Non-Fallot Absent Pulmonary Valve Syndrome in Fetuses: Key Insights for Prenatal Diagnosis and Postnatal Care

Non-Fallot Absent Pulmonary Valve Syndrome in Fetuses: Key Insights for Prenatal Diagnosis and Postnatal Care

Received: 24 July 2025 Revised: 20 October 2025 Accepted: 31 October 2025 Published: 10 November 2025

© 2025 The authors. This is an open access article under the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/).

Graphical Abstract
1. Introduction
Absent pulmonary valve syndrome (APVS) is a rare cardiac malformation characterized by a rudimentary and dysplastic pulmonary valve, which is responsible for significant pulmonary insufficiency. In most cases, this malformation is associated with a Fallot-type ventricular septal defect (VSD), the so-called Tetralogy of Fallot with absent pulmonary valve [1]. A Fallot-type VSD is typically a large malalignment-type VSD with anterior deviation of the outlet septum, often accompanied by overriding of the aorta. More rarely, APVS can occur in isolation with an intact ventricular septum (IVS) or be associated with a non-Fallot type VSD. To date, less than 40 cases of antenatal diagnosis of this second type of APVS have been reported in the medical literature [2,3,4]. Thirty-one cases were reported by Rakha et al on a review of the literature [2], 5 cases were reported by Babaoglu et al. [3] and 2 cases with non-Fallot type VSD by Razavi et al. [4]. The limited number of observations described has hindered our understanding of the physiopathology and prognosis of this malformation when detected antenatally, thus affecting the quality of counselling provided to affected families.
The aim of this study is to report the fetal echocardiographic features and clinical course of 3 new cases diagnosed antenatally in a single institution from 2015 to 2020 and to discuss the characteristics that can assist in counseling families when detected antenatally, incorporating insights gained from our recent comprehensive literature review.
2. Case Reports
2.1. Case 1
A 31-year-old primigravida was referred for fetal echocardiography at 21 weeks of pregnancy because of asymmetry of the ventricles and the great vessels. The first trimester fetal echography was considered normal, and there was no increased nuchal translucency.
