels blocks radiation-induced vesicular VEGF-A release and blood-brain barrier dysfunction. Glia 2024, 72, 34–50. DOI:10.1002/glia.24460 [Google Scholar]
-
Zhang S, Li M, Qiu Y, Wu J, Xu X, Ma Q, et al. Enhanced VEGF secretion and blood-brain barrier disruption: Radiation-mediated inhibition of astrocyte autophagy via PI3K-AKT pathway activation. Glia 2024, 72, 568–587. DOI:10.1002/glia.24491 [Google Scholar]
-
Niranjan R. The Role of Inflammatory and Oxidative Stress Mechanisms in the Pathogenesis of Parkinson’s Disease: Focus on Astrocytes. Mol. Neurobiol. 2014, 49, 28–38. DOI:10.1007/s12035-013-8483-x [Google Scholar]
-
Cabezas R, Baez-Jurado E, Hidalgo-Lanussa O, Echeverria V, Ashrad GM, Sahebkar A, et al. Growth Factors and Neuroglobin in Astrocyte Protection Against Neurodegeneration and Oxidative Stress. Mol. Neurobiol. 2019, 56, 2339–2351. DOI:10.1007/s12035-018-1203-9 [Google Scholar]
-
Sulimai N, Brown J, Lominadze D. Fibrinogen Interaction with Astrocyte ICAM-1 and PrPC Results in the Generation of ROS and Neuronal Death. IJMS 2021, 22, 2391. DOI:10.3390/ijms22052391 [Google Scholar]
-
Elangovan S, Holsinger RMD. Cyclical amyloid beta-astrocyte activity induces oxidative stress in Alzheimer’s disease. Biochimie 2020, 171–172, 38–42. DOI:10.1016/j.biochi.2020.02.003 [Google Scholar]
-
Miyazaki I, Asanuma M. Neuron-Astrocyte Interactions in Parkinson’s Disease. Cells 2020, 9, 2623. DOI:10.3390/cells9122623 [Google Scholar]
-
Baev AY, Vinokurov AY, Novikova IN, Dremin VV, Potapova EV, Abramov AY. Interaction of Mitochondrial Calcium and ROS in Neurodegeneration. Cells 2022, 11, 706. DOI:10.3390/cells11040706 [Google Scholar]
-
Betlazar C, Middleton RJ, Banati RB, Liu G-J. The impact of high and low dose ionising radiation on the central nervous system. Redox Biol. 2016, 9, 144–156. DOI:10.1016/j.redox.2016.08.002 [Google Scholar]
-
Azzam EI, Jay-Gerin J-P, Pain D. Ionizing radiation-induced metabolic oxidative stress and prolonged cell injury. Cancer Lett. 2012, 327, 48–60. DOI:10.1016/j.canlet.2011.12.012 [Google Scholar]
-
Burdak-Rothkamm S, Folkard M, Rothkamm K, Prise K. ATR-dependent radiation-induced cH2AX foci in bystander primary human astrocytes and glioma cells. Oncogene 2007, 26, 993–1002. DOI:10.1038/sj.onc.1209863 [Google Scholar]
-
Tomasello DL, Barrasa MI, Mankus D, Alarcon KI, Lytton-Jean AKR, Liu XS, et al. Mitochondrial dysfunction and increased reactive oxygen species production in MECP2 mutant astrocytes and their impact on neurons. Sci. Rep. 2024, 14, 20565. DOI:10.1038/s41598-024-71040-y [Google Scholar]
-
Wang Q-Q, Yin G, Huang J-R, Xi S-J, Qian F, Lee R-X, et al. Ionizing Radiation-Induced Brain Cell Aging and the Potential Underlying Molecular Mechanisms. Cells 2021, 10, 3570. DOI:10.3390/cells10123570 [Google Scholar]
-
Rosenthal RA, Fish B, Hill RP, Huffman KD, Lazarova Z, Mahmood J, et al. Salen Mn complexes mitigate radiation injury in normal tissues. Anticancer. Agents Med. Chem. 2011, 11, 359–372. DOI:10.2174/187152011795677490 [Google Scholar]
-
Marino N, Bedeschi M, Vaccari ME, Cambiaghi M, Tesei A. Glitches in the brain: The dangerous relationship between radiotherapy and brain fog. Front. Cell Neurosci. 2024, 18, 1328361. DOI:10.3389/fncel.2024.1328361 [Google Scholar]
-