Fetal echocardiography revealed asymmetric cardiomegaly and a leftward deviation of the heart axis by 90°, attributed to right ventricular dilatation (Video S1). The pulmonary valves were rudimentary, and while the main pulmonary trunk was significantly dilated, the pulmonary branches showed only mild dilatation (Figure 1A,B). Doppler studies demonstrated to-and-fro blood flow across the pulmonary orifice with severe pulmonary regurgitation, as well as reverse flow during diastole through a widely patent ductus arteriosus (Figure 1C) (Video S2). There was no tricuspid regurgitation. The aortic arch was normally oriented to the left, and there was no pericardial effusion. The thymus was of normal size.
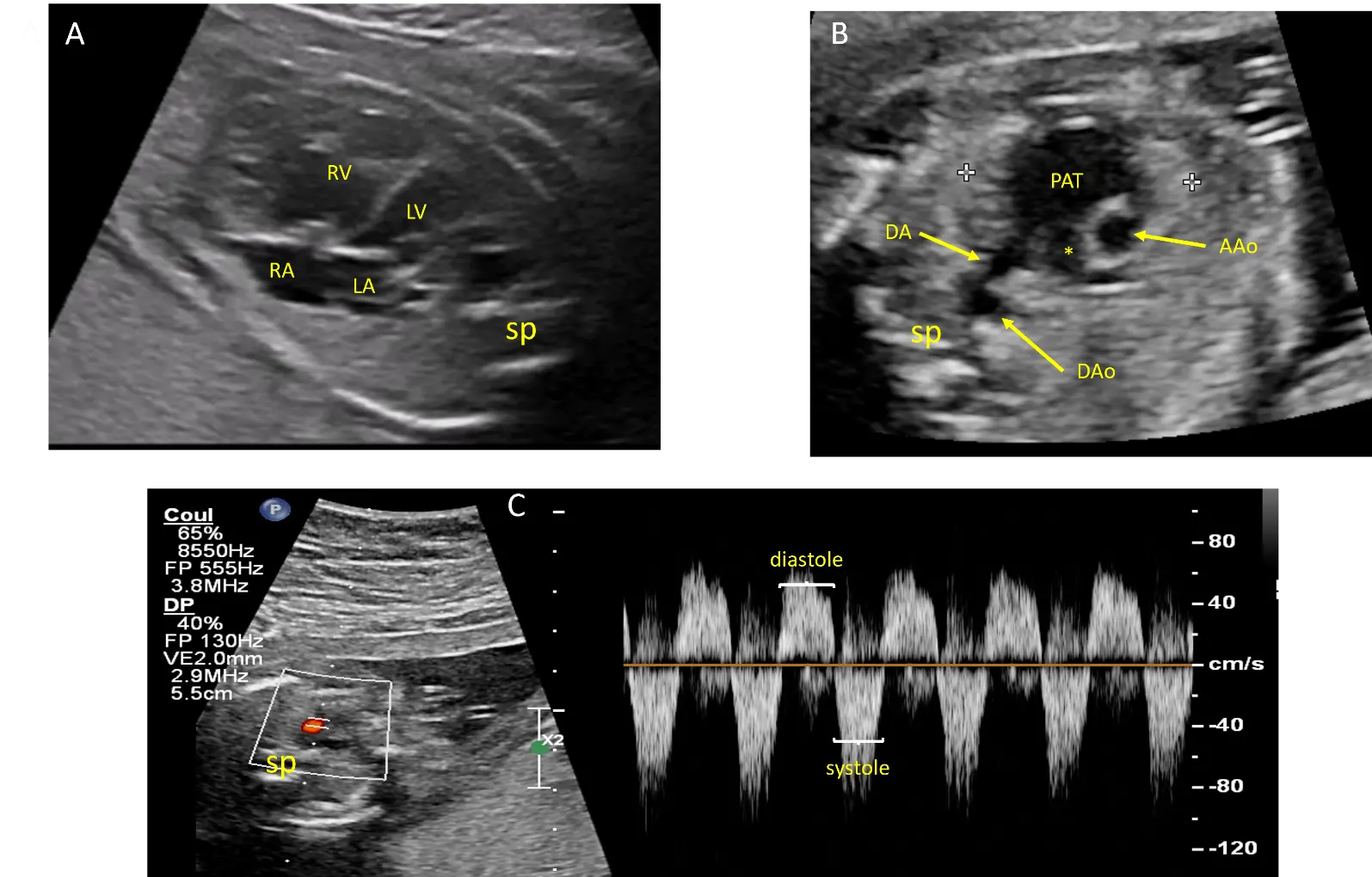
Figure 1. Echocardiographic views from case #1 at 21 weeks of gestation: Four chamber view showing cardiomegaly with dilatation of the right ventricle (RV) and leftward deviation of the heart axis (A). Short axis view of the great arteries showing dilatation of the pulmonary artery trunk (PAT) but not of the right pulmonary artery (*). The 2 calipers (white crosses) delimitate the thymus (B). Pulsed doppler flow on the ductus arteriosus showing an antegrade flow during systole and a reverse flow during diastole (C). AAo: ascending aorta, DA: ductus arteriosus, DAo: descending aorta, LA: Left atrium, LV: left ventricle, RA: right atrium, sp: spine.
Amniocentesis with Comparative Genomic Hybridization (CGH) revealed a 16p13.11 microduplication that was also present in the father. This duplication has been associated with neurodevelopmental disorders and aortic aneurysm [5]. The father underwent normal neurologic and cardiac evaluations, including echocardiography, to exclude the presence of an aortic aneurysm.
Prenatal follow-up until delivery did not reveal significant changes. A male infant weighing 3.2 kg was delivered at term via normal vaginal delivery. Post-natal echocardiography confirmed the diagnosis of APVS with IVS. The ductus arteriosus was large, and there was massive pulmonary regurgitation. There was no bronchial compression. On the second day, the baby experienced significant hemodynamic instability. Surgical ligation of the ductus arteriosus was performed on the third day. Transcutaneous closure was deemed inappropriate due to the large size of the ductus. A breach of the ductus, which was considered aneurysmal with a very thin wall, necessitated the use of extracorporeal circulation. Postoperatively, the patient required several days of ECMO support with progressive stabilization of cardiac function, and weaning was achieved without complication. The recovery was gradual, and at 5 years of age, the child’s development is normal, with significant but well-tolerated pulmonary regurgitation and no airway compression.