Kou J, Shi J, He Y, Hao J, Zhang H, Luo D, et al. Luteolin alleviates cognitive impairment in Alzheimer’s disease mouse model via inhibiting endoplasmic reticulum stress-dependent neuroinflammation. Acta Pharmacol. Sin. 2022, 43, 840–849. DOI:10.1038/s41401-021-00702-8 [Google Scholar]
-
Jung B-K, Park Y, Yoon B, Bae J-S, Han S-W, Heo J-E, et al. Reduced secretion of LCN2 (lipocalin 2) from reactive astrocytes through autophagic and proteasomal regulation alleviates inflammatory stress and neuronal damage. Autophagy 2023, 19, 2296–2317. DOI:10.1080/15548627.2023.2180202 [Google Scholar]
-
Lee JH, Han J-H, Joe E-H, Jou I. Small heterodimer partner (SHP) aggravates ER stress in Parkinson’s disease-linked LRRK2 mutant astrocyte by regulating XBP1 SUMOylation. J. Biomed. Sci. 2021, 28, 51. DOI:10.1186/s12929-021-00747-1 [Google Scholar]
-
Havaki S, Kotsinas A, Chronopoulos E, Kletsas D, Georgakilas A, Gorgoulis VG. The role of oxidative DNA damage in radiation induced bystander effect. Cancer Lett. 2015, 356, 43–51. DOI:10.1016/j.canlet.2014.01.023 [Google Scholar]
-
Hoorelbeke D, Decrock E, Smet MD, Bock MD, Descamps B, Haver VV, et al. Cx43 channels and signaling via IP3/Ca2+, ATP, and ROS/NO propagate radiation-induced DNA damage to non-irradiated brain microvascular endothelial cells. Cell Death Dis. 2020, 11, 194. DOI:10.1038/s41419-020-2392-5 [Google Scholar]
-
Guerrero A, De Strooper B, Arancibia-Cárcamo IL. Cellular senescence at the crossroads of inflammation and Alzheimer’s disease. Trends Neurosci. 2021, 44, 714–727. DOI:10.1016/j.tins.2021.06.007 [Google Scholar]
-
Jiang S-Y, Tian T, Yao H, Xia X-M, Wang C, Cao L, et al. The cGAS-STING-YY1 axis accelerates progression of neurodegeneration in a mouse model of Parkinson’s disease via LCN2-dependent astrocyte senescence. Cell Death Differ. 2023, 30, 2280–2292. DOI:10.1038/s41418-023-01216-y [Google Scholar]
-
Park J-E, Park JW, Sim M-K, Kim SR, Kim KS. Inhibition of DAPK3 Suppresses Radiation-Induced Cellular Senescence by Activation of a PGC1α-Dependent Metabolism Pathway in Brain Endothelial Cells. J. Gerontol. A Biol. Sci. Med. Sci. 2024, 79, glae088. DOI:10.1093/gerona/glae088 [Google Scholar]
-
Melo Dos Santos L, Trombetta-Lima M, Eggen B, Demaria M. Cellular senescence in brain aging and neurodegeneration. Ageing Res. Rev. 2024, 93, 102141. DOI:10.1016/j.arr.2023.102141 [Google Scholar]
-
Zhang W, Sun H-S, Wang X, Dumont AS, Liu Q. Cellular senescence, DNA damage, and neuroinflammation in the aging brain. Trends Neurosci. 2024, 47, 461–474. DOI:10.1016/j.tins.2024.04.003 [Google Scholar]
-
Xu A, Li R, Ren A, Jian H, Huang Z, Zeng Q, et al. Regulatory coupling between long noncoding RNAs and senescence in irradiated microglia. J. Neuroinflamm. 2020, 17, 321. DOI:10.1186/s12974-020-02001-1 [Google Scholar]
-
Qiu O, Zhao J, Shi Z, Li H, Wang S, Liao K, et al. Asparagine endopeptidase deficiency mitigates radiation-induced brain injury by suppressing microglia-mediated neuronal senescence. iScience 2024, 27, 109698. DOI:10.1016/j.isci.2024.109698 [Google Scholar]
-
Kim S-B, Heo J-I, Kim H, Kim KS. Acetylation of PGC1α by Histone Deacetylase 1 Downregulation Is Implicated in Radiation-Induced Senescence of Brain Endothelial Cells. J. Gerontol. A Biol. Sci. Med. Sci. 2019, 74, 787–793. DOI:10.1093/gerona/gly167 [Google Scholar]
-
Luo N, Zhu W, Li X, Fu M, Zhang Y, Yang F, et al. Defective autophagy of pericytes enhances radiation-induced senescence promoting radiation brain injury. Neuro-Oncol. 2024, 26, 2288–2304. DOI:10.1093/neuonc/noae153 [Google Scholar]
-
Cohen J, Torres C. Astrocyte senescence: Evidence and significance. Aging Cell 2019, 18, e12937. DOI:10.1111/acel.12937 [Google Scholar]
-
Turnquist C, Horikawa I, Foran E, Major EO, Vojtesek B, Lane DP, et al. p53 isoforms regulate astrocyte-mediated neuroprotection and neurodegeneration. Cell Death Differ. 2016, 23, 1515–1528. DOI:10.1038/cdd.2016.37 [Google Scholar]
-
Ali FS, Arevalo O, Zorofchian S, Patrizz A, Riascos R, Tandon N, et al. Cerebral radiation necrosis: Incidence, pathogenesis, diagnostic challenges, and future opportunities. Curr. Oncol. Rep. 2019, 21, 66. DOI:10.1007/s11912-019-0818-y [Google Scholar]
-
Zhuang H, Shi S, Yuan Z, Chang JY. Bevacizumab treatment for radiation brain necrosis: Mechanism, efficacy and issues. Mol. Cancer 2019, 18, 21. DOI:10.1186/s12943-019-0950-1 [Google Scholar]
-
Bourbonne V, Ollivier L, Antoni D, Pradier O, Cailleteau A, Schick U, et al. Diagnosis and management of brain radiation necrosis. Cancer Radiother J. Soc. Fr. Radiother. Oncol. 2024, 28, 547–552. DOI:10.1016/j.canrad.2024.07.014 [Google Scholar]
-
Jou M-J. Pathophysiological and pharmacological implications of mitochondria-targeted reactive oxygen species generation in astrocytes. Adv. Drug Deliv. Rev. 2008, 60, 1512–1526. DOI:10.1016/j.addr.2008.06.004 [Google Scholar]
-
Allen NJ, Lyons DA. Glia as architects of central nervous system formation and function. Science 2018, 362, 181–185. DOI:10.1126/science.aat0473 [Google Scholar]
-
Jha MK, Jo M, Kim J-H, Suk K. Microglia-Astrocyte Crosstalk: An Intimate Molecular Conversation. Neuroscientist 2019, 25, 227–240. DOI:10.1177/1073858418783959 [Google Scholar]
-
Noel F, Tofilon PJ. Astrocytes protect against X-ray-induced neuronal toxicity in vitro. NeuroReport 1998, 9, 1133–1137. DOI:10.1097/00001756-199804200-00032 [Google Scholar]
-
Kenigsbuch M, Bost P, Halevi S, Chang Y, Chen S, Ma Q, et al. A shared disease-associated oligodendrocyte signature among multiple CNS pathologies. Nat. Neurosci. 2022, 25, 876–886. DOI:10.1038/s41593-022-01104-7 [Google Scholar]
-
Vrdoljak E, Bill CA, Stephens LC, van der Kogel AJ, Ang KK, Tofilon PJ. Radiation-induced apoptosis of oligodendrocytes in vitro. Int. J. Radiat. Biol. 1992, 62, 475–480. DOI:10.1080/09553009214552361 [Google Scholar]
-
Lu FG, Wong CS. Radiation-induced apoptosis of oligodendrocytes and its association with increased ceramide and down-regulated protein kinase B/Akt activity. Int. J. Radiat. Biol. 2004, 80, 39–51. DOI:10.1080/09553000310001642876 [Google Scholar]
-
Xu T, Liu C, Deng S, Gan L, Zhang Z, Yang G-Y, et al. The roles of microglia and astrocytes in myelin phagocytosis in the central nervous system. J. Cereb. Blood Flow. Metab. 2023, 43, 325–340. DOI:10.1177/0271678X221137762 [Google Scholar]
-
Sanmarco LM, Polonio CM, Wheeler MA, Quintana FJ. Functional immune cell-astrocyte interactions. J. Exp. Med. 2021, 218, e20202715. DOI:10.1084/jem.20202715 [Google Scholar]
-