2.2. Case 2
A 39-year-old woman with type 1 diabetes in her fourth pregnancy was referred for evaluation at 17 weeks of gestation due to an abnormal fetal heart detected during routine obstetric ultrasound screening. The fetal ultrasound performed at 12 weeks was considered normal, with no increased nuchal translucency.
Fetal echocardiography revealed an absent pulmonary valve with to-and-fro flow across the pulmonary orifice and the ductus arteriosus. A muscular VSD was present, but there was no Fallot-type VSD. The pulmonary trunk was significantly dilated, while the pulmonary branches remained unaffected. There was no tricuspid regurgitation. The aortic arch was oriented to the left. The mother declined amniocentesis.
Subsequent serial fetal ultrasounds demonstrated cardiomegaly and pericardial effusion at 28 weeks of gestation (Figure 2). At 34 weeks of gestation, fetal intra uterine demise was found. The parents declined an autopsy. Karyotype analysis of the placenta did not reveal genetic anomalies.
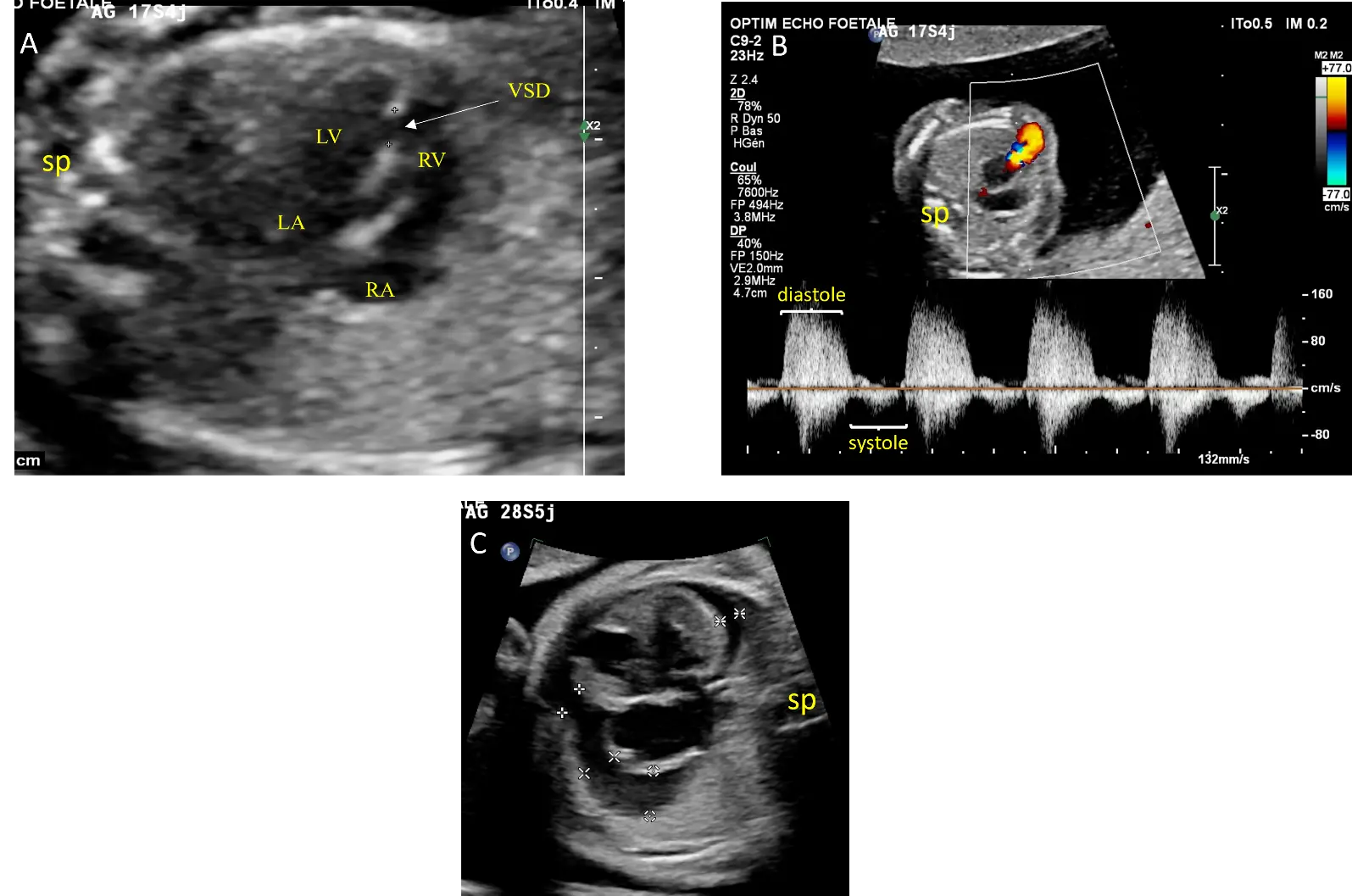
Figure 2. Echocardiographic views from case #2: Four chamber view showing a muscular ventricular septal defect (VSD) delimited by calipers and balanced ventricular and atrial cavities without cardiomegaly at 17 weeks of gestation (A). Doppler interrogation of the pulmonary annulus showing pulmonary insufficiency (B). At 28 weeks of gestation, there is pericardial effusion marked by calipers (C). LA: Left atrium, LV: left ventricle, RA: right atrium, RV: right ventricle, sp: spine.
2.3. Case 3
A 30-year-old woman in her first pregnancy was referred for evaluation at 22 weeks of pregnancy because of cardiomegaly and ventricular asymmetry. An ultrasound study confirmed a significant dilatation of the right ventricle. The absence of the pulmonary valve was characterized by a to-and-fro flow through both the pulmonary orifice and the ductus arteriosus. The pulmonary trunk showed significant dilatation, but the pulmonary branches were unaffected. Additionally, a pericardial effusion was observed (Figure 3). There was no tricuspid regurgitation. Amniocentesis with CGH yielded normal results.
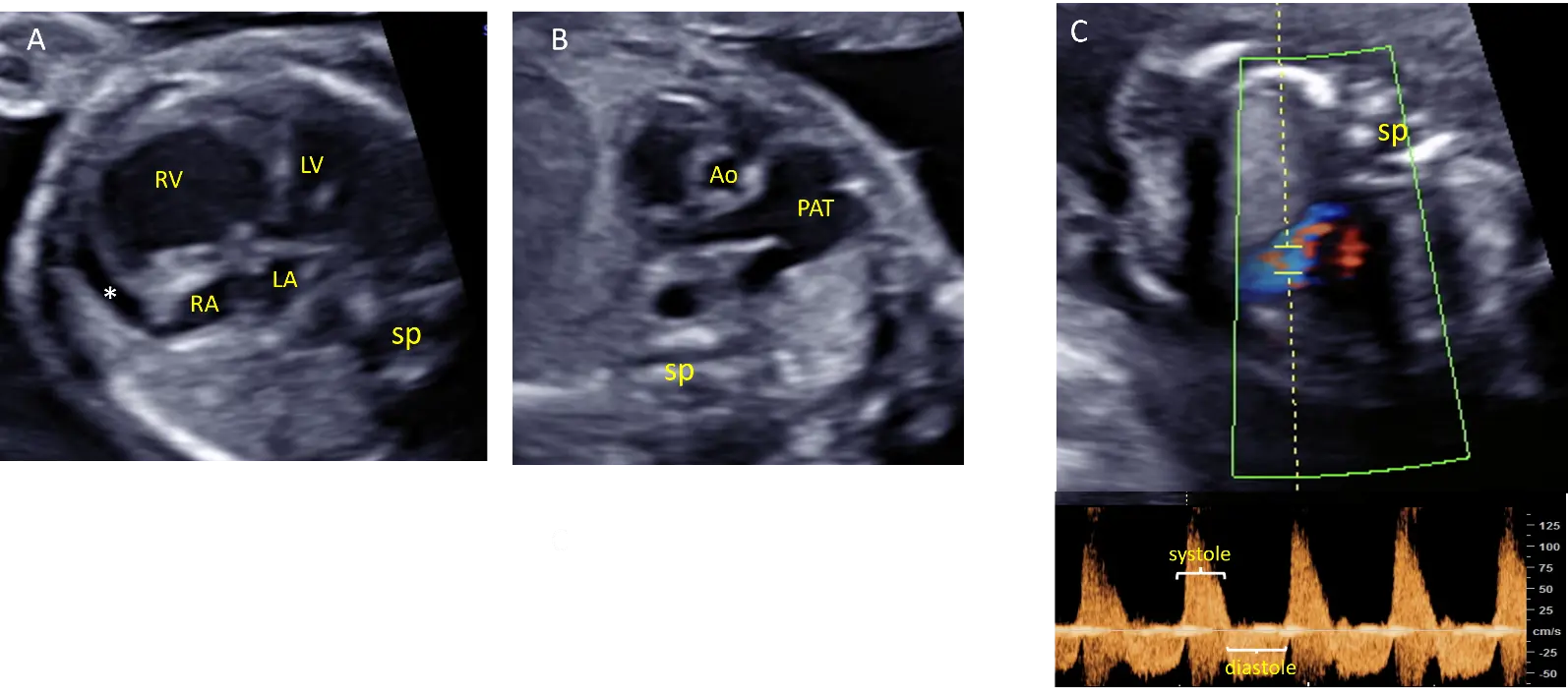
Figure 3. Echocardiographic views from case #3 at 22 weeks of gestation: Four chamber view showing cardiomegaly with dilatation of the right ventricle and pericardial effusion (*) adjacent to dilated right cavities (A). Short axis view of the great arteries showing dilatation of the pulmonary artery trunk (PAT) but not of the pulmonary branches (B). Pulsed doppler showing a to-and-fro flow through the pulmonary annulus demonstrating pulmonary insufficiency (C). Ao: Aorta, LA: Left atrium, LV: left ventricle, RA: right atrium, RV: right ventricle, sp: spine.
The progression of pericardial effusion justified a cesarean section at 35 weeks of gestation. Echocardiography confirmed the AVP with IVS. The right heart chambers were severely dilated, resulting in compression of the left ventricle. Initially, there was a large patent ductus arteriosus with left to right shunting. The pulmonary trunk was significantly dilated, while the pulmonary branches showed only mild dilatation. The aortic arch was normally oriented to the left. The female infant initially presented with congestive heart failure and desaturation at 85%, necessitating positive pressure ventilation. Subsequently, the ductus spontaneously closed on the third day of life, leading to the resolution of congestive heart failure. The baby was discharged from the hospital at 9 days of age. At her most recent evaluation at the age of 6, she is leading a normal life. Echocardiography revealed well-tolerated significant pulmonary insufficiency with dilatation of the pulmonary trunk.