Qian K, Jiang X, Liu Z-Q, Zhang J, Fu P, Su Y, et al. Revisiting the critical roles of reactive astrocytes in neurodegeneration. Mol. Psychiatry 2023, 28, 2697–2706. DOI:10.1038/s41380-023-02061-8 [Google Scholar]
-
Beamish CA, Zawaski JA, Inoue T, Sarkar P, Grosshans DR, Sabek OM, et al. NF-κB Blockade by NEMO Binding Domain Peptide Ameliorates Inflammation and Neurobehavioral Sequelae After Cranial Radiation Therapy in Juvenile Mice. Int. J. Radiat. Oncol. Biol. Phys. 2021, 109, 1508–1520. DOI:10.1016/j.ijrobp.2020.11.067 [Google Scholar]
-
Zhang S, Deng Z, Qiu Y, Lu G, Wu J, Huang H. FGIN-1-27 Mitigates Radiation-induced Mitochondrial Hyperfunction and Cellular Hyperactivation in Cultured Astrocytes. Neuroscience 2023, 535, 23–35. DOI:10.1016/j.neuroscience.2023.10.017 [Google Scholar]
-
Villasana L, Acevedo S, Poage C, Raber J. Sex- and APOE Isoform-Dependent Effects of Radiation on Cognitive Function. Radiat. Res. 2006, 166, 883–891. DOI:10.1667/RR0642.1 [Google Scholar]
-
Ou M, Fan W, Sun F, Li M, Lin M, Yu Y, et al. Nasal Delivery of AntagomiR-741 Protects Against the Radiation-Induced Brain Injury in Mice. Radiat. Res. 2021, 195, 355–365. DOI:10.1667/RADE-20-00070.1 [Google Scholar]
-
Apte RS, Chen DS, Ferrara N. VEGF in Signaling and Disease: Beyond Discovery and Development. Cell 2019, 176, 1248–1264. DOI:10.1016/j.cell.2019.01.021 [Google Scholar]
-
Pérez-Gutiérrez L, Ferrara N. Biology and therapeutic targeting of vascular endothelial growth factor A. Nat. Rev. Mol. Cell Biol. 2023, 24, 816–834. DOI:10.1038/s41580-023-00631-w [Google Scholar]
-
Argaw AT, Asp L, Zhang J, Navrazhina K, Pham T, Mariani JN, et al. Astrocyte-derived VEGF-A drives blood-brain barrier disruption in CNS inflammatory disease. J. Clin. Investig. 2012, 122, 2454–2468. DOI:10.1172/JCI60842 [Google Scholar]
-
Lan G, Wang P, Chan RB, Liu Z, Yu Z, Liu X, et al. Astrocytic VEGFA: An essential mediator in blood-brain-barrier disruption in Parkinson’s disease. Glia 2022, 70, 337–353. DOI:10.1002/glia.24109 [Google Scholar]
-
Kugimiya K, Tsubouchi H, Saito K, Kadota Y, Azuma M, Sakai K, et al. Cerebral radiation necrosis successfully treated with high-dose bevacizumab. Respir. Med. Case Rep. 2025, 58, 102282. DOI:10.1016/j.rmcr.2025.102282 [Google Scholar]
-
Khan M, Zhao Z, Arooj S, Liao G. Bevacizumab for radiation necrosis following radiotherapy of brain metastatic disease: A systematic review & meta-analysis. BMC Cancer 2021, 21, 167. DOI:10.1186/s12885-021-07889-3 [Google Scholar]
-
Jung K, Sivadas SD, Fitzgerald X, Phillips C, Plumridge N, Spain L, et al. Long-term clinical outcomes of bevacizumab for treatment of stereotactic radiosurgery-induced radiation necrosis in patients with brain metastases. J. Neuro-Oncol. 2025, 175, 209–218. DOI:10.1007/s11060-025-05121-x [Google Scholar]
-
New LE, Wang N, Smith HE, Birks R, Afridi SK, Griffiths JC, et al. Insulin evokes release of endozepines from astrocytes of the NTS to modulate glucose metabolism in male rats. Mol. Metab. 2025, 101, 102255. DOI:10.1016/j.molmet.2025.102255 [Google Scholar]
-
Peng Y, Zhang Y, Wang W, Liu B, Zhang Z, Gong Z, et al. Potential role of remimazolam in alleviating bone cancer pain in mice via modulation of translocator protein in spinal astrocytes. Eur. J. Pharmacol. 2024, 979, 176861. DOI:10.1016/j.ejphar.2024.176861 [Google Scholar]
-