3. Discussion
APVS without aorta overriding the IVS, the non-Fallot-type APVS, is rare. In addition to our 3 cases, we found 38 other cases in the medical literature [2,3,4]. This cardiopathy exhibits several characteristics, observed in our three cases and in the cases reported in the medical literature, that distinguish it from Fallot-type APVS, which are summarized in Table 1. The main differences compared to a Fallot-type APVS are as follows: antenatally, the presence of a ductus arteriosus in all cases, less dilatation of the pulmonary branches, absence of 22q11 deletion, and a high incidence of intrauterine death. Postnatally, there is a rarity of respiratory distress caused by compression of the respiratory airways, and a higher frequency of congestive heart failure.
Table 1. Characteristics of APVS according to the presence or absence of a Fallot-like VSD, according to the literature review.
|
Antenatal Characteristics |
APVS with Fallot Type VSD |
APVS without Fallot Type VSD |
|---|---|---|
|
Massive Pulmonary Insufficiency |
Yes |
Yes |
|
Hydrops fetalis |
Uncommon |
Common |
|
Patent Ductus Arteriosus |
30% |
100% |
|
Dilatation of the pulmonary artery trunk |
Yes |
Yes |
|
Dilatation of the pulmonary branches Right ventricular dilatation |
Always significant None or Moderate 10% |
No or moderate Significant in the absence of VSD Never observed |
|
Postnatal Characteristics |
||
|
Respiratory symptoms |
Common (40–50%) |
Rare |
|
Congestive heart failure |
Uncommon |
Common |
|
Genetic Characteristics |
||
|
22q11 deletion |
20% |
Never observed |
|
Other chromosomal anomalies |
Common |
Common |
APVS: Absent pulmonary valve syndrome; VSD: Ventricular septal defect.
In fetuses with APVS, the absence of a VSD results in a significant overload of the right ventricle, resulting in marked right ventricular dilatation, which is the primary antenatal sign that aids in diagnosis. Since the ductus arteriosus is always present, these fetuses are likely to miscarry early in gestation due to massive aortopulmonary shunting and right ventricular overload. It can be inferred that the absence of a ductus in Fallot-type APVS has a protective effect against heart failure and hydrops [6]. When a VSD is present, with or without overriding of the aorta on the IVS, the redistribution of blood flow between the 2 ventricles may account for the absence of asymmetry in their sizes. Similarly, the absence of a patent ductus arteriosus in most cases of the Fallot-type APVS may contribute to the aneurysmal dilatation of the pulmonary branches that receive a high velocity to-and-fro flow through the pulmonary valves. When the ductus is present, the pulmonary branches are relatively spared by the significant increase in flow within the pulmonary trunk, which is partly directed towards the ductus arteriosus. This flow pattern can explain the mild dilatation observed in the pulmonary branches compared to patients without a ductus arteriosus.
After birth, the persistence of a large ductus arteriosus, along with significant pulmonary insufficiency, can account for the development of severe congestive heart failure. This condition may necessitate early occlusion of the ductus, as observed in our first case. Although circular shunt physiology, as described in conditions such as Ebstein’s anomaly, can lead to severe right heart overload, this mechanism has not been reported in association with absent pulmonary valve syndrome. None of our three fetuses showed tricuspid regurgitation or right atrial dilation suggestive of a circular shunt, and the postnatal hemodynamic instability observed in Case 1 was attributed to a large patent ductus arteriosus with massive pulmonary regurgitation rather than to a circular shunt physiology. Additionally, this case was also characterized by a 16p13.11 microduplication, which was also identified in the father. This microduplication is considered a susceptibility locus rather than a directly causal variant, but has been associated with an increased risk of aortic aneurysm [5] (https://www.deciphergenomics.org/syndrome/79/overview, accessed on 30 October 2025). This patient also had an aneurysmal ductus arteriosus, which required the implementation of extracorporeal circulation due to rupture in the ductus wall during surgery. The association between the 16p13.11 microduplication and ductus arteriosus aneurysm is likely not coincidental, as other conditions linked to aortic aneurysm, such as Marfan disease or Loeys-Dietz syndrome, can also predispose individuals to ductus arteriosus aneurysm [7,8]. Other chromosomal anomalies were frequent in patients with non-Fallot-type APVS. Among the 27 patients who underwent genetic analysis, a chromosomal anomaly was identified in 5 cases, including our first case (19%), with 4 of them involving chromosome 18 (2 trisomy, 1 deletion 18p and 1 translocation 18;14).
The outcomes of the 38 fetuses reported in the literature and our 3 cases are presented in Figure 4 (flow chart). Among the reported cases, the evolution was described as “unknown” in several instances. This lack of follow-up information mainly reflects the limited data available in published reports, which often focus on prenatal findings or immediate postnatal management rather than long-term outcomes. Out of the 41 sets of parents, 11 opted for termination of pregnancy. Among the 25 continuing pregnancies with known evolution, 5 (20%) were characterized by intrauterine death. Twenty babies survived until delivery, but the postnatal evolution was not documented in five of the published cases. Among the 15 cases with known outcomes, 5 died after birth (33%), with 2 deaths occurring 5 months and 9 months after neonatal surgeries, though detailed information about these surgeries was not provided. Meanwhile, 10 babies survived, out of which 6 required neonatal ductus arteriosus ligation. Additionally, 4 patients showed improvement without surgery, all of whom experienced spontaneous closure or had restrictive ductus arteriosus. Interestingly, the observations of the 3 patients who died in the neonatal period were published more than 20 years ago, and no neonatal deaths have been reported in the most recent publications. This improvement probably reflects significant progress in neonatal intensive care.