Angeloni E, Germelli L, Costa B, Martini C, Da Pozzo E. Neurosteroids and Translocator Protein (TSPO) in neuroinflammation. Neurochem. Int. 2025, 182, 105916. DOI:10.1016/j.neuint.2024.105916 [Google Scholar]
-
Ceyzériat K, Nicolaides A, Amossé Q, Fossey C, Cailly T, Fabis F, et al. Reactive astrocytes mediate TSPO overexpression in response to sustained CNTF exposure in the rat striatum. Mol. Brain 2023, 16, 57. DOI:10.1186/s13041-023-01041-x [Google Scholar]
-
Boche D, Rodriguez-Vieitez E. Prospects and challenges of imaging neuroinflammation beyond TSPO in Alzheimer’s disease. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 2831–2847. DOI:10.1007/s00259-019-04462-w [Google Scholar]
-
Tournier BB, Tsartsalis S, Ceyzériat K, Fraser BH, Grégoire M-C, Kövari E, et al. Astrocytic TSPO Upregulation Appears Before Microglial TSPO in Alzheimer’s Disease. J. Alzheimer’s Dis. 2020, 77, 1043–1056. DOI:10.3233/JAD-200136 [Google Scholar]
-
Loth MK, Choi J, McGlothan JL, Pletnikov MV, Pomper MG, Guilarte TR. TSPO in a murine model of Sandhoff disease: Presymptomatic marker of neurodegeneration and disease pathophysiology. Neurobiol. Dis. 2016, 85, 174–186. DOI:10.1016/j.nbd.2015.11.001 [Google Scholar]
-
Scholz R, Caramoy A, Bhuckory MB, Rashid K, Chen M, Xu H, et al. Targeting translocator protein (18 kDa) (TSPO) dampens pro-inflammatory microglia reactivity in the retina and protects from degeneration. J. Neuroinflamm. 2015, 12, 201. DOI:10.1186/s12974-015-0422-5 [Google Scholar]
-
Ma L, Zhang H, Liu N, Wang P, Guo W, Fu Q, et al. TSPO ligand PK11195 alleviates neuroinflammation and beta-amyloid generation induced by systemic LPS administration. Brain Res. Bull. 2016, 121, 192–200. DOI:10.1016/j.brainresbull.2016.02.001 [Google Scholar]
-
Tournier BB, Bouteldja F, Amossé Q, Nicolaides A, Duarte Azevedo M, Tenenbaum L, et al. 18 kDa Translocator Protein TSPO Is a Mediator of Astrocyte Reactivity. ACS Omega 2023, 8, 31225–31236. DOI:10.1021/acsomega.3c03368 [Google Scholar]
-
Betlazar C, Middleton RJ, Howell N, Storer B, Davis E, Davies J, et al. Mitochondrial Translocator Protein (TSPO) Expression in the Brain After Whole Body Gamma Irradiation. Front. Cell Dev. Biol. 2021, 9, 715444. DOI:10.3389/fcell.2021.715444 [Google Scholar]
-
Rodina AV, Semochkina YP, Vysotskaya OV, Parfenova AA, Moskaleva EY. Radiation-induced neuroinflammation monitoring by the level of peripheral blood monocytes with high expression of translocator protein. Int. J. Radiat. Biol. 2023, 99, 1364–1377. DOI:10.1080/09553002.2023.2177765 [Google Scholar]
-
Boyd A, Byrne S, Middleton RJ, Banati RB, Liu G-J. Control of Neuroinflammation through Radiation-Induced Microglial Changes. Cells 2021, 10, 2381. DOI:10.3390/cells10092381 [Google Scholar]
-
Porel P, Hunjan G, Singh S, Aran KR. Is the renin-angiotensin system a friend or foe in neurological diseases? Unveiling its role and therapeutic potential. Ageing Res. Rev. 2025, 112, 102854. DOI:10.1016/j.arr.2025.102854 [Google Scholar]
-
Haliga RE, Cojocaru E, Sîrbu O, Hrițcu I, Alexa RE, Haliga IB, et al. Immunomodulatory Effects of RAAS Inhibitors: Beyond Hypertension and Heart Failure. Biomedicines 2025, 13, 1779. DOI:10.3390/biomedicines13071779 [Google Scholar]
-