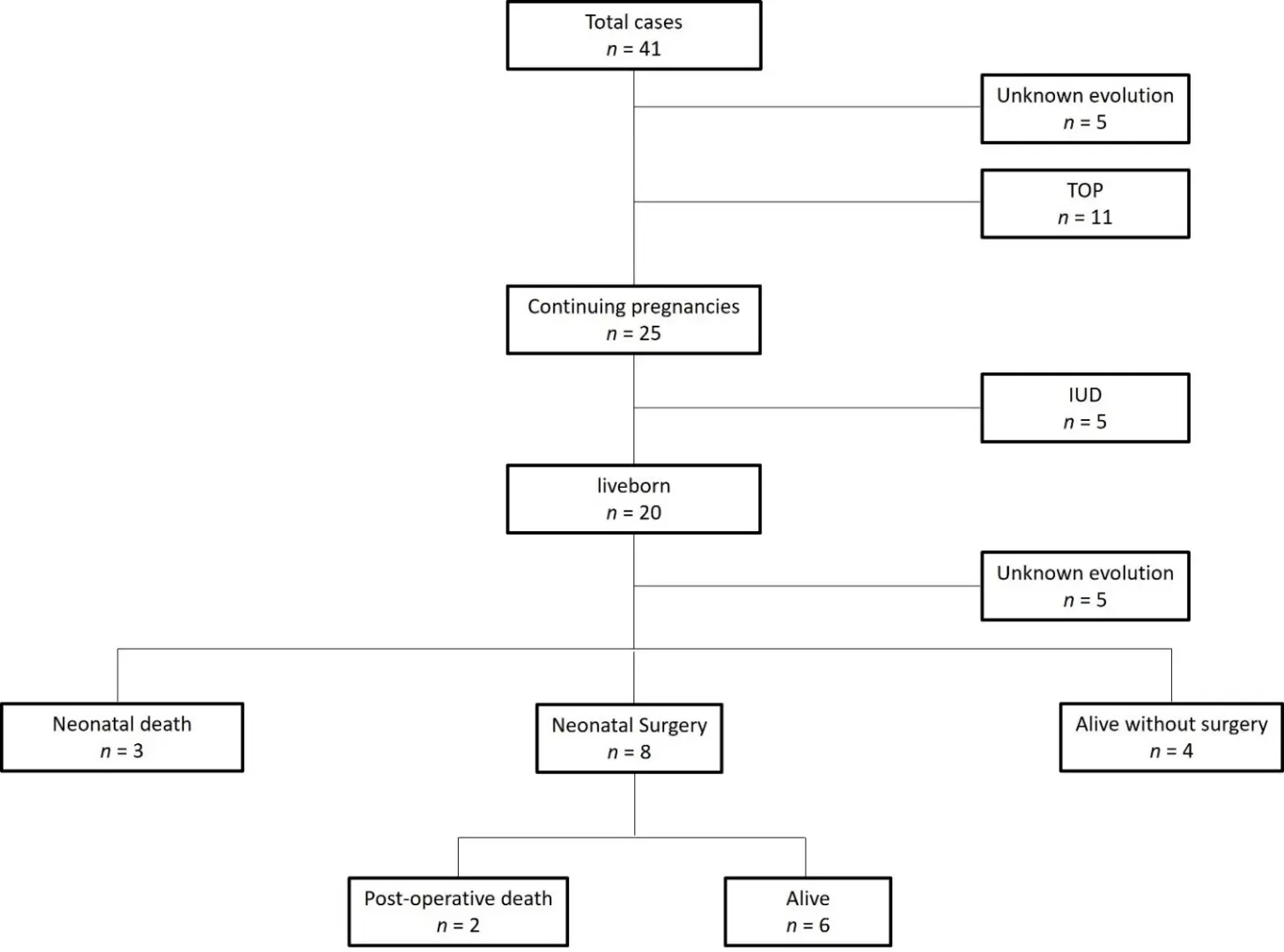
Figure 4. Flowchart of the 41 cases of Absent Pulmonary Valve Syndrome without Fallot-type ventricular septal defect reported in the literature. IUD: Intrauterine death, TOP: termination of pregnancy.