Robbins ME, Zhao W, Garcia-Espinosa MA, Diz DI. Renin-angiotensin System Blockers and Modulation of Radiation-induced Brain Injury. Curr. Drug Targets 2011, 11, 1413–1422. DOI:10.2174/1389450111009011413 [Google Scholar]
-
Jadhav SS, Sharma N, Meeks CJ, Espinoza TB, Roda NR, DiZerega GS, et al. Effects of Combined Radiation and Burn injury on the Renin- Angiotensin System. Wound Repair. Regen. 2014, 21, 131–140. DOI:10.1111/j.1524-475X.2012.00867.x [Google Scholar]
-
Conner KR, Forbes ME, Lee WH, Lee YW, Riddle DR. AT1 Receptor Antagonism Does Not Influence Early Radiation- Induced Changes in Microglial Activation or Neurogenesis in the Normal Rat Brain. Radiat. Res. 2012, 176, 71–83. DOI:10.1667/rr2560.1 [Google Scholar]
-
Santos RAS, Sampaio WO, Alzamora AC, Motta-Santos D, Alenina N, Bader M, et al. The ACE2/Angiotensin-(1-7)/MAS Axis of the Renin-Angiotensin System: Focus on Angiotensin-(1-7). Physiol. Rev. 2018, 98, 505–553. DOI:10.1152/physrev.00023.2016 [Google Scholar]
-
Karahan H, Dabin LC, Tate MD, Kim J. MicroRNAs on the move: microRNAs in astrocyte-derived ApoE particles regulate neuronal function. Neuron 2021, 109, 907–909. DOI:10.1016/j.neuron.2021.02.021 [Google Scholar]
-
Jackson RJ, Meltzer JC, Nguyen H, Commins C, Bennett RE, Hudry E, et al. APOE4 derived from astrocytes leads to blood-brain barrier impairment. Brain 2022, 145, 3582–3593. DOI:10.1093/brain/awab478 [Google Scholar]
-
Fuentes D, Fernández N, García Y, García T, Morales AR, Menéndez R. Age-Related Changes in the Behavior of Apolipoprotein E Knockout Mice. Behav. Sci. 2018, 8, 33. DOI:10.3390/bs8030033 [Google Scholar]
-
Bell RD, Winkler EA, Singh I, Sagare AP, Deane R, Wu Z, et al. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature 2012, 485, 512–516. DOI:10.1038/nature11087 [Google Scholar]
-
Kempf SJ, Janik D, Barjaktarovic Z, Braga-Tanaka I, Tanaka S, Neff F, et al. Chronic low-dose-rate ionising radiation affects the hippocampal phosphoproteome in the ApoE−/− Alzheimer’s mouse model. Oncotarget 2016, 7, 71817–71832. DOI:10.18632/oncotarget.12376 [Google Scholar]
-
Casciati A, Pasquali E, De Stefano I, Braga-Tanaka I, Tanaka S, Mancuso M, et al. Role of Apolipoprotein E in the Hippocampus and Its Impact following Ionizing Radiation Exposure. Cells 2024, 13, 899. DOI:10.3390/cells13110899 [Google Scholar]
-
Knoferle J, Yoon SY, Walker D, Leung L, Gillespie AK, Tong LM, et al. Apolipoprotein E4 produced in GABAergic interneurons causes learning and memory deficits in mice. J. Neurosci. 2014, 34, 14069–14078. DOI:10.1523/JNEUROSCI.2281-14.2014 [Google Scholar]
-
Wieg L, Ciola JC, Wasén CC, Gaba F, Colletti BR, Schroeder MK, et al. Cognitive Effects of Simulated Galactic Cosmic Radiation Are Mediated by ApoE Status, Sex, and Environment in APP Knock-In Mice. Int. J. Mol. Sci. 2024, 25, 9379. DOI:10.3390/ijms25179379 [Google Scholar]
-
Yu T-S, Tensaouti Y, Stephanz EP, Chintamen S, Rafikian EE, Yang M, et al. Astrocytic ApoE underlies maturation of hippocampal neurons and cognitive recovery after traumatic brain injury in mice. Commun. Biol. 2021, 4, 1303. DOI:10.1038/s42003-021-02841-4 [Google Scholar]
-
Fernández-Calle R, Konings SC, Frontiñán-Rubio J, García-Revilla J, Camprubí-Ferrer L, Svensson M, et al. APOE in the bullseye of neurodegenerative diseases: Impact of the APOE genotype in Alzheimer’s disease pathology and brain diseases. Mol. Neurodegener. 2022, 17, 62. DOI:10.1186/s13024-022-00566-4 [Google Scholar]
-
Laskowitz DT, Van Wyck DW. ApoE Mimetic Peptides as Therapy for Traumatic Brain Injury. Neurotherapeutics 2023, 20, 1496–1507. DOI:10.1007/s13311-023-01413-0 [Google Scholar]
-
Fujita K, Mondal AM, Horikawa I, Nguyen GH, Kumamoto K, Sohn JJ, et al. p53 isoforms Delta133p53 and p53beta are endogenous regulators of replicative cellular senescence. Nat. Cell Biol. 2009, 11, 1135–1142. DOI:10.1038/ncb1928 [Google Scholar]
-
Gong L, Gong H, Pan X, Chang C, Ou Z, Ye S, et al. p53 isoform Δ113p53/Δ133p53 promotes DNA double-strand break repair to protect cell from death and senescence in response to DNA damage. Cell Res. 2015, 25, 351–369. DOI:10.1038/cr.2015.22 [Google Scholar]
-
Chen J, Ng SM, Chang C, Zhang Z, Bourdon J-C, Lane DP, et al. p53 isoform D113p53 is a p53 target gene that antagonizes p53 apoptotic activity via BclxL activation in zebrafish. Genes. Dev. 2009, 23, 278–290. DOI:10.1101/gad.1761609 [Google Scholar]
-
Kalluri R, LeBleu VS. The biology, function, and biomedical applications of exosomes. Science 2020, 367, eaau6977. DOI:10.1126/science.aau6977 [Google Scholar]
-
Lu TX, Rothenberg ME. MicroRNA. J. Allergy Clin. Immunol. 2018, 141, 1202–1207. DOI:10.1016/j.jaci.2017.08.034 [Google Scholar]
-
Diener C, Keller A, Meese E. Emerging concepts of miRNA therapeutics: From cells to clinic. Trends Genet. 2022, 38, 613–626. DOI:10.1016/j.tig.2022.02.006 [Google Scholar]
-
Mittelbrunn M, Sánchez-Madrid F. Intercellular communication: Diverse structures for exchange of genetic information. Nat. Rev. Mol. Cell Biol. 2012, 13, 328–335. DOI:10.1038/nrm3335 [Google Scholar]
-
Wang H, Ma Z-W, Ho F-M, Sethi G, Tang FR. Dual Effects of miR-181b-2-3p/SOX21 Interaction on Microglia and Neural Stem Cells after Gamma Irradiation. Cells 2023, 12, 649. DOI:10.3390/cells12040649 [Google Scholar]
-
Sabirzhanov B, Makarevich O, Barrett J, Jackson IL, Faden AI, Stoica BA. Down-Regulation of miR-23a-3p Mediates Irradiation-Induced Neuronal Apoptosis. Int. J. Mol. Sci. 2020, 21, 3695. DOI:10.3390/ijms21103695 [Google Scholar]
-
Cai S, Shi G-S, Cheng H-Y, Zeng Y-N, Li G, Zhang M, et al. Exosomal miR-7 Mediates Bystander Autophagy in Lung after Focal Brain Irradiation in Mice. Int. J. Biol. Sci. 2017, 13, 1287–1296. DOI:10.7150/ijbs.18890 [Google Scholar]
-
Wang C, Wang S, Xue Y, Zhong Y, Li H, Hou X, et al. Intravenous administration of blood-brain barrier-crossing conjugates facilitate biomacromolecule transport into central nervous system. Nat. Biotechnol. 2024, 43, 1783–1789. DOI:10.1038/s41587-024-02487-7 [Google Scholar]
-
Rehman FU, Liu Y, Zheng M, Shi B. Exosomes based strategies for brain drug delivery. Biomaterials 2023, 293, 121949. DOI:10.1016/j.biomaterials.2022.121949 [Google Scholar]
-
Han W, Zhang H, Feng L, Dang R, Wang J, Cui C, et al. The emerging role of exosomes in communication between the periphery and the central nervous system. MedComm 2020, 4, e410. DOI:10.1002/mco2.410 [Google Scholar]
-
Tang R, Yin J, Liu Y, Xue J. FLASH radiotherapy: A new milestone in the field of cancer radiotherapy. Cancer Lett. 2024, 587, 216651. DOI:10.1016/j.canlet.2024.216651 [Google Scholar]
-