4. Conclusions
The non-Fallot type of APVS differs from the Fallot type in several important aspects that should be emphasized during prenatal counseling. Antenatally, it is characterized by the constant presence of a large ductus arteriosus, the absence of aneurysmal pulmonary branches, and a higher frequency of chromosomal anomalies, although the 22q11 microdeletion is typically absent. In-utero demise has been reported in about 20% of cases, particularly in the presence of hydrops or severe right heart failure. After birth, early ductal closure may be required when left-to-right shunting through a large ductus contributes to pulmonary overcirculation and heart failure. Conversely, the prognosis can be favorable when hydrops and chromosomal anomalies are absent.
Supplementary Materials
The following supporting information can be found at: https://www.sciepublish.com/article/pii/751, Video S1: Case #1: Four-chamber view showing a 90° deviation of the cardiac axis and right ventricular hypertrophy and dilation. LA: left atria, LV: left ventricle, RA: right atria, RV right ventricle; Video S2: Case #1: Color Doppler showing a to-and-fro flow between the right ventricle (RV) and the pulmonary artery (PA).
Author Contributions
Conceptualization, G.V., P.G., M.-P.G., A.R. and I.B.; Investigation, G.V., P.G., M.-P.G., A.R. and I.B.; Writing—Original Draft Preparation, G.V.; Review and editing, G.V., P.G., M.-P.G., A.R. and I.B.
Ethics Statement
All patient data were fully anonymized, and individual consent was not required in accordance with institutional and ethical guidelines for retrospective studies. This study was conducted in accordance with the ethical principles outlined in the Declaration of Helsinki.
Informed Consent Statement
Informed consent was obtained from the patients for the publication of this case series.
Data Availability Statement
No datasets were generated or analyzed during the current study.
Funding
The research did not receive external funding.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Recker F, Weber EC, Strizek B, Geipel A, Berg C, Gembruch U. Management and outcome of prenatal absent pulmonary valve syndrome. Arch. Gynecol. Obstet. 2022, 306, 1449–1454. doi:10.1007/s00404-022-06397-4. [Google Scholar]
- Rakha S, Alkhushi N. Fetal diagnosis of isolated absent pulmonary valve with intact interventricular septum. How to counsel the parents? Ann. Pediatr. Cardiol. 2020, 13, 136–140. doi:10.4103/apc.apc_101_19. [Google Scholar]
- Babaoglu K, Dogan Y, Erdem S, Ozbarlas N, Baçar E, Uzun O. Prenatal diagnosis, associations and outcome for fetuses with congenital absence of the pulmonary valve syndrome. Anatol. J. Cardiol. 2022, 26, 702–709. doi:10.5152/anatoljcardiol.2022.1461. [Google Scholar]
- Razavi R, Sharland G, Simpson J. Prenatal diagnosis by echocardiogram and outcome of absent pulmonary valve syndrome. Am. J. Cardiol. 2003, 91, 429–432. doi:10.1016/s0002-9149(02)03238-1. [Google Scholar]
- El Khattabi LA, Heide S, Caberg JH, Andrieux J, Fenzy MD, Vincent-Delorme C, et al. 16p13.11 microduplication in 45 new patients: Refined clinical significance and genotype–phenotype correlations. J. Med. Genet. 2020, 57, 301–307. doi:10.1136/jmedgenet-2018-105389. [Google Scholar]
- Yeager SB, Van Der Velde ME, Waters BL, Sanders SP. Prenatal role of the ductus arteriosus in absent pulmonary valve syndrome. Echocardiography 2002, 19, 489–493. doi:10.1046/j.1540-8175.2002.00489.x. [Google Scholar]
- Dyamenahalli U, Smallhorn JF, Geva T, Fouron JC, Cairns P, Jutras L, et al. Isolated ductus arteriosus aneurysm in the fetus and infant: a multi-institutional experience. J. Am. Coll. Cardiol. 2000, 36, 262–269. doi:10.1016/s0735-1097(00)00707-5. [Google Scholar]
- Jacques F, Grosse-Wortmann L, Hickey EJ, Chitayat D, Van Arsdell G, Bradley TJ. Unexpected contained rupture of a ductus arteriosus aneurysm found at surgical repair in an infant with Loeys-Dietz syndrome. Ann. Thorac. Surg. 2013, 95, 710–711. doi:10.1016/j.athoracsur.2012.06.047. [Google Scholar]