Montay-Gruel P, Acharya MM, Petersson K, Alikhani L, Yakkala C, Allen BD, et al. Long-term neurocognitive benefits of FLASH radiotherapy driven by reduced reactive oxygen species. Proc. Natl. Acad. Sci. USA 2019, 116, 10943–10951. DOI:10.1073/pnas.1901777116 [Google Scholar]
-
He R, Liu Y, Li W, Xie S, Liu J, Cheng G, et al. Time-dependent alterations in brain metabolites and gut microbiota following whole-brain FLASH versus conventional radiotherapy in mice. Brain Res. Bull. 2026, 235, 111755. DOI:10.1016/j.brainresbull.2026.111755 [Google Scholar]
-
Bastos DC de A, Weinberg J, Kumar VA, Fuentes DT, Stafford J, Li J, et al. Laser interstitial thermal therapy in the treatment of brain metastases and radiation necrosis. Cancer Lett. 2020, 489, 9–18. DOI:10.1016/j.canlet.2020.05.014 [Google Scholar]
-
Shah S, Alberts AH, Tillotson S, Jain H, Lucke-Wold B. Recent advances in laser interstitial thermal therapy in the treatment of brain metastases and radiation necrosis. Lasers Med. Sci. 2025, 40, 186–202. DOI:10.1007/s10103-025-04447-2 [Google Scholar]
-
Jansen J, Kimbler A, Drayson O, Lanz B, Mosso J, Grilj V, et al. Ex. vivo brain MRI to assess conventional and FLASH brain irradiation effects. Radiother. Oncol. 2025, 208, 110894. DOI:10.1016/j.radonc.2025.110894 [Google Scholar]
-
Drayson OGG, Melemenidis S, Katila N, Viswanathan V, Kramár EA, Zhang R, et al. A multi-institutional study to investigate the sparing effect after whole brain electron FLASH in mice: Reproducibility and temporal evolution of functional, electrophysiological, and neurogenic endpoints. Radiother. Oncol. 2024, 201, 110534. DOI:10.1016/j.radonc.2024.110534 [Google Scholar]
-
Dickstein DL, Zhang R, Ru N, Vozenin M-C, Perry BC, Wang J, et al. Structural plasticity of pyramidal cell neurons measured after FLASH and conventional dose-rate irradiation. Brain Struct. Funct. 2025, 230, 41. DOI:10.1007/s00429-025-02902-y [Google Scholar]
-
Gao J, Li Y, Chen J, Feng W, Bu J, Lu Z, et al. Emodin ameliorates acute radiation proctitis in mice by regulating AKT/MAPK/NF-κB/VEGF pathways. Int. Immunopharmacol. 2024, 132, 111945. DOI:10.1016/j.intimp.2024.111945 [Google Scholar]
-
Sun M, Song Y, Hu X, Zhang Z, Tan R, Cai Z, et al. Leptin reduces LPS-induced A1 reactive astrocyte activation and inflammation via inhibiting p38-MAPK signaling pathway. Glia 2024, 73, 25–37. DOI:10.1002/glia.24611 [Google Scholar]
-
Deng Y, Duan R, Ding W, Gu Q, Liu M, Zhou J, et al. Astrocyte-derived exosomal nicotinamide phosphoribosyltransferase (Nampt) ameliorates ischemic stroke injury by targeting AMPK/mTOR signaling to induce autophagy. Cell Death Dis. 2022, 13, 1057. DOI:10.1038/s41419-022-05454-9 [Google Scholar]
-
Liu J, Zhu L, Bao Y, Du Z, Shi L, Hong X, et al. Injectable dexamethasone-loaded peptide hydrogel for therapy of radiation-induced ototoxicity by regulating the mTOR signaling pathway. J. Control. Release 2024, 365, 729–743. DOI:10.1016/j.jconrel.2023.12.004 [Google Scholar]
-
Rachamala HK, Madamsetty VS, Angom RS, Nakka NM, Dutta SK, Wang E, et al. Targeting mTOR and survivin concurrently potentiates radiation therapy in renal cell carcinoma by suppressing DNA damage repair and amplifying mitotic catastrophe. J. Exp. Clin. Cancer Res. 2024, 43, 159. DOI:10.1186/s13046-024-03079-8 [Google Scholar]